

Abstract
Background
Materials rich in lignocellulose, such as straw, are abundant, cheap and highly interesting for biogas production. However, the complex structure of lignocellulose is difficult for microbial cellulolytic enzymes to access, limiting degradation. The rate of degradation depends on the activity of members of the microbial community, but the knowledge of this community in the biogas process is rather limited. This study, therefore, investigated the degradation rate of cellulose and straw in batch cultivation test initiated with inoculums from four co-digestion biogas plants (CD) and six wastewater treatment plants (WWTP). The results were correlated to the bacterial community by 454-pyrosequencing targeting 16S rRNA gene and by T-RFLP analysis targeting genes of glycoside hydrolase families 5 (cel5) and 48 (cel48), combined with construction of clone libraries
Results
UniFrac principal coordinate analysis of 16S rRNA gene amplicons revealed a clustering of WWTPs, while the CDs were more separated from each other. Bacteroidetes and Firmicutes dominated the community with a comparably higher abundance of the latter in the processes operating at high ammonia levels. Sequences obtained from the cel5 and cel 48 clone libraries were also mainly related to the phyla Firmicutes and Bacteroidetes and here ammonia was a parameter with a strong impact on the cel5 community. The results from the batch cultivation showed similar degradation pattern for eight of the biogas plants, while two characterised by high ammonia level and low bacterial diversity, showed a clear lower degradation rate. Interestingly, two T-RFs from the cel5 community were positively correlated to high degradation rates of both straw and cellulose. One of the respective partial cel5 sequences shared 100 % identity to Clostridium cellulolyticum.
Conclusion
The degradation rate of cellulose and straw varied in the batch tests dependent on the origin of the inoculum and was negatively correlated with the ammonia level. The cellulose-degrading community, targeted by analysis of the glycoside hydrolase families 5 (cel5) and 48 (cel48), showed a dominance of bacteria belonging the Firmicutes and Bacteriodetes, and a positive correlation was found between the cellulose degradation rate of wheat straw with the level of C. cellulolyticum.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-016-0543-9) contains supplementary material, which is available to authorized users.
Keywords: Biogas, Cellulose, Community composition, Glycoside hydrolases, cel48, cel5, Terminal restriction fragment length polymorphism (T-RFLP), Next generation amplicon sequencing
Background
Anaerobic digestion (AD) transforms biodegradable organic materials into a renewable energy resource, biogas, which can be used for production of vehicle fuel and/or for combined heat and electricity generation [1]. Furthermore, the residue after digestion is rich in nutrients and can be used as a fertiliser in crop production [2]. In a number of studies, production of biogas has been shown to offer significant advantages over other forms of bioenergy production and it has been rated one of the most energy-efficient and environmentally beneficial technologies for bioenergy production [3, 4]. Biogas can be produced from many different types of materials, including various types of waste streams from the food and feed industry, sludge from wastewater treatment plants, plant residues and manure from agriculture and energy crops [5, 6]. Plant-based biomass is very interesting in this regard, with lignocellulosic residues being the most promising material as these do not compete directly with food and feed production [7]. They include residues of agricultural plants, e.g. stalks, straw, husks, cobs, etc. The total amount of lignocellulosic residues accumulated annually in the world is estimated to be at least 10 billion tons [8].
Unfortunately, biogas production from lignocellulose-rich materials poses some challenges, as the complex structure, consisting of cellulose, hemicellulose and lignin cross-linked in a matrix structure, is very resistant to microbial degradation [9–13]. These difficulties can be overcome to some degree by a pre-treatment that breaks up the complex structure and makes the material more accessible for degradation, but the degradation rate and biogas yields are still typically rather low [7]. Moreover, many pre-treatments are energy- and cost-intensive, limiting large-scale application. Alternative less energy-consuming approaches to improve the degradation of lignocellulosic materials include management of the biogas process to ensure growth of microorganisms with efficient degradation capabilities, for example by adjustment of the solid retention time, using a two-stage approach or by a co-digestion approach as reviewed by Sawatdeenarunat and co-workers [7]. Bioaugmentation with efficient cellulose-degrading bacteria and addition of enzymes have also been suggested as promising methods to increase methane production from lignocellulosic materials [14, 15].
