

Xu et al. identify the nuclear receptor liver receptor homolog 1 (LRH-1) as a key regulator in the process of hepatic tumorigenesis through the coordination of a noncanonical glutamine pathway that is reliant on the mitochondrial and cytosolic transaminases glutamate pyruvate transaminase 2 (GPT2) and glutamate oxaloacetate transaminase 1 (GOT1), which fuel anabolic metabolism.
Keywords: Hepatocellular carcinoma, cancer metabolism, nuclear receptor NR5A2, mitochondria, anaplerosis, mTOR, NADPH
Abstract
Various tumors develop addiction to glutamine to support uncontrolled cell proliferation. Here we identify the nuclear receptor liver receptor homolog 1 (LRH-1) as a key regulator in the process of hepatic tumorigenesis through the coordination of a noncanonical glutamine pathway that is reliant on the mitochondrial and cytosolic transaminases glutamate pyruvate transaminase 2 (GPT2) and glutamate oxaloacetate transaminase 1 (GOT1), which fuel anabolic metabolism. In particular, we show that gain and loss of function of hepatic LRH-1 modulate the expression and activity of mitochondrial glutaminase 2 (GLS2), the first and rate-limiting step of this pathway. Acute and chronic deletion of hepatic LRH-1 blunts the deamination of glutamine and reduces glutamine-dependent anaplerosis. The robust reduction in glutaminolysis and the limiting availability of α-ketoglutarate in turn inhibit mTORC1 signaling to eventually block cell growth and proliferation. Collectively, these studies highlight the importance of LRH-1 in coordinating glutamine-induced metabolism and signaling to promote hepatocellular carcinogenesis.
During tumorigenesis, cancer cells usually switch from oxidative metabolism to a highly glycolytic metabolic status (Vander Heiden et al. 2009). While glucose is predominantly metabolized into lactate rather than entering the tricarboxylic acid (TCA) cycle, cancer cells particularly rely on glutamine to replenish TCA cycle intermediates. This process, termed anaplerosis, is accomplished through the conversion of glutamine to α-ketoglutarate (α-KG) via a two-step deamination reaction catalyzed by glutaminases and then by glutamate dehydrogenase 1 (GLUD1) or transaminases (DeBerardinis et al. 2008; Wise et al. 2008; Csibi et al. 2013; Son et al. 2013). Cancer cells therefore critically depend on glutamine as a fuel for proliferation, and abrogation of glutamine metabolism blocks tumorigenesis, indicating an accessible therapeutic window for cancer treatment (Hensley et al. 2013).
Liver receptor homolog 1 (LRH-1; also called NR5A2) is a nuclear receptor that is enriched in enterohepatic tissues, where it has diverse molecular and physiological functions (Stein and Schoonjans 2015). LRH-1 has been linked to cell proliferation and cancer development in the intestine (Botrugno et al. 2004; Schoonjans et al. 2005) and pancreas (Petersen et al. 2010; Benod et al. 2011). In the liver, LRH-1 regulates various metabolic processes, including bile acid synthesis (Mataki et al. 2007; Lee et al. 2008; Out et al. 2011), glucose sensing and processing (Oosterveer et al. 2012), and reverse cholesterol transport (Stein et al. 2014). Although the function of LRH-1 in the liver has been extensively studied, its commanding role in intermediary metabolism has never been connected to tumorigenesis.
In this study, we report that LRH-1 promotes diethylnitrosamine (DEN)-induced hepatocellular carcinoma (HCC) by coordinating glutamine-induced anabolic metabolism. We demonstrate that LRH-1 facilitates the production of NADPH from glutamine by favoring a noncanonical glutamine pathway that optimizes reductive biosynthesis. Importantly, chronic and acute disruption of LRH-1 also impairs glutamine-induced anaplerosis and α-KG availability, ultimately leading to reduced mTORC1 signaling. These results unveil an unexpected role of LRH-1 in cancer intermediary metabolism with broad-ranging implications on mTORC1 signaling.
