

Protein kinase C alpha plays a major role in mediating Epac-dependent enhancement of purinergic P2X3R activity in dorsal root ganglion neurons after inflammation.
Keywords: PKCα, PKCε, DRG, P2X3R, Epac1, Epac2, inflammatory pain
Abstract
Sensitization of purinergic P2X3 receptors (P2X3Rs) is a major mechanism contributing to injury-induced exaggerated pain responses. We showed in a previous study that cyclic adenosine monophosphate (cAMP)–dependent guanine nucleotide exchange factor 1 (Epac1) in rat sensory dorsal root ganglia (DRGs) is upregulated after inflammatory injury, and it plays a critical role in P2X3R sensitization by activating protein kinase C epsilon (PKCε) inside the cells. protein kinase C epsilon has been established as the major PKC isoform mediating injury-induced hyperalgesic responses. On the other hand, the role of PKCα in receptor sensitization was seldom considered. Here, we studied the participation of PKCα in Epac signaling in P2X3R-mediated hyperalgesia. The expression of both Epac1 and Epac2 and the level of cAMP in DRGs are greatly enhanced after complete Freund adjuvant (CFA)–induced inflammation. The expression of phosphorylated PKCα is also upregulated. Complete Freund adjuvant (CFA)–induced P2X3R-mediated hyperalgesia is not only blocked by Epac antagonists but also by the classical PKC isoform inhibitors, Go6976, and PKCα-siRNA. These CFA effects are mimicked by the application of the Epac agonist, 8-(4-chlorophenylthio)-2 -O-methyl-cAMP (CPT), in control rats, further confirming the involvement of Epacs. Because the application of Go6976 prior to CPT still reduces CPT-induced hyperalgesia, PKCα is downstream of Epacs to mediate the enhancement of P2X3R responses in DRGs. The pattern of translocation of PKCα inside DRG neurons in response to CPT or CFA stimulation is distinct from that of PKCε. Thus, in contrast to prevalent view, PKCα also plays an essential role in producing complex inflammation-induced receptor-mediated hyperalgesia.
1. Introduction
P2X3R, activated by ATP, is a major receptor processing nociceptive information in dorsal root ganglia (DRGs).7,35,36,50 Rats with inflammation or nerve injuries, in response to the application of ATP or of the P2X3R agonist, α,β-meATP, display exaggerated nocifensive behaviors, including rapid paw withdrawals, increasing tail flicks, and paw flinching.11,23 This is a result of a large amount of ATP released from damaged skin cells causing rapid activation of P2XRs and triggering robust firing in DRG neurons.13 The electrical responses to ATP or α,β-meATP in DRG neurons are also dramatically enhanced.22,52 We showed that inflammation enhances total P2X3R expression52 and spared nerve injury upregulates membrane expression of P2X3Rs in DRG neurons.11 Chronic compression of the DRG was found to increase total P2X3R expression.51 In addition, prostaglandin2 (PGE2), a lipid mediator synthesized and released from inflamed tissue, greatly increases α,β-meATP–induced nociceptive behavioral responses in rats.47 We showed that under controlled conditions, PGE2 increases ATP-mediated P2X3R responses through a protein kinase A (PKA)-mediated pathway.48 Following complete Freund adjuvant (CFA)–induced chronic inflammation, PGE2 activates protein kinase C epsilon (PKCε) in addition to PKA to produce much enhanced modulatory influence on P2XR-mediated responses.47 These results are consistent with the findings that a variety of inflammatory mediators, such as carrageenan,4 PGE2,41 protease-activated receptor 2 (PAR2),5 cytokines,17,40 and epinephrine,29 activate PKCε to evoke sustained activation of nociceptors.26,39,43
Epac has been found to play a key role in the activation of cyclic adenosine monophosphate (cAMP)–dependent activation of PKCε in DRGs.27,47 Hucho et al.27 showed that a PKCε-specific inhibitor blocks mechanical hyperalgesia induced by the Epac activator, 8-(4-chlorophenylthio)-2 -O-methyl-cAMP (CPT), similar to that observed in epinephrine-induced PKCε-dependent hyperalgesia. We showed that Epac1 is upregulated after inflammation. Activation of Epac by CPT mimics the potentiating effect of PGE2 on P2X3R currents and occludes the enhancing effect of PGE2 on P2X3R-mediated currents after inflammation. These observations suggest that PGE2 potentiation of P2X3R currents and P2X3R-mediated hyperalgesia under inflammation are mediated by Epac–PKCε signaling.47
In contrast to the extensive studies of the effects of PKCε on producing sensitization to thermal, mechanical, and chemical stimuli in DRGs,26 the role of PKCα in receptor sensitization has been largely ignored, even though PKCα was found to be required in phorbol 12,13-dibutyrate (PDBu)–induced activation37 and in capsaicin-evoked upregulation of transient receptor potential cation channel subfamily V1 receptors (TRPV1Rs) in DRG neurons.53 One reason is the observation that PKCα was expressed minimally in cultured DRGs prepared from neonatal rats.9 This assertion is not consistent with the observations that substantial PKCα expression was found in DRGs prepared from embryonic,37 adult,53 and aged rats.32 Here, we study the role of PKCα in Epac signaling and in nociceptor sensitization and provide strong evidence that Epac activates PKCα to produce enhanced P2X3R-mediated hyperalgesia after inflammatory injury.
2. Experimental procedures
2.1. Animals
Animal experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch and performed according to the guidelines of the National Institutes of Health and the International Association for the Study of Pain. Male Sprague–Dawley rats of 4 to 6 weeks of age were used to prepare cultured DRG neurons. Seven- to 9-week-old (weight, 200-250 g) male rats were used in Western, immunocytochemical, and behavioral experiments. To induce inflammation, animals were anesthetized with isoflurane (5%), and CFA (50 μL) was injected into the plantar surface of the rat left hindpaw. Solution of CFA was prepared by mixing Mycobacterium butyricum (10 mg/mL) (Difco, Detroit, MI) in a peanut oil–saline (1:1) emulsion.52 The injected paw showed signs of localized inflammation, that is, redness, swelling, and hyperalgesia a day later. The inflammatory condition reached a steady state 2 days later and persisted for 2 weeks.21,52 Experiments were performed 3 to 14 days after the CFA treatment, during which the enhancement in nociceptive behavioral responses and the increase in P2X3R-mediated currents remained stable. No systematic temporal variations were observed during this period. Rats that developed polyarthritis or could not resume normal activity were euthanized with CO2 asphyxiation.
2.2. Behavioral experiments
Flinching of the rat left hindpaw in response to an intradermal paw injection of the P2X receptor agonist, α,β-meATP, was used to assess nociception elicited by the activation of purinergic receptors.11,23 The nocifensive behavior was assessed 3 to 10 days after CFA treatment and analyzed according to a previously described method.11,47 In brief, 3 to 5 days after CFA injection, saline, an Epac, or PKC inhibitor was injected into the rat paw. Ten minutes later, α,β-meATP was injected into the same paw, and behavioral responses were monitored. In response to α,β-meATP injection, rats not only lifted the injected paw more frequently but also kept the paw in the air for a longer period. Instead of using flinching frequency (ie, number of paw lifts per minute, a parameter commonly used to assess flinching behaviors), paw withdrawal (PW) duration (ie, the accumulative duration that the hindpaw was lifted in the air in a 1-minute time bin) was used. Because PW duration depends on both paw lift frequency and duration, it gives a more accurate measure of nociception. All behavioral studies were performed under blind conditions.
