

Summary
CXCR2 has been suggested to have both tumor-promoting and tumor-suppressive properties. Here we show that CXCR2 signaling is upregulated in human pancreatic cancer, predominantly in neutrophil/myeloid-derived suppressor cells, but rarely in tumor cells. Genetic ablation or inhibition of CXCR2 abrogated metastasis, but only inhibition slowed tumorigenesis. Depletion of neutrophils/myeloid-derived suppressor cells also suppressed metastasis suggesting a key role for CXCR2 in establishing and maintaining the metastatic niche. Importantly, loss or inhibition of CXCR2 improved T cell entry, and combined inhibition of CXCR2 and PD1 in mice with established disease significantly extended survival. We show that CXCR2 signaling in the myeloid compartment can promote pancreatic tumorigenesis and is required for pancreatic cancer metastasis, making it an excellent therapeutic target.
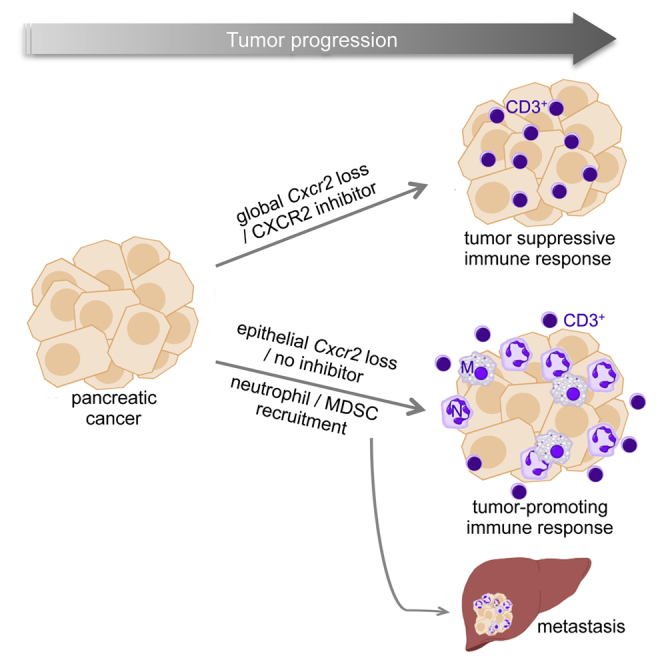
Graphical Abstract
Highlights
-
•
CXCR2 signaling is upregulated in myeloid cells in human pancreatic cancer
-
•
Cxcr2 loss reduces metastasis and inhibition prolongs tumor-free survival in mice
-
•
Neutrophils/MDSCs play a key role in the establishment of the metastatic niche
-
•
CXCR2 inhibition enhances T cell entry and confers sensitivity to anti-PD1 therapy
Steele et al. show that CXCR2 is important in immune modulation of pancreatic cancer and that inhibition of CXCR2 reduces metastasis and improves response to gemcitabine and anti-PD1. Peptide inhibitor, but not germline deletion of Cxcr2, improved survival, revealing differential effects in early and late tumors.
Significance
PDAC is predicted to become the second commonest cause of cancer death in the United States by 2020. Aggressive invasion and early metastases are characteristic of the disease, and even the few patients eligible for potentially curative resection inevitably develop recurrent or metastatic disease. Early indications suggest that immunotherapy will not work in unselected pancreatic cancer patients. Our data highlight two therapeutic opportunities for PDAC: first the use of CXCR2 inhibitors in surgically resected patients, and second the use of CXCR2 inhibition in combination with immunotherapy in surgically unresectable advanced disease. Our data also suggest that therapeutic targets that may cause senescence escape will not have deleterious effects in late-stage disease in PDAC, because tumors have already escaped this checkpoint.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an almost universally lethal malignancy and represents a significant therapeutic challenge (http://info.cancerresearchuk.org/cancerstats). Gemcitabine has been the standard of care for patients since 1997, despite offering only marginal benefit (Burris et al., 1997). Recent improvements in survival using FOLFIRINOX and gemcitabine plus nab-paclitaxel have offered a choice to clinicians for the first time (Conroy et al., 2011, Goldstein et al., 2015). However, the 5-year survival rate remains ∼6%. Surgical resection is the only potential cure; however, only 15% of patients are suitable for surgery, and most die within 2 years of surgery due to recurrence or metastatic disease. Thus, a greater understanding of how key molecular and cellular regulators of tumor progression combine to drive invasion and metastases in PDAC is required.
Pancreatic cancer develops and metastasizes as a result of the accumulation of multiple genetic and epigenetic changes. Activating mutations of the KRAS proto-oncogene occur in >90% of cases (Almoguera et al., 1988), while inactivation of tumor suppressor genes, including CDKN2A, TP53, SMAD4, and BRCA2 accrue throughout disease development (Hruban et al., 2000). Progression is a complex process and is reliant on interactions between the tumor and its microenvironment (Baumgart et al., 2013). Ubiquitous to all PDAC is the dense desmoplastic stroma, consisting of immune cells, stellate cells, fibroblasts, and a dense extracellular collagenous matrix, which surrounds PDAC cells, providing vital signals for survival, tumor cell invasion, and metastasis (Olive et al., 2009). However, two recent studies found that targeting the stroma led to accelerated disease progression (Ozdemir et al., 2014, Rhim et al., 2014), although both approaches provided opportunities for immune-targeting therapies. Indeed, targeting FAP+ fibroblasts in pancreatic tumor-bearing mice can synergize with anti-programmed death 1 (anti-PD1) immunotherapy to cause tumor regression (Feig et al., 2013). Thus, there may be a complex interplay of tumor-promoting and tumor-suppressive consequences when targeting specific pathways.
The relationship between inflammation and PDAC progression is complex. Initially PDAC must overcome immune surveillance; indeed, both human and mouse pancreatic intraepithelial neoplasia (PanINs) and PDAC are characterized by the infiltration of immune suppressor cells, suggesting tumor immunity is blocked early during tumorigenesis (Clark et al., 2007). Once immune surveillance has been bypassed, the net effect of interactions between tumor and immune cells is PDAC progression. However, certain cell types may have the capacity for tumor suppression and promotion in different contexts. For example, inflammatory signaling can promote oncogene-induced senescence (Acosta et al., 2008, Kuilman et al., 2008). On the other hand, inflammation can drive pancreatic tumorigenesis: there is enhanced tumorigenesis in mice subjected to pancreatitis (Guerra et al., 2007), and patients with hereditary pancreatitis have a greatly increased risk of PDAC (Lowenfels et al., 1997). More recently, specific inflammatory signaling pathways, such as STAT3/IL-6 (Baumgart et al., 2014, Corcoran et al., 2011, Fukuda et al., 2011, Lesina et al., 2011), NF-κB (Daniluk et al., 2012, Ling et al., 2012, Maniati et al., 2011), and CXCR2 (Ijichi et al., 2011, Matsuo et al., 2009a) have been implicated in PDAC progression.
