

Abstract
Multiple system atrophy (MSA) is a rare atypical parkinsonian disorder characterized by a rapidly progressing clinical course and at present without any efficient therapy. Neuropathologically, myelin loss and neurodegeneration are associated with α-synuclein accumulation in oligodendrocytes, but underlying pathomechanisms are poorly understood. Here, we analyzed the impact of oligodendrocytic α-synuclein on the formation of myelin sheaths in order to define a potential interventional target for MSA. Post-mortem analyses of MSA patients and controls were performed to quantify myelin and oligodendrocyte numbers. As pre-clinical models, we used transgenic MSA mice, a myelinating stem cell-derived oligodendrocyte-neuron co-culture, and primary oligodendrocytes to determine functional consequences of oligodendrocytic α-synuclein overexpression on myelination. We detected myelin loss accompanied by preserved or even increased numbers of oligodendrocytes in post-mortem MSA brains or transgenic mouse forebrains, respectively, indicating an oligodendrocytic dysfunction in myelin formation. Corroborating this observation, overexpression of α-synuclein in primary and stem cell-derived oligodendrocytes severely impaired myelin formation, defining a novel α-synuclein-linked pathomechanism in MSA. We used the pro-myelinating activity of the muscarinic acetylcholine receptor antagonist benztropine to analyze the reversibility of the myelination deficit. Transcriptome profiling of primary pre-myelinating oligodendrocytes demonstrated that benztropine readjusts myelination-related processes such as cholesterol and membrane biogenesis, being compromised by oligodendrocytic α-synuclein. Additionally, benztropine restored the α-synuclein-induced myelination deficit of stem cell-derived oligodendrocytes. Strikingly, benztropine also ameliorated the myelin deficit in transgenic MSA mice, resulting in a prevention of neuronal cell loss.
In conclusion, this study defines the α-synuclein-induced myelination deficit as a novel and crucial pathomechanism in MSA. Importantly, the reversible nature of this oligodendrocytic dysfunction opens a novel avenue for an intervention in MSA.
Keywords: Multiple system atrophy, Oligodendrocytes, Oligodendrocyte progenitor cells, Myelin, α-synuclein
Introduction
The atypical parkinsonian disorder multiple system atrophy (MSA) is a sporadic, rare, and age-related neurodegenerative disease with rapid progression. From onset of symptoms, the median survival is less than 10 years [32]. Importantly, there are currently no efficient symptomatic or disease-modifying therapies available [13]. MSA patients present with a heterogeneous combination of autonomic dysfunction, parkinsonism, cerebellar ataxia, and pyramidal features as a consequence of pronounced neurodegenerative changes. The predominance of degeneration within the striatonigral or olivopontocerebellar system classifies MSA in a parkinsonian (MSA-P) and a cerebellar (MSA-C) subtype [16]. In addition to widespread axonal and neuronal loss, major neuropathological features of MSA are myelin loss, micro- and astrogliosis [16, 35, 38]. Although MSA etiology is poorly understood, convergent lines of evidence indicate that oligodendrocytes, the myelinating cells of the central nervous system, may be causatively involved in MSA pathogenesis [15, 16, 47].
Myelin sheaths are generated throughout life by preexisting or newly formed oligodendrocytes derived from adult oligodendrocyte progenitor cells (OPCs) [54]. Active myelination in the adult brain mediates a continuous myelin turnover [53] and ensures myelin remodeling required for learning processes [37], but also contributes to myelin repair upon demyelination under pathological conditions [22, 39]. Remarkably, proper remyelination is essentially required to prevent demyelination-associated axonal and neuronal degeneration [23]. In contrast, myelin loss without sufficient remyelination leads to a breakdown of oligodendrocyte-derived metabolic supply at the myelin-axon interface, which consequently results in axonal dysfunction and neuronal loss [21, 46, 51]. Hence and although not yet been considered, targeting myelin regeneration may represent a promising approach to slow or even halt disease progression in MSA.
The key neuropathological feature of MSA is the accumulation of α-synuclein in glial cytoplasmic inclusions within oligodendrocytes, which classifies MSA as synucleinopathy [15]. According to the current consensus guidelines [19], post-mortem detection of abundant α-synuclein-positive glial cytoplasmic inclusions is required for definite MSA diagnosis. The association of genetic variants in the SNCA gene coding for α-synuclein points toward a crucial role of α-synuclein during MSA pathogenesis [43]. Furthermore, most recent studies have shown increased α-synuclein mRNA levels in MSA patients [4, 11], suggesting that pathological α-synuclein accumulation in MSA is the result of excessive α-synuclein gene expression. In line, transgenic mice overexpressing α-synuclein in oligodendrocytes recapitulate the most important neuropathological features of MSA, including myelin loss, neurodegeneration, and microgliosis [24, 45, 52]. Intriguingly, the level of α-synuclein overexpression corresponds to the severity of functional and neurodegenerative features [45]. In fact, transgenic mice with highest oligodendrocytic α-synuclein overexpression die prematurely at 4-6 months of age, resembling the rapidly progressing clinical course of MSA patients [45].
In addition to its pathological overexpression in oligodendrocytes of MSA patients, α-synuclein is physiologically expressed in murine and human OPCs and downregulated upon maturation towards pre-myelinating oligodendrocytes [8, 40]. In this context, we have recently shown that sustained α-synuclein expression in primary rat OPCs delays myelin gene expression [14].
Considering the increasing evidence for a physiological role of α-synuclein in oligodendrocyte maturation, we hypothesized that α-synuclein accumulation within oligodendrocytes impairs myelin maintenance and repair, potentially explaining myelin loss in early stages of MSA. While we detected profound myelin loss in the presence of widespread α-synuclein pathology in the putamen of MSA patients, oligodendrocytes were surprisingly preserved. Corroborating this observation, overexpression of human α-synuclein in mice, in mouse embryonic stem cell-derived oligodendrocytes co-cultured with cortical neurons, and in primary rat oligodendrocytes severely impaired myelin formation. Intriguingly, the deficit in myelin formation linked to α-synuclein accumulation was restored in vitro by the muscarinic acetylcholine receptor antagonist benztropine, which has recently been identified from a high-throughput screen as a potent pro-myelinating small molecule [7]. In addition, benztropine attenuated the myelin deficit in transgenic mice overexpressing α-synuclein in oligodendrocytes, resulting in the prevention of neuronal loss, whereas microglial cell density was not altered.
Our data provide compelling evidence for α-synuclein-induced myelination deficit without oligodendrocyte loss in MSA. Given that the pro-myelinating intervention using benztropine was able to overcome the α-synuclein-induced oligodendrocytic dysfunction in vitro and in vivo, our study provides a possible translation for a novel treatment of MSA.
Methods
Human post-mortem samples
Human post-mortem samples used in this study were obtained from the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam (open access: www.brainbank.nl). MSA was clinically and neuropathologically diagnosed according to current consensus guidelines [19]. Tissue blocks were cut into 5μm sections and processed using standard protocols for hematoxylin and Luxol Fast Blue/Periodic Acid Schiff. α-Synuclein-positive inclusions were detected using a rabbit anti human α-synuclein antibody (clone 5G4; Analytik Jena AG; #847-0102004001). Luxol Fast Blue intensity was quantified by a blinded researcher using ImageJ as mean gray value in at least 10 outlined striae crossing the putamen. Oligodendrocytic cell density was quantified by a blinded researcher using ImageJ in at least five outlined striae.
Animals and treatment
Mice overexpressing human α-synuclein under the control of a murine myelin basic protein (Mbp) promoter (high-expressing line 29; on a BDF1 background) were previously generated [45]. In contrast to moderately expressing lines (e.g., line 1), strongest α-synuclein overexpression in the forebrain of line 29 results in severe neurological deficits, such as tremor, ataxia, and pronounced seizure activity, and premature death at 4-6 months of age [45]. Mice were maintained under standard animal housing conditions with a 12-hour-day/night cycle and free access to food and water. Mice (mixed gender) received daily intraperitoneal injections of vehicle or benztropine mesylate (Alfa Aesar; in saline; 2 mg/kg) starting at the age of 8 weeks for 30 consecutive days. All experiments were carried out in accordance with the guidelines of the National Institutes of Health (NIH) in regard to the care and use of animals for experimental procedures. Mice were transcardially perfused with PBS and brains were collected. Brains were post-fixed in 4% paraformaldehyde (in PBS) overnight and transferred into 30% sucrose (in PBS).
Magnetic resonance imaging of mouse brains
Left hemispheres (n = 3 per group, females) were scanned on a preclinical ultra-high field magnetic resonance imaging system (7 Tesla ClinScan 70/30, Bruker). The imaging protocol consisted of a 3D T2-weighted turbo spin echo sequence for morphological images and 3D multi-echo gradient echo sequence was acquired to calculate voxel-based maps of T2* (Syngo software, Siemens). On T2-weighted images, the corpus callosum was segmented using Chimaera's segmentation tool (Chimeara). As previously described [27], T2* relaxations time were determined to quantify myelin. By transferring the segmentation mask to the T2*-map, T2* relaxation time of the corpus callosum was calculated.