The microbial degradation of organic materials for biogas production requires at least four steps: hydrolysis, fermentation, acetogenesis and methanogenesis [16, 17]. In the first step, various hydrolytic microorganisms degrade complex organic polymers to monomers such as amino acids and sugars. In this step, microbes responsible for cellulose degradation use either free extracellular or cell-anchored enzyme complexes including cellulosomes, the latter more commonly found in anaerobic environments [10, 18]. A recent survey of around 1500 complete bacterial genomes revealed that ~38 % of the sequenced bacterial genomes encoded at least one cellulase gene, with a small fraction containing more than three cellulases, a prerequisite for effective degradation of natural cellulose [19]. The genes necessary for degradation of cellulose have been found in bacteria belonging to several different phyla: Actinobacteria, Fimicutes, Bacteroidetes, Thermotogae, Choloroflexi and Proteobacteria [19–21]. Cellulose-degrading bacterial communities specifically in biogas processes have been investigated by various methods. These include cultivation [22–27] and different molecular techniques targeting bacterial groups involved specifically in hydrolysis/acetogenesis [28] and functional genes, i.e. the glycoside hydrolase [29], or targeting the overall bacterial community [30–34]. The majority of cultivated cellulose degraders isolated from different anaerobic environments mainly belong to the order Clostridiales [10, 20, 21]. Bacteria from this order have also been shown to dominate in various AD processes operating with various lignocellulosic materials, such as wheat straw and cattle manure [31], maize, green rye and chicken manure [35], and maize straw and hay [36]. Clostridiales is also suggested to be of importance, specifically in the hydrolysis step, based on results obtained using a metagenomic approach [30]. In addition to Clostridiales, bacteria belonging to the order Bacteroidales have been suggested to be involved in the degradation of lignocellulose materials, such as straw and hay, in biogas processes [31, 36].
A range of studies have examined different technical/chemical/thermal methods for enhanced degradation of lignocellulosic materials and lately knowledge regarding the microbiological mechanisms, and organisms involved in the degradation of lignocellulose have also increased. However, the correlation between the structure of the cellulose-degrading community in a biogas process and efficient degradation is still unclear and requires further research. The aim of this study was, therefore, to search for correlations between the degradation rate of cellulose and straw and the bacterial community structure, including potentially cellulose-degrading bacteria. An additional aim was to obtain further information about the cellulose-degrading bacterial population in different AD processes. The degradation of straw and cellulose was analysed using a batch culture approach and with inoculums from ten different Swedish biogas plants, operating with different substrates and with different operating parameters. These inoculum samples were analysed both before and after the batch cultivation to determine the composition of potential cellulose-degrading bacteria by targeting the glycoside hydrolase genes of family 5 and family 48 glycoside hydrolases. The total bacterial community in the different biogas plants was also analysed, using next generation amplicon sequencing targeting the 16S rRNA gene.
Results and discussion
Characterisation of inoculum
The inoculum samples investigated in this study originated from four co-digestion plants processing various combinations of substrates (CD01–04) and six wastewater treatment plants processing mixed sludge (WWTP01–06). In general, the co-digesters had a longer hydrolytic retention time (HRT) and higher levels of VFA and total ammonium-nitrogen than the WWTP (Table 1). However, one co-digestion plant (CD03) was more similar to the WWTP in this respect. Moreover, the free ammonia level in the three plants CD01, 02 and 04 was considerably higher (>0.218 g L−1) than in the other plants (<0.063 g L−1).
Table 1.