CXCR2 is a G-protein-coupled receptor for the human CXC chemokines CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8. Mouse CXCR2 has a more limited repertoire of ligands because mice lack CXCL6 and CXCL8 genes. The primary immune function of CXCR2 is the regulation of neutrophil migration, as it controls the egress of these cells from the bone marrow, and their recruitment to sites of inflammation (Cacalano et al., 1994, Eash et al., 2010). CXCR2 also regulates the migration of myeloid-derived suppressor cells (MDSCs) (Highfill et al., 2014). We have previously shown that CXCR2 is fundamental to the process of tumorigenesis in both colon and skin (Jamieson et al., 2012). However, there is growing evidence that CXCR2 is also important for the metastatic process. For example, Cxcr2 deletion can reduce the invasive and metastatic potential of lung cancer cells (Saintigny et al., 2013). In models of breast cancer, CXCR2 signaling is important for attracting Gr-1+CD11b+ MDSCs to the tumor microenvironment, where they drive invasion and metastasis (Yang et al., 2008). Moreover, CXCL1/2 secretion at metastatic sites can promote the establishment of a metastatic niche by enhancing the influx of MDSCs (Acharyya et al., 2012). Indeed, neutrophils have recently been shown to support metastatic colonization by breast cancer cells (Wculek and Malanchi, 2015).
There is emerging evidence for a role of neutrophils, MDSCs, and CXCR2 in pancreatic cancer. Neutrophil infiltration is observed in pancreatic tumors with the poorest prognosis (Reid et al., 2011), while high levels of the CXCR2 ligand CXCL5 are associated with poorer survival and, by implication, metastatic disease (Li et al., 2011). Recent studies have described the contribution of MDSCs to pancreatic cancer progression (Bayne et al., 2012), and the accumulation of MDSCs in patients with advanced pancreatic cancer correlates with disease stage (Diaz-Montero et al., 2009). Moreover, in vitro experiments and transplants in immunodeficient animals have implicated CXCR2 in the regulation of pancreatic tumor cell proliferation, invasion, and angiogenesis (Matsuo et al., 2009a, Matsuo et al., 2009b, Purohit et al., 2016, Wang et al., 2013). CXCR2 inhibition has also been shown to disrupt interactions between tumor cells and fibroblasts to slow tumor progression in a mouse model of pancreatic cancer, although the mice used exhibit high levels of stromal deregulation and are short lived (Ijichi et al., 2011). These tumor-promoting properties of CXCR2 may be counterbalanced by CXCR2-dependent tumor-suppressing activities. Of particular relevance to PDAC, are the elegant studies that show that CXCR2 reinforces RAS-mediated senescence in culture, and regulates the senescence-associated secretory phenotype (Acosta et al., 2008). We therefore sought here to determine the role of CXCR2 signaling in pancreatic tumorigenesis using relevant in vivo models.
Results
CXCR2 Signaling at the Tumor Border Is Associated with Poor Outcome in Human PDAC
To investigate the importance of CXCR2 in human pancreatic cancer we analyzed the expression of CXCR2 and its ligands in samples from a cohort of 44 PDAC patients. RNA was prepared from targeted biopsies of tumor borders, and from the adjacent normal pancreas, and chemokine and CXCR2 expression examined. We found that genes encoding CXCR2, and two of its ligands, CXCL2 and CXCL8, were significantly upregulated compared with adjacent normal pancreas (Figure 1A), and high CXCR2 or CXCL2 expression was associated with significantly poorer prognosis (Figure 1B). High CXCR2 expression was also associated with advanced T stage (p = 0.05), tumor grade (p = 0.037), and resection margin status (p = 0.015). We next examined CXCR2 protein by immunohistochemistry (IHC) on full-face sections taken from the edges of tumors adjacent to normal tissue, and found that high expression was associated with poor outcome (Figure 1C). Rather than being expressed in the tumor, most of the observed CXCR2 staining was within the stroma (Figure 1D). Further analysis of the cells present at these areas revealed high numbers of myeloperoxidase (MPO)-positive cells indicating the presence of neutrophils or their precursors (Figure 1D). There was a significant correlation between expression of MPO and CXCR2 in the stroma at the edge of these tumors adjacent to normal regions (Spearman’s rho 0.907, p = 0.01). There were also high numbers of CD68+ macrophages, but few CD3+ T cells: these cells were, however, observed in the tissue surrounding the tumors (Figure 1D).
Figure 1.

CXCR2 Expression at the Tumor Border Is Associated with Poor Outcome in Human PDAC
(A) Expression of CXCR2 and its ligands in the tumor border compared with adjacent normal pancreas within pancreaticoduodenectomy specimens (n = 44). RNA was prepared from whole targeted biopsies of the edges of tumors, post-resection, and from adjacent normal pancreas. p Values, Mann-Whitney U test.
(B) Kaplan-Meier analysis of survival in terms of low or high CXCR2 and CXCL2 expression from RNA from whole targeted biopsies of the edges of resected tumors. p Values, log rank test.
(C) Kaplan-Meier analysis of survival in terms of low or high CXCR2 expression as assessed by IHC on full-face sections of tumor border regions (n = 11). p Values, log rank test.
(D) IHC staining for CXCR2, MPO, CD68, and CD3 in the stroma at the edge of resected tumors. Arrowheads indicate direction of invasion into either adjacent duodenum or normal pancreas. Scale bars represent 500 μm. Boxplots below show quantification of cells staining positive for each marker in the tumor center versus tumor border (n ≥ 3). p Values, Mann-Whitney U test. The region 1 mm proximal to the adjacent normal tissue was assessed in (C and D). See also Figure S1.
Interestingly, the association of CXCR2-positive cells with prognosis appeared to depend on their location. When we examined tissue from the tumor body (Figure S1A), neither stromal nor tumor epithelial CXCR2 expression was associated with survival (Figure S1B). These data show that the effects of CXCR2 signaling, and recruitment of myeloid cells, differ depending on the site to which these cells are recruited. It is clear, however, that CXCR2 signaling and myeloid cell recruitment at the tumor border are linked to poor outcome in patients. Importantly, very few tumor cells expressed CXCR2.
KPC Mice Recapitulate the Microenvironment and CXCR2 Expression Profile of Human PDAC
In order to further investigate the importance of CXCR2 signaling in PDAC we used a mouse model that both phenotypically and histologically recapitulates the human disease. KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre) mice carry a pancreas-specific Trp53R172H mutation alongside an initiating mutation in KrasG12D, and develop invasive, metastatic tumors that exhibit an extensive stroma (Hingorani et al., 2005) (Figure S2A) with significant collagen deposition (visualized by picrosirius red staining, Figure S2B), macrophage (F4/80, Figure S2C) and neutrophil infiltration (MPO or S100A9, Figures S2D and S2E), few CD3+ T cells (Feig et al., 2013) (Figure S2F), numerous activated α-smooth muscle actin (α-SMA)-positive stellate cells (Figure S2G), abundant levels of the pro-invasive protein tenascin C (Oskarsson et al., 2011) (Figure S2H), and high levels of tumor cell proliferation (marked by Ki67, Figure S2I).
