

Abstract
Background
Multiple sclerosis (MS) is a complex disorder of the central nervous system whose cause is currently unknown. Evidence is increasing that DNA methylation alterations could be involved in inflammatory and neurodegenerative diseases and could contribute to MS pathogenesis. Repetitive elements Alu, LINE-1 and SAT-α, are widely known as estimators of global DNA methylation. We investigated Alu, LINE-1 and SAT-α methylation levels to evaluate their difference in a case–control setup and their role as a marker of disability.
Results
We obtained blood samples from 51 MS patients and 137 healthy volunteers matched by gender, age and smoking. Methylation was assessed using bisulfite-PCR-pyrosequencing. For all participants, medical history, physical and neurological examinations and screening laboratory tests were collected. All repetitive elements were hypermethylated in MS patients compared to healthy controls. A lower Expanded Disability Status Scale (EDSS) score was associated with a lower levels of LINE-1 methylation for ‘EDSS = 1.0’ and ‘1.5 ≤ EDSS ≤ 2.5’ compared to an EDSS higher than 3, while Alu was associated with a higher level of methylation in these groups: ‘EDSS = 1.0’ and ‘1.5 ≤ EDSS ≤ 2.5’.
Conclusions
MS patients exhibit an hypermethylation in repetitive elements compared to healthy controls. Alu and LINE-1 were associated with degree of EDSS score. Forthcoming studies focusing on epigenetics and the multifactorial pathogenetic mechanism of MS could elucidate these links further.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0395-0) contains supplementary material, which is available to authorized users.
Keywords: Multiple sclerosis, Hypermethylation, DNA methylation, Repetitive elements, Epigenetics, Expanded disability status scale
Background
Multiple Sclerosis (MS) is a neurodegenerative disease involving the central nervous system (CNS) in which the infiltration of focal lymphocytes in myelin causes inflammatory lesions leading to axonal damage [1]. MS knows many different disease courses, of which relapsing-remitting is the most common one. This course is typed with attacks of worsening neurological functioning (relapsing), followed by partial or complete improvement of the symptoms (remitting) [2]. Women are more prone to develop the disease than men, and the age of onset of the patient ranges mostly between 20 and 40 years, with a transition to the progressive forms at the age of 40 to 50 [3].
The causal factors of MS are still poorly understood, but they are probably heterogeneous [4]. Recent findings suggest that the interplay between individual genetic susceptibility and external, environmental influences modulate the disease presentation and therapeutic responsiveness [5, 6]. Evidence is increasing that epigenetic mechanisms could be involved in inflammatory and neurodegenerative diseases and some studies have suggested that changes in these mechanisms could contribute to MS pathogenesis, representing a bridge between genetics and environmental causal factors [7]. Epigenetics are stable and heritable patterns that modify the phenotype without altering the genotype. In particular, DNA methylation has been the most extensively studied epigenetic marker. It involves adding a methyl group to the 5′ cytosine located in a CpG site to form 5 methylcytosine (5mC). A recent study from Huynh et al. found that genes important in oligodendrocyte survival were hypermethylated and had a lower expression in MS-affected human brain tissue compared to controls [8]. Furthermore, studies have shown a relation between repetitive elements hypermethylation and adverse health outcomes [9, 10].
Repetitive elements comprise roughly 66–69 % of the human genome [11]. It is estimated that one million Alu repeats are present in the human genome, which accounts for over 10 % of the entire genome [12], while 20 % are long interspersed nuclear element 1 (LINE-1) repeats [13] and 3–5 % comprises of alpha satellite DNA sequences (SAT-α) [14]. SAT-α sequences can be found in centromeres or centromere-adjacent heterochromatin containing several CpG sites [15]. The methylation status of these sequences might be considered a good estimate for global DNA methylation levels, and have been previously investigated in relation to human diseases [16, 17]. Hitherto there is limited information about Alu, LINE-1 and SAT-α methylation in MS. Consequently, this study aims to evaluate the changes in methylation of these repetitive elements using a quantitative approach.
