

SUMMARY
Circuit function in the CNS relies on the balanced interplay of excitatory and inhibitory synaptic signaling. How neuronal activity influences synaptic differentiation to maintain such balance remains unclear. In the mouse spinal cord, a population of GABAergic interneurons, GABApre, forms synapses with the terminals of proprioceptive sensory neurons and controls information transfer at sensory-motor connections through presynaptic inhibition. We show that reducing sensory glutamate release results in decreased expression of GABA-synthesizing enzymes GAD65 and GAD67 in GABApre terminals and decreased presynaptic inhibition. Glutamate directs GAD67 expression via the metabotropic glutamate receptor mGluR1ß on GABApre terminals and regulates GAD65 expression via autocrine influence on sensory terminal BDNF. We demonstrate that dual retrograde signals from sensory terminals operate hierarchically to direct the molecular differentiation of GABApre terminals and the efficacy of presynaptic inhibition. These retrograde signals comprise a feedback mechanism by which excitatory sensory activity drives GABAergic inhibition to maintain circuit homeostasis.
INTRODUCTION
Synapses are key elements in the organization and function of neuronal circuits. The assembly of synapses is a progressive process that depends on the alignment and differentiation of pre- and postsynaptic structures. Initial synaptic associations involve surface recognition and adhesion, with acquired proximity permitting signals from the pre- and postsynaptic structures to induce specialized features of the complementary synaptic element (Akins and Biederer, 2006). This general scheme is supported by molecular studies of vertebrate and invertebrate neuromuscular junctions (Fox et al., 2007; McCabe et al., 2003; Noakes et al., 1995). Once synaptic structures are established, a process of highly dynamic communication between synaptic partners transmits neural information and maintains synaptic homeostasis. However, in the mammalian central nervous system (CNS), it remains unclear what retrograde molecular signals induce presynaptic differentiation and support homeostatic adaptations of synaptic function in local circuits.
Intrinsic neural activity and postsynaptic trophic signals have been implicated in the control of presynaptic differentiation (Andreae and Burrone, 2014). Neuronal activity can act through cell-autonomous mechanisms to activate voltage dependent Ca++ channels and downstream Ca++-triggered signaling pathways (Greenberg et al., 1986). Independently, activity-driven neurotransmitter release can act cell autonomously, signaling through autoreceptors, to influence presynaptic function (Bewick et al., 2005), as well as non-autonomously, to influence the presynaptic differentiation of neighboring synapses via local diffusion (Kullmann and Asztely, 1998). Neuronal activity also regulates expression of trophic factors important for synaptic development (Hong et al., 2008; Poo, 2001). However, how presumed homeostatic signals that influence presynaptic differentiation in the CNS converge to direct the complex and multifaceted biochemical differentiation of a single synaptic terminal remains unclear.
Many studies of synaptic differentiation have focused on inhibitory interneurons given the biochemical and functional diversity of their neuronal subclasses (Ascoli et al., 2008). At a functional level, inhibitory interneurons within the same microcircuit can exhibit distinct synaptic dynamics, reflecting the specialized character of different inhibitory terminals (Glickfeld and Scanziani, 2006). Inhibitory synaptic terminals can also differ in their choice of neurotransmitter, predominantly GABA or glycine. Moreover, even within a single inhibitory neuronal class, multiple genes encoding different transmitter synthetic enzymes, synaptic vesicle proteins and membrane channels can be expressed (Cauli et al., 1997; Lambolez et al., 1992; Monyer and Markram, 2004). Different populations of GABAergic interneurons are characterized by expression of two GABA-synthesizing enzymes, glutamic acid decarboxylase-67 (GAD67) and glutamic acid decarboxylase-65 (GAD65) (Monyer and Markram, 2004; Soghomonian and Martin, 1998). Both enzymes synthesize GABA and thus determine net neuronal GABA expression (Asada et al., 1997). GAD67 is expressed in the cytosol of most GABAergic neurons and produces the majority of basal-level neurotransmitter released at GABAergic synapses (Asada et al., 1997). In contrast, GAD65, which is expressed with GAD67 in a subset of GABAergic neurons, is vesicle bound and ensures GABA synthesis during prolonged synaptic transmission (Esclapez et al., 1994; Jin et al., 2003; Tian et al., 1999). These different molecular characteristics permit individual terminals to respond to the physiological demands of their particular circuit.
In the spinal cord, GABApre neurons form highly specialized axo-axonic synapses with the terminals of primary sensory afferents, providing the anatomical substrate for presynaptic inhibition. GABApre neurons can be distinguished from other spinal inhibitory interneurons in that they co-express GAD65 in conjunction with GAD67 (Betley et al., 2009; Hughes et al., 2005). BDNF released by sensory terminals acts on GABApre terminals to promote synaptic terminal accumulation of GAD65 (Betley et al., 2009). GAD67 levels are not affected in the absence of BDNF, suggesting that these two GABA synthetic enzymes are differentially regulated. More broadly, this distinction raises the possibility that multiple regulatory pathways act in parallel or hierarchically to influence the functional differentiation of GABApre presynaptic terminals.
Here we have explored the roles of activity-evoked transmitter release from sensory afferents in the differentiation and function of GABApre inhibitory terminals. We have used mouse molecular genetics to manipulate glutamate release from proprioceptive sensory terminals and glutamate receptor expression in GABApre neurons, examining the consequences of these manipulations on GAD enzyme expression by GABApre neurons and on presynaptic inhibition. Compromising glutamate release from sensory terminals by inactivating the predominant vesicular glutamate transporter vGluT1 elicits a marked reduction in GABApre GAD67 levels and functionally reduces the strength of presynaptic inhibition. Similarly, inactivating the mGluR1ß receptor, which we show is expressed at GABApre terminals, reduces GAD67 expression and presynaptic inhibition. In addition, we find that depletion of glutamate release lowers spinal cord BDNF levels and GABApre terminal GAD65 levels, implying an upstream hierarchical role for activity-dependent glutamate release in coordinating both GABA synthetic enzymes. Together, these results indicate that distinct sensory-derived signals act together to determine the specialized presynaptic differentiation and function of a defined set of inhibitory interneurons in the mammalian CNS. Functionally, this constellation of signaling mechanisms supports a homeostatic feedback system by which sensory-derived glutamate controls GABA release from GABApre terminals to constrain activity across the sensory-motor synapse.
RESULTS
Decreased glutamate release from proprioceptive synapses reduces strength of sensory-motor transmission
To assess the role of sensory-derived glutamate release in the differentiation and function of GABApre terminals, we used a mouse model in which the major vesicular glutamate transporter, vGluT1, utilized in proprioceptive synapses, was eliminated (Alvarez et al., 2004; Fremeau et al., 2004; Todd et al., 2003). vGluT1 mutant mice survive for three weeks after birth and have compromised motor coordination, consistent with defects in sensory-motor transmission (Akay et al., 2014; Fremeau et al., 2004). We confirmed complete absence of vGluT1 from proprioceptive terminals in postnatal day (p)12 and p21 vGluT1 mutant mice (Figures 1A–1C; Figures S1A, S1B, S1E–F‴, S1K, S1L). We further found that loss of vGluT1 did not lead to up-regulation of the other two vesicular glutamate transporters, vGluT2 and vGluT3 (Figures S1E–S1F‴; and additional data not shown).
Figure 1. Physiological deficits at sensory afferent terminals in vGluT1−/− mice.
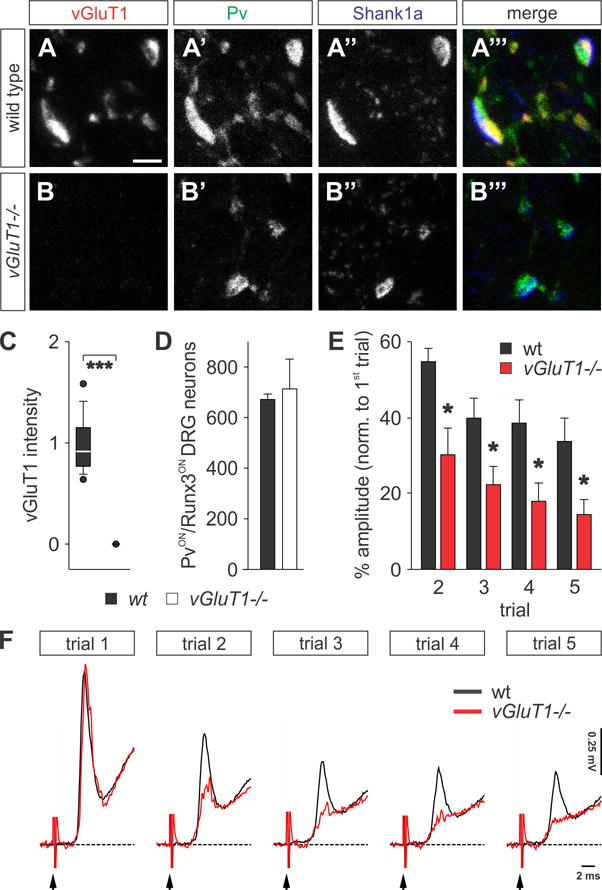
(A–B‴) PvON (green) proprioceptive afferent terminals are juxtaposed to postsynaptic Shank1a (blue) in p12 wt (A–A‴) and vGluT1−/− (B–B‴) mice (similar results obtained at p21 (Figures S1K and S1L). (B) Complete absence of vGluT1 immunoreactivity (red) in sensory terminals of vGluT1−/− mice.
(C) Quantification of vGluT1 fluorescence intensity in proprioceptive afferent terminals of p12 wt (n = 133 boutons, 3 mice) and vGluT1−/− spinal cords (n = 101 boutons, 3 mice). vGluT1 levels are not detectable in vGluT1−/− mice (Mann-Whitney Rank Sum, p < 0.001 ***).