2.3. Pharamacologic agents
The Epac activator, 8-(4-chlorophenylthio)-2 -O-methyl-cAMP (CPT) was purchased from Calbiochem (La Jolla, CA). The P2XR agonists, ATP, and α,β-meATP, the Epac1 antagonist, CE3F4,14 and the Epac2 antagonist, HJC0350,10 were from Tocris (Minneapolis, MN). The classical PKC isoform antagonist, Go6976,34 was purchased from Abcam (Cambridge, MA). The PKCε antagonist ε-V1-26 was purchased from AnaSpec (Fremont, CA). Mycobacterium butyricum (CFA) was from Fisher scientific (Pittsburgh, PA). To reduce PKCα expression, siRNA-PKCα (SC-108089; Santa Cruz, Dallas, TX) was used according to the described method.12,31 Control siRNA-A (SC-37007; Santa Cruz) was used as a negative control.
2.4. Determination of cAMP levels using enzyme-linked immunosorbent assay
Ipsilateral L4 and L5 DRGs were removed from rats 3 to 5 days after intraplantar injection of CFA. L4 and L5 DRGs from normal rats were used as control. The DRGs were rinsed with an oxygenated external solution (in millimoles, 130 NaCl, 5 KCl, 2 KH2PO4, 2.5 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH7.4, 295-300 mosM) 3 times and then homogenized with Cell Lysis Buffer (R&D Systems, Minneapolis, MN) on ice. Following centrifugation, supernatants were collected and frozen at −70°C until enzyme-linked immunospecific assay (enzyme-linked immunosorbent assay [ELISA]) was performed. The cAMP concentration was measured using a cAMP ELISA kit (ENZO Life Science, Farmingdale, NY). The cAMP values were normalized with the protein content in DRGs, which was determined using a bicinchoninic acid assay (BCA) kit (Pierce, Life Technologies, Grand Island, NY).
2.5. DRG cell culture
DRGs were removed from male Sprague–Dawley rats and dissected in an ice-cold, oxygenated, dissecting solution consisting of (millimoles) 135 NaCl, 5KCl, 2 KH2PO4, 1.5 CaCl2, 6 MgCl2, 10 glucose, and 10 HEPES, pH 7.2 (osmolarity, 300-310 mosmol/L). The ganglia were incubated in a dissecting solution containing 1 mg/mL trypsin (T1005; Sigma, St. Louis, MO) and 1 mg/mL collagenase D (11088858001; Roche, Waltham, MA) at 37°C for 1 hour. DRGs were then taken out of the enzyme solution, washed and dissociated by trituration with fire-polished glass pipettes. Isolated cells were plated on glass coverslips and placed in culture dishes and grown with medium containing Dulbecco's Modified Eagle Medium/F12 (GIBCO, Life Technologies, Grand Island, NY) plus 2.5% fetal bovine serum and antibiotics. Experiments were performed on DRG cells cultured for 24 to 48 hours.
2.6. Immunocytochemistry
For immunocytochemical staining, cultured DRG cells were treated with different chemicals for various periods. Immediately after treatment, DRG cells were fixed with 4% paraformaldehyde at room temperature for 20 minutes, washed with phosphate-buffered saline (PBS), and blocked with PBS containing 5% normal goat serum for 30 minutes. The cells were then incubated with a primary antibody (1:500 dilution) at 4°C overnight. The primary antibodies used included rabbit anti-PKCα (SC-208; Santa Cruz) and mouse anti-PKCε (610085; BD Biosciences, San Jose, CA) After washing cells with PBS to remove the primary antibody next morning, cells were incubated with secondary antibody at room temperature for 1 hour. The secondary antibody used was Alexa Fluor 488 goat anti-rabbit IgG (H+L) (green) (1:500, A11034; Invitrogen, Life Technologies, Grand Island, NY) or goat anti-mouse IgG (H+L) (A11001) (A11001; Invitrogen). After sealing the coverslips onto glass slides with nail polish, cells were visualized with a laser scanning confocal microscope. ImageJ was used for analyses. The cell labels were viewed under a Nikon confocal microscope.
For immunocytochemical staining of DRG neurons in slices, rats were perfused with 4% paraformaldehyde 7 to 10 days after an injection of CFA to the left paws. L4 and L5 DRGs were removed and then sectioned (10 μm) in a cryostat. The sections were incubated with a PBS solution containing 10% normal goat or donkey serum and 0.3% triton X-100 for 60 minutes to block nonspecific binding. To test the specificity of primary antibodies, 2 control experiments were performed. In the first control experiment, primary antibody was omitted during tissue staining. In the second control experiment, tissue was incubated with the primary antibody pretreated with a known binding peptide (preabsorb experiments). A primary antibody was considered specific when no detectable labeling was observed in the control experiments.
The sections were then incubated with the polyclonal rabbit anti-pPKCα (1:1000; Santa Cruz), anti-PKCβ1 (SC209), or goat anti-pPKCε (1:1000; Santa Cruz) antibodies diluted in a 0.3% triton X-100 PBS plus 10% normal goat or donkey serums at 4°C overnight. To probe endogenous P2X3R, guinea pig anti-P2X3R antibody (1:5,000; Millipore,Temeculla, CA) was used at room temperature for 48 hours. After washing tissue sections with PBS, the sections were incubated with the secondary antibodies, including Alexa Fluor 488 goat anti-rabbit IgG (H+L), Alexa Fluor 488 donkey anti-goat IgG (H+L), and Alexa Fluor 594 goat anti guinea pig IgG (H+L) (1:500; Life Technologies) at room temperature for 1 hour. The sections were then washed and mounted with mount medium (Vector Laboratories, Burlingame, CA). Labeled cells from 6 tissue sections obtained from each rat were counted, and the results from 3 rats were analyzed. Image J software was used for digital image data analysis.
2.7. Western blots
Rat DRGs were homogenized in radioimmunoprecipitation assay lysis buffer (Fisher). The homogenate was centrifuged at 10,000g at 4°C for 12 minutes to pellet out cellular debris and nuclei. The concentration of proteins in the supernatant was determined using the BCA assay (Pierce). Protein samples were loaded into each well of a 7% sodium dodecyl sulfate polyacrylamide gel and underwent 2-hour electrophoresis. The proteins were then electrotransferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was subsequently incubated in a Tris-buffered saline (TBS) blocking buffer containing 5% wt/vol fat-free dry milk for 1 to 2 hours and immunoblotted with primary antibodies at 4°C, overnight. Antibodies used were mouse anti-Epac1, anti-Epac2 (1:1000; Cell Signaling, Danvers, MA), mouse anti-PKCε (1:1000; Santa Cruz), rabbit anti-pPKCε (1:1000; Santa Cruz), and rabbit anti-P2X3R (1:2000; Alomone Labs). β-Actin, probed with anti-β-actin (1:10,000; Chemicon, Temeculla, CA), or the neuronal marker, β-tubulin, probed with anti β-tubulin antibody (1:2000; Santa Cruz), was used as loading control. The PVDF membranes were then washed with T-TBS solution (1 X TBS and 0.1% Tween 20) and then incubated with HRP-conjugated secondary antibodies at room temperature for 1 hour. The immunoreactive proteins were detected by enhanced chemiluminescence (ECL kit; Amersham Biosciences, Pittsburgh, PA) and visualized by exposing the PVDF membrane using Odyssey FC imaging system from Li-Cor. The intensities of protein bands were determined, quantified, and normalized with β-actin or β-tubulin bands.
2.8. Statistical analysis
All data are expressed as mean ± SEM. Differences between 2 means were analyzed with Student paired or unpaired t test. Comparisons between multiple means were done with 1-way analysis of variance followed by Newman–Keuls post hoc test. A P < 0.05 was considered significant.