Results and Discussion
Hepatic loss of LRH-1 prevents DEN-induced liver carcinogenesis
To investigate the specific contribution of hepatic LRH-1 on HCC formation, we used the well-established DEN method to induce liver cancer (Bakiri and Wagner 2013). Liver-specific Lrh-1-deficient (Lrh-1hep−/−) and wild-type control (Lrh-1hep+/+) mice were injected with DEN on postnatal day 14. Tumor burden was assessed 6 mo (mid-term) or 10 mo (long-term) after injection (Fig. 1A). While long-term DEN-challenged Lrh-1hep+/+ littermates developed multiple hepatic tumors, Lrh-1hep−/− mice were strikingly protected (Fig. 1B,C). The robust reduction of total tumor number and size was not caused by differences in DEN carcinogenicity as evidenced by the equal accumulation of DNA adducts induced by DEN exposure in 14-d-old Lrh-1hep+/+ and Lrh-1hep−/− livers (Fig. 1D; see the Supplemental Material for more details). Furthermore, DEN moderately increased LRH-1 protein abundance but did not affect its nuclear compartmentalization (Fig. 1E). We then performed histological and immunohistochemical analysis on the long-term DEN-treated liver sections. H&E staining of Lrh-1hep−/− liver sections demonstrated fewer microscopic tumor foci, while BrdU and Ki67 staining confirmed reduced cell proliferation in Lrh-1-deficient livers (Supplemental Fig. S1A). Moreover, long-term DEN-treated Lrh-1hep−/− livers were significantly lighter compared with Lrh-1hep+/+ livers, while the body weight did not differ between the two genotypes (Supplemental Fig. S1B–D). Together, these results indicate that LRH-1 is required for efficient HCC induction and progression in response to DEN treatment.
Figure 1.

Hepatic Lrh-1-deficient mice are protected against DEN-induced HCC formation and display reduced glutamine-dependent anaplerosis. (A) Experimental strategies of DEN administration. (DOB) Date of birth. (B) Representative livers of 10-mo DEN-treated Lrh-1hep+/+ and Lrh-1hep−/− mice. (C) Hepatic tumor number (left) and tumor size (right) in the corresponding genotypes. (D) Hepatic O6-ethylguanine DNA adducts 6, 24, and 48 h after DEN injection to 14-d-old Lrh-1hep+/+ and Lrh-1hep−/− mice. n = 5–6 per genotype and time point. (E) LRH-1 protein levels in cytosol and nucleus fractions of livers from untreated control (Ctrl), 6-mo DEN-treated (MT), and 10-mo DEN-treated (LT) Lrh-1hep+/+ and Lrh-1hep−/− mice. (F) Gene set enrichment analysis (GSEA) demonstrates down-regulated pathways that were ranked by normalized enrichment scores (NES) in livers of 6-mo DEN-treated Lrh-1hep−/− (n = 6) mice compared with Lrh-1hep+/+ (n = 7) mice. Specific pathways are indicated. (G) Heat map displaying the core-enriched gene set “metabolism of amino acids and derivatives,” expressed in the livers of the mice described in F. (H) Graphical representation of enzymes involved in glutamine breakdown and metabolism. Enzymes highlighted in red are reduced in Lrh-1hep−/− livers, as shown in I. (I) Hepatic mRNA levels of glutaminase 2 (Gls2), glutamate oxaloacetate transaminase 1 (Got1), glutamate pyruvate transaminase 2 (Gpt2), and malic enzyme 1 (Me1) in livers of mice described in F. Data represent mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 by two-tailed Student's t-test.
Hepatic loss of LRH-1 inhibits noncanonical glutamine processing
LRH-1 coordinates intestinal cell renewal and tumor formation through cross-talk with the β-catenin pathway (Botrugno et al. 2004; Schoonjans et al. 2005). It is also required for hepatic endoplasmic reticulum (ER) stress resolution through transcriptional control of polo-like kinase 3 (Plk3) and subsequent phosphorylation of activating transcription factor 2 (ATF2) (Mamrosh et al. 2014). To understand the robust tumor-suppressive phenotype, we first assessed the β-catenin pathway in mid-term DEN-treated livers in which tumors were not yet developed (Supplemental Fig. S1E). In contrast to the findings in intestinal crypts of germline Lrh-1+/− mice (Botrugno et al. 2004), β-catenin targets c-Myc, Ccnd1, and Ccne1 were not reduced in the unchallenged (Supplemental Fig. S1F) or DEN-challenged (Supplemental Fig. S1G) Lrh-1hep−/− livers. We also evaluated the Plk3–ATF2 cascade in response to acute DEN exposure. Plk3 mRNA levels and ATF2 phosphorylation were not induced by DEN (Supplemental Fig. S1H; data not shown), indicating that, in our model, LRH-1 impacts hepatocarcinogenesis via other mechanisms. We then performed microarray analysis to compare the transcriptomes of mid-term DEN-exposed Lrh-1hep+/+ and Lrh-1hep−/− livers. As expected, gene set enrichment analysis (GSEA) confirmed previously established functions and target pathways of LRH-1, such as synthesis of bile acids (Fig. 1F; Supplemental Fig. S1I,J). Of interest, metabolism of amino acid and derivatives scored among the most significantly