Operating data for the 10 industrial-scale biogas plants investigated in this study
| Digester code | TSa (%) | VSb (%) | HRTc (day) | TMd (°C) | Tot Ne (g L−1 ww) | pH | VFAf (g L−1) | OLRg (vs g L−1 day−1) | TANh (g L−1 ww) | Ammoniai (g L−1 ww) | Major substrate |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CD01 | 5.7 | 3.5 | 45 | 38 | 8.7 | 7.8 | 1.3 | 3.0 | 4.6 | 0.365 | SSMOWj, slaughterhouse waste |
| CD02 | 4.8 | 3.8 | 55 | 38 | 9.1 | 7.8 | 0.8 | 2.9 | 5.1 | 0.408 | Thin stillage |
| CD03 | 2.6 | 2.0 | 30 | 37 | 2.6 | 7.3 | <0.1 | 3.0 | 0.9 | 0.022 | SSMOW |
| CD04 | 7.1 | 1.3 | 70 | 38 | 6.2 | 7.7 | 3.0 | 3.0 | 3.5 | 0.218 | Grass, wheat-based stillage |
| WWTP01 | 2.7 | 1.9 | 17 | 38 | 2.5 | 7.3 | <0.1 | 2.4 | 1.4 | 0.036 | Mixed sludge |
| WWTP02 | 3.5 | 2.5 | 23 | 37 | 2.9 | 7.5 | 0.2 | 3.1 | 1.6 | 0.063 | Mixed sludge |
| WWTP03 | 2.4 | 1.9 | 18 | 38 | 1.8 | 7.3 | 0.1 | 2.0 | 0.9 | 0.022 | Mixed sludge |
| WWTP04 | 3.4 | 2.4 | 30 | 37 | 2.9 | 7.5 | 0.3 | 1.6 | 1.5 | 0.058 | Mixed sludge |
| WWTP05 | 2.5 | 2.1 | 22 | 37 | 2.1 | 7.3 | 0.1 | 2.8 | 1.1 | 0.027 | Mixed sludge |
| WWTP06 | 2.1 | 5.5 | 26 | 34 | 1.8 | 7.3 | <0.1 | 1.1 | 1.1 | 0.023 | Mixed sludge |
CD 01–04 co-digestion plants, WWTP 01–06 wastewater treatment plants
aTotal solids
bVolatile solids
cHydraulic retention time
dTemperature
eTotal nitrogen
fVolatile fatty acids
gOrganic loading rate
hTotal ammonium nitrogen
iFree ammonia, calculated according to Hansen et al. [106]
jSource-separated municipal organic waste
BMP analysis
The final methane potential achieved with inoculum from the different industrial-scale biogas plants varied from 307 ± 54 to 376 ± 8 N mL CH4 g VS−1 for cellulose and from 233 ± 38 to 316 ± 37 N mL CH4 g VS−1 for straw (Table 2). The degradation rate and the time needed to reach the final potential also varied, with a clearly lower degradation rate in the tests initiated with inoculum from CD01 to 02. The time needed to reach 50 % of the final potential with these two inoculum samples was 18–47 days and 28–45 days for cellulose and straw, respectively, while for the other inoculum samples the corresponding values were 5–8 and 8–19, respectively. The methane potential values obtained for cellulose and straw were in line with those reported in previous studies, illustrating that the inoculum samples evaluated in this study came from fully functional biogas plants [37–39]. Some of the tests showed large standard deviation, most likely due to the non-homogeneous character of some inoculum samples making it difficult to divide them evenly between the triplicate bottles. It is also worth noting that the inoculum:substrate ratio inadvertently differed slightly between the different inoculum samples, but was still within the range suggested as optimal for a BMP test, i.e. 2–4. However, this difference could potentially have had an impact on the degradation rate and possibly also on the final potential in the test. The ratio for the inoculum from WWTP04–06 was adjusted to 3.8, 3.2 and 2.4, respectively, while the rest of the tests started with a ratio of 2. Even so, the degradation rate obtained in the tests of CD01–02 was still lower than for CD03–04 and WWTP01–03, despite all these sharing the same ratio.
Table 2.