3. Results
3.1. The expression of Epac1 and Epac2 and the cytosolic cAMP level in DRGs increase after CFA-induced inflammation
Epacs, consisting of Epac1 and Epac2, are found in most tissues.20 The major factors affecting the activation of Epacs are the expression of Epacs and the level of cAMP. We compared the expression level of Epac1 and of Epac2 under normal and inflammatory conditions using Western blot analyses. To reduce the variability among rats, identical DRG protein samples obtained from the same rats were used to probe the expression of both Epacs. In the control rat group, the left paw was injected with saline. In the inflammatory rat group, the left paw was injected with CFA (50 μg/50 μL). Five to 14 days after the injection, L4-5 DRGs were excised. Epac1 and Epac2 expressions were probed with anti-Epac1 and anti-Epac2 antibodies. Both Epac1 and Epac2 were expressed in adult DRGs. The expression of Epac1 in CFA-treated rats was 3.5 times higher than that in control rats (Figs. 1A, B). In the same CFA injected group, the Epac2 expression was enhanced by 3.0 fold.
Figure 1.
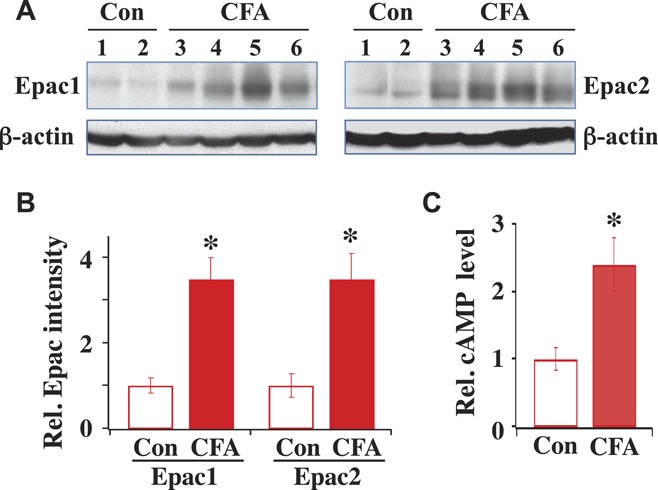
Epac1 and Epac2 expression and cyclic adenosine monophosphate (cAMP) level increase after complete Freund adjuvant (CFA)–induced inflammation. (A) Expression of Epac1 and Epac2 in control (Con) and CFA-treated rats. Western blots of Epac1 and Epac2 expressed in L4 and L5 dorsal root ganglia (DRGs) isolated from control rat 1 and 2 or from inflamed rat 3 to 6. Same rat protein samples were used for both Epac1 and Epac2 measurements. (B) Summary of Epac1 and Epac2 expression. The intensity of each protein band was determined and normalized with its respective β-actin, which was used as a loading control. Relative protein intensity after CFA is expressed as fold-increase by dividing the normalized protein intensity obtained after CFA with respect to that obtained under pre-CFA control condition. Compared to the control, Epac1 expression after CFA was (CFA/Con = 3.5 ± 0.5; n = 4) and Epac2 expression after CFA was (CFA/Con = 3.0 ± 0.6; n = 4). *P < 0.05 compared with Con. (C) The cAMP level in control DRGs was 7.6 ± 1.2 pmole/mg protein (n = 4). After CFA-induced inflammation, the relative level of cytosolic cAMP in DRGs with respect to the average control cAMP level was 2.4 ± 0.4 (n = 4) fold larger (*P < 0.05, compared with Con).
We also determined the change in the cAMP level after inflammation (Fig. 1C). The ELISA test was used to examine the cytosolic cAMP level in DRGs of normal and CFA-treated rats. The cytosolic cAMP level in DRGs isolated from CFA-treated rats was 2.4 fold higher than that in control DRGs. Thus, the cytosolic cAMP level in DRGs is increased substantially after inflammation. The high level of cAMP and increased Epac expression gives rise to an enhanced level of Epac activation after inflammatory injury.
3.2. The enhancement of α,β-meATP–induced flinch responses after inflammation depends on the activation of Epac
We then determined the involvement of Epac subtypes in α,β-meATP–evoked flinch responses in saline control and CFA-treated rats. Following an injection of 50 μL of saline containing 50 nmol α,β-meATP into the paw, rats displayed repeated rapid withdrawals (flinches), a result of activation of purinergic receptors, including P2X3Rs.47 In both rat groups, the flinch responses peaked approximately 2 minutes after α,β-meATP application. The peak flinch duration was approximately 3 seconds for the control and approximately 9 seconds for CFA-treated rats (Fig. 2A). The peak flinch responses are also shown in bar graphs (Figs. 2B, C). The effects of specific Epac antagonists on α,β-meATP–evoked flinching in control and CFA-treated rats were also determined. We applied the Epac1 antagonist, CE3F4,14 or the Epac2 antagonist, HJC0350,10 to the rat paw 10 minutes prior to the α,β-meATP application. Both the Epac1 and Epac2 antagonist inhibited α,β-meATP–evoked flinch responses substantially (Figs. 2A and C).
Figure 2.
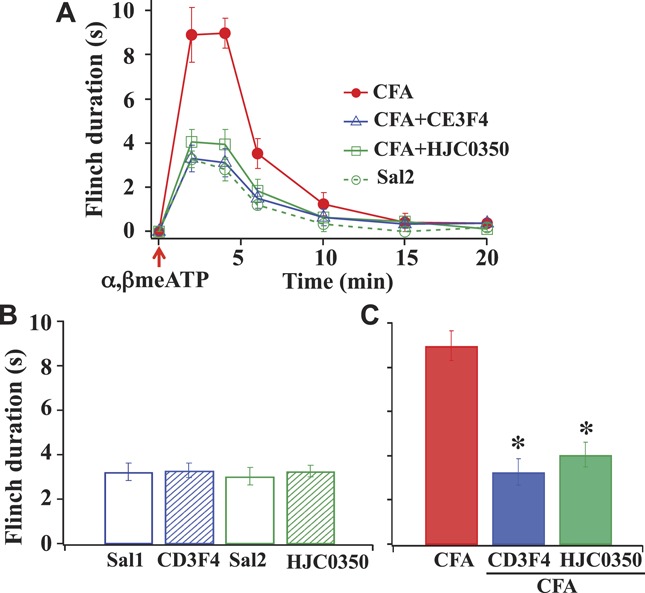
Epac1 and Epac2 affect α,β-meATP- elicited flinch nocifensive responses after inflammation. (A) Time course of nociceptive responses in saline-injected and complete Freund adjuvant–injected rats. In both rat groups, the flinch responses peaked 2 minutes after an injection of α,β-meATP. Paw-injection of the Epac 1 antagonist, CE3F4 (10 nmol in 50-μL saline) or the Epac2 antagonist, HJC0350 (3 nmol in 50-μL saline) 10 minutes before α,β-meATP application inhibited the CFA-induced enhancement. (B) Actions of Epac antagonists on saline-treated control rats (Sal1: 50-μL saline and Sal2: 50-μL saline + 0.2% DMSO). CE3F4 and HJC0350 had no effect on α,β-meATP–elicited flinch responses in control rats. (C) A bar graph summarizes the actions of Epac antagonists on peak flinch durations in CFA-treated rats (n = 6; *P < 0.05 compared with peak flinch duration with CFA treatment).
As a negative control, the same experiments were repeated in control rats (Fig. 2B). We compared α,β-meATP–evoked responses by preinjecting CE3F4 or HJC0350-containing saline into the paw of control rats. In addition, we also injected DMSO-containing saline into the rat paw to test if DMSO, which was used to dissolve Epac antagonists, altered the α,β-meATP–evoked responses. All gave similar flinch responses (∼3 seconds). Thus, DMSO and the Epac antagonists (ie, CE3F4 and HJC0350) at the applied dose did not elicit any nonspecific effect. The results suggest that the level of Epac activation in control rats was minimum. The large enhanced α,β-meATP–evoked flinch responses in CFA-treated rats and the substantial block of the flinch responses by CE3F4 and HJC0350 suggest that Epac1 and Epac2 play an essential role in producing P2X3R-mediated hyperalgesia after inflammatory injury.