(D) Number of PvON and Runx3ON proprioceptive sensory neurons in L4 dorsal root ganglion (DRG) in wt and vGluT1−/− mice (n(wt) = 1343 neurons; n(vGluT1−/−) = 1427 neurons; Mann-Whitney Rank Sum, p = 1.00 NS).
(E) Quantification of the monosynaptic reflex amplitude at different trials as a percentage of the first trial. The reflex is significantly more depressed in vGluT1−/− mice (n(wt) = 5; n(vGluT1−/−) = 4; t-test, p = 0.013 * (2nd trial), 0.043 * (3rd trial), 0.048 * (4th trial), 0.048 * (5th trial)).
(F) Fifth lumbar segment (L5) ventral root responses following repetitive stimulation (5 trials at 1 Hz) of the homonymous dorsal root at supramaximal intensities (5x threshold) in p12 wt (black) and vGluT1−/− (red) mice. Black arrows indicate the stimulus artifact.
Scale bar: 2.5 μm (A–B‴). Error bars represent s.e.m. Lines and whiskers on box diagrams represent data between 9th and 91st percentile, dots show the 5th and 95th percentile. See also Figure S1.
To measure the sensory-motor monosynaptic reflex in vGluT1 mutants, we employed a spinal cord in vitro preparation (Jiang et al., 1999; Mentis et al., 2005; Shneider et al., 2009). In rodents, during the first two weeks after birth, proprioceptive synapses depress readily at frequencies as low as 1 Hz under in vitro conditions (Lev-Tov and Pinco, 1992; Li and Burke, 2001). We therefore compared the efficacy of glutamate release in wild type (wt) and vGluT1 mutant mice by measuring the amplitude of the ventral root reflex following successive dorsal root stimuli at 1Hz. We saw no difference in the amplitude of the mono-synaptically-mediated reflex between vGluT1 mutant and wt mice at the first trial; however, we noticed a significant reduction with successive trials of stimulation in vGluT1 mutants (Figure 1F). The monosynaptic reflex recorded from wt mice was depressed by 45% at the 2nd stimulus, and reached its maximum depression (~60%) by the 5th stimulus, consistent with previous reports (Figures 1E and 1F) (Li and Burke, 2001). In contrast, vGluT1 mutants revealed a more prominent reduction in synaptic depression compared to wt mice from the 2nd trial (~70%), reaching a maximum level of depression (~90%) by the 5th trial (Figures 1E and 1F). These results demonstrate that glutamate neurotransmission in proprioceptive synapses is severely impaired in the absence of vGluT1.
The effects of glutamate release on the differentiation of GABApre terminals
To test the role of glutamate release in the formation of GABApre-sensory contacts we first assessed whether the differentiation of proprioceptive neurons and their synapses on motor neurons occurs normally in vGluT1 mutant mice. We found that dorsal root ganglia in vGluT1 mutants expressed normal numbers of Parvalbumin (Pv)ON and Runt-related transcription factor 3 (Runx3)ON sensory neurons (Figure 1D) (Chen et al., 2006; Inoue et al., 2002; Kramer et al., 2006; Levanon et al., 2002). Also, PvON sensory terminals in vGluT1 mutants normally co-expressed the presynaptic marker SV2B (Gronborg et al., 2010) and aligned with the postsynaptic marker Shank1a (Figures 1A–1B‴; Figures S1K–S1N″). Finally, analysis at the ultrastructural level revealed that proprioceptive terminals showed only a modest decrease in active zone (AZ) length (~17%; Figures S1G–S1I) and a similarly modest decrease in the density of synaptic vesicles near the AZ (~14%; Figures S1G, S1H and S1J). Thus, the early differentiation of proprioceptive synapses appears relatively unaffected by loss of vGluT1.
We next examined whether the loss of vGluT1 from proprioceptive sensory terminals impacts the organization of GABApre terminals. We observed no change in number and size of GABApre contacts on sensory afferent terminals between wild type and vGluT1 mutant mice (Figures S2F and S2G). To analyze GABApre terminal differentiation, we analyzed expression of the GABA-synthetic enzymes GAD65 and GAD67 in p21 GABApre synaptic terminals (Betley et al., 2009). GAD67 expression levels were reduced by 52% in vGluT1 mutants as compared to wt mice (Figures 2A–2C; for similar p12 values see Figures S2A–S2C). To test the possibility that loss of vGluT1 affects GABApre terminal GAD67 expression by diminishing potential direct sensory input onto GABApre interneurons, we assessed GAD67 levels at terminals of GABApost interneurons, which form contacts directly on motor neurons and are known to receive direct sensory afferent input. We found that the level of GAD67 in GABApost terminals was not affected in vGluT1 mutants (Figure 2C), indicating that reduced glutamate release leads to a decrease in GAD67 expression specifically in GABApre terminals.
Figure 2. Reduction of GAD enzyme expression in vGluT1−/− mice.
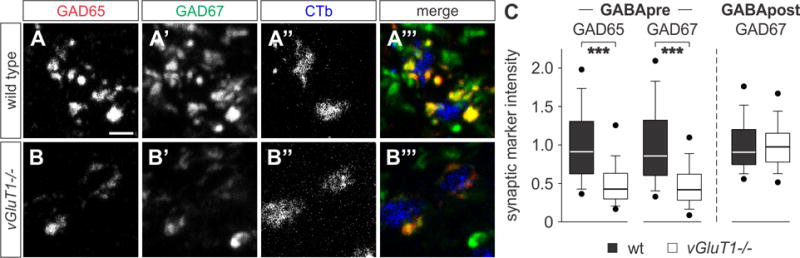
(A–B‴) Immunoreactivity of GAD65 (red) and GAD67 (green) in GABApre terminals contacting CTbON (blue) proprioceptive afferent terminals in p21 wt (A–A‴) and vGluT1−/− (B–B‴) mice.
(C) GAD65 and GAD67 intensity measurements in GABApre and GABApost terminals at p21 (GABApre GAD65: n(wt) = 467 boutons, 3 mice; n(vGluT1−/−) = 315 boutons, 3 mice; Mann-Whitney Rank Sum, p < 0.001 ***; GABApre GAD67: n(wt) = 385 boutons, 3 mice; n(vGluT1−/−) = 173 boutons, 3 mice; Mann-Whitney Rank Sum, p < 0.001 ***; GABApost GAD67: n(wt) = 120 boutons, 3 mice; n(vGluT1−/−) = 119 boutons, 3 mice; Mann-Whitney Rank Sum, p = 0.58 NS). Data shown normalized with respect to wt data.
Scale bar: 2.5 μm (A–B‴). Lines and whiskers on box diagrams represent data between 9th and 91st percentile, dots show the 5th and 95th percentile. See also Figure S2.
We next assessed whether GAD65 was also altered in GABApre terminals of vGluT1 mutant mice. vGluT1 mutant mice exhibited significantly reduced levels of GAD65 in GABApre terminals compared to wt mice (49%; Figures 2A–2C; for similar p12 values see Figures S2A–S2C). Since GAD65 is vesicle-bound, we considered whether the reduction of GAD65 might be due to a reduced number of synaptic vesicles in vGluT1 mutant GABApre terminals. We quantified the density of synaptic vesicles in GABApre terminals in wt and vGluT1 mutant mice. To label synaptic vesicles in GABApre terminals, we intercrossed Gad65-N45GFP mice – previously shown to label ≥ 95% of GABApre terminals (Betley et al., 2009) – with vGluT1 mutants. Analysis at the ultrastructural level (EM single immunolabeling-GFP) revealed no significant difference in the density of synaptic vesicles in GABApre terminals between Gad65-N45GFP and Gad65-N45GFP; vGluT1−/− mice (Figure S2H). This suggests that the observed decrease in the amount of GAD65 localized to the GABApre terminal is not due to a decrease in synaptic vesicle number. Taken together the significant reduction in localization of the GABA-synthesizing enzymes to GABApre terminals seen in vGluT1 mutants suggests that compromised sensory-derived glutamate release affects the synaptic terminal differentiation of GABApre neurons.
Reduced glutamate release from sensory terminals decreases the strength of presynaptic inhibition
Given that GAD levels are reduced in GABApre terminals of vGluT1 mutant mice, we assessed whether loss of vGluT1 has a functional impact on presynaptic inhibition. We used an in vitro conditioning paradigm to measure the efficacy of presynaptic inhibition (see Supplemental Experimental Procedures for details). In this protocol, the amplitude of the monosynaptic reflex evoked by homonymous dorsal root stimulation was compared to that evoked by time-conditioned stimulation of the adjacent dorsal root (Figures 3A–3C; Figures S3A–S3C). At long conditioning intervals (> 300 ms), there was a gradual reduction of the monosynaptically-evoked response, reaching statistical significance at conditioning intervals of 700–900 ms (Figure 3D; Figure S3D). Blockade of GABAA receptors with bicuculline reversed this reduction, but application of strychnine, a glycine receptor blocker, did not (Figures 3E and 3F; Figures S3E and S3F), suggesting that the conditioned reduction in monosynaptically-evoked response was due to GABAergic presynaptic inhibition.
Figure 3. Compromised presynaptic inhibition in vGluT1−/− mice.