enriched pathways (Fig. 1F; Supplemental Fig. S1K). We next analyzed this gene set in more detail. While transcripts of several proteasomal subunits were down-regulated in Lrh-1hep−/− livers, a more striking reduction of several enzymes involved in glutamine catabolism was observed (Fig. 1G). Glutamine plays an essential role in tumor growth to support anaplerosis and reductive biosynthesis (DeBerardinis et al. 2008). Several genes involved in the processing of glutamine were reduced in mid-term DEN-exposed Lrh-1hep−/− livers, including mitochondrial glutaminase 2 (Gls2), cytosolic glutamate oxaloacetate transaminase 1 (Got1), and mitochondrial glutamate pyruvate transaminase 2 (Gpt2) (Fig. 1G,H). This pathway is reminiscent of a noncanonical pathway of glutamine breakdown that was earlier reported in human glioma (Wise et al. 2008) and pancreatic ductal adenocarcinoma (PDAC) cells as an alternative mechanism to support NADPH production via malic enzyme (Son et al. 2013). Not only these genes but also malic enzyme 1 (Me1) were significantly blunted, as confirmed by quantitative RT–PCR (qRT–PCR) (Fig. 1I). Many cancer cells typically rely on GLUD1 to fuel the TCA cycle through repleting α-KG (DeBerardinis et al. 2008). Transcript levels of Glud1, however, remained unchanged upon hepatic loss of function (LOF) of LRH-1 (Supplemental Fig. S1L). Moreover, mRNA expression of Gls1, Got2, and TCA cycle-related genes was not altered between the two genotypes (Supplemental Fig. S1L,M). Collectively, these data indicate that an alternative pathway involved in hepatic glutamine processing is most likely compromised in Lrh-1hep−/− mice.
LRH-1 regulates reductive biosynthesis fueled by glutamine processing
We previously showed that LRH-1 coordinates glucose intermediary metabolism via glucokinase (GCK) activation and subsequent carbohydrate response element-binding protein (ChREBP) nuclear translocation (Oosterveer et al. 2012). Consistent with this study, the ChREBP pathway was significantly enriched between both genotypes (Supplemental Fig. S2A,B). Because Me1 is a known ChREBP target gene (Iizuka et al. 2004; Chambers et al. 2013), we first analyzed whether the reduction of our candidate genes (Fig. 1I) results from impaired GCK–ChREBP signaling. GCK reconstitution in Lrh-1hep−/− livers restored Chrebpβ and Me1 (Fig. 2A), but not Gls2, Got1, or Gpt2 expression (Supplemental Fig. S2C), indicating that LRH-1 regulates only Me1 via the GCK–ChREBP axis. In parallel to the reduced Me1 expression, NADPH/NADP+ levels were significantly reduced in unchallenged (Fig. 2B) or DEN-challenged (Fig. 2C) Lrh-1hep−/− livers and was accompanied by a corresponding reduction of the GSH/GSSG ratio in DEN-treated livers (Fig. 2D). Although Me1 was readily rescued upon GCK reconstitution (Fig. 2A), normalization of NADPH/NADP+ levels was still incomplete (Fig. 2B), supporting the notion that the generation of NADPH from glutamine is also attenuated in Lrh-1hep−/− livers.
Figure 2.

Gls2 is a direct transcriptional target of LRH-1. (A,B) Hepatic mRNA levels of Gck, Chrebpβ, and Me1 (A) and NADPH/NADP+ levels (B) in control virus-infected Lrh-1hep+/+ and Lrh-1hep−/− mice and AAV8-GCK virus-infected Lrh-1hep−/− mice. n = 4–5 per group. Data represent mean ± SEM. (*) P < 0.05 versus Lrh-1hep+/+; (#) P < 0.05 versus Lrh-1hep−/− by one-way ANOVA and Tukey's post-hoc test. (C,D) Relative NADPH/NADP+ (C) and GSH/GSSG (D) levels in livers of 6-mo DEN-treated Lrh-1hep+/+ (n = 7) and Lrh-1hep−/− (n = 6) mice. (E,F) mRNA levels of Gls2, Got1, and Gpt2 (E) and protein levels of GLS2 (F) in Hepa 1.6 cells transfected with control or Lrh-1 expression plasmids. n = 3 per group. (G,H) mRNA levels of Gls2, Got1, and Gpt2 (G) and protein levels of GLS2 (H) in Hepa 1.6 cells transfected with scrambled or Lrh-1 targeted siRNAs. n = 3 per group. Data represent mean ± SEM. (**) P < 0.01; (***) P < 0.001 by two-tailed Student's t-test. (I) Hepatic protein levels of GLS2 in mice described in C. (J) University of California at Santa Cruz (UCSC) genome browser (mm9) view displaying the occupancy of mouse Gls2 by IgG and LRH-1 (Chong et al. 2012). (K) Schematic representation of the five putative LRH-1 response elements in the proximal mouse Gls2 promoter. (L) ChIP-qPCR (chromatin immunoprecipitation [ChIP] combined with qPCR) assay to evaluate the relative LRH-1 binding to the mouse Gls2 promoter. Amplified regions (a, b and c) are depicted in Figure 1K. (M) Luciferase activities in HEK293A cells after cotransfection of a Lrh-1 expression vector and with empty luciferase reporter (pGL4) and long or short Gls2 promoter constructs with or without the indicated mutations. Data represent mean ± SEM. (***) P < 0.001 versus empty reporter (pGL4); (#) P < 0.05 versus long Gls2 promoter construct by one-way ANOVA and Tukey's post-hoc test.