In the present study, we estimated repetitive element methylation levels in a population of 51 MS patients and 137 healthy controls matched for gender, age, and smoking status. These matching phenotypes were selected as they have been linked with differential methylation [18]. Methylation of Alu, LINE-1 and SAT-α was evaluated in association with MS course markers (i.e. Multisystem Deficits at disease onset, presence of oligoclonal bands in cerebrospinal fluid, CSF) and Expanded Disability Status Scale (EDSS). Quantitative bisulfite-polymerase chain reaction (PCR)-pyrosequencing was applied to determine methylation levels of Alu, LINE-1 and SAT-α.
Methods
Study design
Adult patients with MS (n = 51) were recruited at the Dino Ferrari Center, Fondazione Ca’ Granda, IRCCS Ospedale Maggiore Policlinico in Milan, from January to December 2010. Clinical diagnosis was performed using the McDonald criteria and their subsequent revisions [19–21]. All patients received standard clinical examinations, including medical history check, physical and neurological examinations, screening laboratory tests and a brain magnetic resonance imaging (MRI). Healthy control subjects (n = 137), matched for age, gender, smoking habits and ethnic background, were enrolled at the Department of Preventive Medicine, IRCCS Ospedale Maggiore Policlinico in Milan. Written informed consent was signed by each participant.
Healthy volunteers donated a blood sample at time of recruitment, while the MS patients donated blood at least one month after the completion of their steroid treatment following neurological symptoms. MS patients in remitting phase donated a single blood sample, while patients that were in the relapse phase during the first collection donated a second blood sample one month after an acute phase. Genomic DNA from 3 ml whole-blood was extracted using a FlexiGene DNA Kit (Qiagen Inc., Hilden, Germany), following manufacturer’s instructions. The DNA concentration in each sample was determined measuring the optical density (OD) at 260 nm wavelength on a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). DNA samples were aliquoted and stored at −20 °C until further measurements.
Analysis of DNA methylation
DNA methylation was quantified using bisulfite-PCR and pyrosequencing. In short, each sample (concentration 50 ng/μl) was treated using the EZ DNA Methylation Gold™ kit (Zymo Research, Orange, CA, USA). Final elution was performed in 30 μl of M-Elution Buffer. Bisulfite treated samples were used to assess DNA methylation of repetitive elements according to Yang et al. [16]. PCR primers were designed towards a consensus Alu, LINE-1, and SAT-α, and allowed the amplification of a representative pool of repetitive elements as a surrogate for global DNA methylation changes. PCR was carried out in 50 μl of Go-Taq Green Master Mix (Promega, Madison, WI, USA) with 1 pmol of the forward primer, 1 pmol of the reverse primer, 50 ng of bisulfite-treated DNA and water. Per primer pair (Table 1) either the forward or reverse primer was biotin labelled to purify the final PCR product by binding them to Streptavidin Sepharose HP beads (Amersham Biosciences, Uppsala, Sweden). The beads containing the bound PCR product were purified using the Pyrosequencing Vacuum Prep Tool (Pyrosequencing Inc., Westborough, MA, USA). The degree of methylation (%5mC) for each DNA locus is reported as a percentage of methylated cytosines divided by the sum of methylated and unmethylated cytosines. Samples were tested in triplicate for each marker to confirm reproducibility of the results. Concordance correlation coefficient obtained from duplicate runs was 0.518, 0.349 and 0.473 for Alu, LINE-1 and SAT-α respectively.
Table 1.