3.3. PKCα is involved in CFA-induced enhancement of flinch responses
We next determined if PKCα, in addition to PKCε, plays a role in the increase in α,β-meATP–induced flinch responses after inflammation. The experiments described in Figure 2 were repeated, except that the classical, that is, Ca2+-dependent and diacylglycerol-dependent, PKC isoform inhibitor, Go6976, was applied prior to the α,β-meATP application. To minimize the possibility that the inhibitor affects the activity of PKCε, a low concentration (0.23 μM) of Go6976 was used. It has been shown that Go6976, even at 3 μM, does not alter the activity of novel, ie, Ca2+-independent and diacylglycerol-dependent, PKC isoform δ, ε, or ζ.34 Behaviorally, Go6976 did not affect α,β-meATP–induced flinch activity in control rats but significantly diminished the flinch activity in inflamed rats (Fig 3A), suggesting the involvement of the classical PKC isoforms in the enhancement of P2X3R-mediated responses after CFA-induced inflammation. In addition to blocking PKCα, Go6976 also inhibits another classical PKC isoform, ie, PKCβ1.34 However, PKCβ1 is not likely to participate in the modulation of the α,β-meATP–induced flinch responses because PKCβ1 is expressed only in large diameter (>25 μm) DRG neurons.44 We determined PKCβ1 distribution in DRG neurons and found that indeed it was expressed in large neurons, whereas P2X3Rs were expressed exclusively in small and medium diameter (≤25 μm) neurons (Fig. 3B). These observations suggest that PKCα, which is sensitive to the Go6576 block, rather than PKCβ1 is involved in the enhanced P2X3R-mediated flinch responses after CFA.
Figure 3.
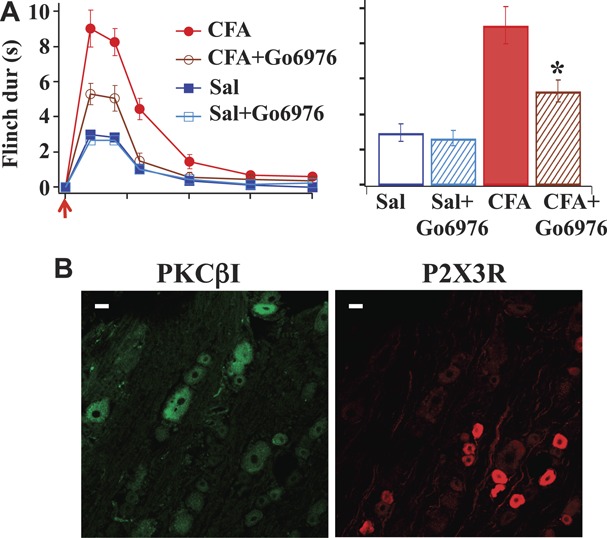
Go6976 reduces α,β-meATP–elicited flinch nocifensive responses in complete Freund adjuvant–treated rats, and PKCβ1 is not expressed in P2X3R-containing dorsal root ganglia (DRG) neurons. (A) In saline control rats, Go6976 (11.5 pmol = 4.35 ng in 50-μL saline) had no effect on α,β-meATP–elicited flinch activity. In complete Freund adjuvant–treated rats, Go6976 inhibited the enhanced α,β-meATP–induced flinch responses (n = 6; *P < 0.05 compared with peak flinch duration in CFA rats). (B) PKCβ1 labels were found only in large DRG neurons (>25 μm), whereas P2X3R labels were found in small or medium (≤25 μm) DRG neurons. Scale bar = 25 μm.
To further confirm the involvement of PKCα in the purinergic receptor–mediated flinch responses, we studied the effect of siRNA-PKCα on α,β-meATP–induced flinch responses and found that the flinch responses were significantly reduced in siRNA-PKCα–treated rats (Fig. 4A). This observation further supports the conclusion that PKCα is involved in CFA-induced enhancement of α,β-meATP–induced flinch responses. For comparison purposes, the effect of the PKCε inhibitor, ε-V1-2, on α,β-meATP–induced flinch duration was also examined. We found that ε-V1-2 significantly inhibited CFA-induced enhancement of flinch responses (Fig. 4B).
Figure 4.
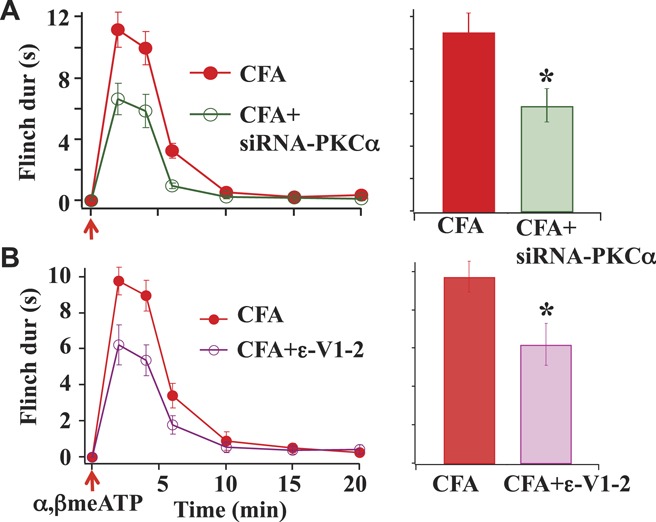
Effects of siRNA-PKCα and ε-V1-2. (A) Reducing PKCα levels with siRNA-PKCα (20 μM) significantly decreased the α,β-meATP–induced flinch responses in complete Freund adjuvant (CFA)–treated rats. (B) The PKCε inhibitor, ε-V1-2 (0.89 nmol = 0.75 μg in10 μL saline), potently reduced CFA-induced flinch responses (n = 3) (*P < 0.05 compared with CFA-treated rats).
3.4. PKCα is well expressed in DRGs of neonatal rats
Cesare et al.9 concluded that PKCα expression is undetectable in neonatal DRGs. The result is in contrast to the later findings that PKCα is well expressed in adult DRGs.32,37,53 We therefore determined if PKCα expression in DRGs is age dependent. The total PKCα expression in DRGs isolated from 5-day-old neonatal rats was measured and compared with those obtained from adult rats (Fig. 5A). We found that PKCα is well expressed in neonatal DRGs, similar to that in adult DRGs—a result disagreeing with the assertion of Cesare et al.9
Figure 5.
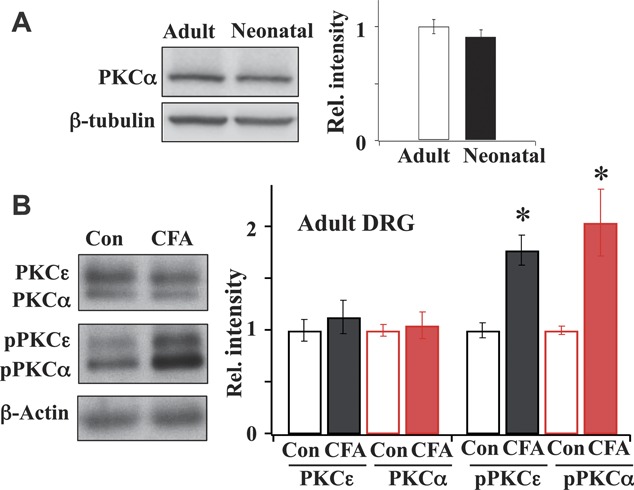
PKCα expression in neonatal and adult dorsal root ganglia (DRGs). (A) The level of expression of PKCα in neonatal DRGs was comparable with that in adult DRGs (adult = 1.00 ± 0.07; n = 5; neonatal = 0.91 ± 0.06; n = 4). (B) Phosphorylated PKCα and phosphorylated PKCε expression increased in adult DRGs after inflammation. Expressions of total and phosphorylated PKCα and PKCε in DRGs were measured in saline-treated and complete Freund adjuvant–treated adult rats. Inflammation did not alter total PKCα and PKCε expression but significantly enhanced the expression of phosphorylated PKCα and phosphorylated PKCε (n = 4; *P < 0.05, Student t test).