We next investigated the molecular mechanism through which LRH-1 regulates glutamine metabolism. Overexpression of LRH-1 in mouse hepatoma Hepa 1.6 cells resulted in an increase of GLS2 transcripts and protein, while Got1 and Gpt2 transcripts were unchanged (Fig. 2E,F). Conversely, siRNA-mediated silencing of LRH-1 exclusively reduced the expression of GLS2 mRNA and protein (Fig. 2G,H). In Lrh-1hep−/− mice, reduced hepatic Gls2 mRNA expression (Fig. 1H) translated into lower GLS2 protein levels (Fig. 2I). Of interest, Gls2 is highly expressed in the liver compared with Gls1 (Supplemental Fig. S2D). GLS2 deaminates mitochondrial glutamine, thus controlling a major anaplerotic step for hepatic glutamine utilization (Hensley et al. 2013). We then asked whether Gls2 is subjected to direct transcriptional regulation by LRH-1. Analysis of a genome-wide hepatic LRH-1 ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) data set (Chong et al. 2012) revealed LRH-1 recruitment at the Gls2 promoter (Fig. 2J), and computational analysis identified five putative LRH-1 response elements within the Gls2 promoter under the LRH-1 ChIP-seq peak (Fig. 2K). Site-specific ChIP assays using DNA from mid-term DEN-treated Lrh-1hep+/+ and Lrh-1hep−/− livers revealed LRH-1 recruitment to putative binding sites 1, 2, and 3 (Fig. 2L). Mutation of these binding sites in mouse Gls2-luciferase reporter constructs further mapped site 3, which is conserved in the human Gls2 promoter (Supplemental Fig. S2E), as the major site that confers LRH-1 responsiveness (Fig. 2M). Accordingly, silencing of LRH-1 in human hepatoma HepG2 cells also led to a significant reduction of Gls2 transcripts (Supplemental Fig. S2F).
LRH-1 regulates GLS2 to promote glutamine-induced anaplerosis
Given the marked reduction of GLS2 in Lrh-1hep−/− mice, we hypothesized that hepatic loss of LRH-1 blunts the conversion of glutamine to glutamate. To test the flux through GLS2 in vivo, we performed 13C nuclear magnetic resonance (13C MR) spectroscopy measurements following hyperpolarized [5-13C]glutamine injection (Cabella et al. 2013; Cheng et al. 2013). [5-13C]glutamine was hyperpolarized using dissolution dynamic nuclear polarization (DNP) and rapidly injected into DEN-treated Lrh-1hep−/− and Lrh-1hep+/+ mice followed by real-time recording of its conversion to [5-13C]glutamate (Fig. 3A,B). As expected, Lrh-1hep−/− showed a strong decrease in hepatic [5-13C]glutamate content compared with Lrh-1hep+/+ mice (Fig. 3C). Unlike the expression levels of glutamine transporters Slc1a5 and Slc7a5, which were unchanged (Supplemental Fig. S3A), hepatic α-KG levels were diminished in Lrh-1hep−/− mice (Fig. 3D), indicating that LRH-1 LOF may attenuate glutamine-fueled anaplerosis. To further explore the direct roles of LRH-1 and GLS2 in maintaining glutaminolysis and intracellular α-KG pools, we examined the effect of glutamine metabolism on α-KG levels. Hepa 1.6 cells were starved of glutamine for 6 h, and removal of glutamine significantly reduced the intracellular levels of α-KG (Supplemental Fig. S3B), demonstrating that glutamine sustains glutaminolysis. We then acutely modulated LRH-1 or GLS2 expression in Hepa 1.6 cells. In line with the reduced α-KG abundance in Lrh-1hep−/− livers, overexpression of LRH-1 or GLS2 increased, while siRNA-mediated silencing of LRH-1 or GLS2 decreased, α-KG levels in Hepa 1.6 cells (Fig. 3E–H). Together, these results demonstrate that LRH-1 promotes glutamine-induced anaplerosis via the induction of GLS2.