It has been previously shown that KPC tumor cells express the ligands CXCL1 and CXCL2 (Stromnes et al., 2014). When we examined the expression of CXCR2 and its ligands in tumors from KPC mice we found that, similar to human tumors, Cxcl2 and Cxcr2 were significantly upregulated in PDAC compared with normal pancreas, while there was also increased expression of Cxcl1 and Cxcl5 in a proportion of the tumors (Figure 2A). To determine the source of ligand production we performed qPCR on RNA prepared from laser-capture micro-dissected tumor epithelium and stroma. Cxcl1, Cxcl2, Cxcl5, and Cxcr2 transcripts were quantified in these samples relative to wild-type (WT) pancreas. This demonstrated increased expression of Cxcl2 and Cxcl5 by tumor cells (Figure 2B). Similar to human tumors, Cxcr2 was highly expressed by stromal cells, likely neutrophils, and some tumor cells (Figure 2B), and this was confirmed by IHC (Figure 2C). Indeed, previously published work has shown that there is significant infiltration of granulocytes into KPC tumors (Clark et al., 2007), and these are likely the CXCR2-expressing cells that we observe. In addition, we found that KPC tumor cells in culture secreted substantial amounts of CXCL1 and CXCL5, and smaller amounts of CXCL2 (Figure 2D). Finally we analyzed expression of Cxcl1, Cxcl2, Cxcl5, and Cxcr2 by RNA sequencing (RNA-seq) of FAP+ fibroblasts isolated from normal pancreas, PanIN, and PDAC from KPC mice (Feig et al., 2013). Expression of Cxcl1, Cxcl2, and Cxcl5 was increased in FAP+ fibroblasts from PanIN and PDAC compared with WT (Figure 2E). CXCL1 expression by stromal fibroblasts was confirmed by co-IHC for CXCL1 and α-SMA (Figure 2F). CXCR2 was expressed at negligible levels in FAP+ fibroblasts, and, compared with normal controls, was unchanged in PanIN and PDAC (Figure 2E). The expression we did observe was likely due to contamination by a very small number of neutrophils, given that we observe similar levels of MPO (Figure 2E). Our results show that KPC mice recapitulate the microenvironment and upregulated CXCR2 signaling seen in human PDAC, and represent an ideal model in which to investigate the role of CXCR2 signaling in pancreatic cancer.
Figure 2.

KPC Mice Recapitulate the Microenvironment and CXCR2 Expression of Human PDAC
(A) Expression of Cxcr2 and its ligands in KPC PDAC (n = 6) compared with normal WT pancreas. p Values, Mann-Whitney U test.
(B) Expression of Cxcl1, Cxcl2, Cxcl5, and Cxcr2 from pooled (n = 3) laser-capture micro-dissected stroma or tumor epithelium compared with WT pancreas. Expression normalized to Gapdh. p Value, ANOVA. Error bars are ±SEM.
(C) Representative IHC for CXCL2 and CXCR2 in PDAC from KPC mice. Scale bars represent 200 μm.
(D) Cytokine array analysis of CXCR2 ligands produced by KPC cell lines compared with control pancreatic duct epithelial cells (n = 6). p Values, Mann-Whitney U test.
(E) RNA-seq expression of Cxc1, Cxcl2, Cxcl5, Cxcr2, and Mpo in FAP+ fibroblasts from normal pancreas, PanIN, or PDAC from KPC mice (n = 2). Error bars are ±SD.
(F) Dual IHC for CXCL1 (red) and CK19, α-SMA, or MPO (brown) in KPC tumors. See also Figure S2.
Cxcr2 Deletion Abrogates Metastasis in KPC Mice
We generated KPC Cxcr2−/− mice, which were born at the expected Mendelian ratios and exhibited normal pancreatic pathology. There was no difference in overall or tumor-free survival between KPC Cxcr2−/− and KPC mice (Figure 3A), which initially suggested that CXCR2 was neither tumor suppressive nor tumor promoting in this system. To investigate whether inhibition of CXCR2 signaling might affect response to chemotherapy in pancreatic cancer, we treated mice with gemcitabine from 10 weeks of age. At this time mice have widespread advanced pancreatic neoplasia (Hingorani et al., 2005, Morton et al., 2010), and are more likely to mimic non-metastatic surgically resectable disease. Using this timepoint also allowed us to monitor effects of drugs or combinations over a longer period of time. This was crucial given recent studies that have generated contrasting results depending on the timing of intervention (Olive et al., 2009, Rhim et al., 2014). However, it is important to note that pancreata at this stage will exhibit mostly pre-invasive disease with occasional progression. Nevertheless, we did not detect any significant impact of gemcitabine treatment on survival in either KPC or KPC Cxcr2−/− mice (Figure 3A).
Figure 3.

Cxcr2 Deletion Inhibits Metastasis in KPC Mice
(A) Kaplan-Meier survival analysis of KPC and KPC Cxcr2−/− mice untreated, or treated from 10 weeks of age with 100 mg/kg gemcitabine, n = 21, 24, 10, 13, respectively (not significant, log rank test).
(B) Table comparing incidence of metastases in KPC and KPC Cxcr2−/− mice treated as indicated. p Values, chi-square test.
(C and D) H&E staining of representative primary tumors from (C) KPC and (D) KPC Cxcr2−/− mice.
(E and F) H&E staining of representative (E) liver and (F) diaphragm metastasis from KPC mice.
(G and H) IHC for MPO, F4/80, CD3, and tenascin C (TNC) in tumors from (G) KPC and (H) KPC Cxcr2−/− mice.
(I and J) Boxplots of signature scores (I) upregulated and (J) downregulated, in KPC Cxcr2−/− mice, stratified by human PDAC class. p Values, Kruskall-Wallis test. See also Figure S3.
There were no clear histological differences between tumors in KPC and KPC Cxcr2−/− mice; however, we found that Cxcr2 deletion was sufficient to almost completely abrogate metastasis (Figures 3B–3F). Unsurprisingly, gemcitabine had no significant effect on metastases in the KPC model (Figure 3B). IHC of immune cell infiltrate in pancreatic tumors from KPC and treated and untreated KPC Cxcr2−/− mice showed that, as expected, given the role of CXCR2 in neutrophil homing, there was a significant reduction in the number of MPO+ cells infiltrating tumors lacking CXCR2 (Figures 3G, 3H, and S3A). Interestingly, this was accompanied by a significant increase in F4/80+ macrophages and CD3+ T cells in the KPC Cxcr2−/− tumors (Figures 3G, 3H, S3B, and S3C), and a significant decrease in expression of the pro-invasive protein tenascin C (Figures 3G, 3H, and S3D). There were also fewer proliferative cells in the KPC Cxcr2−/− tumors, but no difference in the levels of apoptosis, as assessed by cleaved caspase 3 IHC (Figures S3E and S3F). We also found a decrease in picrosirius red staining indicating a reduction in collagen I expression (Figure S3G).
Recently, we performed integrated genomic analysis of 456 human PDAC that defined four subtypes of PC that are associated with distinct histopathological characteristics and differential survival (Bailey et al., 2016). Based on a number of key molecular characteristics, these subtypes have been named: (1) squamous; (2) pancreatic progenitor; (3) immunogenic; and (4) aberrantly differentiated endocrine exocrine (ADEX). The squamous subtype is an independent prognostic factor and is associated with poor outcomes. To further investigate the role of CXCR2 in PDAC progression we set out to assess whether loss of CXCR2 was significantly associated with a specific PDAC subtype. This analysis clearly demonstrated that CXCR2 loss is associated with an apparent switch from the poorly prognostic squamous identity commonly observed in KPC tumors, and an enrichment of gene expression that defines the pancreatic progenitor, immunogenic, and ADEX subtypes (Figures 3I and 3J). These data are in line with our finding that Cxcr2 deletion inhibits metastasis, and provide further evidence of a role for CXCR2 signaling in promoting aggressive pancreatic cancer.