3.5. Inflammation enhances the expression of phosphorylated PKCα and PKCε in adult DRGs
We further determined how inflammation affected the total expression and phosphorylation of PKCα and PKCε in adult DRGs (Fig. 5B). Treatment with CFA did not alter the levels of total PKCα and PKCε. In contrast, the phosphorylated PKCα (pPKCα) and phosphorylated PKCε (pPKCε) expression levels in CFA-treated rats were significantly upregulated.
3.6. CPT-elicited enhancement of α,β-meATP–induced flinching responses in control rats is Epac and PKCα mediated
To further confirm the involvement of Epac under inflammatory injury conditions, we determined if the functional consequence of Epac activation in response to CFA-induced inflammation could be mimicked by studying the effect of the Epac agonist, CPT, on P2X3R-mediated hyperalgesia in control rats (Fig. 6A). Ten minutes after paw application of CPT, α,β-meATP injection produced approximately 2-fold increase in flinch duration. To determine if the increase is Epac mediated, the Epac antagonist (ie, either CE3F4 or HJC0350) was applied to the paw 10 minutes prior to the CPT application. The CPT-induced increase was inhibited by the Epac antagonists. Thus, treatment with CPT in control rats gives rise to an Epac antagonist–sensitive enhanced hyperalgesia, which mimics the effect of α,β-meATP–induced nocifensive behavior in CFA-treated rats.
Figure 6.
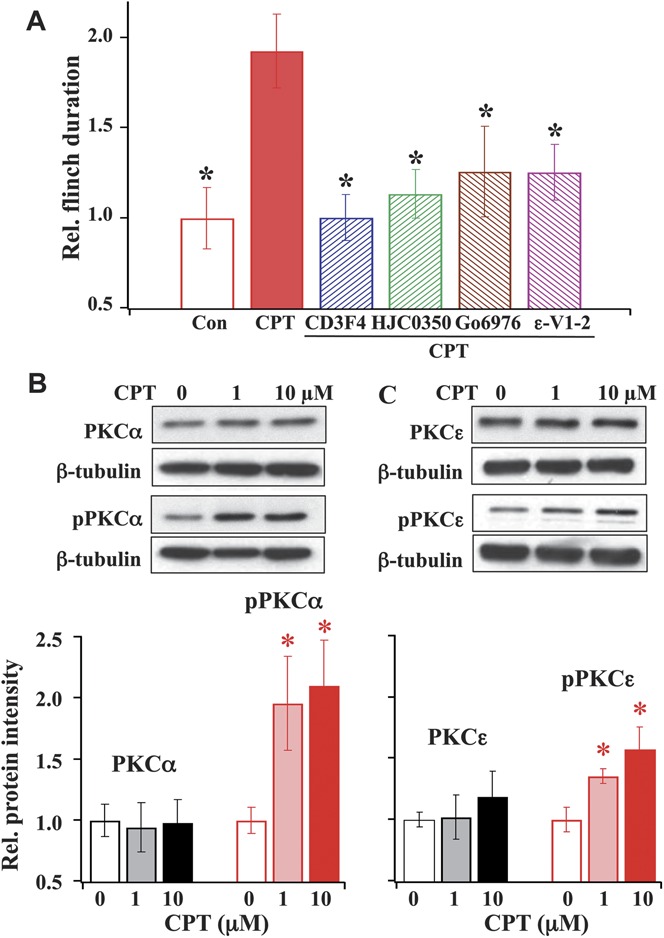
Effects of CPT on α,β-meATP–induced behavioral responses and PKCα or PKCε expression in dorsal root ganglia. (A) CPT-induced enhancement in α,β-meATP–induced flinching responses is inhibited by Epac, PKCα, and PKCε antagonists. Paw injection of the Epac-agonist, CPT (20.25 nmole = 10 μg in 50-μL saline) enhanced α,β-meATP–induced flinch duration by 2-fold. The Epac1 antagonist, CE3F4, the Epac2 antagonist, HJC0350, and the PKC inhibitors, Go6976 and ε-V1-2, significantly blocked the enhanced flinch responses induced by CPT (n = 6; *P < 0.05 compared with CPT). (B and C) Epac activation increases the expression of phosphorylated PKCα (pPKCα) and phosphorylated PKCε (pPKCε). Western blots showed that treating cultured dorsal root ganglia cells with 1 or 10 μM CPT did not change the expression of total PKCα (B) or PKCε (C) but increased pPKCα and pPKCε expression. The pPKCα expression was increased by 1.96 ± 0.38 fold with 1 μM CPT and by 2.10 ± 0.37 fold at 10 μM. The pPKCε expression was increased by 1.36 ± 0.06 with 1 μM CPT and by 1.58 ± 0.18 with 10 μM CPT (n = 3) (*P < 0.05 compared with the expression of PKCα, pPKCα, PKCε, or pPKCε prior to CPT treatment).
We then determined if the CPT-induced enhancement of α,β-meATP responses is PKCα mediated (Fig. 6A). Instead of using an Epac antagonist, Go6976 was applied 10 minutes prior to the application of CPT. Ten minutes later, the α,β-meATP–induced hyperalgesic responses were assessed. The CPT-induced increase was also inhibited by the preapplication of Go6976. This result suggests that PKCα is downstream of Epac and is essential for mediating Epac signaling of enhanced α,β-meATP–induced hyperalgesia. For comparison, we also measured the effect of the PKCε inhibitor, ε-V1-2, on the CPT-induced increase in flinch responses and found that ε-V1-2 similarly blocked the CPT effect.
3.7. CPT-induced Epac activation promotes the expression of pPKCα
To determine the mechanism underlying Epac modulation of P2X3R-mediated hyperalgesia, we studied the effect of CPT on the expression of PKCα and pPKCα in cultured DRG neurons. Following 30-minute incubation of 1 or 10 μM CPT at 37°C, DRG proteins were isolated, and Western blot analyses were performed. We found that CPT treatment did not change the total PKCα expression but increased pPKCα expression in DRG neurons (Fig. 6B) similar to those observed in CFA-induced rats (Fig. 5B). For comparison, the expression of PKCε and pPKCε following CPT stimulation was also measured. Similarly, CPT was found to increase the pPKCε expression without altering the expression of total PKCε (Fig. 6C).
3.8. CPT and CFA induce the translocation of PKCα and PKCε in DRG neurons
To further confirm that the activation of Epac affects the activity of PKCα, in addition to that of PKCε, we studied the CPT-induced changes in the translocation of both PKCα and PKCε in cultured DRG neurons. In the absence of CPT, there was seldom translocation of either PKC subtypes (Figs. 7A, B). Following treatment of CPT (100 μM), translocations of both PKCα and PKCε in DRG neurons were observed.
Figure 7.