Sequences of the primers for Alu, LINE-1 and SAT-α from left to right are from 5′ to 3′. Biotinilation (BIO) of the primers occurred at the 5′ end. Forward and reverse primers were utilized in the PCR step while the sequencing primer was used during pyrosequencing
| Primer (5′ to 3′) | Alu | LINE-1 | SAT-α |
|---|---|---|---|
| Forward | BIO-TTTTTATTAAAAATATAAAAATT | TTTTGAGTTAGGTGTGGGATATA | BIO-TGGATATTTGGATTATTGG |
| Reverse | CCCAAACTAAAATACAATAA | BIO-AAAATCAAAAAATTCCCTTTC | TTTCCAAAAAAATCTTCAAAAAAAT |
| Sequencing | AATAACTAAAATTACAAAC | AGTTAGGTGTGGGATATAGT | CTCAAAAATTTCTAAAAATACTTCTC |
Statistical analysis
A descriptive analysis was performed on demographic and clinical characteristics. Mean and standard deviation were reported for continuous variables, count and percentage for categorical variables. Mean age and mean levels of methylation markers: Alu, LINE-1 and SAT-α were compared between MS patients and healthy controls by means of t-test.
Characteristics of MS patients and healthy controls were compared using the Chi-Square test or Fisher’s exact test for categorical variables and for continuous variables by the t-test. Correlation between three methylation markers in all participants was assessed with the Pearson correlation coefficient test. The same test was used to evaluate this correlation in controls and in MS patients. We computed odds ratios (ORs) and 95 % confidence intervals (CI) for the association of methylation levels with case/control status using multivariable logistic regression models, adjusted for age, gender and smoking status.
In order to investigate the potential association between disability status and the repetitive elements in MS patients, study participants were distributed equally between three groups according to their EDSS score: ‘EDSS = 1.0’, ‘1.5 ≤ EDSS ≤ 2.5’ and ‘3.0 ≤ EDSS ≤ 7.5’. Adjusted multivariable linear regression analyses was carried out to evaluate the relationship between the level of methylation in repetitive elements and EDSS score. The assumptions underlying the linear regression model (i.e. linearity, normality and homoscedasticity) were satisfied for all the independent continuous variables. Coefficients (ß) and 95 % CI were calculated firstly using ‘3.0 ≤ EDSS ≤ 7.5’ as reference for the other two groups and secondly using ‘EDSS = 1.0’ as the reference. Adjusted means and 95 % CI were also calculated and compared for each methylation marker.
Linear regression (adjusted for sex, age and smoking status) was applied to investigate the relationship between methylation marker level and disease activity, year of neurological symptoms onset, multisystem disorder, presence of oligoclonal bands, presence of multiple bands in CSF and spinal cord relapse. Statistical analysis was carried out using SAS software (version 9.2, SAS Institute, Milan, Italy). A two-sided p-value of less than 0.05 was considered statistically significant.
Results
Characteristics of the study population
Table 2 shows the characteristics of the 51 MS patients and 137 healthy control subjects. The mean age of the patients and control group was 39.6 and 41.4 years, respectively. Both groups had more female participants than male (25.5 % and 27.0 % for the patients and controls, respectively) while most of the subjects declared to be non-smokers (62.5 % of the MS patients and 62.0 % of the control group). Among MS patients, 58.8 % have had only one episode of neurological symptoms, while 41.2 % had at least two episodes. Thirty-eight percent of the patients had an EDSS score of 1 (38 %), 34 % a score between 1.5 and 2.5, and 30.0 % had a severe EDSS score of 3.5 to 7.
Table 2.