Depletion of Ly6G+ Cells Recapitulates the Effects of Cxcr2 Deletion
Ly6G+ cells, including neutrophils and MDSCs, are the most prominent source of CXCR2 in mice (Cacalano et al., 1994). Thus, we considered whether depletion of these cells would recapitulate the phenotypes that arise as a consequence of Cxcr2 deletion. The anti-Ly6G antibody 1A8, has been routinely used to deplete Ly6G+ cells, and is well-tolerated and effective long term (Jamieson et al., 2012). KPC mice were treated from 10 weeks of age with 1A8 or 2A3 isotype control. Compared with 2A3-treated mice, and like Cxcr2 deletion, 1A8 treatment had no effect on survival (Figure 4A), but did result in strong suppression of metastasis (Figure 4B), indicating that the CXCR2-dependent recruitment of Ly6G+ cells is indeed important in the establishment of secondary disease. As expected, the primary tumors of 1A8-treated mice contained fewer MPO+ neutrophils than tumors from 2A3-treated mice and, importantly, 1A8+ cells were also reduced even at endpoint in both tumor and spleen (Figures 4C–4F), suggesting that treatment remains effective even if some antibody neutralization may occur. F4/80+ macrophage infiltration did not change, but, like Cxcr2 deletion, the depletion of Ly6G+ cells resulted in a marked increase in the number of infiltrating CD3+ T cells (Figures 4C–4E), supporting previous work (Stromnes et al., 2014). Thus, depletion of Ly6G+ cells, the dominant cell type expressing CXCR2, has a similar effect to Cxcr2 deletion, namely inhibition of metastasis, and substantial changes in the immune cell profile of tumors, most notably a loss of MPO+ neutrophils and a marked increase in CD3+ T cells.
Figure 4.

Neutrophil Ablation Also Inhibits Metastasis in the KPC Model
(A) Kaplan-Meier analysis of survival of KPC mice treated from 10 weeks of age with 2A3 isotype-control antibody (n = 11) or 1A8, anti-Ly6G neutrophil-ablating antibody (n = 15). p Values, log rank test.
(B) Table comparing incidence of metastases in KPC mice treated with 2A3 or 1A8. p Value, chi-square test.
(C and D) IHC on tumors from (C) 2A3- or (D) 1A8-treated mice, for MPO, 1A8, F480, and CD3.
(E) Boxplots showing quantification of IHC in (C) and (D). p Values, Mann-Whitney U test.
(F) IHC for 1A8 on spleens from 2A3- and 1A8-treated mice, quantified on right. p Values, Mann-Whitney U test.
(G) Kaplan-Meier survival analysis of KPC (n = 19, median = 157 days) and KPC Cxcr2fl/fl mice (n = 28, median = 141 days). p Values, log rank test.
(H) Table comparing incidence of metastases in KPC and KPC Cxcr2fl/fl mice. p Values, chi-square test.
(I) Boxplots showing quantification of CD3 IHC in tumors from KPC and KPC Cxcr2fl/fl mice. p Value, Mann-Whitney U test.
Although these data suggest that Ly6G+ cells mediate the effects of Cxcr2 deletion, others have reported a pro-tumorigenic role for CXCR2 signaling in pancreatic tumor cells (Purohit et al., 2016, Wang et al., 2013). To confirm that the effects of Cxcr2 deletion were not mediated by effects on autocrine tumor cell signaling we deleted Cxcr2 specifically from the pancreatic epithelium of KPC mice using a conditional floxed Cxcr2 allele (KPC Cxcr2fl/fl). Importantly, we did not see any effect on survival (Figure 4G), and we no longer observed any effects on metastasis or CD3+ T cell infiltration (Figures 4H and 4I), confirming that the effects of Cxcr2 deletion are not dependent on the loss of expression on the tumor cells.
CXCR2 Inhibition Reduces Metastases and Prolongs Survival in KPC Mice
Although anti-metastatic therapies may not inhibit growth of the primary tumor, they may be useful in patients where the primary tumor can be resected. Thus, we wanted to determine if pharmacological inhibition of CXCR2 signaling could inhibit metastasis in the KPC model. To inhibit CXCR2, we first used a short peptide CXCR2 “pepducin” (1/2i-pal), which inhibits CXCR2 signaling by interfering with its ability to couple to intracellular signal-transduction molecules (Jamieson et al., 2012, Kaneider et al., 2005). Control mice received a scrambled pepducin. Other groups of KPC mice received gemcitabine alone, or gemcitabine along with the CXCR2 pepducin. All treatments were started when the mice were 10 weeks old. CXCR2 pepducin treatment resulted in a significant increase in survival compared with controls and appeared to reduce metastasis (Figures 5A and 5B). Gemcitabine alone had no effect on survival or the development of metastasis, but when CXCR2 pepducin and gemcitabine were combined, survival was significantly extended and we were unable to detect any metastases in these animals (Figures 5A and 5B). We did not see significant changes in neutrophil, macrophage, or CD3 infiltration in pepducin-treated mice (Figures 5C, 5D, and S4A–S4C), although the number of infiltrating neutrophils was significantly higher in mice treated with the combination of CXCR2 pepducin and gemcitabine (Figure S4A), which may be due in part to necrosis within these tumors. Indeed, tenascin C, the expression of which is induced by hypoxia (Lal et al., 2001), was upregulated in pepducin-treated tumors (Figures 5C, 5D, and S4D). There was no significant change in either tumor cell proliferation or apoptosis (Figures S4E and S4F), but again we saw a decrease in picrosirius red staining indicating a reduction in collagen I (Figure S4G).
Figure 5.

Cxcr2 Inhibition Inhibits Metastasis and Prolongs Survival in KPC Mice
(A) Kaplan-Meier analysis of KPC mice treated from 10 weeks of age with scrambled pepducin (n = 15), gemcitabine (n = 14), CXCR2-inhibiting pepducin (n = 20), or CXCR2-inhibiting pepducin + gemcitabine (n = 11). p Values, log rank test.
(B) Table comparing incidence of metastases in KPC mice treated as indicated. p Values, chi-square test.
(C and D) H&E staining and IHC for MPO, F4/80, CD3, and tenascin C (TNC) in tumors in response to (C) scrambled pepducin and (D) CXCR2-targeting pepducin. See also Figure S4.
CXCR2 Inhibition Results in Failure to Set up a Metastatic Niche
Given the profound effect that inhibiting CXCR2 signaling has on metastasis in our model, we sought to examine potential mechanisms for this phenomenon, particularly in distant metastatic sites. MDSCs are immature bone marrow cells typified by the expression of CD11b, Gr1, and Ly6G/Ly6C (Youn and Gabrilovich, 2010). They have been shown to play a role in establishing the metastatic niche in different metastatic tumor models (Acharyya et al., 2012, Yan et al., 2010). Therefore, we investigated the levels of these, and other immune cells, in the pre-metastatic livers of KPC mice (Figure S4H), in KPC liver metastases (Figure S4I), in the livers of KPC mice treated with pepducin and gemcitabine from 10 weeks old (Figure S4J), and in liver metastases from KPC mice treated with CXCR2 pepducin and gemcitabine when symptomatic (Figure S4K). We included this latter group because of the lack of metastases in KPC Cxcr2−/− mice and mice with CXCR2 inhibited from 10 weeks.
We found that there were a substantial number of F4/80+ macrophages, NIMP1+ neutrophils, and cells staining positively with the MDSC marker, S100A9, in the pre-metastatic liver (Figure S4H), and in established liver metastases where there were also CXCR2-expressing cells (Figure S4I). CXCR2 inhibition with pepducin considerably decreased the number of myeloid cells in the livers of KPC mice (Figure S4J) and, even in mice with late-stage tumors, reduced the number of neutrophils and S100A9+ cells infiltrating liver metastases, although had no effect on monocyte/macrophage recruitment (Figure S4K). Interestingly, the number of CD3+ T cells infiltrating these metastases was increased compared with untreated mice, in line with our observations in primary tumors. The changes we observe in primary tumors and metastases following CXCR2 inhibition suggest that the migration of myeloid lineage cells to the tumor microenvironment is impaired when CXCR2 signaling is suppressed. This reduction in chemotaxis may also result in a failure of myeloid cells to migrate to the liver and establish a niche for tumor cells. These findings suggest a key role for CXCR2 signaling and immune cell migration in metastatic progression in this model.