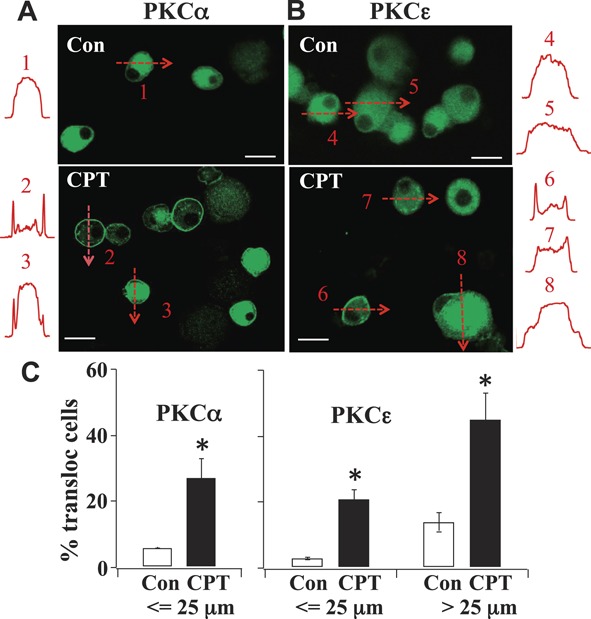
PKC isoform translocation in CPT-treated cultured dorsal root ganglia neurons. (A and B) Neurons were exposed to CPT (100 μM) for 5 minutes. The distribution of PKCα or PKCε was monitored by fluorescent intensity along the drawn lines. PKCα was expressed only in small or medium cells. PKCε was expressed in both small or medium cells and large cells. Bar scale = 25 μm. (C) Percentage of neurons with translocated PKCα or PKCε. Under controlled conditions, 5.9 ± 0.1% of 221 small or medium neurons contained translocated PKCα. After CPT, 27.0 ± 5.9% of 424 small or medium neurons contained translocated PKCα. None of the large dorsal root ganglia neurons were labeled with PKCα. In contrast, 3.0 ± 0.3% of 299 small or medium control neurons contained translocated PKCε. After CPT, 21.0 ± 3.0% of 424 small or medium neurons contained translocated PKCε. Under controlled conditions, 14.0 ± 3.0% of 41 large neurons contained translocated PKCε. Following CPT treatment, 45.0 ± 8% of 126 large neurons contained translocated PKCε (*P < 0.05).
The translocation of PKCα and PKCε were both time and cell dependent. In order to compare the data obtained from cultured neurons with DRG slices obtained from CFA-treated rats, we chose to analyze the distribution of PKCα and PKCε in cultured neurons after the application of CPT for 5 minutes, during which the translocation had largely reached a steady state. The characteristics and the extent of translocation of PKCα and PKCε were different. DRG neurons were divided into small or medium (≤25 μm in diameter) and large (>25 μm) size groups according to the nerve fiber conduction velocity.30 In the control and CPT-treated cultured DRG neurons, PKCα was expressed only in small or medium cells (Fig. 7A). In the control condition, PKCα was expressed uniformly (eg, cell 1). After 5 minutes of CPT treatment, PKCα was translocated in 27% of small or medium–cultured DRG neurons (Figs. 7A and C). In some neurons, PKCα was translocated to the cell membrane (eg, cell 2). In other neurons (eg, cell 3), PKCα was found both in the cell membrane and cytoplasm.
On the other hand, PKCε labels exhibited different changes in response to CPT treatment. Generally, PKCε was found in both small or medium and large cell populations. (Fig. 7B) In control neurons, PKCε were expressed uniformly (eg, cells 4 and 5). After CPT, translocated PKCε labels appeared in 21% of small or medium cells. In some neurons (eg, cell 6), PKCε was translocated to the cell membrane. Other neurons (eg, cell 7) contained punctate PKCε labels in the cytoplasm. Following CPT treatment, translocated PKCε was found in 45% of large neurons. Most of them (eg, cell 8) contained patchy PKCε labels in the cytoplasm (Figs. 7B and C).
We also inspected the distribution of pPKCα, pPKCε, and P2X3Rs in DRG slices prepared from CFA-treated rats (Fig. 8). The purposes were to determine the distribution of pPKCα and pPKCε in DRG neurons in vivo and the colocalization of P2X3Rs with the PKC subtypes. Seven days after CFA injection into the left rat paw, DRGs slices were prepared. As observed in CPT-treated cultured cells, pPKCα was found exclusively in small or medium cells in slices. Labels of pPKCα were found either in the cell membrane alone or in both the cell membrane and cytoplasm. P2X3Rs were found in the cytoplasm of 27.0 ± 3.0% of small or medium DRG neurons (Fig. 8). All of P2X3R-labeled small or medium neurons contained pPKCα labels. Phosphorylated PKCα was also seen in some small neurons that did not express P2X3Rs (Fig. 8).
Figure 8.
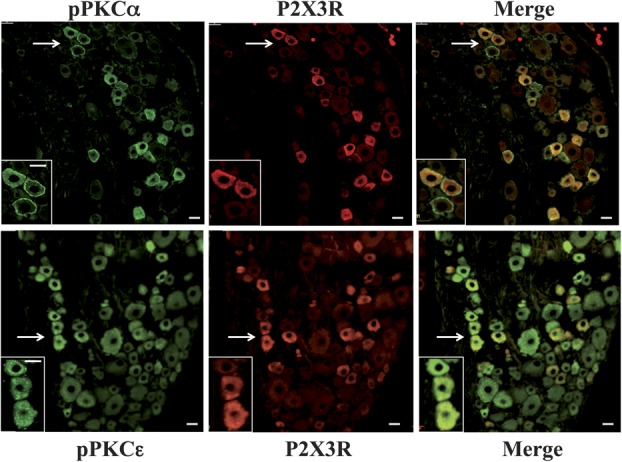
Phosphorylated PKC (pPKC) isoform and P2X3R expression in dorsal root ganglia slices prepared from complete Freund adjuvant–treated rats. Upper: pPKCα and P2X3Rs were expressed only in small or medium cells. pPKCα labels were found both at the cell membrane and in the cytoplasm. P2X3Rs were colocalized with pPKCα in small or medium cells. Enlarged views of cells (indicated by arrows) are shown in the lower left corners. Lower: pPKCε was expressed in both small or medium and large cells, most contained punctate labels. pPKCε labels were colocalized with P2X3Rs in small or medium cells (bar scale = 25 μm).
Similar to the observations in cultured DRGs, pPKCε was found in both small or medium and large DRG neurons in slices. Rarely, PKCε was (8 of 732 cells) found in both the cell membrane and cytoplasm. Most pPKCε labels appeared as punctate structures in the cytoplasm; pPKCε was expressed in 75 ± 5.0% of 354 P2X3R-labeled small or medium neurons (Fig. 8).
4. Discussion
One major finding of this study is that CFA-induced inflammation increases the expression of both Epac1 and Epac2 in DRGs by 2 to 3 fold (Fig. 1). The increased Epac expression contributes to purinergic receptor–mediated hyperalgesia (Fig. 2). Our result differs from a recent study by Vasko et al.46 who found that only Epac2, not Epac1, is upregulated in CFA-treated rat DRGs. To make sure that our results were not caused by experimental variations, 2 precautions were taken in our study. First, we used the same DRG sample to probe both Epac1 and Epac2 expression to diminish sampling variation. Second, we used the antibody purchased from Cell Signaling Technology, the same source as used by Vasko's group, to avoid varying cross-reactivity of antibodies. We found that Epac1 is upregulated after CFA-induced inflammation, as described in our previous study.47 The reason for the discrepancy in the results of Epac1 upregulation between our study and that by Vasko et al. is unclear. In CFA-treated mice, the expression of Epac1 was also found to be increased,49 similar to our findings. Studying the functional consequence of Epac1 and Epac2 upregulation, we found that the enhancement in purinergic receptor–mediated hyperalgesia following CFA-induced inflammation is inhibited by the Epac1 and Epac2 antagonist, CE3F4 and HJC0350, respectively (Fig. 2). These results further support our conclusion that hyperalgesia is mediated by both Epacs.
Epac1 and Epac2 can have varying functions in 1 cell type. For example, both Epacs were found in cardiac myocytes. Epac1 is expressed at the nuclear envelope to regulate gene transcription, whereas Epac2 is concentrated in T-tubules in myocytes to affect cardiac contraction.42 In DRGs, Epac1-mediated and Epac2-mediated nocifensive hyperalgesia induced by α,β-meATP show similar characteristics (Fig. 2). Before we can conclude that the 2 Epacs have the same functions in DRGs, additional experiments need to be performed to differentiate Epac1-mediated and Epac2-mediated α,β-meATP responses.