Histology and electron microscopy
Left hemispheres (n = 5 per group, males) were cut on a sliding microtome into 40μm coronal sections. For Luxol Fast Blue staining, one section per animal (at Bregma +1) was used to group all samples on a single glass slide, ensuring optimal comparability. Sections were stained in 0.1% Luxol Fast Blue solution and destained using 0.05% lithium carbonate and 70% ethanol until white matter became apparent dark blue. Images were taken with equal exposure time using StereoInvestigator software (MicroBrightFlield) on an Imager.M2 microscope (Zeiss). ImageJ was used to calculate the mean gray value of inverted images. Immunofluorescent analyses were performed as described previously [36] using following primary antibodies: rabbit anti Olig2 (Millipore; AB9619; 1:500), goat anti platelet-derived growth factor receptor-α (Pdgfrα) (R&D Systems; AF1062; 1:250), mouse anti glutathione-s-transferase-π (Gstπ) (BD; 610718; 1:100), mouse anti NeuN (Millipore; MAB377; 1:200), rabbit anti Iba1 (WAKO; 019-19741; 1:1000), and rat anti human α-synuclein (15G7, ENZO; ALX-804-258; 1:200). For quantification of cell density, 3 coronal sections per animal were analyzed. Three images per section were taken using ApoTome technology (Zeiss). The corpus callosum and the motor cortex (at Bregma -1 to +1) were outlined and quantified using ImageJ by a blinded researcher.
Ultrastructural analysis of myelin was performed as previously described [33]. Briefly, vibratome sections (n = 6) were post-fixed in 1% glutaraldehyde, treated with osmium tetroxide, embedded in epon araldite and sectioned with an ultramicrotome (Leica). Electron micrographs were randomly obtained from three grids at a magnification of 5,000× and 20,000× using a Zeiss OM 10 electron microscope. Myelinated axons and myelin layers were quantified in the corpus callosum in areas of similar axon size.
Lentiviral vectors
Third-generation lentiviral vectors coding for human α-synuclein and/or an internal ribosomal entry site (IRES) followed by the enhanced green fluorescent protein (Gfp) sequence were generated as previously described [14]. For transducing myelinating co-cultures, the identical Mbp promoter [20] as used for transgene expression in MBP29 mice was cloned into the lentiviral vector using the restriction enzymes Cla1 and Age1, resulting in the α-synuclein expression vector MBP-SYN-IRES-GFP (MBP-SYN-IG) and the control vector MBP-IRES-GFP (MBP-IG). For primary oligodendrocyte monocultures, an elongation factor 1-α promoter was used to constitutively drive transgene expression (α-synuclein expression vector: EF1a-SYN-IG; control vector: EF1a-IG) [14]. Lentiviral titers were determined by biological titration in HEK293T cells using flow cytometry.
Myelinating co-culture
The protocol to obtain a myelinating stem cell-derived co-culture has recently been described [25]. In this study, the functionality of stem cell-derived oligodendrocytes in terms of myelination has been confirmed by demonstrating the formation of physiological myelin on an ultrastructural level. Briefly, E14Tg2a mouse embryonic stem cells were differentiated into cortical neurons [17]. To obtain mature oligodendrocytes, neural progenitor cells were derived from mouse D3 embryonic stem cells as described elsewhere [34] and underwent a two-step differentiation/maturation protocol [25]. OPCs were differentiated from neural progenitor cells using differentiation medium (DMEM/F12-GlutaMax, N2, B27, 10ng/ml insulin-like growth factor-1, 10 ng/ml PDGF-aa) for 7 days before switching to maturation medium for another 7 days (DMEM/F12-GlutaMax, N2, B27, 10 ng/ml ciliary neurotrophic factor, 5 ng/ml neurotrophin-3, 40 ng/ml triiodothyronine (T3)). Medium change was conducted every other day.
Twelve hours prior to starting the co-culture, oligodendrocytes were lentivirally transduced using MBP-SYN-IG or MBP-IG. Oligodendrocytes were detached using accutase and seeded on cortical neurons at a density of 50,000 per well. Additionally, oligodendrocytes were plated in poly-ornithine and laminin-coated 8-well chamber slides to control for specificity of lentiviral transduction. Co-cultures were maintained for 14 days in neuron medium (1:1 DDM and Neurobasal/B27, 20 ng/ml glial cell-derived neurotrophic factor, 500 μg/ml 3′5′-cyclic adenosine monophosphate, 0.2 μM ascorbic acid) supplemented with T3 at 40 ng/ml. Benztropine was added at 1 μM to assess its effects on myelination. Medium was changed every other day.
Immunocytochemistry
Myelinating co-cultures and D3 embryonic stem cell-derived oligodendrocytes were stained as recently described [25]. The following primary antibodies were used in combination for overnight incubation: chicken anti Gfp (Aves Labs, GFP-1020; 1:250), rat anti Mbp (AbD Serotec; MCA409S; 1:50), mouse anti beta-III-tubulin (Tuj1, Covance, MMS-435P; 1:1000), rat anti human α-synuclein (15G7, ENZO; ALX-804-258; 1:200), rabbit anti Pdgfrα (Santa Cruz; sc-338; 1:400), rabbit anti cleaved Caspase 3 (Asp175; Cell Signaling; #9661; 1:500). For O4 staining, the primary antibody (mouse anti O4; MAB1326; R&D Systems; 1:40) was added to the supernatant 30 min prior to fixation and permeabilization. Images were acquired on a laser scanning microscope LSM710 (Zeiss) using ZEN black software.
Quantification of myelination in vitro
Myelination in the oligodendrocyte-neuron co-culture was semi-automatically quantified using the Computer-assisted Evaluation of Myelin formation (CEM) tool [25]. Myelination was assessed by analyzing individual oligodendrocytes. Upon lentiviral transduction, Gfp/Mbp-positive oligodendrocytes were randomly selected from at least 8 different wells per experiment and condition by a blinded researcher. Images were processed using ImageJ (generation of binary images; calculation of Mbp-positive, Tuj1-positive, and Mbp/Tuj1-positive pixels; pictures with less than 200,000 Tuj1 pixels were excluded) and MATLAB (MathWorks; cell body removal). Myelin pixels (Mbp/Tuj1-positive) relative to total Mbp-positive pixels were calculated and considered as myelination (in percentage) of an individual oligodendrocyte.
Primary OPC culture
Primary rat OPCs were isolated, maintained, and transduced as previously described [14]. Briefly, mixed glial cultures were obtained from P0-P2 neonatal Wistar rats and kept in DMEM supplemented with 10% FCS. After 14 days in vitro, OPCs were separated by overnight orbital shaking at 37°C and plated on poly-ornithine coated plates or coverslips. OPCs were allowed to recover by culturing in SATO medium [6] supplemented with Pdgf-aa and fibroblast growth factor-2 at 10 ng/ml. Twenty-four hours after seeding, OPCs were lentivirally transduced at a multiplicity of infection of 2. Maturation was initiated 48 h after seeding by withdrawal of growth factors. To assess the effects of benztropine on OPC maturation, benztropine mesylate (Alfa Aesar) dissolved in deionized water was added at the initiation of maturation and compared to vehicle control. Medium was completely replaced after 2 days of maturation. Oligodendrocyte maturity and the transcriptome were analyzed after 4 days.
Western blot
Western blot analysis was performed according to standard protocols. The NuPage® gel electrophoresis system (Invitrogen) was used according to the manufacturer's protocol. Primary antibodies: rat anti Mbp (Abd Serotec; MCA409S; 1:500) and mouse anti glyceraldehyde 3-phosphate dehydrogenase (Gapdh; Millipore; MAB374; 1:100000).
Quantitative PCR (qPCR)
Total RNA of primary oligodendrocytes was extracted using the RNeasy mini kit according to the manufacturer's instructions (Qiagen). CDNA was generated using GoScriptTM Reverse Transcription System (Promega). qPCRs were performed on a Light Cycler 480 (Roche) using the Sso Fast EvaGreen Supermix (Biorad). Primers: Mbp (5′-ACTACGGCTCCCTGCCCCAG-3′, 5′-GGGATGGAGGGGGTGTACGAGG-3′; detecting all five Mbp transcript variants in Rattus norvegicus), Gapdh (5′-CACAGTCAAGGCTGAGAATGGGAAG-3′, 5′-GTGGTTCACACCCATCACAAACATG-3′)
Transcriptome analysis
Before and during library preparation, RNA quality was ascertained using a 2100 Bioanalyzer system (Agilent Technologies). Barcoded strand-specific whole transcriptome sequencing libraries were prepared from 100 ng of DNase digested total RNA using Ovation Human FFPE RNA-Seq System (NuGEN) according to the manufacturer's instructions. Rat rRNA and tRNA were depleted using custom-designed oligonucleotides in strand selection. Pooled libraries were sequenced on a HiSeq 2500 platform (Illumina) generating on average 86 million 101bp single-end reads.
After alignment against the Rattus norvegicus reference genome Rnor5.0 using STAR v.2.4.0i [9] absolute read counts for all Ensembl genes (version 75) were determined with HTSeq count v.0.6.1 [3]. Differential expression analysis accounting for the paired design with respect to primary cell cultures was performed using the DESeq2 package v.1.6.3 [30]. Gene ontology (GO) term enrichment of differentially expressed genes against background of expressed genes after independent filtering was performed using the Gene Ontology Enrichment Analysis And Visualization (GOrilla) tool [12]. P-values were corrected using the false discovery rate method.
Statistics
Data were analyzed using GraphPad Prism®. Graphs are presented as mean ± standard error of mean. One-way analysis of variation (ANOVA) with post-hoc Dunnett's multiple comparison test was used when comparing to control. Two-tailed, unpaired student's t-test was performed for comparing two groups. P values <0.05 were considered significant.