Clinically Relevant Targeting of CXCR2 Reduces Metastases and Prolongs Survival in KPC Mice
The finding that CXCR2 inhibition but not constitutive knockout could slow tumorigenesis suggested that CXCR2 might play opposing roles in early and late tumorigenesis. Loss during initiation might allow pre-neoplastic PanIN lesions to progress beyond senescence, while loss at late stages might inhibit the growth and metastatic potential of PDAC. Thus, we wanted to further test the therapeutic effects of CXCR2 inhibition at a later stage of neoplasia, using a clinically relevant CXCR2 inhibitor. Therefore, we tested the effects of AZ13381758, a small-molecule inhibitor of CXCR2 (referred to as CXCR2 SM) that is related to AZD5069 (Nicholls et al., 2015). AZ13381758 is a potent inhibitor of both murine and human CXCR2 (Figures S5A–S5F, Table S1), the efficacy of which was confirmed by full blood-count analysis showing increased circulating neutrophils due to their inability to home (Figure 6A).
Figure 6.

Therapeutic Targeting of CXCR2 Inhibits Metastasis and Prolongs Survival in KPC Mice
(A) Boxplot showing circulating neutrophils in CXCR2 SM-treated mice (n = 4). p Values, Mann-Whitney U test.
(B) Kaplan-Meier survival analysis of KPC mice treated from 10 weeks of age with vehicle (n = 11), gemcitabine (n = 14), CXCR2 SM (n = 15), or CXCR2 SM + gemcitabine (n = 12). p Values, log rank test.
(C) Table comparing incidence of metastases in KPC mice treated as indicated. p Values, chi-square test.
(D–F) H&E staining and IHC for MPO, F4/80, CD3, and tenascin C (TNC) in tumors in response to (D) vehicle, (E) CXCR2 SM, and (F) CXCR2 SM + gemcitabine. See also Figure S5 and Table S1.
Similar to CXCR2 pepducin, treatment with CXCR2 SM alone from 10 weeks of age prolonged the survival of KPC mice, and was most effective in combination with gemcitabine (Figure 6B). In addition, mice treated with CXCR2 SM were significantly protected from metastasis (Figure 6C). We did not observe significant changes in the number of intra-tumoral neutrophils or macrophages (Figures 6D, 6F, S5G, and S5H), similar to pepducin treatment; however, we did observe an increase in infiltrating T cells (Figures 6D, 6F, and S5I), similar to the results seen in KPC Cxcr2−/− mice. We also saw a reduction of tenascin C (Figures 6D, 6F, and S5J), a reduction in the number of proliferative cells (Figure S5K), no change in apoptosis (Figure S5L), and a reduction in stromal collagen (Figure S5M) in CXCR2 SM-treated tumors. Thus, like Cxcr2 deletion or Ly6G+ cell depletion, pharmacological inhibitors of CXCR2 can reduce the metastatic spread of pancreatic tumors in KPC mice and may provide a survival advantage over control animals. More significantly, the combination of CXCR2 inhibition and gemcitabine treatment extends the life span of KPC mice by 50–70 days and substantially suppresses metastasis.
CXCR2 Inhibition Substantially Enhances Sensitivity to Anti-PD1 Immunotherapy
There is a growing awareness that immunosuppression by infiltrating immune cells plays an important role in the resistance of tumors to endogenous anti-tumor immune responses as well as therapeutic interventions. Pancreatic cancer cells themselves actively contribute to immune suppression through production of cytokines (Beatty et al., 2011), so that although a systemic anti-tumor immune response is elicited, it is ineffective (Dodson et al., 2011). The efficacy of therapy in PDAC would likely be improved by overcoming this immune suppression. Indeed, immunotherapy aimed at harnessing endogenous anti-tumor immunity has shown promise in multiple tumor types (Sharma and Allison, 2015). Given that Cxcr2 deletion or inhibition increased the number of CD3+ T cells in pancreatic tumors in KPC mice, we wanted to know if CXCR2 inhibitors could enhance sensitivity to therapies aimed at de-repressing T cells. We chose to use anti-PD1 antibodies, which block T cell suppression by preventing interactions between PD1 and its ligands (Barber et al., 2006, Fife et al., 2009), but which have previously been found to be ineffective as a single agent in this model (Winograd et al., 2015).
In this experiment, treatment was only started once the KPC mice had developed palpable pancreatic tumors. One group of mice then received CXCR2 SM for 2 weeks to increase T cell infiltration into the tumor. Control mice received either vehicle alone or gemcitabine alone. After this initial priming phase, animals in the CXCR2 SM and vehicle groups received anti-PD1 antibody while continuing on the CXCR2 SM or vehicle treatments. Unsurprisingly, given that they are carrying late-stage tumors, few vehicle-treated animals survived long enough (2 weeks) to allow commencement of PD1 treatment (Figure 7A). Remarkably, however, even in these late-stage tumors, treatment with CXCR2 SM and anti-PD1 significantly extended survival beyond that of the mice treated with vehicle plus anti-PD1, with two mice living 100 days beyond the start of treatment before succumbing to pancreatic tumors (Figure 7A).
Figure 7.

CXCR2 Blockade Promotes T Cell Infiltration into Tumors and Sensitivity to Immunotherapy
(A) Kaplan-Meier survival analysis of tumor-bearing KPC mice treated with either gemcitabine, CXCR2 SM alone for 2 weeks, and then in combination with anti-PD1, vehicle alone for 2 weeks, and then combined with anti-PD1, CXCR2 SM alone (censors on pink line), or vehicle alone (censors on cyan line). Few mice on vehicle alone survived for 2 weeks to allow PD1 treatment as shown in the table below. p Values, chi-square test.
(B and C) IHC for Ki67 in tumors from KPC mice treated with (B) vehicle + PD1 or (C) CXCR2 SM + PD1.
(D and E) IHC for cleaved caspase 3 in tumors from KPC mice treated with (D) vehicle + PD1 or (E) CXCR2 SM + PD1.
(F) FACS analysis of intratumoral CD3+ cells in mice treated as indicated.
(G) Boxplot showing quantification of IHC for CD4+ and CD8+ T cells in tumors from KPC mice treated as indicated.
(H) FACS analysis of intratumoral CD4+, CD8+, CD4+CD25+, and NK1.1+ cells (% of CD3+ cells) in mice treated as indicated.
(I) FACS profile of CD4+ and CD8+ T cells isolated from tumors in mice treated with either vehicle + anti-PD1 or CXCR2 SM + anti-PD1. (F–I) n = 3. p Values, Mann-Whitney U test.
When tumors from mice at endpoint were examined by IHC for Ki67 and cleaved caspase 3 (CC3) we observed a reduction in proliferation following CXCR2 SM + anti-PD1 treatment (Figures 7B and 7C), but no change in apoptosis (Figures 7D and 7E), although both tumor cells and cells of the microenvironment may contribute to this phenotype. Analysis of the tumors by flow cytometry confirmed the presence of an increased percentage of CD3+ T cells in CXCR2 SM + anti-PD1-treated tumors following 2 weeks of treatment (Figure 7F). We were able to show by both IHC (Figure 7G) and fluorescence-activated cell sorting (FACS) analysis (Figure 7H) that the number of both CD4+ and CD8+ T cells was increased in CXCR2 SM-treated mice and, importantly, the number of inhibitory regulatory T cells was actually reduced (Figure 7H). Interestingly, in vehicle-treated mice a substantial proportion of both CD4+ and CD8+ T cells exhibited an effector memory phenotype (CD62L−CD44+). In contrast, in CXCR2 SM-treated mice, this population was less abundant, and a greater proportion of the CD4+ and CD8+ T cells expressed a naive T cell phenotype (CD62L+CD44-) perhaps allowing for increased intratumoral T cell priming (Figure 7I).