Figure 3.

LRH-1 controls glutamine-induced anaplerosis and regulates mTORC1 activity. (A) GLS2-mediated biochemical reaction with hyperpolarized [5-13C]glutamine. Red dots indicate the labelling of C5. (B–D) Representative in vivo 13C MR spectra showing hyperpolarized [5-13C]glutamate production with the by-product signal of hyperpolarized [5-13C]pyroglutamate (B), the mean signal intensity of the hyperpolarized [5-13C]glutamate formed via glutaminase (C), and intracellular α-KG levels (D) in the livers of DEN-treated Lrh-1hep+/+ and Lrh-1hep−/− mice. (E–H) Intracellular α-KG levels in Hepa 1.6 cells transfected with either control or Lrh-1 expression plasmids (n = 3 per group (E) or scrambled or Lrh-1 targeted siRNAs (n = 3 per group) (F), transduced with either AdGFP or AdGLS2 viruses (n = 3 per group) (G), or transfected with scrambled or Gls2 targeted siRNAs (n = 3 per group) (H). (I) Phosphorylation states of S6K and 4EBP1 in the livers of mice described in B. (J–M) Phosphorylation states of S6K and 4EBP1 in Hepa 1.6 cells transfected as in E (J), transduced as in G (K), transfected as in F (L), or transfected as in H (M) with or without dimethyl-KG (DM-KG) supplementation (L,M). Data represent mean ± SEM. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 by two-tailed Student's t-test.
LRH-1 modulates the mTORC1 pathway in an α-KG-dependent manner
Glutamine is metabolized through glutaminolysis to produce α-KG. Previous studies showed that increased glutamine (Duran et al. 2012; Bar-Peled and Sabatini 2014) or α-KG (Duran et al. 2012) availability stimulates the mTORC1 signaling pathway. Of note, a robust reduction of mTORC1 activation was observed in Lrh-1hep−/− livers, as evidenced by the decreased phosphorylation of 4EBP1 and S6K (Fig. 3I). We then investigated the importance of glutamine in the activation of mTORC1 in Hepa 1.6 cells. Depletion of glutamine for 6 h reduced α-KG levels (Supplemental Fig. S3B) and inhibited mTORC1 activity (Supplemental Fig. S3C). Moreover, supplementation of a cell-permeable α-KG analog, dimethyl-KG (DM-KG), restored the activation of mTORC1 signaling upon glutamine deprivation (Supplemental Fig. S3D), indicating that intracellular glutamine and its derived α-KG are essential to stimulate mTORC1. Based on these results, we overexpressed LRH-1 or GLS2 in Hepa 1.6 cells. In both settings, mTORC1 activity was induced in the presence of glutamine (Fig. 3J,K). These effects were reversed upon glutamine starvation (Supplemental Fig. S3E,F). Furthermore, RNAi-mediated suppression of LRH-1 or GLS2 interfered with phosphorylation of 4EBP1 and S6K in the presence of glutamine, while addition of DM-KG or overexpression of GLS2 or LRH-1, respectively, rescued mTORC1 activities (Fig. 3L,M; Supplemental Fig. S3G,H). These data hence suggest that the LRH-1–GLS2 axis increases α-KG levels and consequently activates mTORC1.