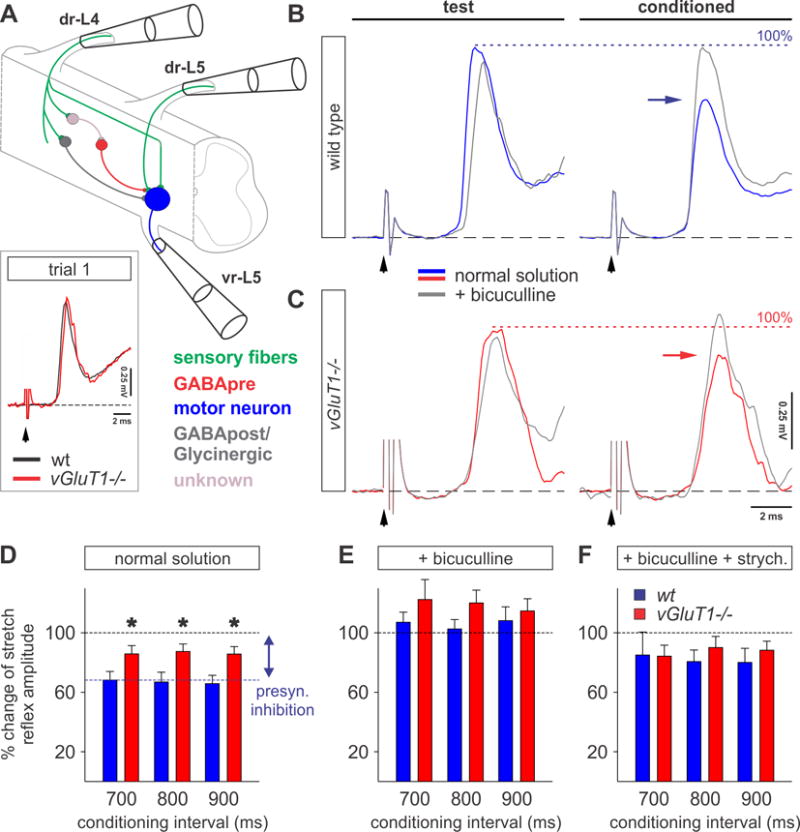
(A) Experimental protocol to test presynaptic inhibition in the in vitro spinal cord preparation. The dorsal root-to-ventral root reflex in the L5 segment was conditioned by stimulation of the L4 dorsal root (dr-L4) at various time intervals. For simplicity, the key neurons activated in this pathway are color-coded. Insert shows the monosynaptic reflex recorded extracellularly from the L5 ventral root following stimulation of the homonymous dorsal root in p12 wt (black) and vGluT1−/− (red) mice. Black arrows indicates stimulus artifact.
(B and C) Averaged (n = 5) response of the monosynaptic reflex recorded from L5 ventral root following dr-L5 stimulation (test stimulus) and after the conditioning stimulus from dr-L4 (conditioned; 800 ms) in wt (blue traces in B) and vGluT1−/− (red traces in C) mice. Frequency of stimulation: 0.1 Hz. The level of presynaptic inhibition of the monosynaptic reflex is highlighted by horizontal arrows in wt (blue) and vGluT1−/− (red) mice. Exposure to bicuculline (grey traces) restored the amplitude of the monosynaptic reflex to test stimulus level (dotted line: assigned as 100% of test response).
(D–F) Percentage change of the monosynaptic reflex amplitude during 700–900 ms conditioning intervals (grey boxed region in Figures S3D–S3F) in normal solution (D), under 10 μM bicuculline (E) and under 10 μM bicuculline and 10 μM strychnine (F) in wt (blue, n = 5, p12) and vGluT1−/− (red, n = 4, p12) mice. The level of presynaptic inhibition is significantly reduced in vGluT1−/− preparations under normal solution (D: n(wt) = 4; n(vGluT1−/−) = 4; t-test, p = 0.044 * (700 ms), 0.026 * (800 ms), 0.043 * (900 ms); E: n(wt) = 4; n(vGluT1−/−) = 4; t-test, p = 0.33 NS (700 ms), 0.17 NS (800 ms), 0.57 NS (900 ms); F: n(wt) = 5; n(vGluT1−/−) = 3; t-test, p = 0.97 NS (700 ms), 0.45 NS (800 ms), 0.57 NS (900 ms)). For details see Figures S3D–S3F.
Error bars represent s.e.m. See also Figure S3.
To assess presynaptic inhibition in vGluT1 mutant mice, we took advantage of the observation that with a single stimulus, the monosynaptically-mediated reflex in vGluT1 mutant mice was comparable to that of wt mice (inset in Figure 3A). The specific AMPA receptor blocker, NBQX, abolished this monosynaptic response (Figures S1C and S1D), confirming that neurotransmission at the proprioceptive synapses in vGluT1 mutant mice was glutamatergic in nature. We assessed the strength of presynaptic inhibition in vGluT1 mutant mice using the in vitro conditioning paradigm. In vGluT1 mutants, the strength of presynaptic inhibition was ~50% reduced as compared to wt mice (wt: ~35%, vGluT1−/−: ~18%; cond. interval: 700–900 ms; Figures 3D–3F). These results suggest that the reduction of GAD synthesizing enzymes seen in vGluT1 mutants has significant functional consequences for the strength of presynaptic inhibition.
Group I mGluR expression in GABApre terminals
We next examined whether glutamate influences the level of GAD65 and GAD67 in GABApre terminals. To assess whether glutamate might signal to GABApre terminals directly, we examined the expression of glutamate receptors on GABApre terminals. Presynaptic glutamate receptors have been shown to influence synaptic neurotransmitter release through a variety of mechanisms (Pinheiro and Mulle, 2008). Unlike fast acting ionotropic glutamate receptors, activation of metabotropic receptors (mGluRs) can last for long periods of time (Conn and Pin, 1997). Since proprioceptive activity is relatively high even at quiescent periods (Prochazka, 1999), we hypothesized that mGluRs are strong candidates to be acting as sensors for sustained glutamate release (Eccles et al., 1962). To identify mGluR gene expression in postnatal spinal neurons we carried out in situ hybridization analysis in p6 lumbar spinal cord, an age when GABApre neurons have formed clearly defined boutons on proprioceptive afferent terminals (Betley et al., 2009). Transcripts of group I, II and III mGluR family members were expressed at detectable levels by various cells throughout the spinal cord (data not shown) consistent with previous reports (Alvarez et al., 2000; Berthele et al., 1999). To test whether any of the mGluRs are expressed in GABApre neurons, we analyzed their expression immunochemically in Gad65-N45GFP mice (Betley et al., 2009). We detected a group I mGluR, mGluR1β, in putative N45GFP-positive (N45GFPON) GABApre neurons in the intermediate spinal cord (Figure S4A).
We next asked whether mGluR1β is localized to GABApre terminals. We used both light and EM immunocytochemistry to assess the expression of mGluR1β in N45GFPON GABApre terminals in the ventral spinal cord. Pre-embedding EM immunoperoxidase (ImP; N45GFP) and immunogold (ImG; mGluR1β) cytochemistry (Pierce et al., 2009) revealed mGluR1β ImG particles predominantly associated with the plasma membrane of N45GFPON terminals (Figures 4A–4D). We observed mGluR1β immunogold particles associated with synthetic pathway endomembranes in somata (Figure S4B), but did not observe mGluR1β expression at the plasma membranes of GABApre somata. We next quantified the extent of mGluR1β and N45GFP co-expression by light microscopy using the endogenous expression of GAD67 and Synaptotagmin-1 (Syt1) as additional markers of GABApre terminals (Betley et al., 2009). We found that mGluR1β co-localized with 40% of N45GFPON GABApre terminals on vGluT1ON sensory terminals (Figures 4E–4E‴). Since we previously showed that N45GFP is expressed in over 95% of GABApre terminals on sensory terminals (Betley et al., 2009), we infer that 40% of GABApre terminals express mGluR1β. A subset of GAD67ON/N45GFPON and Syt1ON/N45GFPON GABApre boutons similarly expressed mGluR1β (Figures 4F–4G‴; Figures S4C–S4C‴). This mosaic expression of mGluR1β is reminiscent of our previous finding that sensory terminals receive input from more than one GABApre neuron (Betley et al., 2009) and suggests that this group I metabotropic glutamate receptor is expressed presynaptically in a subset of GABApre neurons.
Figure 4. mGluR1β expression in GABApre terminals.
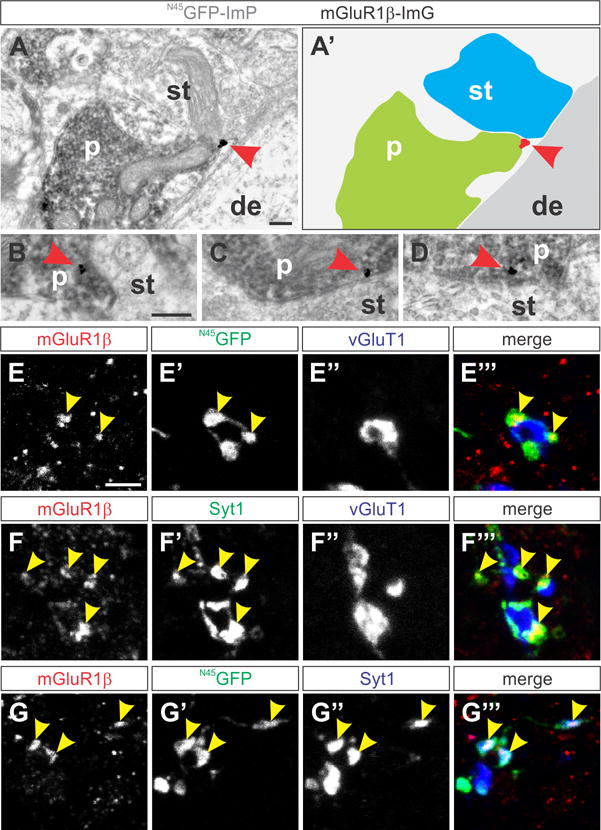
(A–D) N45GFP immunoperoxidase (N45GFP-ImP; grey immunoreactivity) and mGluR1β immunogold (mGluR1β-ImG; gold particle) double-labeled in GABApre presynaptic terminals (p). mGluR1β ImG particle (red arrowhead) is membrane-associated and in close proximity to sensory afferent terminals (st) on p21 motor neuron dendrites (de). (A′) Color-coded outline of terminals and mGluR1β labeling shown in a.