Methane potential and time for degradation of straw and cellulose obtained in biochemical methane potential tests using inoculum from different biogas plants co-digesting different substrates (CD) or sludge from wastewater treatment plants (WWTP)
| Inoculum | Cellulose | Straw | ||||||
|---|---|---|---|---|---|---|---|---|
| Days to reach % of the final potential | Final potential | Days to reach % of the final potential | Final potential | |||||
| 100 % | 80 % | 50 % | 100 % | 80 % | 50 % | |||
| CD01 | 41 | 24 | 18 | 319 ± 24 | 110 | 56 | 28 | 258 ± 47 |
| CD02 | 110 | 71 | 47 | 307 ± 54 | 75 | 62 | 45 | 233 ± 38 |
| CD03 | 12 | 6 | 5 | 347 ± 15 | 60 | 23 | 8 | 316 ± 37 |
| CD04 | 20 | 10 | 8 | 348 ± 24 | 26 | 10 | 8 | 274 ± 17 |
| WWTP01 | 57 | 15 | 7 | 350 ± 7 | 110 | 44 | 19 | 290 ± 9 |
| WWTP02 | 36 | 13 | 7 | 314 ± 34 | 57 | 24 | 14 | 240 ± 38 |
| WWTP03 | 27 | 11 | 6 | 322 ± 7 | 75 | 25 | 13 | 310 ± 76 |
| WWTP04 | 29 | 15 | 8 | 325 ± 8 | 60 | 20 | 11 | 277 ± 11 |
| WWTP05 | 45 | 9 | 6 | 376 ± 8 | 59 | 30 | 10 | 281 ± 32 |
| WWTP06 | 49 | 13 | 8 | 324 ± 13 | 135 | 39 | 18 | 296 ± 8 |
Bacterial communities
Diversity indices
Analysis of the bacterial communities in the ten industrial-scale biogas plants by 454 pyrosequencing resulted in 36,523 sequences after quality trim and chimera check, with a range from 2573 to 4915 sequences per sample. Unique barcode was assigned to each replicate of one sample. The triplicate sequencing analysis was evaluated with unweighted UniFrac principal coordinate analysis (PCoA), where no outlier was observed (data not shown). The triplicates were then pooled in silico and randomly subsampled according to the sample having the lowest number of sequences (2500 sequences per sample). The number of OTUs per sample ranged from 52 to 258, with a comparatively lower value for the CD plants (Table 3). The rarefaction curve revealed the same general trend, i.e. a lower number of OTUs in the co-digestion plants compared with the WWTP (Table 3; Fig. 1). Based on the observed species and the Chao1 index, the sequencing covered 64.7–89.7 % of the total bacterial community. The three co-digestion plants CD01, 02 and 04 had low diversity and evenness of the bacterial community, as indicated by low values of species richness, Shannon and Simpson index. However, WWTP04 had similarly low values in this regard (Table 3).
Table 3.
Summary of observed OTUs, Chao1, Shannon and Simpson index in 10 industrial-scale biogas plants
| Sample | Chao 1 | OTUs | Shannon | Simpson |
|---|---|---|---|---|
| CD01 | 58 | 52 | 1.937 | 0.456 |
| CD02 | 94 | 69 | 3.111 | 0.763 |
| CD03 | 147 | 120 | 5.042 | 0.947 |
| CD04 | 109 | 96 | 3.619 | 0.767 |
| WWTP01 | 294 | 227 | 5.851 | 0.956 |
| WWTP02 | 215 | 187 | 4.737 | 0.861 |
| WWTP03 | 304 | 258 | 5.823 | 0.930 |
| WWTP04 | 209 | 135 | 3.088 | 0.609 |
| WWTP05 | 354 | 244 | 5.899 | 0.955 |
| WWTP06 | 280 | 242 | 5.857 | 0.930 |
CD 01-04 co-digestion plants, WWTP 01-06 wastewater treatment plants
Fig. 1.