Collectively, our data show that in KPC mice, Cxcr2 deficiency, Ly6G+ cell depletion, or pharmacological inhibition of CXCR2 suppresses metastasis in PDAC, and that CXCR2 inhibitors enhance response to chemotherapeutics and immunotherapy to prolong survival. These results suggest that CXCR2 targeting might have therapeutic efficacy in the pre-metastatic setting, and may provide an opportunity for immunotherapy in pancreatic cancer.
Discussion
The outcome for patients suffering from PDAC remains dismal (Siegel et al., 2015), and it is clear that improvements in pancreatic cancer treatment are required. Here, we show that CXCR2 signaling in the myeloid compartment is tumor promoting and required for pancreatic cancer metastasis, and in the notoriously therapy-resistant KPC model of pancreatic cancer we highlight therapeutic opportunities: Not only does inhibition of CXCR2 prevent metastasis; it also augments the efficacy of immune checkpoint inhibitors by allowing T cell infiltration. Indeed, when we compare our mouse tumors with human tumors, we find that Cxcr2 deletion is associated with a switch away from the poorly prognostic squamous identity (Bailey et al., 2016).
Our data support other studies linking inflammation and metastasis (Colotta et al., 2009, Kim et al., 2009) and suggest that inflammatory signaling molecules may be excellent targets in the neoadjuvant treatment of pancreatic cancer. A number of key inflammatory pathways, including IL-6/STAT3, NF-κB, and COX2 pathways, have already been shown to be key in the process of PDAC progression and metastasis (Corcoran et al., 2011, Daniluk et al., 2012, Fukuda et al., 2011, Lesina et al., 2011, Ling et al., 2012). Indeed, patients with significant tumor-associated inflammation have a poor prognosis following surgery (Jamieson et al., 2005).
It has been suggested that pancreatic tumors metastasize prior to the development of a detectable mass in the pancreas (Haeno et al., 2012, Rhim et al., 2012). However, we found that whether CXCR2 signaling was targeted by genetic or pharmacological means, alone, or in combination with chemotherapy, metastasis was significantly inhibited. The ameliorated recruitment of immature myeloid cells to metastatic sites suggests that CXCR2 functions at multiple stages of the metastatic process, hence the striking effect on metastases seen following genetic knockout. In addition, given that CXCR2 inhibition can restrict pancreatitis, targeting CXCR2 signaling in cancer may have additional benefit in terms of ameliorating symptoms (Steele et al., 2015).
Predicting the outcome of targeting stromal elements within PDAC has been difficult. Combination of anti-stromal agents and chemotherapy represents a promising approach to therapy in this disease, with encouraging findings in studies targeting the extracellular matrix glycosaminoglycan or hyaluronic acid (Jacobetz et al., 2013). On the other hand, recent studies targeting tumor-associated fibroblasts and hedgehog signaling at different stages of tumorigenesis produced differing results, with accelerated tumorigenesis if treatment was early and improved survival for late-stage treatment (Olive et al., 2009, Ozdemir et al., 2014, Rhim et al., 2014). The results we present here suggest that CXCR2 has stage- and also cell-type-specific roles in PDAC cancer. During early carcinogenesis, CXCR2 can reinforce senescence in epithelial cells (Acosta et al., 2008); however, once the senescence pathway is abrogated, MDSC/neutrophil CXCR2 drives tumor progression and metastasis. We propose this as the reason that Cxcr2 deletion had no overall effect on survival of KPC mice. These findings are consistent with the expression of CXCR2 in human cancer, which is expressed in PanINs and more rarely in epithelial tumor cells, but is highly expressed in neutrophils/MDSCs at tumor fronts.
We do find that a subset of human tumors show high epithelial CXCR2 expression; however, it is unclear what role CXCR2 is playing in those tumors. Many studies have shown that CXCR2 expression on cancer cells can drive proliferation, invasion, and migration (Matsuo et al., 2009a, Matsuo et al., 2009b, Purohit et al., 2016, Wang et al., 2013). However, our data showing that epithelial cell-specific Cxcr2 loss has no effect on tumor-free survival or metastasis suggests that the effects we observe in the case of Cxcr2 deletion or inhibition are not mediated by tumor cell expression. Together, these findings highlight an intriguing idea, namely, that tumor cells develop a requirement for autocrine CXCR2 signaling once explanted, as a result of loss of the paracrine signaling that exists in vivo. This hypothesis would further underscore the importance of CXCR2 signaling in pancreatic cancer.
Given that most of the mutations that accompany KRAS mutation in PDAC have been shown to abrogate growth arrest/senescence, for example CDKN2A, TP53, and TGFβ pathway-targeting mutations, and the fact that at the time of presentation tumors are highly proliferative and not senescent, concerns over treatment of patients with PDAC with CXCR2 inhibitors are negated. Any remaining concerns are alleviated given the good efficacy we have seen with combination with gemcitabine or anti-PD1. This clearly differentiates CXCR2 inhibition from other stromal targeting agents such as hedgehog inhibitors.
Among the stromal changes we observed following CXCR2 inhibition, one of the most striking was enhanced T cell infiltration. This enhanced T cell infiltration may be responsible for the increased efficacy of gemcitabine, given that enhanced T cell accumulation in KPC tumors can induce stromal remodeling (Stromnes et al., 2015), which in turn can increase the efficacy of gemcitabine (Olive et al., 2009, Provenzano et al., 2012). The infiltration of T cells also rendered tumors sensitive to immunotherapy with PD1-blocking antibody. There are a number of possible mechanisms by which CXCR2 inhibition enables T cell infiltration. For example, we observed a decrease in monocyte/macrophage tumor infiltration in treated mice, and a role for macrophages in the exclusion of T cells from pancreatic tumors has been described (Beatty et al., 2015).
Immunotherapy to reactivate anti-tumor immunity has delivered promising results in several tumor types (Sharma and Allison, 2015), but not, as yet, in pancreatic cancer. Dose scheduling will be important in future clinical trials, given that CXCR2 targeting may be most effective when used to prime the tumor microenvironment. Indeed, the anti-metastatic effect we observe was uncovered when treating mice at an early time point with mostly pre-invasive disease and, as such, may not truly reflect the clinical situation. Nevertheless, our results suggest that inhibiting CXCR2 signaling, and thus recruitment of myeloid cells, may offer the opportunity for immunotherapy in pancreatic cancer, even in patients not eligible for resection. In addition, neoadjuvant targeting of CXCR2 in combination with standard chemotherapy could provide hope for effective targeting of disease progression and metastases in resectable PDAC.
Experimental Procedures
Human Pancreatic Cancer Tissue
All tissue was collected prospectively following informed patient consent. West of Scotland Research Ethics Committee 4 approved the study. For further information see Supplemental Experimental Procedures.
Animal Experiments
All animal experiments were performed under UK Home Office license and approved by the University of Glasgow Animal Welfare and Ethical Review Board. For further information see Supplemental Experimental Procedures.