The LRH-1–GLS2 axis promotes cell proliferation
Activation of mTORC1 inhibits autophagy (Kim et al. 2011), activates protein translation (Ma and Blenis 2009), and promotes cell growth (Schmelzle and Hall 2000). To investigate the importance of the LRH-1–GLS2–mTORC1 pathway, we first assessed autophagy in mid-term DEN-treated livers. As expected, disruption of LRH-1 induced autophagy, as evidenced by reduced phosphorylation of ULK-1 at Ser757, blunted P62, and increased LC3-II levels (Supplemental Fig. S4A). Silencing of LRH-1 or GLS2 decreased global protein translation as measured by incorporation of 35S-labelled methionine in Hepa 1.6 cells (Fig. 4A,B), while their overexpression enhanced translation (Supplemental Fig. S4B,C). We then evaluated the link between LRH-1, α-KG, and cell proliferation. As expected, LRH-1 or GLS2 overexpression promoted cell proliferation, while additional glutamine deprivation prevented the increase in cell proliferation (Fig. 4C,D). Conversely, inhibition of glutaminolysis by LRH-1 or GLS2 silencing inhibited cell proliferation, while overexpression of LRH-1 or GLS2 rescued this effect (Supplemental Fig. S4D,E). Moreover, diminished cell proliferation upon LRH-1 or GLS2 suppression could also be rescued by addition of DM-KG (Fig. 4E,F), indicating that the LRH-1–GLS2 axis activates cell proliferation in an α-KG-dependent manner. It has been shown that GLS2-catalyzed deamination of glutamine is also essential for the control of intracellular reactive oxygen species (ROS) levels (Hu et al. 2010). Supplementation with the antioxidant N-acetyl-cysteine (NAC), however, could not rescue the inhibited cell proliferation upon LRH-1 or GLS2 silencing in Hepa 1.6 cells (Supplemental Fig. S4F,G), suggesting that reduced mTORC1 signaling rather than induced oxidative stress accounts for the reduction in cell proliferation. Furthermore, Hepa 1.6 cells silenced for LRH-1 or GLS2 induced significantly less tumor growth after propagation in athymic nude mice (Fig. 4G). Taken together, these findings highlight that LRH-1 promotes cell proliferation through glutaminolysis and mTORC1 signaling.
Figure 4.
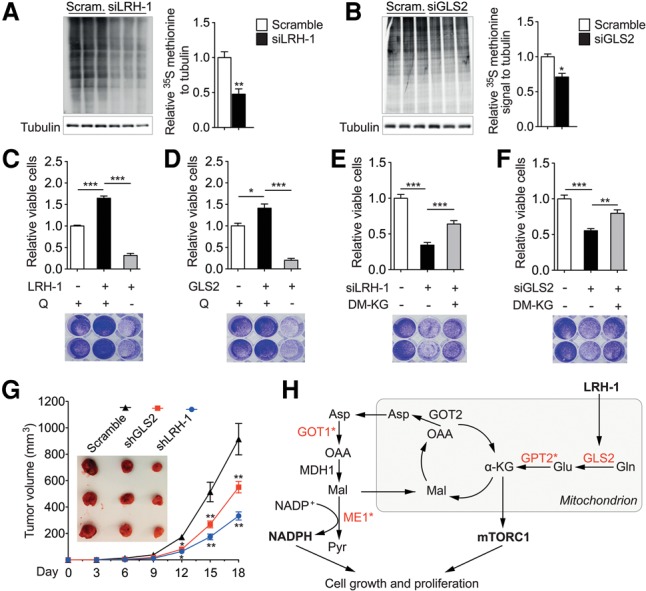
The LRH-1–GLS2 axis promotes protein translation and cell proliferation. (A,B) Global protein synthesis measured by 35S-labelled methionine incorporation in Hepa 1.6 cells transfected with either scrambled or Lrh-1 targeted siRNAs (n = 3 per group) (A) or scrambled or Gls2 targeted siRNAs (n = 3 per group) (B). Relative 35S methionine signals were normalized to Tubulin. Data represent mean ± SEM. (*) P < 0.05; (**) P < 0.01 by two-tailed Student's t-test. (C–F) Relative viable cells and representative Crystal Violet staining images of Hepa 1.6 cells transfected with either control or Lrh-1 expression plasmids (n = 3 per group) (C) or transduced with either AdGFP or AdGLS2 viruses (n = 3 per group) (D) with or without glutamine deprivation or scrambled or Lrh-1 targeted siRNAs (n = 3 per group) (E) or scrambled or Gls2 targeted siRNAs (n = 3 per group) (F) with or without DM-KG supplementation. Data represent mean ± SEM. (*) P < 0.05; (***) P < 0.001 by one-way ANOVA and Tukey's post-hoc test. (G) Comparison of tumor growth and volume of mice subcutaneously injected with Hepa 1.6 cells that were transduced with scrambled, LRH-1 targeted, or GLS2 targeted shRNA. n = 6 per group. (H) Graphical summary illustrating how LRH-1 promotes glutamine-induced anaplerosis and reductive biosynthesis in hepatic cancer cells. Enzymes highlighted in red are reduced in Lrh-1hep−/− livers, and an asterisk indicates indirect regulation by LRH-1.
In conclusion, our study assigns a critical role to LRH-1 in hepatic fuel metabolism with a striking impact on hepatic tumorigenesis. Unlike the role of LRH-1 in the intestine and pancreas, the oncogenic potential of hepatic LRH-1 is independent from the β-catenin/Wnt signaling pathway and is instead driven by the regulation of specific gene programs involved in mitochondrial glutamine catabolism (Fig. 4H). The enhanced mTORC1 signaling upon LRH-1-induced glutaminolysis indicates that the effect of LRH-1 on glutamine processing also impinges on established kinases in cell growth and cancer, thereby further amplifying the overall growth-stimulating effect. These observations, together with our previous findings linking LRH-1 to glucose-dependent fatty acid biosynthesis via ChREBP activation (Oosterveer et al. 2012), support the notion that LRH-1 confers a protumorigenic status to hepatocytes by promoting the metabolism of the principal fuel substrates of cancer cells. Further studies are warranted to fully understand its role in human HCC and explore its potential as a drug target.