Characteristics of Multiple Sclerosis (MS) patients and healthy controls
| Characteristics | MS patients | Healthy controls | p-value |
|---|---|---|---|
| n = 51 | n = 137 | ||
| Age (years) | |||
| Mean ± SD | 39.6 ± 8.1 | 41.4 ± 9.1 | >0.1 |
| Gender | |||
| Female | 38 (74.5) | 100 (73.0) | >0.1 |
| Male | 13 (25.5) | 37 (27.0) | - |
| Smoking habita | |||
| Yes | 15 (35.3) | 52 (38.0) | >0.1 |
| No | 33 (64.7) | 85 (62.0) | - |
| Neurological episodes | |||
| Only one | 30 (58.8) | - | |
| At least two | 21 (41.2) | - | |
| Disabilityb | |||
| EDSS = 1.0 | 18 (36.0) | - | |
| 1.5 ≤ EDSS ≤ 2.5 | 17 (34.0) | - | |
| 3.0 ≤ EDSS ≤ 7 | 15 (30.0) | - | |
| Onset of MS | |||
| 1981 – 1999 | 18 (35.3) | - | |
| 2000 – 2004 | 15 (29.4) | - | |
| 2005 – 2010 | 18 (35.3) | - | |
| Multisystem deficits at onset | |||
| Yes | 14 (27.5) | - | |
| No | 37 (72.5) | - | |
| Oligoclonal Bands in CSFc | |||
| Yes | 44 (89.8) | - | |
| No | 5 (10.2) | - | |
| Methylation markers (%5mC) | |||
| Alu d | 25.3 ± 0.6 | 24.5 ± 1.1 | <0.001 |
| LINE-1 | 85.1 ± 1.4 | 82.6 ± 2.8 | <0.001 |
| SAT- α | 80.3 ± 2.6 | 78.9 ± 2.8 | 0.014 |
Data is presented as Mean ± SD or number (%). Disability is defined as ‘expanded disability status scale’ (EDSS)
Statistical comparison: student t-test between MS patients versus healthy controls: age and methylation markers. Chi-Square, total population: gender (50 males versus 138 females) and smoking habit (67 smokers versus 118 non-smokers)
aData available for: 48, b50 and c49 MS patients and d135 healthy controls
Internal correlations of repetitive elements
Considering the entire study population, Alu methylation level was positively correlated with the methylation level of both LINE-1 (ρ = 0.614, p < 0.0001) and SAT-α (ρ = 0.259, p = 0.0003). LINE-1 methylation was also positively correlated with SAT-α (ρ = 0.553, p < 0.0001). After case/control stratification, these correlations were confirmed in the control group (Alu and LINE-1: ρ = 0.627, p < 0.0001; Alu and SAT-α: ρ = 0.253, p = 0.0003; LINE-1 and SAT-α: ρ = 0.559, p < 0.0001). Conversely, in the MS patients group (Fig. 1), only LINE-1 methylation was correlated with the degree of methylation in SAT-α (ρ = 0.380, p < 0.0059). Alu methylation levels were not correlated with the methylation of either LINE-1 (ρ = −0.015, p = 0.285) or SAT-α (ρ = −0.025, p = 0.861).
Fig. 1.
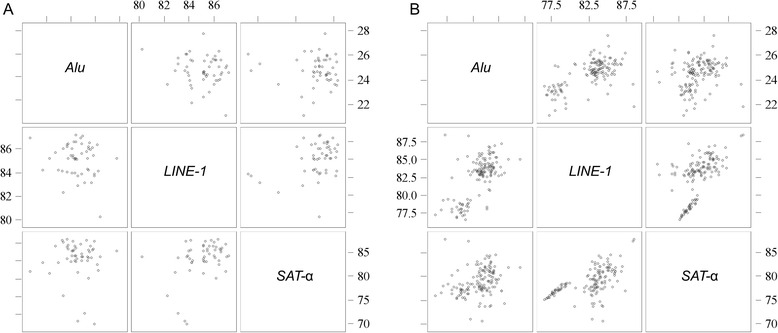
Scatter plot comparing Alu, LINE-1 and SAT-α methylation levels. In panel a, the correlation is represented for the MS patients, in panel b for the healthy controls. For the latter group, all repetitive elements were significantly correlated with each other in terms of methylation (Alu and LINE-1, ρ = 0.627; Alu and SAT-α, ρ = 0.253; LINE-1 and SAT-α, ρ = 0.559). In the MS patients, only LINE-1 methylation was significantly correlated with SAT-α methylation (ρ = 0.380). Significance level was set at 0.05
Differences in methylation levels in MS patients and healthy control subjects
As shown in Table 1, all methylation markers showed a significant increase in methylation in the MS patients group compared to the healthy control group. Alu methylation was 25.3 % 5mC in MS patients and 24.5 % 5mC in healthy controls (p < 0.0001). LINE-1 methylation was 85.1 %5mC in MS patients and 82.6 % 5mC in the healthy controls (p < 0.0001). SAT-α methylation was 80.3 % 5mC in MS patients and 78.9 %5mC in healthy controls (p < 0.003). The difference between cases and controls remained significant after adjusting for age, gender and smoking status in a multivariate logistic regression analysis for all markers (Alu: p = 0.0029; LINE-1: p = 0.0003; SAT-α: p = 0.0456), respectively the OR for Alu, LINE-1 and SAT-α were 2.137, 1.619 and 1.155. CpG site specific analyses are shown in Additional file 1: Figure S1 and their characteristics are presented in Additional file 2: Table S1.