The second major finding of this study is that PKCα, in addition to PKCε (Fig. 6),27,47 is involved in the Epac signaling. To determine if Epac-activated PKC subtypes are involved in P2X3R-flinched hyperalgesia, the effects of PKC antagonists in CFA-treated rats were studied. A potent classical PKC isoform inhibitor, Go6976, was used to determine the contribution of PKCα to P2X3R-medilated hyperalgesia. Even though Go6976 also inhibits PKCβ1,34 the contribution of PKCβ1 to P2X3R-mediated responses in DRG neurons should be minimal because PKCβ1 is not expressed in P2X3R-containing small DRG neurons (Fig. 3B). We further confirmed that the effect of Go6976 is PKCα-mediated by showing that Go6976 reduces CFA-induced hyperalgesia (Fig. 3A) similar to that of siRNA-PKCα (Fig. 4A). These observations suggest that PKCα is involved in the Epac signaling.
In another set of experiments, we treated control rats with the Epac activator, CPT, to mimic the Epac activation after inflammation. This approach allowed us to determine the actions of PKCα and PKCε on solely Epac-induced hyperalgesia. Similar to their effects on CFA-induced hyperalgesia (Fig. 2), the Epac antagonists, CE3F4 and HJC0350, also block CPT-induced hyperalgesia (Fig. 6A), further confirming the importance of Epacs in producing inflammatory hyperalgesia. Go6976 applied prior to CPT application can still inhibit CPT-mediated hyperalgesic responses (Fig. 6A). This result strongly suggests that PKCα is downstream of Epac activation to participate in the Epac signaling of purinergic receptor–mediated hyperalgesia. Because injection of CPT directly into the paw of control rats to activate Epacs in subcutaneous tissue evokes enhanced α,β-meATP responses similar to those in CFA-treated rats (Figs. 2-4), the observations confirm that Epac–PKC signaling occurs at primary afferent terminals. In our previous studies, we found that CFA treatment enhanced P2X3R-mediated ATP currents (IATP) in dissociated DRG neurons.47 This enhancement is dependent on the activation of Epac and PKC, suggesting that Epac-PKC signaling also plays an important role in the modulation of P2X3R activities in the somata of the DRG neurons. Taken together, the studies suggest that inflammation-induced Epac–PKC signaling can lead to both P2X3R-mediated hyperalgesic responses at afferent terminals and enhanced purinergic currents in DRG neurons.
Studying the expression level of the downstream effectors of Epac (ie, PKCα and PKCε), we showed that both PKC subtypes are expressed robustly in DRGs prepared from adult rats (Fig. 5B). We also measured the PKCα expression in DRGs isolated from neonatal rats and found that PKCα is also well expressed (Fig. 5A). These observations support the conclusion that PKCα is expressed in DRG neurons of rats of various age.32,37,53 Thus, the minimal expression of PKCα in neonatal DRGs described by Cesare et al.,9 is not likely a result of age-dependent PKCα expression in DRGs.
The involvement of PKCε in pain responses has been extensively explored. As stated before, inflammatory mediators upregulate PKCε to give rise to thermal hyperalgesia.26,39,43 In addition, other substances, such as the signaling molecule, lysophosphatidic acid,38 alcohol,15 and the chemotherapeutic drug, paclitaxel,16,24 are also found to activate PKCε and produce mechanical allodynia. In contrast, the participation of PKCα in producing and maintaining chronic pain in response to inflammation and nerve injury has not been well defined. Deletion of PKCα in mutant mice does not change CFA-induced heat hyperalgesia, but surprisingly, it enhances mechanical allodynia following spared nerve ligation.54 Paclitaxel-induced peripheral neuropathy was found to be mediated by PKCβ, δ, and ε, but it is independent of PKCα.24 There is evidence that PDBu activates vanilloid-activated TRPV1 receptors in 7- to 15-day cultured DRGs prepared from embryonic rats.37 Behavioral consequences of the PDBu-activated PKCα have not been determined. Our studies clearly show that CFA activates both PKCα and PKCε in DRG neurons and exaggerates α,β-meATP–induced flinch responses (Figs. 3A and 4).
PKCε activation has been shown to affect the activity of a number of ion and receptor channels, including TTX-resistant Na+ channels,8,29 P2X3Rs,47 TRPV1,33 TRPV4,3,18 TRPC1 and TRPC6,2 and Kv7.2 channels.25 Studies of the effectors of PKCα activation in the nociceptive system are limited. Olah et al.37 found that PKCα, not PKCε, is necessary for PDBu-induced activation of TRPV1 receptors in cultured DRGs. TRPV4 can be activated by acetylcholine-induced PKCα in endothelial cells.1 It has yet to be established that PKCα is involved in TRPV4 activation in DRGs. In this study, we found that PKCα alters P2X3R activity to increase flinch responses (Figs. 3 and 4). Because of our interests in Epac signaling of P2X3R-mediated responses, this study is limited to the examination of nocifensive responses in response to α,β-meATP stimulation. It is of interest to determine if the participation of PKCα activation alters the activity of other receptors to produce thermal and mechanical hyperalgesia.
Another interesting observation is that Epac activation resulting from inflammation gives rise to upregulation of multiple PKC subtypes with distinct patterns of translocation (Figs. 7 and 8). Studying PKC subtype translocation in CPT-treated cultured DRG neurons and DRG slices isolated from CFA-treated rats, we found that CPT- and CFA-induced Epac activation stimulates both PKCα and PKCε (Figs. 7 and 8). PKCα translocation occurs almost exclusively in small or medium DRG neurons. On the other hand, PKCε translocates in both small or medium neurons and large neurons. In CPT-treated cultured neurons, PKCα labels were mostly found in the cell membrane and cytoplasm. The intensity of PKCε labels increases in the cytoplasmic structure after CPT treatment, most with patchy or punctate appearance. In DRG slices obtained from CFA-treated rats, pPKCα also appears exclusively in small cells, most of them expressing P2X3Rs. However, pPKCε was found to be expressed in small or medium neurons expressing P2X3Rs and in large cells without P2X3R expression. These differential translocation patterns suggest that the PKCα and PKCε are located at different subcellular sites to exert varying influences on target proteins.19,45 Studying the translocation of PKCα and another novel PKC (ie, PKCδ) in response to P2Y11 receptor activation in HEK cells, Hui et al.28 showed that PKCδ translocation depends on Epac-mediated production of diacylglycerol at the membrane of endoplasmic reticulum. We do not know if PKCε uses the same pathway as PKCδ for its activation in DRGs. Our observed Epac-dependent PKCα translocation in DRG neurons (Figs. 7 and 8) demonstrates the importance of PKCα in addition to PKCε in modulating P2X3R activity in response to CFA treatment. These results suggest that CFA elicits Epac-induced activation of both PKCα and PKCε, thus producing complex modulation of DRG neuronal activity after tissue injury.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Acknowledgements
The authors thank Dr Frank Lezoualc'h from Inserm for providing the Epac1 antagonist, CE3F4. The work was supported by grants from NINDS NS030045, NIDCR DE017813, National Institutes of Health.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Adapala RK, Talasila PK, Bratz IN, Zhang DX, Suzuki M, Meszaros JG, Thodeti CK. PKCalpha mediates acetylcholine-induced activation of TRPV4-dependent calcium influx in endothelial cells. Am J Physiol Heart Circ Physiol 2011;301:H757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Alessandri-Haber N, Dina OA, Chen X, Levine JD. TRPC1 and TRPC6 channels cooperate with TRPV4 to mediate mechanical hyperalgesia and nociceptor sensitization. J Neurosci 2009;29:6217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alessandri-Haber N, Dina OA, Joseph EK, Reichling D, Levine JD. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci 2006;26:3864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci 2000;20:4680–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, Karanjia R, Barajas-Lopez C, Vanner S, Vergnolle N, Bunnett NW. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 2006;575(pt 2):555–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brandman R, Disatnik MH, Churchill E, Mochly-Rosen D. Peptides derived from the C2 domain of protein kinase C epsilon (epsilon PKC) modulate epsilon PKC activity and identify potential protein-protein interaction surfaces. J Biol Chem 2007;282:4113–23. [DOI] [PubMed] [Google Scholar]