Results
Myelin loss despite preserved oligodendrocyte numbers in MSA and in mice overexpressing α-synuclein in oligodendrocytes
While myelin loss has previously been reported as pathological feature present already in early stages of MSA [35, 38, 47], underlying causes for this loss have not yet been elucidated. To assess whether demyelination is a result of oligodendrocyte degeneration or rather an oligodendrocytic dysfunction in terms of myelin formation, we quantified myelin and the density of oligodendrocytic cells in post-mortem tissue of MSA patients. Selection criteria for MSA patients were the parkinsonian phenotype (predominance of striatonigral degeneration, MSA-P) based on clinical history and the abundant presence of α-synuclein-positive inclusions within the putamen (Fig. 1; for details of the cohort see Suppl. Table T1). A combined Luxol Fast Blue/Periodic Acid Schiff staining was used to visualize myelin (Fig. 1a). Oligodendrocytes were identified by morphology in hematoxylin stained sections according to Salvesen and colleagues [41] (Fig. 1a, b). While myelin was reduced by 30±5% in MSA patients relative to age- and gender-matched controls, no significant change in the density of oligodendrocytes was detected within fiber tracts crossing the putamen (Fig. 1c). To confirm this dichotomy, we compared myelin with oligodendrocyte density in mice overexpressing α-synuclein in oligodendrocytes under the control of a murine Mbp promoter (high-expressing line 29; hereafter referred to as MBP29). In this well-established preclinical MSA mouse model, we analyzed the corpus callosum, which represents a prototypical and - in contrast to the striatum - pure white matter region, showing most abundant α-synuclein expression in the forebrain and allowing a comprehensive quantification of myelin and oligodendrocytes (Suppl. Fig. S1a, b). Furthermore, mature oligodendrocytes strongly expressed α-synuclein in MBP29 mice, whereas OPCs did not show α-synuclein immunoreactivity (Suppl. Fig. S1c). In similarity to MSA patients, we observed a reduction of myelin by 44±5% accompanied by an 1.7 fold-increased oligodendrocyte density in MBP29 mice compared to non-transgenic littermates (Fig. 2).
Fig. 1.
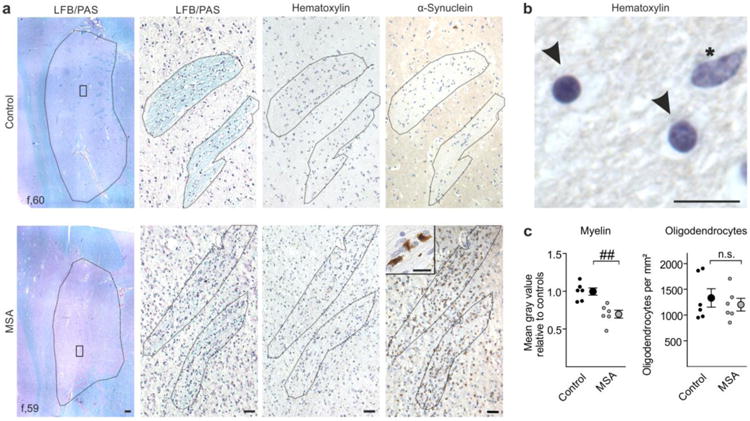
Putaminal myelin loss in multiple system atrophy (MSA) without alterations in oligodendrocyte density. a Representative Luxol Fast Blue/Periodic Acid Schiff (LFB/PAS) stained images (left) depict the putamen (encircled) of a control (upper panel) and a MSA patient (lower panel). The putamen of the MSA patient showed a reduced Luxol Fast Blue staining intensity reflecting severe loss of myelin. Scale bar: 500μm. Boxed areas within the left images are magnified showing LFB/PAS-, hematoxylin-, and α-synuclein-stained striae. Note abundant α-synuclein-positive inclusions in the putamen of the MSA patient (magnified as insert). Scale bars: 50μm, insert 10μm. b Oligodendrocytes (arrowheads; round-shaped, dense chromatin) were distinguished by morphology from astrocytes (asterisk; larger and more elongated in size, loose chromatin) in the hematoxylin stained-sections. Scale bar: 10μm. c While myelin was significantly reduced in MSA patients (gray dots) compared to controls (black dots), oligodendrocyte density was unaltered (n = 6). Data are shown as mean ± standard error of mean. T-test: ##p < 0.01, n.s. (not significant) p > 0.05. f = female.
Fig. 2.
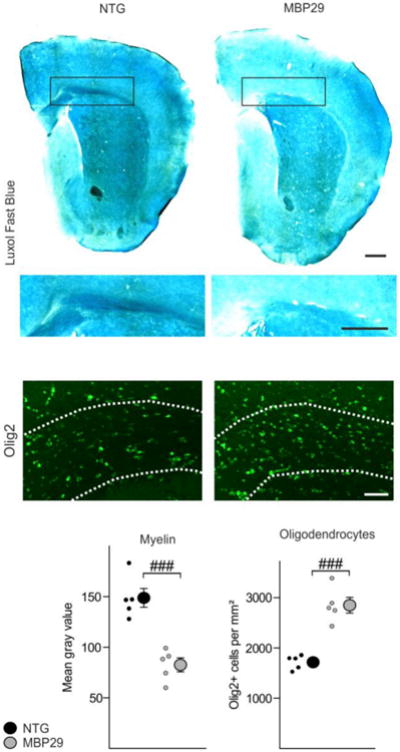
Severe myelin deficit despite increased oligodendrocyte density in mice overexpressing human α-synuclein under a myelin basic protein-promoter (MBP29). MBP29 mice (3 months of age) showed a profound myelin deficit (illustrated by gray scale images of a Luxol Fast Blue staining and quantified within the corpus callosum; scale bars: 500μm). Density of callosal oligodendrocytes (identified by Olig2; scale bar: 50μm) was increased by 1.7 fold. Data are shown as mean ± standard error of mean. T-test (n = 5): ###p < 0.001. NTG = non-transgenic mice.
Impaired myelin formation upon oligodendrocytic α-synuclein overexpression in an oligodendrocyte-neuron co-culture
The intriguing dichotomy of a profound myelin deficit despite preserved or even increased oligodendrocyte numbers in MSA patients (Fig. 1) and MBP29 mice (Fig. 2), respectively, suggests that an oligodendrocytic dysfunction may underlie myelin loss in MSA. Thus, we next asked whether oligodendrocytic α-synuclein accumulation impacts the process of myelin formation. To analyze functional consequences of α-synuclein on myelination, we took advantage of a recently described approach to assess myelin sheath formation in vitro by co-culturing mouse embryonic stem cell-derived oligodendrocytes and cortical neurons [25] (Fig 3a). We adapted the protocol to quantify myelination of individual oligodendrocytes. Withdrawal of the pro-myelinating thyroid hormone T3 from the culture medium significantly decreased myelin formation, demonstrating that this method is suitable to quantify myelination of individual oligodendrocytes (Suppl. Fig. S2). To study the effect of intraoligodendrocytic α-synuclein on myelin formation, lentiviral vectors with the Mbp promoter as a regulatory element were generated (α-synuclein expression vector: MBP-SYN-IG; control vector: MBP-IG), yielding a high proportion of α-synuclein-overexpressing oligodendrocytes (Fig. 3b). The usage of the Mbp promoter resulted in α-synuclein and/or Gfp expression in mature oligodendrocytes, but not in OPCs (Fig. 3c). Upon transduction, there was no difference in the numbers of apoptotic, activated Caspase 3-positive oligodendrocytes between α-synuclein-overexpressing and control oligodendrocytes (Suppl. Fig. S3). A comprehensive and computer-assisted quantification on a single cell level revealed that α-synuclein overexpression significantly reduced myelin sheath formation by 21±3% compared to oligodendrocytes transduced with the control vector lacking the α-synuclein-coding sequence (Fig 3d, e). Importantly, Tuj1- and Mbp-positive pixels were not altered upon α-synuclein overexpression (Suppl. Fig. S4), indicating that there was no effect of Mbp promoter-driven α-synuclein overexpression on neuronal and oligodendrocytic survival in the co-culture.
Fig. 3.