Methylation levels in EDSS in MS patients
When we compared EDSS classes, using the highest EDSS class as reference, Alu methylation was elevated in both ‘EDSS = 1.0’ (ß = 0.47; 95 % CI = 0.04 to 0.90; p = 0.034) and ‘1.5 ≤ EDSS ≤ 2.5’ (ß = 0.63; 95 % CI = 0.18 to 1.08; p = 0.007). LINE-1 methylation was lower in both ‘EDSS = 1.0’ (ß = −0.95; 95 % CI = −1.89 to 0.003; p = 0.050) and ‘1.5 ≤ EDSS ≤ 2.5’ (ß = −1.07; 95 % CI = −2.06 to −0.09; p = 0.033). SAT-α methylation was not significantly different in the EDSS groups. Results are shown in Fig. 2.
Fig. 2.

Methylation levels of Alu, LINE-1 and SAT-α in MS patients, grouped by Expanded Disability Status Scale (EDSS), are represented as box plots. Data were adjusted for age, sex and smoking habit. All groups had a similar number of individuals: 18 participants for ‘EDSS = 1.0’, 17 for ‘1.5 ≤ EDSS ≤ 2.5’ and 15 for ‘3.0 ≤ EDSS ≤ 7.5’. For both Alu and LINE-1 the highest EDSS group (the reference) was significantly different from the lower two groups. An absolute difference in methylation of 0.47 %5mC can be observed for ‘EDSS = 1’ (p = 0.034) and 0.63 %5mC for ‘1.5 ≤ EDSS ≤ 2.5’ (p = 0.007) compared with the reference group for Alu. In LINE-1, methylation decreased with 0.95 %5mC for ‘EDSS = 1’ (p = 0.050) and 1.07 % for ‘1.5 ≤ EDSS ≤ 2.5’ (p = 0.033) compared to the reference group. * p < 0.05
Methylation levels and MS course
No significant differences were observed in the methylation markers between subjects who experienced just one neurological episode or at least two episodes. There was no significant association between methylation and years of onset. Furthermore, no differences were observed between methylation and multisystem disorders. The presence or amount of oligoclonal bands in CSF was also not associated with repetitive element methylation. Finally, no significant differences were found between patients with and without spinal cord relapse. Data is presented in Additional file 3: Table S2.
Discussion
In the present study, we evaluated methylation levels of Alu, LINE-1 and SAT-α in 51 MS patients and 137 healthy volunteers. MS patients showed hypermethylation in all repetitive elements compared to healthy controls. Furthermore, the correlations between these global methylation markers in the control group were lost in MS patients. Moreover, we demonstrated that worsening disability score was associated with hypomethylation in Alu and hypermethylation in LINE-1. These findings suggest that MS patients have an altered methylation of repetitive elements in blood leukocytes compared to healthy individuals. To our knowledge, this is the first study to investigate associations between methylation of Alu, LINE-1 and SAT-α, and MS.