(E–E‴) N45GFPON (green) GABApre terminals contacting vGluT1ON sensory terminals (blue) express mGluR1β (red) in p21 Gad65-N45GFP mice (yellow arrowheads indicate mGluR1β and GFP co-labeling).
(F–F‴) mGluR1β (red) overlaps with Syt1 (green) on vGluT1ON sensory terminals (blue) in wt spinal cords (yellow arrowheads indicate mGluR1β and Syt1 co-labeling).
(G–G‴) mGluR1β (red) and the GABApre presynaptic marker Syt1 (blue) co-label with N45GFPON (green) GABApre terminals (yellow arrowheads indicate mGluR1β, GFP and Syt1 co-labeling).
Scale bars: 200 nm (A, B–D), 2.5 μm (E–G‴). See also Figure S4.
mGluR1 directs GABApre terminal differentiation
To assess whether mGluR1β plays a role in GABApre terminal differentiation, we analyzed spinal cords of constitutive p21 mGluR1 knockout mice (Conquet et al., 1994). mGluR1 mutants are severely ataxic and both mGluR1α and mGluR1β are dysfunctional (Conquet et al., 1994). We first addressed whether sensory-motor connectivity forms normally in these mice. We found no differences in the overall pattern of vGluT1ON proprioceptive afferent terminals on motor neurons in mGluR1 mutant mice (Figures 5A and 5B). Furthermore, the synaptic density of proprioceptive afferent terminals contacting motor neuron cell bodies and proximal dendrites was unchanged compared to wt mice (Figures S5A and S5B). Finally, the percentage reduction of the sensory-motor reflex amplitude in mGluR1 mutant mice was largely similar compared to wt mice (Figure S5C).
Figure 5. Reduction of GAD67 levels in GABApre terminals in mGluR1−/− mice.
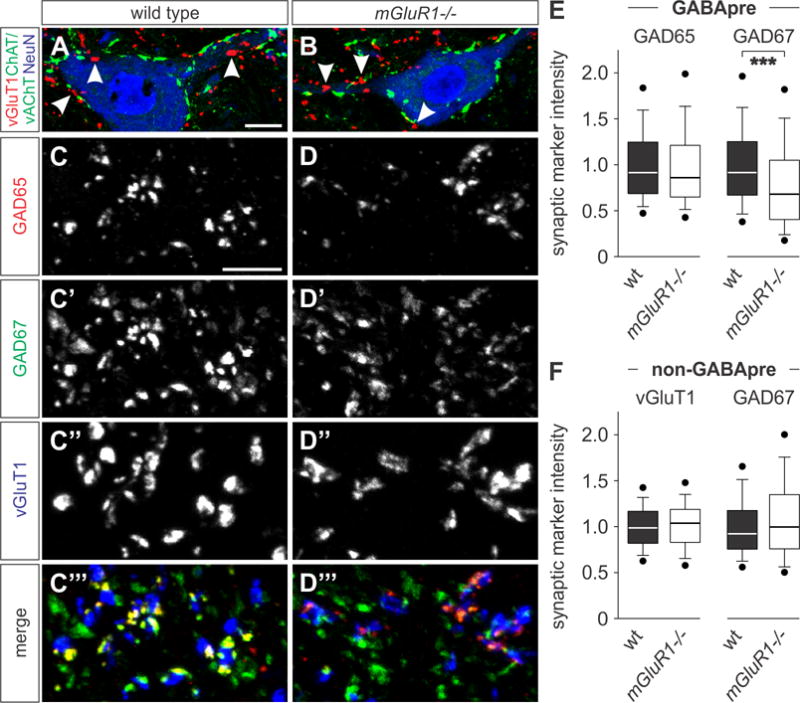
(A and B) Images of vGluT1ON (red) proprioceptive afferent terminals (white arrowheads) and cholinergic terminals (vesicular acetylcholine transporter (vAChT) and ChAT (green)) in p21 wt (A) and mGluR1−/− (B) mice. Motor neurons labeled for neuronal nuclei (NeuN, blue), and ChAT (green). ChAT and vAChT visualized in green.
(C–D‴) GAD65ON (red) and GAD67ON (green) GABApre terminals contacting vGluT1ON proprioceptive afferent terminals (blue) in p21 wt and mGluR1−/− mice.
(E and F) Quantification of GABApre and non-GABApre synaptic marker intensities. GAD67 levels are significantly reduced in mGluR1−/− GABApre terminals as compared to wt mice (E; n(wt) = 564 boutons, 5 mice; n(mGluR1−/−) = 647 boutons, 6 mice; Mann-Whitney Rank Sum, p < 0.001 ***). No significant change in GAD65 protein levels (E; n(wt) = 777 boutons, 5 mice; n(mGluR1−/−) = 898 boutons, 6 mice; Mann-Whitney Rank Sum, p = 0.065 NS). vGluT1 and GAD67 levels in (non-GABApre) terminals on motor neurons are unaffected in mGluR1−/− mice (F; wt (non-GABApre vGluT1): n = 256 boutons, 3 mice; mGluR1−/− (non-GABApre vGluT1): n = 248 boutons, 4 mice; Mann-Whitney Rank Sum, p = 0.29 NS; wt (non-GABApre GAD67): n = 262 boutons, 3 mice; mGluR1−/− (non-GABApre GAD67): n = 226 boutons, 4 mice; Mann-Whitney Rank Sum, p = 0.10 NS).
Scale bars: 10 μm (A, B), 2.5 μm (C–D‴). Lines and whiskers on box diagrams represent data between 9th and 91st percentile, dots show the 5th and 95th percentile. See also Figure S5.
We next examined whether loss of mGluR1 affects differentiation of GABApre terminals. We found that expression of GAD67 was reduced by 21% with respect to wt controls (Figures 5C–5E), but levels of GAD65 remained unchanged (Figures 5C–5E; for similar p12 and p43 values see Figures S5D). The number and size of GABApre contacts was the same between wt and mGluR1 mutant mice, as was GABApre vesicle density (Figure S2H; data not shown). Loss of mGluR1 did not result in any significant differences in vGluT1 fluorescent intensity in proprioceptive terminals, or GAD67 in pre-motor GABApost terminals (Figure 5F). These results provide strong evidence that GAD67 is specifically reduced in GABApre terminals in mGluR1 mutant mice.
Loss of mGluR1 decreases the strength of presynaptic inhibition
We next examined whether the specific decrease in GAD67 levels seen in mGluR1 mutant mice also correlated with deficits in presynaptic inhibition. We measured the strength of presynaptic inhibition in mGluR1 mutant mice and found that it was reduced by 50% compared to wt counterparts (wt: ~34%, mGluR1−/−: ~16%; cond. interval: 700–900 ms; Figures 6A–6C). These results affirm that compromised sensory-derived glutamate signaling to GABApre terminals has a functional consequence on the efficacy of presynaptic inhibition. Given that GAD67, but not GAD65, is reduced in mGluR1 mutants, the observed deficit further suggests that GAD67 plays a more prominent role than GAD65 in mediating presynaptic inhibition.
Figure 6. Loss of mGluR1 results in reduced presynaptic inhibition.
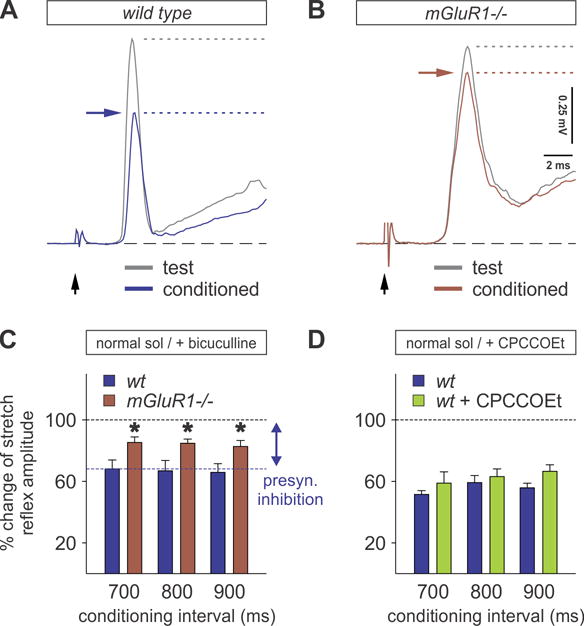
(A and B) Averaged responses of the monosynaptic reflex recorded from vr-L5 following dr-L5 stimulation (test stimulus) and after conditioning stimulus from dr-L4 (conditioned) in wt (A) and mGluR1−/− (B) mice. Horizontal arrows (blue for wt and brown for mGluR1−/− mice) indicate the level of presynaptic inhibition.
(C) Percent change of monosynaptic reflex amplitude at three conditioning intervals (700–900 ms), in which presynaptic inhibition in mGluR1−/− mice (brown bars) was significantly reduced compared to wt mice (blue bars) in normal solution (n = 4 for all groups; t-test compared to wt, p = 0.048 * (700 ms), 0.046 * (800 ms), 0.050 * (900 ms)).
(D) In the presence of 50 μM CPCCOEt, the strength of presynaptic inhibition is not significantly reduced (green bars) (n = 3 in both groups; t-test, p = 0.393 NS (700 ms), 0.585 NS (800 ms), 0.107 NS (900 ms)).
Error bars represent s.e.m.
To further explore the function of presynaptic mGluR1β, we measured presynaptic inhibition physiologically in wt mice, using the in vitro spinal cord preparation, following bath application of the mGluR1 antagonist CPCCOEt (Bonnot et al., 2009). There was a trend towards a reduction in presynaptic inhibition, which did not reach statistical significance (Figure 6D), suggesting that the effect of pharmacological block of mGluR1 signaling on GAD67 requires longer duration exposure.