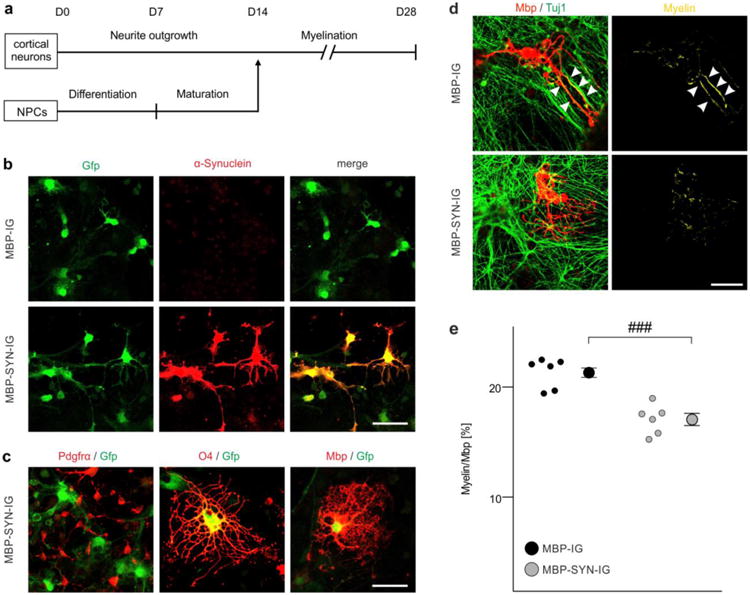
Impaired myelination of α-synuclein-overexpressing mouse embryonic stem cell-derived oligodendrocytes. a The experimental paradigm to analyze myelination of individual oligodendrocytes in the mouse embryonic stem cell-derived oligodendrocyte-neuron co-culture is schematically illustrated according to [25]. b Stem cell-derived oligodendrocytes expressed human α-synuclein (when transduced with MBP-SYN-IG expression vector only; lower panel) and green fluorescent protein (Gfp) (also when transduced with the MBP-IG control vector; upper panel). Expression of α-synuclein overlapped with GFP expression in MBP-SYN-IG transduced oligodendrocytes. Scale bar: 20μm. c Co-labeling with stage-specific oligodendrocytic markers revealed the specificity of myelin basic protein (Mbp) promoter-driven transgene expression for mature oligodendrocytes (positive for the early maturation marker O4 and Mbp as a marker for terminal maturation), whereas transgene expression was not detectable in platelet-derived growth factor receptor-α (Pdgfrα)-positive oligodendrocyte progenitor cells. Scale bar: 20μm. d Representative images of control (transduced with MBP-IG prior to starting the co-culture) and α-synuclein-overexpressing (transduced with MBP-SYN-IG) Mbp-positive oligodendrocytes co-cultured with beta-III-tubulin (Tuj1)-positive cortical neurons are depicted. Myelination (yellow; arrowheads) was regularly detected under control condition, whereas α-synuclein overexpression impaired myelin formation. Scale bar: 20μm. e Myelination of individual oligodendrocytes (altogether: MBP-IG: 235 cells, MBP-SYN-IG: 215 cells) in the stem cell-derived co-culture was quantified (n = 6) by calculating the ratio of Mbp/Tuj1-positive pixels over total Mbp-positive pixels and is shown as percentage. α-Synuclein-overexpressing oligodendrocytes (gray dots) formed significantly less myelin than controls (black dots). Data are shown as mean ± standard error of mean. T-test: ###p < 0.001.
Restored myelination by benztropine in vitro
Next, we asked whether the interference of α-synuclein with myelin formation is reversible and thus, might represent a novel interventional target for MSA. For this purpose, we chose benztropine, which has recently been described as a potent small molecule to enhance myelination [7]. First, we validated the effect of benztropine on myelin gene expression in non-modified primary OPC monoculture. Compared to vehicle, benztropine dose-dependently increased Mbp protein in primary OPCs (Fig. 4a). Gene expression analysis revealed an effective dose in the range of 0.1 to 1.0 μM, peaking at 0.5 μM with an increase in Mbp expression by 2.4±0.2 fold (Fig. 4b). Immunofluorescence against Mbp confirmed that benztropine at its most effective dose (0.5 μM) strongly increased the number of Mbp-positive cells (Fig. 4c). We next evaluated the effect of benztropine on Mbp expression in primary oligodendrocytes overexpressing human α-synuclein. For this purpose, primary oligodendrocytes were lentivirally transduced with a constitutively active α-synuclein expression vector (EF1a-SYN-IG) or the control vector lacking the α-synuclein-coding sequence (EF1a-IG). As previously described [14], α-synuclein overexpression significantly reduced Mbp protein (Fig. 4d) and transcription (Fig. 4e) compared to control condition. Addition of benztropine increased Mbp expression in α-synuclein-overexpressing oligodendrocytes, restoring it to the level control cells. On a morphological level, vehicle-treated α-synuclein-overexpressing oligodendrocytes were bi- or tripolar indicative for an immature morphology, whereas benztropine induced the formation of multiple and highly arborized branches in α-synuclein-overexpressing oligodendrocytes reflecting an advanced maturation stage comparable to control cells (Fig. 4f).
Fig. 4.
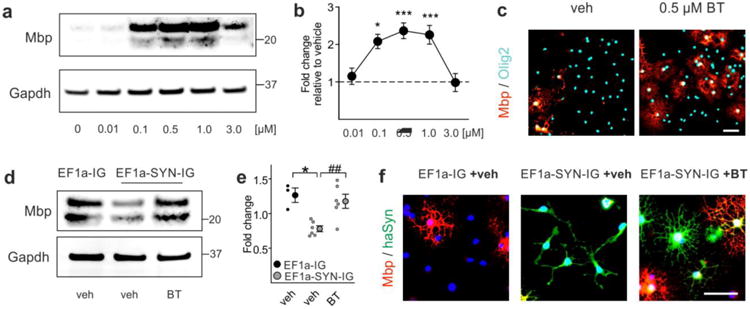
Benztropine restores myelin basic protein (Mbp) expression in α-synuclein-overexpressing primary oligodendrocyte progenitor cells (OPCs). a The dose-dependent effect of benztropine on Mbp expression in primary OPCs is illustrated using Western blot. Gapdh signal served as control. b Benztropine significantly increased Mbp gene expression in a range of 0.1-1.0 μM as quantified by real time PCR (n = 3-6). c Immunocytochemistry for Mbp and Olig2 illustrates the effect of 0.5 μM benztropine (BT) on OPC maturation in comparison to vehicle- (veh) treated cells. Scale bar: 20μm. d The restoration of Mbp expression in α-synuclein-overexpressing cells (EF1a-SYN-IG +veh) upon treatment with 0.5 μM benztropine (EF1a-SYN-IG +BT) to control levels (EF1a-IG +veh) is demonstrated by Western blot. Gapdh signal served as control. e Benztropine restored Mbp gene expression in α-synuclein-overexpressing OPCs as quantified by real time PCR (n = 3-6). f α-Synuclein-overexpressing cells exhibited the typical morphology of OPCs (bi-/tripolarity), whereas treatment with benztropine induced Mbp expression and formation of multiple processes as signs of advanced maturity comparable to control condition. Scale bar: 20μm. Data are shown as mean ± standard error of mean. ANOVA: *p < 0.05, ***p < 0.001. T-test: ##p < 0.01.
To globally analyze the effects of α-synuclein overexpression and benztropine treatment on the myelinogenic capacity of oligodendrocytes, transcriptome profiles of vehicle- or benztropine-treated primary OPCs transduced with the α-synuclein expression vector (EF1a-SYN-IG) or the respective control vector (EF1a-IG) were determined by RNA sequencing (Fig. 5). The genes, which were differentially regulated by both α-synuclein and benztropine compared to respective controls, were subjected to a GO analysis (Fig 5a, b). Enriched GO terms were implicated in myelin formation (GO:0042552 myelination, GO:0007272 ensheathment of neurons, GO:0008366 axon ensheathment), membranogenesis (GO:0006695 cholesterol biosynthetic process, GO:0044283 small molecule biosynthetic process, GO:0061024 membrane organization), and the maintenance of oligodendrocyte immaturity (GO:0050793 regulation of developmental process, GO:0051093 negative regulation of developmental process, GO:0021782 glial cell development). Intriguingly, transcripts related to myelin formation and membranogenesis were significantly underrepresented in α-synuclein-overexpressing cells and likewise upregulated by benztropine (e.g., Mbp, Plp1, Hmgcr, and Ank3) (Fig. 5c). In contrast, transcripts involved in maintaining the immaturity of OPCs were significantly enriched upon α-synuclein overexpression and accordingly downregulated by benztropine treatment (e.g., Nkx2.2, Sox9, and Tcf7l1).
Fig. 5.
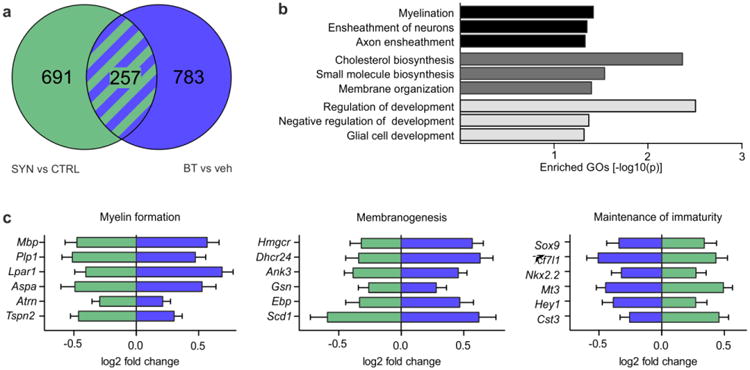
Restored myelinogenic capacity of α-synuclein-overexpressing primary rat oligodendrocyte progenitor cells (OPCs) by benztropine. a A Venn diagram depicts differentially regulated genes revealed by RNA sequencing of primary rat OPCs (n = 3). Green circle: α-synuclein-overexpressing (SYN) versus control cells (CTRL). Blue circle: benztropine (BT) versus vehicle (veh)-treated α-synuclein-overexpressing cells. b Genes differentially regulated by both α-synuclein overexpression and benztropine treatment classified into gene ontologies (GOs) implicated among others in myelin formation (black), membranogenesis (dark gray), and maintenance of oligodendrocyte immaturity (light gray). c Expression dynamics of representative genes identified by GO term enrichment are listed. Benztropine (blue) oppositely regulated genes differentially expressed upon α-synuclein overexpression (green). Myelin formation: Mbp = myelin basic protein, Plp1 = proteolipid protein 1, Lpar1 = lysophosphatidic acid receptor 1, Aspa = aspartoacylase, Atrn = attractin, Tspn2 = tetraspanin 2; Membranogenesis: Hmgcr = 3-hydroxy-3-methylglutaryl-CoA reductase, Dhcr24 = 24-dehydrocholesterol reductase, Ank3 = ankyrin 3, Gsn = gelsolin, Ebp = emopamil binding protein (sterol isomerase), Scd1 = stearoyl-CoA desaturase-1; Maintenance of immaturity: Sox9 = sex determining region Y-box 9, Tcf7l1 = transcription factor 7-like 1, Nkx2.2 = Nk2 homeobox 2, Mt3 = metallothionein 3, Hey1 = hes-related family bHLH transcription factor with YRPW motif 1, Cst3 = cystatin C.