Cytokine Array on Medium Conditioned by KPC Cells
Murine pancreatic cancer cell lines have been described previously (Morton et al., 2010). AAM-CYT-G2000-4 Ray Biotech slides (Holzel Diagnostics) were used according to the manufacturer's protocol. Laser scanning using the Cy3 channel was used for detection of protein expression.
In Vivo Treatment Experiments
For drug treatments, mice were randomly assigned to cohorts. Treatments used were: CXCR2 pepducin (X1/2pal-i3, Genscript) or scrambled pepducin at 2.5 mg/kg by subcutaneous injection daily; gemcitabine (LC Laboratories) at 100 mg/kg by intraperitoneal (i.p.) injection twice weekly; CXCR2 SM (AstraZeneca) at 100 mg/kg per os (p.o.) twice daily; vehicle p.o. twice daily; anti-PD1 (Biolegend) or isotype control at 10 mg/kg by i.p. injection twice weekly; 1A8 antibody or 2A3 isotype control (BioXcell) at 10 mg/kg by i.p. injection thrice weekly. Efficacy testing of CXCR2 SM is described in Supplemental Experimental Procedures.
Statistical Analysis
Kaplan-Meier survival analysis was performed using log rank tests. Assessment of differences in counts between different mice was performed using non-parametric Mann-Whitney U tests. For all boxplots in the paper, boxes depict the middle 50% of the records and the line indicates the median. Whiskers show the highest and lowest values that are no greater than 1.5 times the interquartile (IQ) range, and asterisks show outliers (cases with values between 1.5 and 3 times the IQ range). Chi-square tests were used to assess the statistical differences in metastasis rate between categorical groups. Spearman's rho correlation coefficient method was used to assess correlation. ANOVA test was used for analysis of qPCR data.
Author Contributions
C.W.S., study design, data acquisition, data analysis, and drafting of manuscript; S.A.K., J.D.G.L., R.U.-G., L.R., M.F., S.B., K.M., Z.W., C.E., J.B.C., M.C., C.N., J.C., N.B., D.K.C., and D.S., data acquisition and analysis; P.B., data acquisition and analysis and drafting of the manuscript; F.B., C.R.C., T.R.J.E., and A.V.B., study supervision; S.B. and R.B.J.N., study concept and design, data analysis, and drafting of manuscript; O.J.S., study concept and design, study supervision, data analysis, and drafting of manuscript; J.P.M., study concept and design, study supervision, data acquisition, data analysis, and drafting of manuscript. All authors read and agreed on the final manuscript.
Acknowledgments
The authors would like to thank the Cancer Research UK Glasgow Centre and the BSU facilities and Histology Service at the Cancer Research UK Beatson Institute. We would also like to thank Jane Hair for curation of the NHSGCC Biorepository and Steve Connolly (AstraZeneca, Molndal) for providing AZ13381758. This work was funded by Cancer Research UK (C596/A18076, C596/A17196) and by a Wellcome Trust Research Training Fellowship (096021, C.W.S.).
Published: June 2, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2016.04.014.
Accession Numbers
RNA-seq data are available in the ArrayExpress database (www.ebi.ac.uk/arrayexpress) under accession number ArrayExpress: E-MTAB-4659.
Supplemental Information
References
- Acharyya S., Oskarsson T., Vanharanta S., Malladi S., Kim J., Morris P.G., Manova-Todorova K., Leversha M., Hogg N., Seshan V.E. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta J.C., O'Loghlen A., Banito A., Guijarro M.V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Almoguera C., Shibata D., Forrester K., Martin J., Arnheim N., Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J., Quinn M.C. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Barber D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Baumgart S., Ellenrieder V., Fernandez-Zapico M.E. Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis. Gut. 2013;62:310–316. doi: 10.1136/gutjnl-2011-301008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart S., Chen N.M., Siveke J.T., Konig A., Zhang J.S., Singh S.K., Wolf E., Bartkuhn M., Esposito I., Hessmann E. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014;4:688–701. doi: 10.1158/2159-8290.CD-13-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne L.J., Beatty G.L., Jhala N., Clark C.E., Rhim A.D., Stanger B.Z., Vonderheide R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty G.L., Chiorean E.G., Fishman M.P., Saboury B., Teitelbaum U.R., Sun W., Huhn R.D., Song W., Li D., Sharp L.L. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty G.L., Winograd R., Evans R.A., Long K.B., Luque S.L., Lee J.W., Clendenin C., Gladney W.L., Knoblock D.M., Guirnalda P.D., Vonderheide R.H. Exclusion of T Cells from pancreatic carcinomas in mice is regulated by Ly6C F4/80 extra-tumor macrophages. Gastroenterology. 2015;149:201–210. doi: 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris H.A., 3rd, Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- Cacalano G., Lee J., Kikly K., Ryan A.M., Pitts-Meek S., Hultgren B., Wood W.I., Moore M.W. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- Clark C.E., Hingorani S.R., Mick R., Combs C., Tuveson D.A., Vonderheide R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- Colotta F., Allavena P., Sica A., Garlanda C., Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- Conroy T., Desseigne F., Ychou M., Bouche O., Guimbaud R., Becouarn Y., Adenis A., Raoul J.L., Gourgou-Bourgade S., de la Fouchardiere C. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- Corcoran R.B., Contino G., Deshpande V., Tzatsos A., Conrad C., Benes C.H., Levy D.E., Settleman J., Engelman J.A., Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–5029. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniluk J., Liu Y., Deng D., Chu J., Huang H., Gaiser S., Cruz-Monserrate Z., Wang H., Ji B., Logsdon C.D. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Montero C.M., Salem M.L., Nishimura M.I., Garrett-Mayer E., Cole D.J., Montero A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson L.F., Hawkins W.G., Goedegebuure P. Potential targets for pancreatic cancer immunotherapeutics. Immunotherapy. 2011;3:517–537. doi: 10.2217/imt.11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eash K.J., Greenbaum A.M., Gopalan P.K., Link D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C., Jones J.O., Kraman M., Wells R.J., Deonarine A., Chan D.S., Connell C.M., Roberts E.W., Zhao Q., Caballero O.L. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife B.T., Pauken K.E., Eagar T.N., Obu T., Wu J., Tang Q., Azuma M., Krummel M.F., Bluestone J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A., Wang S.C., Morris J.P., Folias A.E., Liou A., Kim G.E., Akira S., Boucher K.M., Firpo M.A., Mulvihill S.J., Hebrok M. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–455. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein D., El-Maraghi R.H., Hammel P., Heinemann V., Kunzmann V., Sastre J., Scheithauer W., Siena S., Tabernero J., Teixeira L. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015;107 doi: 10.1093/jnci/dju413. [DOI] [PubMed] [Google Scholar]