Glutamate release controls expression of BDNF
While GAD67 levels in GABApre terminals depend on glutamate signaling via mGluR1, the lack of change in GAD65 expression in mGluR1 mutants – despite its reduction in vGluT1 mutants – suggests an alternative mechanism of glutamate-dependent regulation. The localization of GAD65 in GABApre terminals is dependent on retrograde BDNF emanating from sensory terminals (Betley et al., 2009). Since GAD65 levels are reduced in vGluT1 mutants, we assessed the expression level of BDNF in vGluT1 mutant spinal cords. We found that BDNF protein levels were ~50% reduced in vGluT1 mutant spinal cords compared to wt mice (Figure 7A). In comparison, BDNF levels were not significantly decreased in mGluR1 mutant spinal cords (Figure 7B).
Figure 7. GAD65 in GABApre terminals is regulated by sensory-derived BDNF.
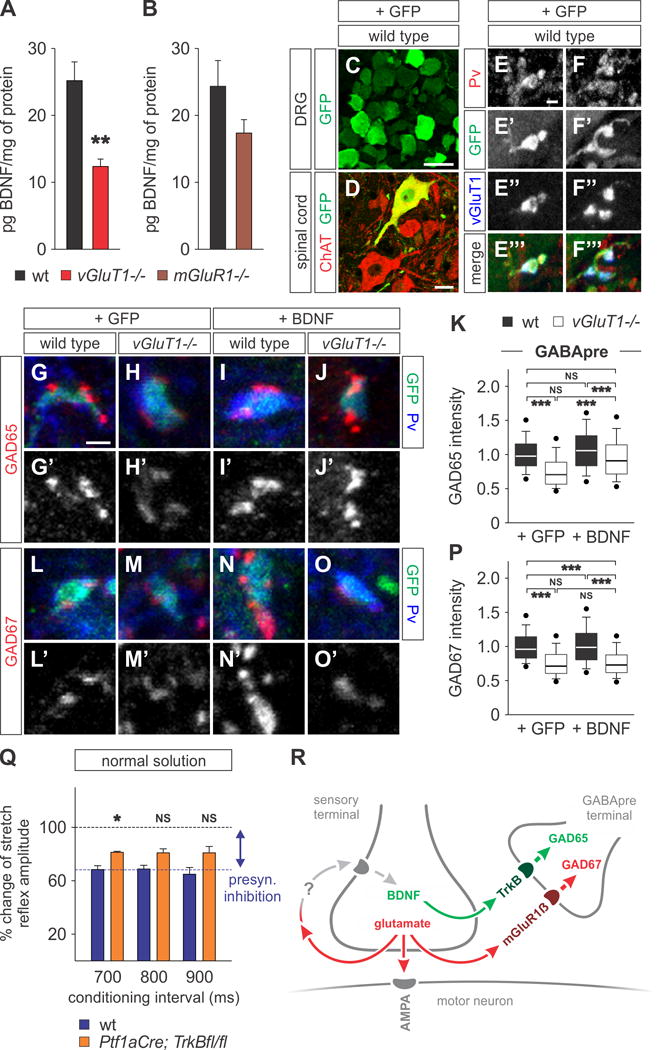
(A and B) BDNF ELISA of L3–L5 mouse spinal cords at p12 shows significantly reduced BDNF in vGluT1−/− as compared to wt mice (A; n = 4 in both groups; t-test, p = 0.005 **). BDNF levels are not significantly reduced in mGluR1 mutant mice (B; n = 4 in both groups; t-test, p = 0.157 NS).
(C–F‴) ICV injection of AAV2/9-CMV-GFP (+GFP) results in GFP labeling of sensory neurons in the DRG (C) and a subset of motor neurons (D). GFP is detectable in terminals of proprioceptive sensory afferents labeled with Pv and vGluT1 (E–F‴). GAD expression levels were analyzed in GABApre terminals on GFPON sensory terminals.
(G–P) GAD65 and GAD67 expression in wt and vGluT1−/− mice injected with AAV2/9-CMV-GFP (+GFP) or AAV2/9-CMV-GFP-BDNF (+BDNF). Reduced GAD65 levels are rescued in vGluT1−/− mice overexpressing BDNF (G–K). GAD67 levels remain unchanged (L–P). Data shown normalized with respect to [wt +GFP] data (GAD65: n = 300 boutons, 3 mice for all conditions; One-way ANOVA with Tukey post test, [wt +GFP] vs [vGluT1−/− +GFP] p < 0.001 ***; [wt +GFP] vs [wt +BDNF] NS; [wt +GFP] vs [vGluT1−/− +BDNF] NS; [wt +BDNF] vs [vGluT1−/− +GFP] p < 0.001 ***; [wt +BDNF] vs [vGluT1−/− +BDNF] p < 0.001 ***; [vGluT1−/− +GFP] vs [vGluT1−/− +BDNF] p < 0.001 ***; GAD67: n = 300 boutons, 3 mice for all conditions; One-way ANOVA with Tukey post test, [wt +GFP] vs [vGluT1−/− +GFP] p < 0.001 ***; [wt +GFP] vs [wt +BDNF] NS; [wt +GFP] vs [vGluT1−/− +BDNF] p < 0.001 ***; [wt +BDNF] vs [vGluT1−/− +GFP] p < 0.001 ***; [wt +BDNF] vs [vGluT1−/− +BDNF] p<0.001 ***; [vGluT1−/− +GFP] vs [vGluT1−/− +BDNF] NS).
(Q) Percentage change of monosynaptic reflex amplitude at three conditioning intervals (700 – 900 ms) in Ptf1aCre; TrkBflox/flox mice (orange bars) resulted in moderate reduction in presynaptic inhibition compared with wt mice (blue bars) (n = 3 for all groups; t-test, p = 0.01 * (700 ms), 0.11 NS (800 ms), 0.08 NS (900 ms)).
(R) The model proposed for regulation of presynaptic inhibition, whereby glutamate (red) release at sensory terminals controls GAD65 and GAD67 levels in GABApre terminals via two distinct pathways. Glutamate controls the level of GAD67 (red) directly via mGluR1β on GABApre terminals. Glutamate also regulates in an autocrine manner sensory terminal expression of BDNF (green), which acts via TrkB receptors on GABApre terminals to control presynaptic levels of GAD65 (green). Consequently, in vGluT1−/− mice, reduced glutamate release from proprioceptive afferent terminals leads to decreased levels of both GAD65 and GAD67. In mGluR1−/− spinal cords, GABApre synapses are impaired in sensing glutamate release, resulting in reduced levels of GAD67 expression, while GAD65 levels remain normal.
Scale bars: 50 μm (C), 20 μm (D), 2 μm (E–J′ and L–O′). Lines and whiskers on box diagrams represent data between 9th and 91st percentile, dots show the 5th and 95th percentile. Error bars represent s.e.m. See also Figure S6.
To assess whether the GAD65 phenotype in vGluT1 mutant mice might be mediated by a reduction in sensory BDNF release, we studied the overexpression of BDNF in the spinal cord in vivo. To do so, we used a recombinant adeno-associated virus (AAV2/9) vector encoding mBDNF under the CMV promoter (AAV2/9-CMV-GFP-BDNF). The virus was delivered by an intracerebroventricular injection one day after birth (p1) in wt and vGluT1 mutant mice (see Supplemental Experimental Procedures for details). Control experiments utilized virus expressing GFP only (AAV2/9-CMV-GFP). At p19. analysis of GFP expression revealed extensive labeling in the dorsal root ganglia (DRG) and in the terminals of PvON/vGluT1ON proprioceptive sensory neurons, as well as co-expression in choline acetyl transferase (ChAT)ON motor neurons and a few select glia (Figures 7C–7F‴; data not shown). Following injection of AAV-GFP-BDNF, levels of BDNF in the DRG of vGluT1 mutant mice were elevated ~5-fold compared to endogenous wt levels (Figure S6A). We next examined whether elevating BDNF levels in vGluT1 mutant mice affects the differentiation of GABApre terminals. Assessing GAD65 expression in GABApre terminals synapsing upon transfected GFP-BDNF expressing sensory terminals revealed a rescue of GAD65 expression to wt levels, while GAD67 levels remained low (Figures 7G–7P). Since viral transfection was incomplete, we assessed whether mosaic restoration of sensory terminal BDNF expression rescued GABApre GAD65 broadly throughout the animal, or only in local circuits. Analysis of GABApre boutons synapsing on non-transfected sensory terminals that did not express extra BDNF revealed no rescue of GAD65 level ([vGluT1−/− +GFP]: 74.4%, n = 300 boutons, 3 mice vs [vGluT1−/− +BDNF (−)]: 79.5%, n = 150 boutons, 3 mice; ANOVA with Tukey post test, p = NS), in marked contrast to those GABApre boutons in the same animal that synapse upon virally-transfected sensory terminals ([vGluT1−/− +BDNF (+)]: 94.2%, n = 300 boutons, 3 mice vs [vGluT1−/− +BDNF (−)]: 79.5%, n = 150 boutons, 3 mice; ANOVA with Tukey post test, p < 0.001 ***; see also Figure 7K). These data strongly support a direct, local-circuit effect of sensory terminal BDNF on GABApre terminal GAD65 expression, and thus suggests against diffuse or indirect impact of BDNF on GABApre differentiation.