To extend the findings from primary oligodendrocyte monocultures, we next analyzed whether benztropine is also able to restore the myelination deficit of α-synuclein-overexpressing stem cell-derived oligodendrocytes in co-culture with cortical neurons. First, the myelinogenic of benztropine effect was validated in non-modified stem cell-derived oligodendrocytes (Suppl. Fig. S5). Strikingly, benztropine treatment also increased myelin sheath formation of α-synuclein-overexpressing oligodendrocytes by 29±4% to the level of control cells (Fig. 6).
Fig. 6.
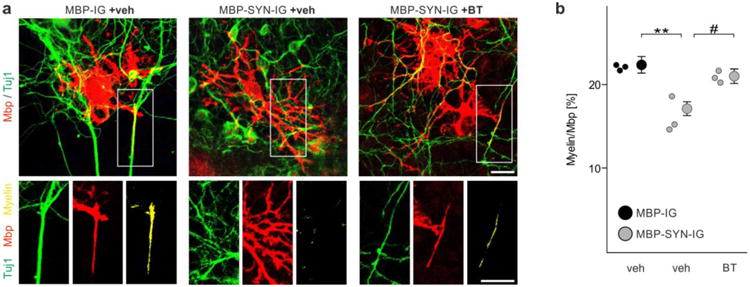
Restored myelination deficit of α-synuclein-overexpressing mouse embryonic stem cell-derived oligodendrocytes by benztropine. a Representative images show that overexpression of α-synuclein (transduced with MBP-SYN-IG prior to starting the co-culture) impaired myelin formation of stem cell-derived oligodendrocytes (stained for myelin basic protein, Mbp) co-cultured with cortical neurons (beta-III-tubulin, Tuj1). Addition of benztropine enhanced myelin formation of α-synuclein-overexpressing cells (comparable to vehicle (veh)-treated control cells, which were transduced with MBP-IG). Mbp-positive pixels co-localizing with Tuj1-positive pixels were considered as myelin (shown in yellow). Scale bar: 20μm. b Myelination of individual oligodendrocytes (in total 90-110 cells per condition) was quantified (n = 3). α-Synuclein-overexpressing oligodendrocytes (gray dots) in the presence of vehicle (veh) formed significantly less myelin than controls (black dots, veh). Benztropine (BT) restored the myelination deficit of α-synuclein overexpressing oligodendrocytes. Data are shown as mean ± standard error of mean. ANOVA: ***p < 0.001. T-test: ##p < 0.01.
Attenuated myelin deficit in MBP29 mice upon benztropine administration
After observing the restoration of the α-synuclein-induced myelination deficit by benztropine in vitro (Fig. 4 to 6), we next administered benztropine at 2 mg/kg to MBP29 mice to assess whether benztropine is also able to enhance myelination in the presence of oligodendrocytic α-synuclein accumulation in vivo. T2-weighted magnetic resonance imaging revealed a reduced gray-white matter contrast in vehicle-treated MBP29 mice compared to non-transgenic littermates, which corresponds to a myelin reduction (Fig. 7a). A significant gain of contrast was observed in benztropine-treated MBP29 mice. For quantification, T2*-weighted magnetic resonance imaging was performed because of its previously demonstrated sensitivity in detecting myelin loss in murine brains [27]. Compared to controls, vehicle-treated MBP29 mice showed a significant increase by 19±3% in the T2* relaxation time within the corpus callosum, confirming the myelin deficit (Fig. 7b). Treatment with benztropine significantly reduced the callosal T2* relaxation time in MBP29 mice by 9±1%, thus restoring the myelin deficit by almost 50%. Matching this imaging approach, benztropine treatment significantly increased the mean Luxol Fast Blue signal intensity in the corpus callosum of MBP29 mice, ameliorating the myelin deficit by more than 50% (Fig. 2 and Fig. 7c, d). Results were further confirmed by ultrastructural analysis of myelin in the corpus callosum using electron microscopy (Fig. 8). In vehicle- and benztropine-treated non-transgenic mice, myelin showed a highly organized multilaminar structure. In contrast, myelin in vehicle-treated MBP29 mice appeared disorganized and substantially reduced. Both density of myelinated axons and myelin layers per axon were significantly reduced. In benztropine-treated MBP29, numbers of myelinated axons and myelin layers per axon were comparable to non-transgenic controls.
Fig. 7.
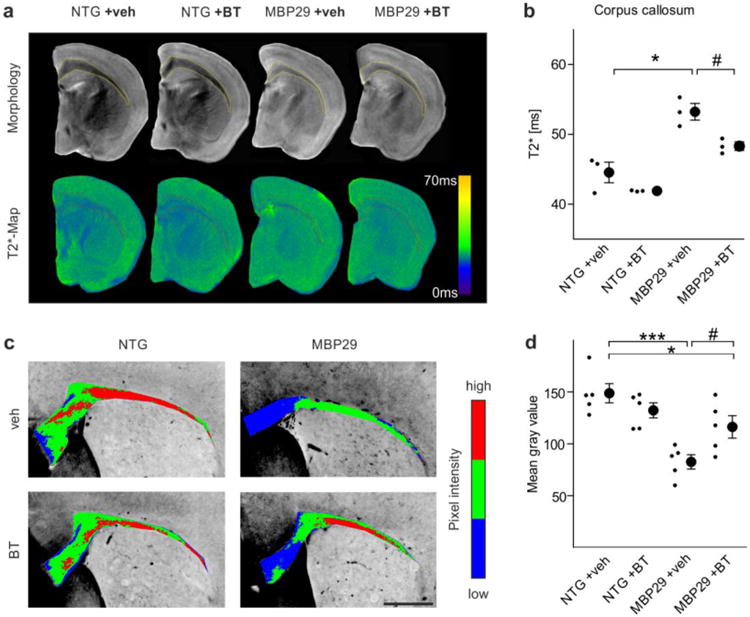
Amelioration of the myelin deficit in mice with oligodendrocytic overexpression of α-synuclein (MBP29) upon benztropine treatment. a Callosal myelin levels of non-transgenic (NTG) and MBP29 mice treated with vehicle (veh) or benztropine (BT, 2mg/kg) were analyzed using magnetic resonance imaging. Upper panel illustrates morphology as determined by T2-weighted magnetic resonance imaging, whereas lower panel depicts T2*-weighted magnetic resonance imaging (shown as color-coded maps), which was used for myelin quantification. b MBP29 mice (n = 3) showed a reduction in myelin levels detected by increased T2*-relaxation times within the corpus callosum. Benztropine attenuated the myelin deficit in MBP29 confirmed by a reduction of T2*. c Representative pictures of a Luxol Fast Blue staining (color-coded for signal intensity: blue low, green intermediate, red high intensity), confirming the effects of α-synuclein overexpression and benztropine treatment on myelination. Scale bar: 500μm. d MBP29 mice (n = 5) exhibited a reduction in Luxol Fast Blue intensity, which was attenuated, but not completely restored by benztropine. Data are shown as mean ± standard error of mean. ANOVA: *p < 0.05, ***p < 0.001. T-test: #p < 0.05.
Fig. 8.
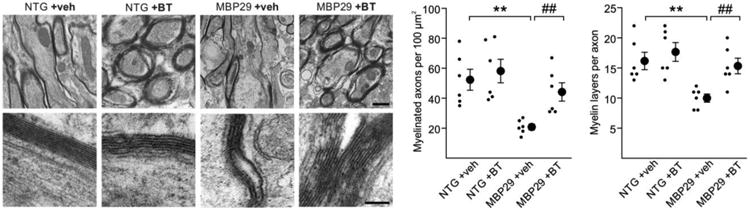
Structural restoration of myelin by benztropine in mice overexpressing α-synuclein in oligodendrocytes (MBP29). Representative electron micrographs of the corpus callosum at low (upper panel, scale bar: 1μm) and high (lower panel, scale bar: 0.2μm) magnification depict myelin sheaths of non-transgenic (NTG) and MBP29 mice treated with either vehicle (veh) or benztropine (BT). In vehicle-treated MBP29 mice only, myelin appeared strongly reduced and disorganized. Quantification of myelinated axons and myelin layers per axon revealed a significant myelin deficit in vehicle-treated MBP29 mice, which was restored by benztropine treatment to control level. Data are shown as mean ± standard error of mean. ANOVA (n = 6): **p < 0.01. T-test: ##p < 0.01.
Taken together, these data demonstrate that the pro-myelinating activity of benztropine attenuates the severe myelin deficit upon α-synuclein overexpression in MBP29 mice.