Materials and methods
Animal studies
Hepatocyte-specific LRH-1 knockout (Lrh-1hep−/−) and wild-type (Lrh-1hep+/+) mice were previously reported (Oosterveer et al. 2012). Congenic neonatal mice at 14 d old were intraperitoneally injected with DEN at a dose of 25 mg per kilogram of body weight to initiate tumor formation. Six months (mid-term DEN) or 10 mo (long-term DEN) after injection, mice were sacrificed, and liver tissue was collected. The experiments with the AAV8 viruses have been described previously (Oosterveer et al. 2012). Five-week-old male BALB/c nu/nu mice were purchased from Charles River and maintained in the animal facilities. All animal procedures were approved by the Swiss authorities (Canton of Vaud, animal protocol IDs 2375 and 2768) and performed in accordance with our institutional guidelines.
ChIP
ChIP analysis was performed as described previously with minor modifications (Stein et al. 2014). DNA was purified using the PCR clean-up extraction kit (Macherey-Nagel), after which qRT-PCR was performed as described previously (Mataki et al. 2007). Data were normalized to the input [fold differences = 2−(Ct sample − Ct input)]. ChIP primer sequences are listed in Supplemental Table 1.
Measurements of metabolites
For NADPH/NADP+ and GSH/GSSG ratios, liver biopsies were extracted with 70% ethanol, and biomass was separated by centrifugation at 4000 rpm for 10 min. Liquid extracts were then dried by vacuum centrifugation, resuspended in 10 µL of water per milligram of wet weight, and analyzed by targeted liquid chromatography-tandem mass spectrometry on a Thermo Quantum Ultra instrument equipped with a Waters Acquity ultra high performance liquid chromatographer (UPLC). Intracellular α-KG levels were determined using commercial kits (Abcam, ab83431) according to the manufacturer's instructions.
In vivo hyperpolarized 13C MR measurements
DEN-treated Lrh-1hep+/+ and Lrh-1hep−/− mice were anesthetized with ∼1.8% isoflurane, 0.5% O2, and 0.5% air. A 750-µL bolus containing a dose of 0.57 mmol/kg ± 0.02 mmol/kg hyperpolarized [5-13C]glutamine was administered in 9 sec. A series of 30°C BIR4 adiabatic RF excitation pulses were applied using a custom-built dual 1H/13C probe (two 1H surface coils placed in quadrature on top of a 13C single-loop surface coil) placed under the animal on the shaved skin located above the mouse's liver. In vivo 13C MR measurements were respiratory-gated and triggered with simulated cardiac signal with a repetition time of 1 sec. Acquisitions were performed with an INOVA spectrometer (Varian/Magnex). The peak integrals were obtained from summed spectra analyzed using VNMRJ.
Allograft tumor study
Hepa 1.6 cells suspended in phosphate-buffered saline were injected subcutaneously into the left flanks of nude mice (4 × 106 cells per flank). The diameters of the tumors were measured every 3 d, and tumor volumes (V) were calculated using the formula V = L×W2/2, where L is length, and W is width.
Statistical analysis
Data represent mean ± SEM. Comparison of differences between two groups was assessed using two-tailed Student's t-tests. Multiple group comparisons were assessed by one-way ANOVA and Tukey's post-hoc test. Differences under P < 0.05 were considered statistically significant (P < 0.05 [*], P < 0.01 [**], and P < 0.001 [***]).
More experimental Materials and Methods are included in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank V. Lemos for assistance with in vivo experiments, and H.A.I. Yoshihara for hyperpolarized 13C data analysis. This study was supported by École Polytechnique Fédérale de Lausanne funding, the Swiss Cancer League (KFS-2809-08-2011, KFS-3082-02-2013, and KFS-3444-08-2014), and the Swiss National Science Foundation (CRSII3_160798/1). S.S. is supported by a post-doctoral fellowship from the Novartis Consumer Health Foundation, and J.A. is the Nestle Chair in Energy Metabolism.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.277483.116.
Freely available online through the Genes & Development Open Access option.