Our mGluR1 mutant analysis suggests that GAD67 is the major GAD isoform controlling presynaptic inhibition. To assess the contribution of GAD65 to presynaptic inhibition, we next disrupted BDNF signaling. Sensory-derived BDNF signals via GABApre-localized TrkB receptor to induce the synaptic localization of GAD65 (Betley et al., 2009). We measured presynaptic inhibition in mice in which we conditionally deleted TrkB from GABApre neurons. To do so, we intercrossed a Ptf1aCre mouse line, which directs Cre expression to GABAergic interneurons (Betley et al., 2009; Glasgow et al., 2005; Kawaguchi et al., 2002) with a conditional TrkBflox/flox mouse line (He et al., 2004) to generate Ptf1aCre; TrkBflox/flox mice (Betley et al., 2009). We found that presynaptic inhibition in Ptf1aCre; TrkBflox/flox mice was affected modestly compared to wt mice (Figure 7Q). This result is consistent with a primary role for GAD67 in determining the efficacy of GABApre-mediated presynaptic inhibition. Taken together our results support a mechanism whereby proprioceptive sensory-derived glutamate and BDNF collaborate in a hierarchical manner to control the presynaptic differentiation of GABApre terminals and the efficacy of presynaptic inhibition (Figure 7R).
DISCUSSION
The inhibitory gating of sensory-motor neurotransmission in the spinal cord is mediated by a specialized set of GABApre interneurons that form contacts directly with sensory terminals. In this study we have explored how distinct retrograde signals – mediated by an excitatory transmitter and a trophic peptide – act together to regulate the specialized features of presynaptic differentiation with consequence for inhibitory gating. Our findings reveal that glutamate release from sensory terminals acts via a metabotropic glutamate receptor expressed on GABApre terminals to control both the expression of GAD67 and the efficacy of presynaptic inhibition. Our results further suggest that glutamate release controls the expression of GAD65 in GABApre terminals indirectly via an autocrine influence on sensory BDNF expression.
Retrograde signaling and the regulation of GAD isoforms
Physiological and genetic studies of GAD67 expression in the CNS suggest strongly that GAD67 mRNA and protein are increased in response to neuronal activity (Esclapez and Houser, 1999; Lau and Murthy, 2012; Liang et al., 1996; Ramirez and Gutierrez, 2001). In contrast, GAD65 levels in GABApre terminals are controlled via protein trafficking in response to retrograde sensory-derived BDNF signaling via TrkB receptors (Betley et al., 2009). Whether the decrease in GAD67 we observe when sensory glutamate signaling is impaired reflects deficits in new mRNA or protein synthesis or altered trafficking of existing protein will require further study. We have performed Gad65 and Gad67 in situ hybridization on vGluT1 mutant and wt spinal cords and observe no qualitative differences (Figures S2D–S2E′). The decrease in GAD67 in the absence of mGluR1 would be consistent with the longstanding model that group I metabotropic glutamate receptor signaling leads to protein synthesis (Weiler and Greenough, 1993). Studies of long term synaptic depression (LTD) and Fragile X syndrome have implicated group I metabotropic glutamate receptors mGluR1 and mGluR5 in directing local translation of synaptic mRNA (Krueger and Bear, 2011). Furthermore, published PAR-CLIP analysis suggests that the transcript encoding GAD67, but not GAD65, binds the Fragile X mental retardation associated RNA binding protein FMRP (Ascano et al., 2012), suggesting local translation as a possible means by which GABApre terminal GAD67 levels are controlled. The decrease in GAD67 we observe in the absence of mGluR1 indicates that sensory-derived glutamate acts directly on GABApre terminals to influence GAD67 levels. However, the incomplete replication of the vGluT1 phenotype in mGluR1 mutants is consistent with our observation that mGluR1β expression defines only a subset of GABApre neurons. This raises the possibility that other mGluRs might be involved in directing GAD67 expression in GABApre terminals or that other changes in GABAergic signaling machinery may contribute to the observed effects of vGluT1 or mGluR1 mutation on presynaptic inhibition. In line with this possibility, we detect RNA expression of mGluR5 in the dorsal intermediate region of spinal cord in which GABApre cell bodies are found (data not shown). Attempts to explore genetic interaction between combined mutations of vGluT1 and mGluR1 were not possible due to embryonic lethality of double mutant mice (data not shown).
How do retrograde signals collaborate to control presynaptic differentiation?
Given the specific architecture of the presynaptic inhibitory microcircuit, in which GABApre terminals directly contact sensory afferent terminals, sensory-derived signals are well positioned to regulate the presynaptic differentiation of associated GABApre boutons. In this work we implicate both sensory derived BDNF and glutamate as signals directing presynaptic differentiation, yet we also suggest these operate in a hierarchical manner, with alterations in glutamate release influencing expression of BDNF. BDNF expression is known to be activity dependent with transcription regulated by membrane depolarization and signaling downstream of calcium influx (Hong et al., 2008; West et al., 2002). Glutamate release from synaptic-like vesicles at the peripheral mechano-sensory endings of sensory afferents has been suggested to act in an autocrine fashion to enhance terminal excitability during sustained stretch, via an atypical phospholipase-D coupled glutamate receptor (Bewick et al., 2005). We propose a similar mechanism at central sensory afferent terminals whereby sensory afferent glutamate release acts cell-autonomously to regulate BDNF expression (Figure 7R). The reported expression of mGluR receptors at central sensory terminals would lend credence to such a model (Ohishi et al., 1995), supporting a local control mechanism at sensory afferent terminals. An alternative possibility is that the decrease in BDNF expression in the CNS resulting from vGluT1 mutation might reflect impaired autocrine glutamate signaling from synaptic-like vesicles in the periphery. If peripheral autocrine glutamate signaling increases terminal excitability in response to stretch, then vGluT1 mutation could decrease overall sensory afferent activity and thereby decrease central terminal BDNF expression. Regardless of the precise mechanism, our results support a model in which glutamate and BDNF act as trans-synaptic retrograde signals in parallel, but also hierarchically, with autocrine glutamate signaling regulating sensory afferent BDNF expression. They further provide a functional context for understanding how activity-dependent regulation of BDNF may ‘fine tune’ synaptic differentiation in a neuronal microcircuit. Indeed, that the retrograde signaling mechanism we describe functions locally to “fine tune” individual circuits is supported by our finding that virally-mediated mosaic increase in sensory BDNF expression in a vGluT1 mutant background leads to tightly correlated mosaic rescue of GABApre terminal GAD65 levels.
Implications of functional presynaptic inhibition for local circuit feedback
Presynaptic mGluRs have previously been suggested to play a role in feed-forward amplification of excitatory signals (Mitchell and Silver, 2000; Semyanov and Kullmann, 2000). Here we demonstrate a novel presynaptic function for an mGluR class I receptor and describe a circuit arrangement in which presynaptic mGluR functions in a feed-back manner to modulate GABAergic inhibition of a proximate sensory afferent glutamate source. This scaling of GABA release would allow for the strength of transmission across the sensory-motor connection to be held constant even when activity levels of the incoming proprioceptive sensory afferent are altered, reflected in changes of vGluT1 expression (Figure 7R). Fink and colleagues (Fink et al., 2014) recently described features of the GABApre interneuron population responsible for scaling presynaptic inhibition to widely variable frequency and intensity of sensory afferent activity during movement: these include high frequency firing in response to proprioceptive input, and the selective expression of GAD65. Our work describes a novel and distinct, but complementary means by which GABApre inhibition is scaled to match sensory afferent output, over a time course involving modulation of synaptic protein levels and in a manner that supports homeostatic “self-tuning” of circuit excitability (Turrigiano and Nelson, 2004). The decrease in presynaptic inhibition we observe in the absence of mGluR1 appears to reflect alterations in the level of GAD67, rather than GAD65. The predominant role of GAD67 is further supported by our observation that disrupting BDNF signaling via targeted TrkB deletion, which prevents GABApre-terminal accumulation of GAD65, has limited impact on measured presynaptic inhibition. The co-expression of GAD67 and GAD65 in GABApre neurons thus may support the interneurons’ capacity to scale inhibition to meet sensory afferent demands dynamically both in the immediate temporal context of a particular behavior but also over a more extended period of adaptive development. The observation that both the 21% decrease in GABApre terminal GAD67 seen in mGluR1 mutants and the 50% decrease seen in vGluT1 mutants lead to similar decrements in measured presynaptic inhibition suggests that the relationship between visualized GAD67 level and presynaptic inhibitory strength is non-linear, and that the reduction in presynaptic inhibition may be maximal in mGluR1 mutants. This non-linearity may reflect the kinetics of GAD67 activity and its relationship to inhibitory function, or it may reflect other, unknown mechanisms of compensation to loss of mGluR1 and/or vGluT1.
In essence, our findings support a model whereby the inhibitory potency of GABAergic interneurons is increased in response to excitatory activity, such that a homeostatic balance of excitation and inhibition is maintained. Deficits in such homeostatic regulation of network activity have been proposed to underlie numerous mental retardation and autistic spectrum disorders (Ramocki and Zoghbi, 2008). Our experiments emphasize the function of GAD67 in homeostatic adaptation of GABApre interneurons to sensory activity, which is congruent with GAD67’s role as the predominant form of the GABA synthesizing enzymes in the CNS. Our model may be specifically relevant to the pathophysiology of schizophrenia, bipolar disorder and epilepsy, in which altered GAD67 levels have been observed (Akbarian et al., 1995; Heckers et al., 2002; Lewis et al., 2005). More broadly, the model we present in which hierarchically organized retrograde signals direct the distinct molecular differentiation of a GABAergic terminal, provides insights into the adaptive mechanisms by which neuronal circuits maintain an appropriate balance of excitation and inhibition.
EXPERIMENTAL PROCEDURES
Mouse Strains
The following mouse strains were used in this study: Gad65-N45GFP (Lopez-Bendito et al., 2004), mGluR1 (Conquet et al., 1994), Ptf1aCre (Kawaguchi et al., 2002), TrkBflox/flox (He et al., 2004) and vGluT1 (Slc17a7tm1Edw) (Fremeau et al., 2004). mGluR1 genotyping information is provided in Supplemental Experimental Procedures. Both male and female mice were used. All procedures were carried out in accordance with Memorial Sloan Kettering and Columbia University IACUC guidelines.