Unchanged oligodendrocyte dynamics in MBP29 mice upon benztropine administration
We next examined whether benztropine impacts the dynamics of oligodendrocytic populations in MBP29 mice. To this end, we histologically identified distinct oligodendrocytic subpopulations within the corpus callosum of vehicle- and benztropine-treated MBP29 mice and non-transgenic littermates using maturation stage-specific markers (Fig. 9a, b): OPCs (Pdgfrα/Olig2-positive) were distinguished from mature oligodendrocytes (Gstπ/Olig2-positive), whereas a minority of Olig2-positive cells co-labeled for neither Pdgfrα nor Gstπ (Olig2 only). Mbp promoter-driven α-synuclein overexpression in MBP29 mice resulted in a 1.5- to 2.2-fold increased density of all distinct subpopulations compared to non-transgenic control animals (Fig. 9c). In contrast to α-synuclein overexpression, it is remarkable that benztropine did not affect oligodendrocyte density. Given that the proportional distribution of oligodendrocytic subpopulations was unchanged in all groups (Suppl. Fig. S6), these findings suggest that treatment with benztropine directly acts on myelin formation, but does not increase OPC maturation in MBP29 mice.
Fig. 9.
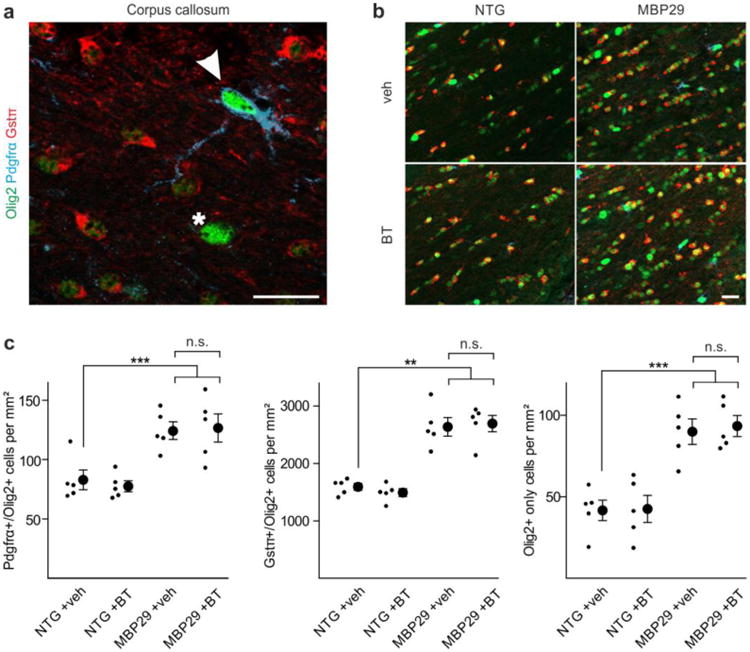
No alteration of oligodendrocyte density by benztropine. a Oligodendrocytic cells within the corpus callosum were identified using stage specific markers (entire population: Olig2; oligodendrocyte progenitor cells: platelet-derived growth factor receptor-α (Pdgfrα)/Olig2strong (arrowhead), mature oligodendrocytes: glutathione-s-transferase-π (Gstπ)/Olig2weak, as well as cells staining for Olig2strong only (asterisk)). Scale bar: 20μm. b A profound increase of oligodendrocytic cells within the corpus callosum of mice overexpressing α-synuclein in oligodendrocytes (MBP29) compared to non-transgenic controls (NTG) was observed. Scale bar: 20μm. c While MBP29 mice exhibited an almost 2-fold increased density of the entire spectrum of oligodendrocytic subpopulations, benztropine (BT) had no effect on oligodendrocytic cell density as compared to vehicle (veh)-treatment. Data are shown as mean ± standard error of mean. ANOVA (n = 5): **p < 0.01, ***p < 0.001. T-test: not significant (n.s.) p > 0.05.
Prevention of motor cortical neuronal loss without affecting microgliosis in MBP29 mice upon benztropine treatment
In addition to the severe loss of myelin, MBP29 mice recapitulate crucial neurodegenerative and inflammatory features of MSA [45]. Thus, we next examined whether benztropine treatment had an impact on neuronal and microglial cell densities in MBP29 mice. To this end, we first quantified the density of NeuN-positive neurons within the motor cortex (Fig. 10). While vehicle-treated MBP29 mice showed a moderate (14%), but significant reduction in neuronal cell density, benztropine treatment prevented neuronal loss in MBP29 animals. We next assessed the effect of benztropine on motor cortical and callosal microglial cells of MBP29 mice by determining the density of Iba1-positive microglia (Suppl. Fig. S7). Microgliosis was apparent in the corpus callosum of vehicle-treated MBP29 mice (2.5-fold increase in Iba1-positive cells), whereas the motor cortex showed no apparent signs of microgliosis when compared to control mice (1.1-fold increase). Benztropine treatment neither affected callosal nor motor cortical Iba1 cell density in MBP29 mice.
Fig. 10.
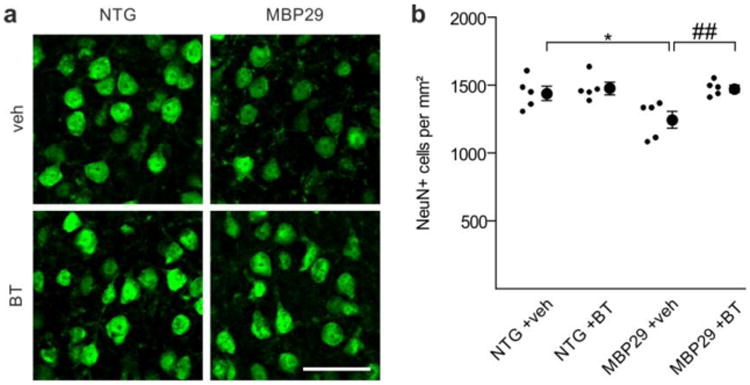
Prevention of motor cortical neuronal cell loss in mice overexpressing α-synuclein in oligodendrocytes by benztropine. a Representative images depict NeuN-positive neuronal cells within the motor cortex of non-transgenic controls (NTG) and α-synuclein transgenic mice (MBP29) treated with vehicle (veh) or benztropine (BT). Scale bar: 20μm. b Densities of motor cortical neurons are shown (n = 5). In vehicle-treated MBP29 mice, a significant reduction in neuronal cell density was observed. Benztropine treatment prevented the loss of cortical neurons in MBP29 mice. Data are shown as mean ± standard error of mean. ANOVA: *p < 0.05. T-test: ##p < 0.01.
Taken together, these findings suggested that benztropine treatment has a beneficial effect on survival of motor cortical neurons, but does not alter microglial cell densities in MBP29 mice.
Discussion
Cellular pathomechanisms that underlie profound myelin loss and neurodegeneration in MSA are poorly understood. In this regard, our study provides compelling evidence for impaired myelin formation in MSA, which is directly associated with the oligodendrocytic accumulation of α-synuclein and not accompanied by loss of oligodendrocytes. Considering that oligodendrocytic α-synuclein accumulation is the primary event during MSA pathogenesis, the deficit in myelin formation may be an early and crucial pathomechanism during disease progression. Given that efficient interventional strategies for MSA do not exist and by demonstrating that the interference of α-synuclein with myelin formation is reversible, our in vitro and in vivo data imply pro-myelinating strategies as a novel approach for an urgently needed therapy in MSA.
Using histological analyses of MSA post-mortem tissue, we showed that myelin loss is a widespread pathological feature in MSA. Loss of myelin, downregulation of myelin proteins [1, 35, 38, 47], and widespread lipid alterations [5, 11] have previously been reported in post-mortem brains of MSA patients, supporting our finding of a severe myelin deficit in MSA. Despite strong evidence for myelin degeneration in MSA, the underlying pathomechanisms of this myelin loss are not well understood. In the MSA cohort analyzed in the present study, the putamen exhibited extensive oligodendrocytic α-synuclein pathology within white matter tracts, suggesting that oligodendrocytic α-synuclein accumulation underlies loss of myelin. This is supported by the fact that a substantial myelin deficit was also present upon transgenic overexpression of α-synuclein in oligodendrocytes of MBP29 mice. In line, a similar myelin reduction has also been observed in transgenic mice with α-synuclein overexpression under alternative myelin gene promoters, namely the proteolipid protein and the 2′,3′-cyclic-nucleotide 3′-phosphodiesterase promoter [48, 52]. Notably, structural alterations, including nuclear shrinkage, cytosolic re-localization of myelin proteins, and oligodendrocytic swelling, were also observed in oligodendrocytes without α-synuclein accumulation and possibly contribute to myelin loss in MSA [47, 49]. Moreover, we selectively used post-mortem material of MSA patients with striatonigral degeneration, limiting the interpretation of the present study to the parkinsonian variant of MSA (MSA-P). Myelin loss in MSA patients and in transgenic MSA mice may be linked to either oligodendrocyte loss or impaired ability of oligodendrocytes to form new myelin sheaths. The observation of an unchanged or even increased oligodendrocyte density in human MSA post-mortem tissue and MBP29 mice suggests that rather a dysfunction of oligodendrocytes in terms of myelin sheath formation than pronounced loss of oligodendrocytes underlies myelin loss in MSA. A recent stereological analysis, focusing on basal ganglia, has indicated that oligodendrocytes are by far more preserved than neurons in MSA [41]. In line, we and others have observed increased numbers of OPCs in the striatum and the cerebellum of MSA patients, supporting the notion of preserved oligodendrocytic cells in MSA [2, 36]. Similarly, we have shown that mice moderately overexpressing α-synuclein driven by the Mbp promoter (line 1) exhibit an increased number of OPCs accompanied by myelin loss [36, 45]. Given that the Mbp promoter did not drive α-synuclein overexpression in OPCs, the observed increase in OPC density of MBP29 mice indicates enhanced OPC proliferation to compensate the myelin deficit.