Previous research found associations of global DNA hypermethylation with neurodegenerative and neurological disorders like Alzheimer’s disease [22, 23] and post-traumatic stress disorder [24]. However, the pathogenesis for MS is different from these disorders. Furthermore, a recent case–control study concerning genome-wide DNA methylation in MS patients finds immune cells to experience hypermethylation [25]. A second epigenetic mechanism involved in MS are micro RNAs (miRNA). These miRNAs are involved in gene silencing by degrading target mRNA sequences to prevent their translation into proteins. Literature suggest that certain miRNA (e.g. miR-155 and miR-326) are highly upregulated in active MS lesions [26]. This upregulation can in turn lead to macrophage activation, myelin degradation and could drive the MS progression [7, 27]. Furthermore, dysregulation of miR-155 and miR-326 has been observed in peripheral blood mononuclear cells and CD4+ T cells of MS patients respectively [7]. We found all repetitive elements to be hypermethylated in MS patients compared to the healthy controls. Since these repetitive elements are a marker for global DNA methylation levels, our results suggest that in MS the genome exhibits a higher degree of methylation. While a hypermethylation would indicate a downregulation of miR-155 and miR-326, no hypermethylation was observed in CD4+ T cells of MS patients, only in CD8+ T cells [28].
Healthy controls showed significant correlations among the degree of methylation for all repetitive elements, while in patients with MS only LINE-1 and SAT-α methylation levels remained significantly correlated. We speculate that for healthy subjects the upkeep mechanisms for these elements are able to maintain the normal methylation levels, while in MS patients these upkeep regulations might be partially lost. This could in turn lead to an abnormal maintenance of repetitive element methylation levels causing their reciprocal correlations to diminish.
We observed different methylation levels in Alu and LINE-1 among the three different EDSS groups. Methylation of Alu decreased with increasing EDSS scores. Although this tendency was also observed in SAT-α, it was not significant. In contrast, LINE-1 methylation was positively associated with EDSS score. These results could be counterintuitive as both Alu and LINE-1 are markers of global DNA methylation and are positively correlated with each other. However, as mentioned earlier, in MS patients this correlation disappears and they tend to be inversely (but not significantly) associated. Prior studies find that EDSS value is strongly correlated with axonal damage and neurodegeneration [29, 30]. Furthermore, a possible mechanism for MS progression and axonal loss could be via oxidative stress, caused by a decrease in antioxidant levels, which might lead to DNA damage [31]. A possible mode of action is through DNA methyl-transferase (DNMT), which are recruited to sites of DNA damage and have previously been suggested as being directly involved in DNA damage repair [32]. These DNMTs have been reported as important and essential elements in development and are responsible for genomic integrity due to their methylating capabilities [32, 33]. As Bollati et al. proposed, a LINE-1 hypermethylation could be the consequence of this DNMT upregulation [22]. The effect of DNMT overexpression was observed in brain tissue of mice, where they associated this upregulation with an increase in methylation in motor neuron cells [34].
Although we found MS patients to have hypermethylated repetitive elements, no distinct methylation differences were observed in different clinical MS groups (i.e. disease activity, phase of MS, days since relapse, year of onset, multisystem disorder, spinal cord relapse and the presence of oligoclonal bands in CSF). Our data had a limited amount of MS patients in different clinical groups, which might contribute to non-significant results.
Conclusion
As MS has a multifactorial pathology, hypotheses focusing solely on environmental or genetic components are missing key components. However, epigenetics are able to bridge the gap between these theories and appears to be promising. In summary, we found that 1) Alu, LINE-1 and SAT-α repetitive elements were hypermethylated in MS patients, 2) Alu, LINE-1 and SAT-α are positively correlated with each other in healthy controls, while only LINE-1 and SAT-α are correlated in MS patients and 3) EDSS values were associated with differential methylation in Alu and LINE-1 elements. We suggest that forthcoming investigations should include a higher number of MS patients to increase statistical power. Future studies focusing on epigenetics and both disease course and clinical prognostic markers could further elucidate the understanding of the multifactorial pathology of MS.
Abbreviations
5mC, 5-methylcytosine; CSF, cerebrospinal fluid; DNMT, DNA methyl-transferase; EDSS, expanded disability status scale; LINE-1, long interspersed nuclear elements; MS, multiple sclerosis; OR, odds ratio; PCR, polymerase chain reaction; SAT-a, satellite DNA alpha
Acknowledgments
This work was supported by Lombardy Region Research Contracts UniMi 31557/2010. Prof. Bollati received support from the EU Programme “Ideas” (ERC-2011-StG 282413).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Availability of data and materials
Not applicable for our dataset. Data will not be shared.