Immunohistochemistry
Antibodies are listed in Supplemental Experimental Procedures. Immunohistochemistry was performed either on 12 μm cryostat sections or 75 μm Vibratome sections (depending on the experimental paradigm) of L4–L5 spinal segments using primary and fluorophore-conjugated secondary antibodies (Jackson Labs) as previously described (Betley et al., 2009).
Histochemistry
Probe information is listed in Supplemental Experimental Procedures. In situ hybridization was performed on 12 μm cryostat sections as described previously (Arber et al., 2000; Betley et al., 2009).
Labeling of sensory-motor connections
CTb labeling
Motor neurons and proprioceptive sensory afferent projections of the Gastrocnemius muscle were labeled by intramuscular injection of 2 μl 1% Alexa488-conjugated attenuated Cholera Toxin b-subunit (CTb; Invitrogen; (Shneider et al., 2009)). Injections were carried out on wt and vGluT1 mutant mice at p8 and p12 (n ≥ 3 for each genotype and developmental time point) and spinal cords were harvested at p12 and p21, respectively. CTb staining was enhanced with anti-CTb antibody, 1:1000 (List Biological Laboratories Inc.).
Dextran backfills and proprioceptive synaptic density on motor neurons
Spinal cords dissected from p12 mice were perfused with cold (~16 °C), oxygenated (95% O2, 5% CO2) aCSF (containing 128.35 mM NaCl, 4 mM KCl, 0.58 mM NaH2PO4.H20, 21 mM NaHCO3, 30 mM D-Glucose, 0.1 mM CaCl2.H20, and 2 mM MgSO4.7H20) and were hemisected for better oxygenation. Motor neurons were retrogradely-labeled by placing the L5 ventral root in a suction electrode and allowing 30–40 mM Cascade Blue dextran (10,000 MW, Invitrogen) diluted in 0.01 M phosphate buffer saline (PBS, pH 7.4) to passively diffuse into the somata and dendrites of motor neurons overnight. Cords were immersion-fixed in 4% paraformaldehyde for 12 hours and stored in 0.01 M PBS. Proprioceptive fibers were visualized with an antibody against Pv as follows: 75 μm serial vibratome-cut L5 sections were blocked with 10% normal donkey serum in 0.01 M PBS with 0.1% Triton X-100 (PBS-T; pH 7.4). Sections were incubated overnight in rabbit anti-Pv 1:1000 (Swant), washed in PBS-T and incubated for 3 hours in DyLight 488-goat anti-rabbit secondary antibody, 1:250 (Jackson Labs). All antibody incubations were performed at room temperature. Sections were washed in PBS and mounted onto slides.
Confocal microscopy
PvON synapses were quantified on the entire soma and proximal dendrites (0–50 and 50–100 μm dendritic segments from the soma) on confocal images in the z-axis. A Leica SP5 confocal microscope equipped with 4 single laser lines was used. Analysis was performed with the Leica software (LAS AF) as we described in detail in Mentis et al. (2011) (Mentis et al., 2011).
Synaptic measurements
Density measurements
Images were acquired on a Leica SP5 confocal microscope and analyzed off-line. The number of PvON synapses contacting motor neuron cell bodies was counted using LAS AF software.
Staining intensity measurements
Images of synaptic terminals were acquired on a Leica SP5 confocal microscope using either a 40× objective with 3× digital zoom or a 100× objective with 4× digital zoom at 1024×1024 optical resolution. Images from wt and mutant mice were acquired using identical microscope settings for excitation and fluorescence detection parameters. Further details on density and staining intensity measurements are provided in Supplemental Experimental Procedures.
EM immunohistochemistry and analysis
All mice were anaesthetized with sodium pentobarbital (150 mg/kg i.p.) and sequentially perfused transcardially and sequentially with 2% heparin-saline, 3.75% acrolein and 2% paraformaldehyde (PFA) in phosphate buffer (PB). Spinal cords were extracted by ventral laminectomy and postfixed for 30 minutes in 2% PFA. 40 μm vibratome sections of L4–L5 spinal cords from CTb-injected mice (p12 or p21) or from Gad65-N45GFP mice (p21) were processed for pre-embedding EM single or dual immunolabeling, respectively, and examined ultrastructurally (Pierce et al., 2009). Further information is provided in Supplemental Experimental Procedures.
Electrophysiology
Detailed methods have been previously described (Mentis et al., 2005; Shneider et al., 2009) and are outlined in Supplemental Experimental Procedures.
BNDF ELISA
Mature BDNF (mBDNF) protein levels were quantified using the BDNF Emax ImmunoAssay System kit (catalog # G7611; Promega, Madison, WI, USA) with recombinant mBDNF as a standard (Kabir et al., 2012). Protein was extracted and quantitated following the manufacturer’s protocol. Further technical details are provided in Supplemental Experimental Procedures.
Real time RT-PCR
Total RNA was extracted from frozen DRG tissue using the mirVana miRNA isolation kit (Ambion, Foster City, CA). cDNA was synthesized by reverse transcription of total RNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA). The relative amount of bdnf exon IX transcript present in the sample was measured by quantitative real-time PCR using SYBR Green detection (Applied Biosystems, Foster City, CA) following parameters as in Schierberl et al (2011) (Schierberl et al., 2011). All samples were measured in triplicates and mRNA levels were normalized to GAPDH mRNA levels. Primer information is provided in Supplemental Experimental Procedures.
AAV experiment
Wt or vGluT1 mutant mice were anaesthetized by isoflurane (by inhalation) and injected intracerebroventricularly (ICV) with 2 μl of 2.3×1012 GC/ml AAV2/9-CMV-GFP virus (Vector BioLabs) or 10 μl of 7.5×1013 GC/ml AAV2/9-CMV-GFP-2A-mBDNF virus (Vector BioLabs) at p1, using a modified Hamilton syringe (Robbins et al., 2014). Pups were allowed to recover from anesthesia for 30 min before returned to the cage. Mice were sacrificed at p19 and spinal cord was harvested following transcardial perfusion with 4% paraformaldehyde.
Statistics
Staining intensity measurements
For each experimental condition > 100 synapses from ≥ 3 mice were analyzed. Staining intensity measurements were normalized with respect to dataset from control tissue.
All values reported are mean +/− standard error (s.e.m.). Statistical differences were established by Student’s t-test or ANOVA. If a data set failed normal distribution (assessed through Shapiro-Wilk test) statistical differences were determined by Mann-Whitney Rank Sum test. The level of statistical significance was assigned as p ≤ 0.05 (*), p ≤ 0.01 (**) and p ≤ 0.001 (***). NS signifies not significant. Box diagrams include data points between lower and upper quartile, with the line representing the median value. Lines and whiskers represent data between 9th and 91st percentile, dots show the 5th and 95th percentile.
Supplementary Material
Acknowledgments
We are grateful to Drs. Francisco Alvarez, Nicholas Betley, Thomas Jessell, Anna Kalinovsky and Michael O’Donovan and members of the Mentis and Kaltschmidt labs for helpful discussions and comments on the manuscript. We thank Jeffrey Russ for critical assistance with in situ hybridization, Richard DiCasoli, Kim Kridsada and Sarah Qamar for technical assistance, Thomas Jessell, Susan Morton and Ryuichi Shigemoto for providing antibodies, Miranda Karson and Bradley Alger for mGluR1::LacZ mice, Lily Erdy and Kirsten Hively for help with preparing the manuscript. This work was supported by NIH grants DA08259, HL096571 and HL098351 (T.A.M.); NIH grants T32 DA007274 (K.C.S) and 5R01DA029122 (A.M.R); NIH grant MH082870 and a Brain & Behavior Research Foundation NARSAD award (A.F.); NIH grant 5R01-NS078375-01, an Audrey Lewis Young Investigator award from Families of SMA and a Department of Defense (10235006) grant (G.Z.M.); and by NIH grants 2 T32 HD 60600-6 (J.L.B.) and 5R01-NS083998, Memorial Sloan-Kettering start-up funds, MSK Cancer Center Support Grant/Core Grant (P30 CA008748), a Whitehall Foundation Research Grant and a Louis V. Gerstner, Jr. Young Investigators Award (J.A.K.).
Footnotes
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
M.M., E.V.F., J.L.B., J.P.P., P.K.B., J.A.W., Z.D.K., K.C.S, J.G.P., A.I.M., A.F., T.A.M., G.Z.M., and J.A.K. performed and analyzed experiments. M.M., A.M.R., P.v.R., G.Z.M., and J.A.K. designed the study and interpreted results. A.F. and R.H.E. provided reagents. M.M., P.v.R., G.Z.M. and J.A.K. wrote the paper.