- Guerra C., Schuhmacher A.J., Canamero M., Grippo P.J., Verdaguer L., Perez-Gallego L., Dubus P., Sandgren E.P., Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Haeno H., Gonen M., Davis M.B., Herman J.M., Iacobuzio-Donahue C.A., Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148:362–375. doi: 10.1016/j.cell.2011.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highfill S.L., Cui Y., Giles A.J., Smith J.P., Zhang H., Morse E., Kaplan R.N., Mackall C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl Med. 2014;6:237ra267. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hruban R.H., Wilentz R.E., Kern S.E. Genetic progression in the pancreatic ducts. Am. J. Pathol. 2000;156:1821–1825. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi H., Chytil A., Gorska A.E., Aakre M.E., Bierie B., Tada M., Mohri D., Miyabayashi K., Asaoka Y., Maeda S. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Invest. 2011;121:4106–4117. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobetz M.A., Chan D.S., Neesse A., Bapiro T.E., Cook N., Frese K.K., Feig C., Nakagawa T., Caldwell M.E., Zecchini H.I. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson N.B., Glen P., McMillan D.C., McKay C.J., Foulis A.K., Carter R., Imrie C.W. Systemic inflammatory response predicts outcome in patients undergoing resection for ductal adenocarcinoma head of pancreas. Br. J. Cancer. 2005;92:21–23. doi: 10.1038/sj.bjc.6602305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson T., Clarke M., Steele C.W., Samuel M.S., Neumann J., Jung A., Huels D., Olson M.F., Das S., Nibbs R.J., Sansom O.J. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 2012;122:3127–3144. doi: 10.1172/JCI61067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneider N.C., Agarwal A., Leger A.J., Kuliopulos A. Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat. Med. 2005;11:661–665. doi: 10.1038/nm1245. [DOI] [PubMed] [Google Scholar]
- Kim S., Takahashi H., Lin W.W., Descargues P., Grivennikov S., Kim Y., Luo J.L., Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Vredeveld L.C., Douma S., van Doorn R., Desmet C.J., Aarden L.A., Mooi W.J., Peeper D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Lal A., Peters H., St Croix B., Haroon Z.A., Dewhirst M.W., Strausberg R.L., Kaanders J.H., van der Kogel A.J., Riggins G.J. Transcriptional response to hypoxia in human tumors. J. Natl. Cancer Inst. 2001;93:1337–1343. doi: 10.1093/jnci/93.17.1337. [DOI] [PubMed] [Google Scholar]
- Lesina M., Kurkowski M.U., Ludes K., Rose-John S., Treiber M., Kloppel G., Yoshimura A., Reindl W., Sipos B., Akira S. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Li A., King J., Moro A., Sugi M.D., Dawson D.W., Kaplan J., Li G., Lu X., Strieter R.M., Burdick M. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am. J. Pathol. 2011;178:1340–1349. doi: 10.1016/j.ajpath.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J., Kang Y., Zhao R., Xia Q., Lee D.F., Chang Z., Li J., Peng B., Fleming J.B., Wang H. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenfels A.B., Maisonneuve P., DiMagno E.P., Elitsur Y., Gates L.K., Jr., Perrault J., Whitcomb D.C. Hereditary pancreatitis and the risk of pancreatic cancer. International hereditary pancreatitis study group. J. Natl. Cancer Inst. 1997;89:442–446. doi: 10.1093/jnci/89.6.442. [DOI] [PubMed] [Google Scholar]
- Maniati E., Bossard M., Cook N., Candido J.B., Emami-Shahri N., Nedospasov S.A., Balkwill F.R., Tuveson D.A., Hagemann T. Crosstalk between the canonical NF-kappaB and Notch signaling pathways inhibits Pparγ expression and promotes pancreatic cancer progression in mice. J. Clin. Invest. 2011;121:4685–4699. doi: 10.1172/JCI45797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y., Ochi N., Sawai H., Yasuda A., Takahashi H., Funahashi H., Takeyama H., Tong Z., Guha S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer. 2009;124:853–861. doi: 10.1002/ijc.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y., Raimondo M., Woodward T.A., Wallace M.B., Gill K.R., Tong Z., Burdick M.D., Yang Z., Strieter R.M., Hoffman R.M., Guha S. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int. J. Cancer. 2009;125:1027–1037. doi: 10.1002/ijc.24383. [DOI] [PubMed] [Google Scholar]
- Morton J.P., Karim S.A., Graham K., Timpson P., Jamieson N., Athineos D., Doyle B., McKay C., Heung M.Y., Oien K.A. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292–303. doi: 10.1053/j.gastro.2010.03.034. [DOI] [PubMed] [Google Scholar]
- Nicholls D.J., Wiley K., Dainty I., MacIntosh F., Phillips C., Gaw A., Mardh C.K. Pharmacological characterization of AZD5069, a slowly reversible CXC chemokine receptor 2 antagonist. J. Pharmacol. Exp. Ther. 2015;353:340–350. doi: 10.1124/jpet.114.221358. [DOI] [PubMed] [Google Scholar]
- Olive K.P., Jacobetz M.A., Davidson C.J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M.A., Caldwell M.E., Allard D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsson T., Acharyya S., Zhang X.H., Vanharanta S., Tavazoie S.F., Morris P.G., Downey R.J., Manova-Todorova K., Brogi E., Massague J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P.P., Cuevas C., Chang A.E., Goel V.K., Von Hoff D.D., Hingorani S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit A., Varney M., Rachagani S., Ouellette M.M., Batra S.K., Singh R.K. CXCR2 signaling regulates KRAS(G12D)-induced autocrine growth of pancreatic cancer. Oncotarget. 2016;7:7280–7296. doi: 10.18632/oncotarget.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid M.D., Basturk O., Thirabanjasak D., Hruban R.H., Klimstra D.S., Bagci P., Altinel D., Adsay V. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod. Pathol. 2011;24:1612–1619. doi: 10.1038/modpathol.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim A.D., Mirek E.T., Aiello N.M., Maitra A., Bailey J.M., McAllister F., Reichert M., Beatty G.L., Rustgi A.K., Vonderheide R.H. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim A.D., Oberstein P.E., Thomas D.H., Mirek E.T., Palermo C.F., Sastra S.A., Dekleva E.N., Saunders T., Becerra C.P., Tattersall I.W. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintigny P., Massarelli E., Lin S., Ahn Y.H., Chen Y., Goswami S., Erez B., O'Reilly M.S., Liu D., Lee J.J. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 2013;73:571–582. doi: 10.1158/0008-5472.CAN-12-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2015. CA Cancer J. Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Steele C.W., Karim S.A., Foth M., Rishi L., Leach J.D., Porter R.J., Nixon C., Jeffry Evans T.R., Carter C.R., Nibbs R.J. CXCR2 inhibition suppresses acute and chronic pancreatic inflammation. J. Pathol. 2015;237:85–97. doi: 10.1002/path.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes I.M., Brockenbrough J.S., Izeradjene K., Carlson M.A., Cuevas C., Simmons R.M., Greenberg P.D., Hingorani S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63:1769–1781. doi: 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes I.M., Schmitt T.M., Hulbert A., Brockenbrough J.S., Nguyen H.N., Cuevas C., Dotson A.M., Tan X., Hotes J.L., Greenberg P.D., Hingorani S.R. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell. 2015;28:638–652. doi: 10.1016/j.ccell.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Wu Y., Hou Y., Guan X., Castelvetere M.P., Oblak J.J., Banerjee S., Filtz T.M., Sarkar F.H., Chen X. CXCR2 macromolecular complex in pancreatic cancer: a potential therapeutic target in tumor growth. Transl Oncol. 2013;6:216–225. doi: 10.1593/tlo.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek S.K., Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–417. doi: 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winograd R., Byrne K.T., Evans R.A., Odorizzi P.M., Meyer A.R., Bajor D.L., Clendenin C., Stanger B.Z., Furth E.E., Wherry E.J., Vonderheide R.H. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol. Res. 2015;3:399–411. doi: 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H.H., Pickup M., Pang Y., Gorska A.E., Li Z., Chytil A., Geng Y., Gray J.W., Moses H.L., Yang L. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70:6139–6149. doi: 10.1158/0008-5472.CAN-10-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Huang J., Ren X., Gorska A.E., Chytil A., Aakre M., Carbone D.P., Matrisian L.M., Richmond A., Lin P.C., Moses H.L. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn J.I., Gabrilovich D.I. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010;40:2969–2975. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.