References
- Bakiri L, Wagner EF. 2013. Mouse models for liver cancer. Mol Oncol 7: 206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Sabatini DM. 2014. Regulation of mTORC1 by amino acids. Trends Cell Biol 24: 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benod C, Vinogradova MV, Jouravel N, Kim GE, Fletterick RJ, Sablin EP. 2011. Nuclear receptor liver receptor homologue 1 (LRH-1) regulates pancreatic cancer cell growth and proliferation. Proc Natl Acad Sci 108: 16927–16931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, et al. 2004. Synergy between LRH-1 and β-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell 15: 499–509. [DOI] [PubMed] [Google Scholar]
- Cabella C, Karlsson M, Canape C, Catanzaro G, Colombo Serra S, Miragoli L, Poggi L, Uggeri F, Venturi L, Jensen PR, et al. 2013. In vivo and in vitro liver cancer metabolism observed with hyperpolarized [5-13C]glutamine. J Magn Reson 232: 45–52. [DOI] [PubMed] [Google Scholar]
- Chambers KT, Chen Z, Lai L, Leone TC, Towle HC, Kralli A, Crawford PA, Finck BN. 2013. PGC-1β and ChREBP partner to cooperatively regulate hepatic lipogenesis in a glucose concentration-dependent manner. Mol Metab 2: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Mishkovsky M, Bastiaansen JA, Ouari O, Hautle P, Tordo P, van den Brandt B, Comment A. 2013. Automated transfer and injection of hyperpolarized molecules with polarization measurement prior to in vivo NMR. NMR Biomed 26: 1582–1588. [DOI] [PubMed] [Google Scholar]
- Chong HK, Biesinger J, Seo YK, Xie X, Osborne TF. 2012. Genome-wide analysis of hepatic LRH-1 reveals a promoter binding preference and suggests a role in regulating genes of lipid metabolism in concert with FXR. BMC Genomics 13: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, et al. 2013. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 153: 840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. 2008. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20. [DOI] [PubMed] [Google Scholar]
- Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. 2012. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 47: 349–358. [DOI] [PubMed] [Google Scholar]
- Hensley CT, Wasti AT, DeBerardinis RJ. 2013. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 123: 3678–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. 2010. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci 107: 7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. 2004. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci 101: 7281–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Schmidt DR, Cummins CL, Choi M, Peng L, Zhang Y, Goodwin B, Hammer RE, Mangelsdorf DJ, Kliewer SA. 2008. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol Endocrinol 22: 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10: 307–318. [DOI] [PubMed] [Google Scholar]
- Mamrosh JL, Lee JM, Wagner M, Stambrook PJ, Whitby RJ, Sifers RN, Wu SPO, Tsai MJ, Demayo FJ, Moore DD. 2014. Nuclear receptor LRH-1/NR5A2 is required and targetable for liver endoplasmic reticulum stress resolution. Elife 3: e01694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataki C, Magnier BC, Houten SM, Annicotte JS, Argmann C, Thomas C, Overmars H, Kulik W, Metzger D, Auwerx J, et al. 2007. Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol Cell Biol 27: 8330–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterveer MH, Mataki C, Yamamoto H, Harach T, Moullan N, van Dijk TH, Ayuso E, Bosch F, Postic C, Groen AK, et al. 2012. LRH-1-dependent glucose sensing determines intermediary metabolism in liver. J Clin Invest 122: 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Out C, Hageman J, Bloks VW, Gerrits H, Sollewijn Gelpke MD, Bos T, Havinga R, Smit MJ, Kuipers F, Groen AK. 2011. Liver receptor homolog-1 is critical for adequate up-regulation of Cyp7a1 gene transcription and bile salt synthesis during bile salt sequestration. Hepatology 53: 2075–2085. [DOI] [PubMed] [Google Scholar]
- Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, et al. 2010. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet 42: 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzle T, Hall MN. 2000. TOR, a central controller of cell growth. Cell 103: 253–262. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, Geboes K, Auwerx J. 2005. Liver receptor homolog 1 contributes to intestinal tumor formation through effects on cell cycle and inflammation. Proc Natl Acad Sci 102: 2058–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. 2013. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Schoonjans K. 2015. Molecular basis for the regulation of the nuclear receptor LRH-1. Curr Opin Cell Biol 33: 26–34. [DOI] [PubMed] [Google Scholar]
- Stein S, Oosterveer MH, Mataki C, Xu P, Lemos V, Havinga R, Dittner C, Ryu D, Menzies KJ, Wang X, et al. 2014. SUMOylation-dependent LRH-1/PROX1 interaction promotes atherosclerosis by decreasing hepatic reverse cholesterol transport. Cell Metab 20: 603–613. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. 2008. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci 105: 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.