References
- Akay T, Tourtellotte WG, Arber S, Jessell TM. Degradation of mouse locomotor pattern in the absence of proprioceptive sensory feedback. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:16877–16882. doi: 10.1073/pnas.1419045111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Archives of general psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Akins MR, Biederer T. Cell-cell interactions in synaptogenesis. Curr Opin Neurobiol. 2006;16:83–89. doi: 10.1016/j.conb.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Carr PA, Grandes P, Somohano PM. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in the rat spinal cord. The Journal of comparative neurology. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Alvarez FJ, Villalba RM, Zerda R, Schneider SP. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. The Journal of comparative neurology. 2004;472:257–280. doi: 10.1002/cne.20012. [DOI] [PubMed] [Google Scholar]
- Andreae LC, Burrone J. The role of neuronal activity and transmitter release on synapse formation. Curr Opin Neurobiol. 2014;27C:47–52. doi: 10.1016/j.conb.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. doi: 10.1016/s0092-8674(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascoli GA, Alonso-Nanclares L, Anderson SA, Barrionuevo G, Benavides-Piccione R, Burkhalter A, Buzsaki G, Cauli B, Defelipe J, Fairen A, et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nature reviews Neuroscience. 2008;9:557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthele A, Boxall SJ, Urban A, Anneser JM, Zieglgansberger W, Urban L, Tolle TR. Distribution and developmental changes in metabotropic glutamate receptor messenger RNA expression in the rat lumbar spinal cord. Brain Res Dev Brain Res. 1999;112:39–53. doi: 10.1016/s0165-3806(98)00156-4. [DOI] [PubMed] [Google Scholar]
- Betley JN, Wright CV, Kawaguchi Y, Erdelyi F, Szabo G, Jessell TM, Kaltschmidt JA. Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell. 2009;139:161–174. doi: 10.1016/j.cell.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick GS, Reid B, Richardson C, Banks RW. Autogenic modulation of mechanoreceptor excitability by glutamate release from synaptic-like vesicles: evidence from the rat muscle spindle primary sensory ending. The Journal of physiology. 2005;562:381–394. doi: 10.1113/jphysiol.2004.074799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnot A, Chub N, Pujala A, O’Donovan MJ. Excitatory actions of ventral root stimulation during network activity generated by the disinhibited neonatal mouse spinal cord. Journal of neurophysiology. 2009;101:2995–3011. doi: 10.1152/jn.90740.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Audinat E, Lambolez B, Angulo MC, Ropert N, Tsuzuki K, Hestrin S, Rossier J. Molecular and physiological diversity of cortical nonpyramidal cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:3894–3906. doi: 10.1523/JNEUROSCI.17-10-03894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AI, de Nooij JC, Jessell TM. Graded activity of transcription factor Runx3 specifies the laminar termination pattern of sensory axons in the developing spinal cord. Neuron. 2006;49:395–408. doi: 10.1016/j.neuron.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, et al. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Kostyuk PG, Schmidt RF. Central pathways responsible for depolarization of primary afferent fibres. The Journal of physiology. 1962;161:237–257. doi: 10.1113/jphysiol.1962.sp006884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. The Journal of comparative neurology. 1999;412:488–505. [PubMed] [Google Scholar]
- Esclapez M, Tillakaratne NJ, Kaufman DL, Tobin AJ, Houser CR. Comparative localization of two forms of glutamic acid decarboxylase and their mRNAs in rat brain supports the concept of functional differences between the forms. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1994;14:1834–1855. doi: 10.1523/JNEUROSCI.14-03-01834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink AJ, Croce KR, Huang ZJ, Abbott LF, Jessell TM, Azim E. Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature. 2014;509:43–48. doi: 10.1038/nature13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fassler R, Hudson BG, John SW, Ninomiya Y, Pedchenko V, Pfaff SL, et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science (New York, NY) 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Glasgow SM, Henke RM, Macdonald RJ, Wright CV, Johnson JE. Ptf1a determines GABAergic over glutamatergic neuronal cell fate in the spinal cord dorsal horn. Development. 2005;132:5461–5469. doi: 10.1242/dev.02167. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nature neuroscience. 2006;9:807–815. doi: 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB, Greene LA. Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science (New York, NY) 1986;234:80–83. doi: 10.1126/science.3749894. [DOI] [PubMed] [Google Scholar]
- Gronborg M, Pavlos NJ, Brunk I, Chua JJ, Munster-Wandowski A, Riedel D, Ahnert-Hilger G, Urlaub H, Jahn R. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2–12. doi: 10.1523/JNEUROSCI.4074-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Archives of general psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, Mackie M, Nagy GG, Riddell JS, Maxwell DJ, Szabo G, Erdelyi F, Veress G, Szucs P, Antal M, Todd AJ. P boutons in lamina IX of the rodent spinal cord express high levels of glutamic acid decarboxylase-65 and originate from cells in deep medial dorsal horn. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9038–9043. doi: 10.1073/pnas.0503646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N, Iseda T, Kawaguchi S, Ogawa M, Bae SC, et al. Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nature neuroscience. 2002;5:946–954. doi: 10.1038/nn925. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Carlin KP, Brownstone RM. An in vitro functionally mature mouse spinal cord preparation for the study of spinal motor networks. Brain research. 1999;816:493–499. doi: 10.1016/s0006-8993(98)01199-8. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D, Floor E, Hsu CC, Kopke RD, Wu JY. Demonstration of functional coupling between gamma-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4293–4298. doi: 10.1073/pnas.0730698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir ZD, Lourenco F, Byrne ME, Katzman A, Lee F, Rajadhyaksha AM, Kosofsky BE. Brain-derived neurotrophic factor genotype impacts the prenatal cocaine-induced mouse phenotype. Dev Neurosci. 2012;34:184–197. doi: 10.1159/000337712. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nature genetics. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- Kramer I, Sigrist M, de Nooij JC, Taniuchi I, Jessell TM, Arber S. A role for Runx transcription factor signaling in dorsal root ganglion sensory neuron diversification. Neuron. 2006;49:379–393. doi: 10.1016/j.neuron.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Krueger DD, Bear MF. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annual review of medicine. 2011;62:411–429. doi: 10.1146/annurev-med-061109-134644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- Lambolez B, Audinat E, Bochet P, Crepel F, Rossier J. AMPA receptor subunits expressed by single Purkinje cells. Neuron. 1992;9:247–258. doi: 10.1016/0896-6273(92)90164-9. [DOI] [PubMed] [Google Scholar]
- Lau CG, Murthy VN. Activity-dependent regulation of inhibition via GAD67. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:8521–8531. doi: 10.1523/JNEUROSCI.1245-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Tov A, Pinco M. In vitro studies of prolonged synaptic depression in the neonatal rat spinal cord. The Journal of physiology. 1992;447:149–169. doi: 10.1113/jphysiol.1992.sp018996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R, Bernstein Y, Goldenberg D, Xiao C, Fliegauf M, et al. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J. 2002;21:3454–3463. doi: 10.1093/emboj/cdf370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nature reviews Neuroscience. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Li Y, Burke RE. Short-term synaptic depression in the neonatal mouse spinal cord: effects of calcium and temperature. Journal of neurophysiology. 2001;85:2047–2062. doi: 10.1152/jn.2001.85.5.2047. [DOI] [PubMed] [Google Scholar]
- Liang F, Isackson PJ, Jones EG. Stimulus-dependent, reciprocal up- and downregulation of glutamic acid decarboxylase and Ca2+/calmodulin-dependent protein kinase II gene expression in rat cerebral cortex. Experimental brain research. 1996;110:163–174. doi: 10.1007/BF00228548. [DOI] [PubMed] [Google Scholar]
- Lopez-Bendito G, Sturgess K, Erdelyi F, Szabo G, Molnar Z, Paulsen O. Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cerebral cortex (New York, NY : 1991) 2004;14:1122–1133. doi: 10.1093/cercor/bhh072. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O’Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Alvarez FJ, Bonnot A, Richards DS, Gonzalez-Forero D, Zerda R, O’Donovan MJ. Noncholinergic excitatory actions of motoneurons in the neonatal mammalian spinal cord. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7344–7349. doi: 10.1073/pnas.0502788102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O’Donovan MJ. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Monyer H, Markram H. Interneuron Diversity series: Molecular and genetic tools to study GABAergic interneuron diversity and function. Trends Neurosci. 2004;27:90–97. doi: 10.1016/j.tins.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature. 1995;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Nomura S, Ding YQ, Shigemoto R, Wada E, Kinoshita A, Li JL, Neki A, Nakanishi S, Mizuno N. Presynaptic localization of a metabotropic glutamate receptor, mGluR7, in the primary afferent neurons: an immunohistochemical study in the rat. Neuroscience letters. 1995;202:85–88. doi: 10.1016/0304-3940(95)12207-9. [DOI] [PubMed] [Google Scholar]
- Pierce JP, Kievits J, Graustein B, Speth RC, Iadecola C, Milner TA. Sex differences in the subcellular distribution of angiotensin type 1 receptors and NADPH oxidase subunits in the dendrites of C1 neurons in the rat rostral ventrolateral medulla. Neuroscience. 2009;163:329–338. doi: 10.1016/j.neuroscience.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nature reviews Neuroscience. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nature reviews Neuroscience. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Prochazka A. Quantifying proprioception. Progress in brain research. 1999;123:133–142. [PubMed] [Google Scholar]
- Ramirez M, Gutierrez R. Activity-dependent expression of GAD67 in the granule cells of the rat hippocampus. Brain research. 2001;917:139–146. doi: 10.1016/s0006-8993(01)02794-9. [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins KL, Glascock JJ, Osman EY, Miller MR, Lorson CL. Defining the therapeutic window in a severe animal model of spinal muscular atrophy. Human molecular genetics. 2014;23:4559–4568. doi: 10.1093/hmg/ddu169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q, Garraway SM, Hofmann F, Moosmang S, Striessnig J, et al. Cav1.2 L-type Ca(2)(+) channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Ca(v)1.3 channels. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:13562–13575. doi: 10.1523/JNEUROSCI.2315-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM. Modulation of GABAergic signaling among interneurons by metabotropic glutamate receptors. Neuron. 2000;25:663–672. doi: 10.1016/s0896-6273(00)81068-5. [DOI] [PubMed] [Google Scholar]
- Shneider NA, Mentis GZ, Schustak J, O’Donovan MJ. Functionally reduced sensorimotor connections form with normal specificity despite abnormal muscle spindle development: the role of spindle-derived neurotrophin 3. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:4719–4735. doi: 10.1523/JNEUROSCI.5790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ, Hughes DI, Polgar E, Nagy GG, Mackie M, Ottersen OP, Maxwell DJ. The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur J Neurosci. 2003;17:13–27. doi: 10.1046/j.1460-9568.2003.02406.x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nature reviews Neuroscience. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT. Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7168–7171. doi: 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nature reviews Neuroscience. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.