Supporting these observations in MSA post-mortem and transgenic mouse forebrains, our in vitro data provide further evidence, that oligodendrocytic α-synuclein impairs myelin formation: α-synuclein-overexpressing oligodendrocytes derived from mouse embryonic stem cells exhibited a reduced capacity to form myelin sheaths around axons of cortical neurons. Moreover, transcriptome profiles of primary rat OPC monocultures showed that α-synuclein overexpression in maturating oligodendrocytes impaired expression of gene families essentially involved in myelin sheath formation. The temporal dynamic of α-synuclein expression in cultured murine and human OPCs during maturation toward pre-myelinating oligodendrocytes suggests that α-synuclein is physiologically restricted to immature oligodendrocytes [8, 40]. In addition, we have previously demonstrated that the sustained presence of intracellular α-synuclein delays the upregulation of myelin gene expression in monocultures of primary rat oligodendrocytes [14], further supporting that α-synuclein expression needs to be downregulated for timed and physiological oligodendrocyte maturation and myelination. Thus, our data further strengthen the view on α-synuclein as an inhibitor of oligodendrocyte maturation and myelination and suggest that pathologic oligodendrocytic α-synuclein accumulation in MSA impairs myelin formation.
In recent years, the view on myelin dynamics in the adult brain has fundamentally changed. The fact, that myelin is physiologically formed in the adult murine and human central nervous system throughout life, has only lately been recognized [18, 37, 42, 53, 54]. In mice, adaptive myelination in the adult forebrain is mediated mostly by newly formed oligodendrocytes derived from OPCs, occurs upon neuronal activity, and is essentially required for motor skill learning [18, 37]. In line, motor training has also induced a remodeling of white matter structures in humans [42]. Importantly, a continuous physiological myelin turnover during adulthood, which is mostly mediated by pre-existing oligodendrocytes, has been detected in the human corpus callosum by determining the integration of nuclear bomb test-derived (14)C [53]. Therefore, impaired myelination in the presence of α-synuclein may result not only in compromised remyelination, but also in reduced myelin turnover. In this context, it is important to note that disturbances in myelin homeostasis result in axonal and neuronal degeneration [51]. The impaired capacity to form myelin sheaths may thus represent a fundamental and early pathological mechanism in MSA, favoring myelin loss and ultimately triggering widespread axonal dysfunction and neuronal loss.
To restore oligodendrocyte functionality, we used the small molecule benztropine, which has recently been identified by a high-throughput screen to accelerate OPC maturation and directly promote myelin formation, thereby ameliorating functional deficits in pre-clinical mouse models of multiple sclerosis [7]. We chose this small molecule to target α-synuclein-induced effects on oligodendrocyte function because i) it is clinically used for the treatment of tremor in patients diagnosed with sporadic Parkinson's disease and thus promises a fast translation into clinics; and ii) benztropine has a dual mode of action as it promotes myelin gene expression in pre-myelinating oligodendrocytes and directly enhances myelin formation in mature oligodendrocytes [7]. Both processes are essentially required for myelination and are compromised by α-synuclein overexpression. While the effect of benztropine on myelin gene expression has been attributed to its antagonistic activity at the muscarinic acetylcholine receptors M1 and M3 [7], the mechanism by which benztropine induces myelin sheath formation is not yet known. Our observation, that benztropine increased myelination without changing the density of OPCs and mature oligodendrocytes in MBP29 mice, favors rather a direct action of benztropine on myelin sheath formation in this model. Importantly, benztropine even ameliorated the myelination deficit in MBP29 mice by approximately 50%, although used at a low dose (2 mg/kg in comparison to the most efficient dose of 10 mg/kg according to Deshmukh and colleagues [7]). Higher dosages (5 and 10 mg/kg) were not tolerated due to the severe phenotype of MBP29 mice with a premature death at 4 to 6 months of age [45]. Moreover, our experiments using the stem cell-derived oligodendrocyte-neuron co-culture also point toward a direct pro-myelinating effect of benztropine. Our transcriptome analysis suggests that benztropine readjusts disturbed lipid metabolism in oligodendrocytes with α-synuclein accumulation, thereby enhancing myelin formation. While not in the focus of the present work, future studies need to dissect the complex pharmacology and define molecular targets underlying the pro-myelinating activity of benztropine.
Benztropine treatment also prevented motor cortical neuronal cell loss in MBP29 mice, suggesting that myelin regeneration supports neuronal survival. Although we cannot exclude a direct effect of benztropine on neuronal cells, the preservation of neurons might be attributed to oligodendrocyte-derived factors and their importance for axonal maintenance and neuronal survival [29]. In line, protection of cortical neurons was also achieved upon immunotherapeutic prevention of myelin loss in transgenic mice expressing α-synuclein in oligodendrocytes [33]. In this study, oligodendrocytic α-synuclein pathology was reduced via specific antibodies against α-synuclein. Importantly, benztropine injections did not alter microglial cell numbers in MBP29 mice, supporting that neuronal preservation is not linked to decreased neuroinflammation. In line, Deshmukh and colleagues have demonstrated that benztropine has no immunomodulatory effect in experimental mouse models for multiple sclerosis [7].
The surprising dichotomy of a substantial myelin deficit despite a preserved oligodendrocyte density opens novel avenues for restorative interventional strategies in MSA. Up to now, experimental and clinical trials for disease-modifying therapies in MSA have focused on direct protection of neurons (e.g., riluzole [44]), prevention or clearance of α-synuclein aggregates (e.g., rifampicin [31]), reduction of neuroinflammation (e.g., minocycline [10]), and neurorestorative approaches (e.g., systemic mesenchymal stem cell delivery [28]). However, all of these strategies showed only very moderate – if any – clinical efficacy (for review see [26]). We propose the myelination deficit of dysfunctional oligodendrocytes as a novel and promising interventional target for MSA because of two reasons: firstly, α-synuclein accumulation within oligodendrocytes is considered as the primary and pathognomonic event during MSA pathogenesis [50]; and secondly, we showed that oligodendrocytes are preserved in MSA and – albeit being dysfunctional – their ability to form myelin was not irreversibly lost in the presence of α-synuclein. Consequently, restoring the capacity to form new myelin may provide a new basis for a therapeutic strategy even in early stages of MSA. It should be emphasized that – although we observed prevention of neuronal cell loss in our model – we cannot state whether a pro-myelinating monotherapy will lead to a functional recovery in the context of MSA, given that the usage of the most severe pre-clinical mouse model for MSA, the MBP29 mouse line, did not allow a comprehensive behavioral assessment. Thus, additional pre-clinical studies have to explore whether a myelinogenic monotherapy or a synergistic combination of a pro-myelinating intervention with strategies targeted against other pathological hallmarks of MSA, including neuronal degeneration and neuroinflammation, may be the basis for treating MSA patients. To conclude, our findings shed new light into α-synuclein-linked pathomechanisms in MSA. Intriguingly, the observed reversibility of the α-synuclein-induced myelination deficit in primary rat OPCs, in mouse embryonic stem cell-derived oligodendrocytes, and in MBP29 mice identified a new direction to treat MSA.
Supplementary Material
Acknowledgments
This work was supported by the Interdisciplinary Center for Clinical Research (IZKF Erlangen, TP E18), the Bavarian State Ministry of Education and Culture, Science and Arts in the framework of the Bavarian Research Network Induced Pluripotent Stem Cells (ForIPS), the Deutsche Forschungsgemeinschaft (DFG grant INST 410/45-1 FUGG), and the NIH (AG5131, AG18440, NS092803). The study was supported in part by the G.Harold & Leila Y. Mathers Charitable Foundation, the JPB Foundation, the Leona M. and Harry B. Helmsley Charitable Trust. BE is an IZKF PhD-student and was supported by the IZKF Erlangen to conduct experiments involving stem cells in the Laboratory of Genetics at the Salk Institute for Biological Studies, La Jolla, CA, USA. The authors greatly acknowledge the NBB for providing human post-mortem tissue. Excellent technical assistance was provided by Holger Meixner, Someya Salem, Arianna Mei, Jazmin Florio, Maria Hirblinger, Petra Rothe, Angelika Diem, and Heike Friebel-Stange. We thank Beate Winner and Chichung Lie for scientific discussion and comments on the manuscript. Mary Lynn Gage is greatly acknowledged for editorial comments.
Footnotes
Compliance with ethical standards: Human brain samples used in this study were obtained from the NBB and have been collected from donors for or from whom a written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the NBB. All animal procedures were conducted with approval of the animal care and use committees of the University of California San Diego, the Friedrich-Alexander-Universität Erlangen-Nürnberg, and the state of Bavaria.
Conflicts of interest: The authors declare that they have no conflict of interest.