- [7].Burnstock G. P2X receptors in sensory neurones. Br J Anaesth 2000;84:476–88. [DOI] [PubMed] [Google Scholar]
- [8].Cang CL, Zhang H, Zhang YQ, Zhao ZQ. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol Pain 2009;5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron 1999;23:617–24. [DOI] [PubMed] [Google Scholar]
- [10].Chen H, Tsalkova T, Chepurny OG, Mei FC, Holz GG, Cheng X, Zhou J. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J Med Chem 2013;56:952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen Y, Li GW, Wang C, Gu Y, Huang LY. Mechanisms underlying enhanced P2X receptor-mediated responses in the neuropathic pain state. PAIN 2005;119:38–48. [DOI] [PubMed] [Google Scholar]
- [12].Chen Y, Zhang X, Wang C, Li G, Gu Y, Huang LY. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proc Natl Acad Sci U S A 2008;105:16773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cook SP, McCleskey EW. Cell damage excites nociceptors through release of cytosolic ATP. PAIN 2002;95:41–7. [DOI] [PubMed] [Google Scholar]
- [14].Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, Blondeau JP, Lezoualc'h F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J Biol Chem 2012;287:44192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dina OA, Barletta J, Chen X, Mutero A, Martin A, Messing RO, Levine JD. Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J Neurosci 2000;20:8614–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dina OA, Chen X, Reichling D, Levine JD. Role of protein kinase Cepsilon and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience 2001;108:507–15. [DOI] [PubMed] [Google Scholar]
- [17].Dina OA, Joseph EK, Levine JD, Green PG. Mechanisms mediating vibration-induced chronic musculoskeletal pain analyzed in the rat. J Pain 2010;11:369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fan HC, Zhang X, McNaughton PA. Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem 2009;284:27884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem 2006;281:30947–56. [DOI] [PubMed] [Google Scholar]
- [20].Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 2010;50:355–75. [DOI] [PubMed] [Google Scholar]
- [21].Guo H, Huang LY. Alteration in the voltage dependence of NMDA receptor channels in rat dorsal horn neurones following peripheral inflammation. J Physiol 2001;537(pt 1):115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hamilton SG, McMahon SB, Lewin GR. Selective activation of nociceptors by P2X receptor agonists in normal and inflamed rat skin. J Physiol 2001;534(pt 2):437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hamilton SG, Wade A, McMahon SB. The effects of inflammation and inflammatory mediators on nociceptive behaviour induced by ATP analogues in the rat. Br J Pharmacol 1999;126:326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].He Y, Wang ZJ. Nociceptor beta II, delta, and epsilon isoforms of PKC differentially mediate paclitaxel-induced spontaneous and evoked pain. J Neurosci 2015;35:4614–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hendrich J, Alvarez P, Joseph EK, Chen X, Bogen O, Levine JD. Electrophysiological correlates of hyperalgesic priming in vitro and in vivo. PAIN 2013;154:2207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron 2007;55:365–76. [DOI] [PubMed] [Google Scholar]
- [27].Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci 2005;25:6119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hui X, Reither G, Kaestner L, Lipp P. Targeted activation of conventional and novel protein kinases C through differential translocation patterns. Mol Cell Biol 2014;34:2370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron 1999;24:253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lawson SN, Waddell PJ. Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. J Physiol 1991;435:41–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li G, Ma F, Gu Y, Huang LY. Analgesic tolerance of opioid agonists in mutant mu-opioid receptors expressed in sensory neurons following intrathecal plasmid gene delivery. Mol Pain 2013;9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ma W, Zheng WH, Belanger S, Kar S, Quirion R. Effects of amyloid peptides on cell viability and expression of neuropeptides in cultured rat dorsal root ganglion neurons: a role for free radicals and protein kinase C. Eur J Neurosci 2001;13:1125–35. [DOI] [PubMed] [Google Scholar]
- [33].Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, Roufogalis BD, Tominaga M. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. PAIN 2006;123:106–16. [DOI] [PubMed] [Google Scholar]
- [34].Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem 1993;268:9194–7. [PubMed] [Google Scholar]
- [35].Nakatsuka T, Gu JG. P2X purinoceptors and sensory transmission. Pflugers Arch 2006;452:598–607. [DOI] [PubMed] [Google Scholar]
- [36].North RA. P2X3 receptors and peripheral pain mechanisms. J Physiol 2004;554(pt 2):301–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Olah Z, Karai L, Iadarola MJ. Protein kinase C(alpha) is required for vanilloid receptor 1 activation. Evidence for multiple signaling pathways. J Biol Chem 2002;277:35752–9. [DOI] [PubMed] [Google Scholar]
- [38].Pan HL, Zhang YQ, Zhao ZQ. Involvement of lysophosphatidic acid in bone cancer pain by potentiation of TRPV1 via PKCepsilon pathway in dorsal root ganglion neurons. Mol Pain 2010;6:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. PAIN 2005;113:185–90. [DOI] [PubMed] [Google Scholar]
- [40].Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci 2003;17:1847–52. [DOI] [PubMed] [Google Scholar]
- [41].Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience 2003;120:219–26. [DOI] [PubMed] [Google Scholar]
- [42].Pereira L, Rehmann H, Lao DH, Erickson JR, Bossuyt J, Chen J, Bers DM. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc Natl Acad Sci U S A 2015;112:3991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci 2009;32:611–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Roivainen R, Koistinaho J, Hervonen A. Localization of protein kinase C-beta-like immunoreactivity in the rat dorsal root ganglion. Neurosci Res 1990;7:381–4. [DOI] [PubMed] [Google Scholar]
- [45].Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol 2010;11:103–12. [DOI] [PubMed] [Google Scholar]
- [46].Vasko MR, Habashy Malty R, Guo C, Duarte DB, Zhang Y, Nicol GD. Nerve growth factor mediates a switch in intracellular signaling for PGE2-induced sensitization of sensory neurons from protein kinase A to Epac. PLoS One 2014;9:e104529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang C, Gu Y, Li GW, Huang LY. A critical role of the cAMP sensor Epac in switching protein kinase signalling in prostaglandin E2-induced potentiation of P2X3 receptor currents in inflamed rats. J Physiol 2007;584(pt 1):191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang C, Li GW, Huang LY. Prostaglandin E2 potentiation of P2X3 receptor mediated currents in dorsal root ganglion neurons. Mol Pain 2007;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang H, Heijnen CJ, van Velthoven CT, Willemen HL, Ishikawa Y, Zhang X, Sood AK, Vroon A, Eijkelkamp N, Kavelaars A. Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J Clin Invest 2013;123:5023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wirkner K, Sperlagh B, Illes P. P2X3 receptor involvement in pain states. Mol Neurobiol 2007;36:165–83. [DOI] [PubMed] [Google Scholar]
- [51].Xiang Z, Xiong Y, Yan N, Li X, Mao Y, Ni X, He C, LaMotte RH, Burnstock G, Sun J. Functional up-regulation of P2X 3 receptors in the chronically compressed dorsal root ganglion. PAIN 2008;140:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Xu GY, Huang LY. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J Neurosci 2002;22:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xu X, Wang P, Zou X, Li D, Fang L, Gong K, Lin Q. The effects of sympathetic outflow on upregulation of vanilloid receptors TRPV(1) in primary afferent neurons evoked by intradermal capsaicin. Exp Neurol 2010;222:93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhao C, Leitges M, Gereau RWt. Isozyme-specific effects of protein kinase C in pain modulation. Anesthesiology 2011;115:1261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]