Authors’ contributions
KN and CF carried out the epigenetic studies. FC performed clinical marker assessment. KN, MP and VB drafted the manuscript. MP and DG recruited study participants and collected biological samples. LA performed the statistical analysis. ACP, ES and VB conceived the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Individual data is not published.
Ethics and consent to participate
This research was approved by the institutional review board (Fondazione IRCCS Ca’Granda, Ospedale Maggiore Policlinico, Milan. 2009). Informed consent was received from all the study participants.
Additional files
CpG specific odds ratios were calculated between MS (n = 51) patients and Healthy controls (n = 137 for LINE-1 and SAT-α; n = 135 for Alu). Methylation was assessed in 3 CpG sites for Alu and SAT-α and 4 CpG sites in LINE-1. Estimates are presented as odds ratios, adjusted for age, gender and smoking status, and were calculated using the multivariate logistic regression analysis. (DOCX 564 kb)
Characteristics of Multiple Sclerosis (MS) patients and healthy controls for each methylation marker. Methylation is subdivided as ‘mean’ (i.e. average of the separate positions) and the individual positions of the markers. (DOCX 66 kb)
Not significant data is presented as the estimate (ß) and their respective p-value (p). Disease activity is presented as ‘annualized relapse rate’ (ARR). The presence and amount of oligoclonal bands was measured in cerebrospinal fluid (CSF). (DOCX 18 kb)
Contributor Information
K. Y. Neven, Email: kristof.neven@student.uhasselt.be
M. Piola, Email: mirkopiola@yahoo.it
L. Angelici, Email: laura.angelici@unimi.it
F. Cortini, Email: francesca.cortini@guest.unimi.it
C. Fenoglio, Email: chiara.fenoglio@guest.unimi.it
D. Galimberti, Email: daniela.galimberti@unimi.it
A. C. Pesatori, Email: angela.pesatori@unimi.it
E. Scarpini, Email: elio.scarpini@unimi.it
V. Bollati, Phone: +39 02 503 20127, Email: valentina.bollati@unimi.it
References
- 1.Compston A, Coles A. Multiple Sclerosis. Lancet. 2008;372(9648):1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Weinshenker BG. Natural history of multiple sclerosis. Ann Neurol. 1994;36:6–11. doi: 10.1002/ana.410360704. [DOI] [PubMed] [Google Scholar]
- 3.Milo R, Kahana E. Multiple Sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev. 2010;9(5):387–94. doi: 10.1016/j.autrev.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Steinman L. Multiple Sclerosis: a two-stage disease. Nat Immunol. 2001;2(9):762–4. doi: 10.1038/ni0901-762. [DOI] [PubMed] [Google Scholar]
- 5.O’Gorman C, Lucas R, Taylor B. Environmental risk factors for multiple sclerosis: a review with a focus on molecular mechanisms. Int J Mol Sci. 2012;13(9):11718–52. doi: 10.3390/ijms130911718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldenberg MM. Multiple Sclerosis Review. Pharm Ther. 2012;37(3):175–84. [PMC free article] [PubMed] [Google Scholar]
- 7.Huynh JL, Casaccia P. Epigenetic mechanisms in Multiple Sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 2013;12(2):195–206. doi: 10.1016/S1474-4422(12)70309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, et al. Epigenome-wide differences in pathology-free regions of Multiple Sclerosis-affected brains. Nat Neurosci. 2014;17(1):121–30. doi: 10.1038/nn.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walters RJ, Williamson EJ, English DR, Young JP, Rosty C, Clendenning M, et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Epigenetics. 2013;8(7):748–55. doi: 10.4161/epi.25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neale RE, Clark PJ, Fawcett J, Fritschi L, Nagler BN, Risch HA, et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and pancreatic cancer. Cancer Epidemiol. 2014;38(5):576–82. doi: 10.1016/j.canep.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 11.de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384. doi:10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed]