References
- 1.Ahmed Z, Asi YT, Sailer A, Lees AJ, Houlden H, Revesz T, Holton JL. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012;38:4–24. doi: 10.1111/j.1365-2990.2011.01234.x. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed Z, Asi YT, Lees AJ, Revesz T, Holton JL. Identification and quantification of oligodendrocyte precursor cells in multiple system atrophy, progressive supranuclear palsy and Parkinson's disease. Brain Pathol. 2013;23:263–73. doi: 10.1111/j.1750-3639.2012.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, Houlden H, Holton JL. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014;62:964–70. doi: 10.1002/glia.22653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bleasel JM, Hsiao JH, Halliday GM, Kim WS. Increased expression of ABCA8 in multiple system atrophy brain is associated with changes in pathogenic proteins. J Parkinsons Dis. 2013;3:331–9. doi: 10.3233/JPD-130203. [DOI] [PubMed] [Google Scholar]
- 6.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci U S A. 1979;76:514–7. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, et al. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502:327–32. doi: 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S, Frisen J, Deierborg T, Roybon L. Alpha-Synuclein Expression in the Oligodendrocyte Lineage: an In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Reports. 2015;5:174–84. doi: 10.1016/j.stemcr.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N, Trenkwalder C, Sixel-Doring F, Herting B, Kamm C, et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial) Mov Disord. 2010;25:97–107. doi: 10.1002/mds.22732. [DOI] [PubMed] [Google Scholar]
- 11.Don AS, Hsiao JH, Bleasel JM, Couttas TA, Halliday GM, Kim WS. Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy. Acta Neuropathol Commun. 2014;2:150. doi: 10.1186/s40478-014-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eschlbock S, Krismer F, Wenning GK. Interventional trials in atypical parkinsonism. Parkinsonism Relat Disord. 2016;22 Suppl 1:S82–92. doi: 10.1016/j.parkreldis.2015.09.038. [DOI] [PubMed] [Google Scholar]
- 14.Ettle B, Reiprich S, Deusser J, Schlachetzki JC, Xiang W, Prots I, Masliah E, Winner B, Wegner M, Winkler J. Intracellular alpha-synuclein affects early maturation of primary oligodendrocyte progenitor cells. Mol Cell Neurosci. 2014;62:68–78. doi: 10.1016/j.mcn.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ettle B, Schlachetzki JC, Winkler J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372:1375–6. doi: 10.1056/NEJMc1501657. [DOI] [PubMed] [Google Scholar]
- 17.Gaspard N, Bouschet T, Herpoel A, Naeije G, van den Ameele J, Vanderhaeghen P. Generation of cortical neurons from mouse embryonic stem cells. Nat Protoc. 2009;4:1454–63. doi: 10.1038/nprot.2009.157. [DOI] [PubMed] [Google Scholar]
- 18.Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344:1252304. doi: 10.1126/science.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Durr A, Fowler CJ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gow A, Friedrich VL, Jr, Lazzarini RA. Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol. 1992;119:605–16. doi: 10.1083/jcb.119.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–3. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 22.Groves AK, Barnett SC, Franklin RJ, Crang AJ, Mayer M, Blakemore WF, Noble M. Repair of demyelinated lesions by transplantation of purified O-2A progenitor cells. Nature. 1993;362:453–5. doi: 10.1038/362453a0. [DOI] [PubMed] [Google Scholar]
- 23.Irvine KA, Blakemore WF. Remyelination protects axons from demyelination-associated axon degeneration. Brain. 2008;131:1464–77. doi: 10.1093/brain/awn080. [DOI] [PubMed] [Google Scholar]
- 24.Kahle PJ, Neumann M, Ozmen L, Haass C. Physiology and pathophysiology of alpha-synuclein. Cell culture and transgenic animal models based on a Parkinson's disease-associated protein. Ann N Y Acad Sci. 2000;920:33–41. doi: 10.1111/j.1749-6632-2000.tb06902.x. [DOI] [PubMed] [Google Scholar]
- 25.Kerman BE, Kim HJ, Padmanabhan K, Mei A, Georges S, Joens MS, Fitzpatrick JA, Jappelli R, Chandross KJ, August P, et al. In vitro myelin formation using embryonic stem cells. Development. 2015;142:2213–25. doi: 10.1242/dev.116517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzdas-Wood D, Stefanova N, Jellinger KA, Seppi K, Schlossmacher MG, Poewe W, Wenning GK. Towards translational therapies for multiple system atrophy. Prog Neurobiol. 2014;118:19–35. doi: 10.1016/j.pneurobio.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Shmueli K, Kang BT, Yao B, Fukunaga M, van Gelderen P, Palumbo S, Bosetti F, Silva AC, Duyn JH. The contribution of myelin to magnetic susceptibility-weighted contrasts in high-field MRI of the brain. Neuroimage. 2012;59:3967–75. doi: 10.1016/j.neuroimage.2011.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee PH, Lee JE, Kim HS, Song SK, Lee HS, Nam HS, Cheong JW, Jeong Y, Park HJ, Kim DJ, et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol. 2012;72:32–40. doi: 10.1002/ana.23612. [DOI] [PubMed] [Google Scholar]
- 29.Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–8. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Low PA, Robertson D, Gilman S, Kaufmann H, Singer W, Biaggioni I, Freeman R, Perlman S, Hauser RA, Cheshire W, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:268–75. doi: 10.1016/S1474-4422(13)70301-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Low PA, Reich SG, Jankovic J, Shults CW, Stern MB, Novak P, Tanner CM, Gilman S, Marshall FJ, Wooten F, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol. 2015;14:710–9. doi: 10.1016/S1474-4422(15)00058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandler M, Valera E, Rockenstein E, Mante M, Weninger H, Patrick C, Adame A, Schmidhuber S, Santic R, Schneeberger A, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener. 2015;10:10. doi: 10.1186/s13024-015-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–57. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Matsuo A, Akiguchi I, Lee GC, McGeer EG, McGeer PL, Kimura J. Myelin degeneration in multiple system atrophy detected by unique antibodies. Am J Pathol. 1998;153:735–44. doi: 10.1016/S0002-9440(10)65617-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.May VE, Ettle B, Poehler AM, Nuber S, Ubhi K, Rockenstein E, Winner B, Wegner M, Masliah E, Winkler J. alpha-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol Aging. 2014;35:2357–68. doi: 10.1016/j.neurobiolaging.2014.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, Richardson WD. Motor skill learning requires active central myelination. Science. 2014;346:318–22. doi: 10.1126/science.1254960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510X(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 39.Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Bruck W, Lucchinetti C, Lassmann H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129:3165–72. doi: 10.1093/brain/awl217. [DOI] [PubMed] [Google Scholar]
- 40.Richter-Landsberg C, Gorath M, Trojanowski JQ, Lee VM. alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J Neurosci Res. 2000;62:9–14. doi: 10.1002/1097-4547(20001001)62:1<9∷AID-JNR2>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 41.Salvesen L, Ullerup BH, Sunay FB, Brudek T, Lokkegaard A, Agander TK, Winge K, Pakkenberg B. Changes in total cell numbers of the basal ganglia in patients with multiple system atrophy - A stereological study. Neurobiol Dis. 2015;74:104–13. doi: 10.1016/j.nbd.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Scholz J, Klein MC, Behrens TE, Johansen-Berg H. Training induces changes in white-matter architecture. Nat Neurosci. 2009;12:1370–1. doi: 10.1038/nn.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, Melchers A, Paudel R, Gibbs JR, Simon-Sanchez J, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–4. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seppi K, Peralta C, Diem-Zangerl A, Puschban Z, Mueller J, Poewe W, Wenning GK. Placebo-controlled trial of riluzole in multiple system atrophy. Eur J Neurol. 2006;13:1146–8. doi: 10.1111/j.1468-1331.2006.01452.x. [DOI] [PubMed] [Google Scholar]
- 45.Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–99. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simons M, Nave KA. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb Perspect Biol. 2015 doi: 10.1101/cshperspect.a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Hojrup P, Otzen D, Halliday GM, Jensen PH. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol. 2007;171:1291–303. doi: 10.2353/ajpath.2007.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stefanova N, Kaufmann WA, Humpel C, Poewe W, Wenning GK. Systemic proteasome inhibition triggers neurodegeneration in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. Acta Neuropathol. 2012;124:51–65. doi: 10.1007/s00401-012-0977-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uyama N, Uchihara T, Mochizuki Y, Nakamura A, Takahashi R, Mizutani T. Selective nuclear shrinkage of oligodendrocytes lacking glial cytoplasmic inclusions in multiple system atrophy: a 3-dimensional volumetric study. J Neuropathol Exp Neurol. 2009;68:1084–91. doi: 10.1097/NEN.0b013e3181b67678. [DOI] [PubMed] [Google Scholar]
- 50.Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol. 2008;64:239–46. doi: 10.1002/ana.21465. [DOI] [PubMed] [Google Scholar]
- 51.Wilkins A, Kondo Y, Song J, Liu S, Compston A, Black JA, Waxman SG, Duncan ID. Slowly progressive axonal degeneration in a rat model of chronic, nonimmune-mediated demyelination. J Neuropathol Exp Neurol. 2010;69:1256–69. doi: 10.1097/NEN.0b013e3181ffc317. [DOI] [PubMed] [Google Scholar]
- 52.Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–59. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- 53.Yeung MS, Zdunek S, Bergmann O, Bernard S, Salehpour M, Alkass K, Perl S, Tisdale J, Possnert G, Brundin L, et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell. 2014;159:766–74. doi: 10.1016/j.cell.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 54.Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–85. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.