- 12.International Human Genome Sequencing C Finishing the euchromatic sequence of the human genome. Nature. 2004;431(7011):931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 13.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 14.Horvath JE, Viggiano L, Loftus BJ, Adams MD, Archidiacono N, Rocchi M, et al. Molecular structure and evolution of an alpha satellite/non-alpha satellite junction at 16p11. Hum Mol Genet. 2000;9(1):113–23. doi: 10.1093/hmg/9.1.113. [DOI] [PubMed] [Google Scholar]
- 15.Lee C, Wevrick R, Fisher RB, Ferguson-Smith MA, Lin CC. Human centromeric DNAs. Hum Genet. 1997;100(3–4):291–304. doi: 10.1007/s004390050508. [DOI] [PubMed] [Google Scholar]
- 16.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38. doi:10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed]
- 17.Peluso M, Bollati V, Munnia A, Srivatanakul P, Jedpiyawongse A, Sangrajrang S, et al. DNA methylation differences in exposed workers and nearby residents of the Ma Ta Phut industrial estate, Rayong, Thailand. Int J Epidemiol. 2012;41(6):1753–60. doi: 10.1093/ije/dys129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaghlool SB, Al-Shafai M, Al Muftah WA, Kumar P, Falchi M, Suhre K. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenetics. 2015;7(1):6. doi: 10.1186/s13148-014-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121–7. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 20.Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58(6):840–6. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 22.Bollati V, Galimberti D, Pergoli L, Dalla Valle E, Barretta F, Cortini F, et al. DNA methylation in repetitive elements and Alzheimer disease. Brain Behav Immun. 2011;25(6):1078–83. doi: 10.1016/j.bbi.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry. 2012;2. [DOI] [PMC free article] [PubMed]
- 24.Rusiecki JA, Chen L, Srikantan V, Zhang L, Yan L, Polin ML, et al. DNA methylation in repetitive elements and post-traumatic stress disorder: a case–control study of US military service members. Epigenomics. 2012;4(1):29–40. doi: 10.2217/epi.11.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maltby VE, Graves MC, Lea RA, Benton MC, Sanders KA, Tajouri L, et al. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics. 2015;7:118. doi: 10.1186/s13148-015-0152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain. 2009;132(Pt 12):3342–52. doi: 10.1093/brain/awp300. [DOI] [PubMed] [Google Scholar]
- 27.Koch MW, Metz LM, Kovalchuk O. Epigenetic changes in patients with multiple sclerosis. Nat Rev Neurol. 2013;9(1):35–43. doi: 10.1038/nrneurol.2012.226. [DOI] [PubMed] [Google Scholar]
- 28.Bos SD, Page CM, Andreassen BK, Elboudwarej E, Gustavsen MW, Briggs F, et al. Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS One. 2015;10(3) doi: 10.1371/journal.pone.0117403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brass SD, Narayanan S, Antel JP, Lapierre Y, Collins L, Arnold DL. Axonal damage in multiple sclerosis patients with high versus low expanded disability status scale score. Can J Neurol Sci. 2004;31(2):225–8. doi: 10.1017/S0317167100053877. [DOI] [PubMed] [Google Scholar]
- 30.Guzel I, Mungan S, Oztekin ZN, Ak F. Is there an association between the Expanded Disability Status Scale and inflammatory markers in multiple sclerosis? J Chin Med Assoc. 2016;79(2):54–7. doi: 10.1016/j.jcma.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Ferretti G, Bacchetti T, Principi F, Di Ludovico F, Viti B, Angeleri VA, et al. Increased levels of lipid hydroperoxides in plasma of patients with multiple sclerosis: a relationship with paraoxonase activity. Mult Scler. 2005;11(6):677–82. doi: 10.1191/1352458505ms1240oa. [DOI] [PubMed] [Google Scholar]
- 32.Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 34.Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31(46):16619–36. doi: 10.1523/JNEUROSCI.1639-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable for our dataset. Data will not be shared.