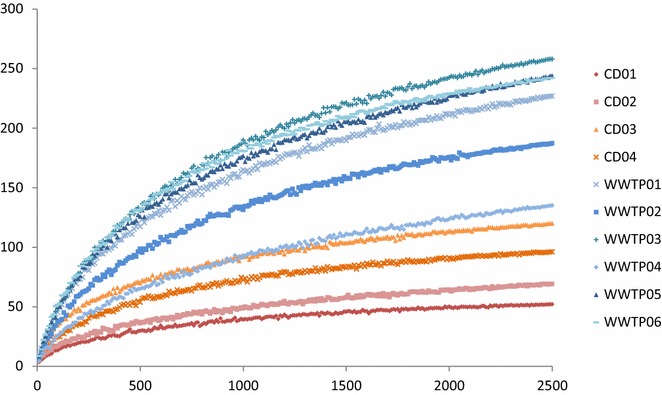
Rarefaction analysis of bacterial communities in 10 industrial-scale biogas plants. CD 01–04 co-digestion plants, WWTP 01–06 wastewater treatment plants
The low diversity in CD01, 02 and 03 correlated with the comparably higher level of ammonia in these digesters (two-sample t test and nonparametric Monte Carlo permutations, n = 999, P < 0.01). In biogas plants processing protein-rich materials, the ammonia released during the AD process is known to have a strong impact on both the community structure and the diversity, most probably because of the inhibitory effects of ammonia [40–42]. High evenness and richness have been suggested to be associated with good conversion efficiency of fatty acids to methane [43]. High initial evenness has also been shown to be important for preserving functional stability in microbial communities in general [44]. In the present study, CD01, 02, 04 and WWTP04 had lower evenness and diversity than the other digesters, implying that these biogas plants might have a comparatively lower potential to reach high degradation efficiencies. Indeed in the BMP tests started with inoculum from CD01 to CD02 a comparably lower methane production rates was seen for both substrates (Table 2). However, both CD04 and WWTP04 showed higher degradation rates, similar to the rates obtained with inoculums having a higher bacterial diversity (CD03, WWTP01–3 and WWTP05–6).
Phylogenetic analysis across samples
The phylogenetic composition, as determined by PCoA of unweighted UniFrac matrices (Fig. 2a), clearly revealed a clustering of the WWTPs, which suggests close phylogenetic distance within these different plants, while the CDs were more separated from each other and from the WWTPs. Considering the relative abundance, as revealed by the weighted UniFrac matrices, the plant CD03 was more closely related to the WWTPs, while the other three CDs plants were still distantly separated from each other and from the WWTPs (Fig. 2b). This confirms findings by Sundberg et al. [45] of two separate clusters distinguishing co-digestion plants and plants processing sewage sludge. In general, Firmicutes and Bacteroidetes were identified as predominant phyla in the industrial-scale biogas plants studied here, but other phyla were also present (Fig. 3). For example, the phyla Chloroflexi, Proteobacteria and OP8 were more associated with plants processing sewage sludge (Welch’s t-test, P < 0.01), while the co-digesting CD01 and 02 biogas plants contained a large fraction of sequences belonging to a unclassified cluster at phylum level. Bacteroidetes and Firmicutes are the two dominant phyla commonly found in various AD processes [21, 28, 31, 35, 36, 46, 47].
Fig. 2.
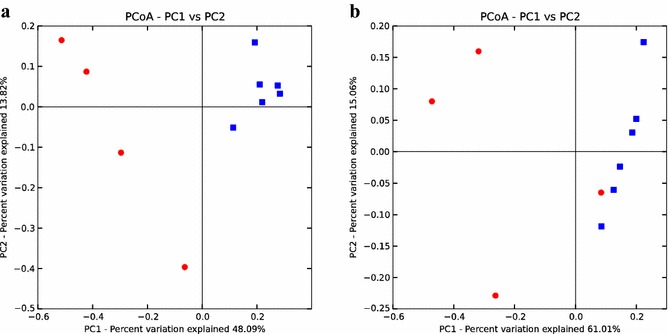
Phylogenetic distance between samples as determined by a unweighted and b weighted UniFrac principal coordinate analysis (PCoA) (red co-digestion plants, blue wastewater treatment plants)
Fig. 3.
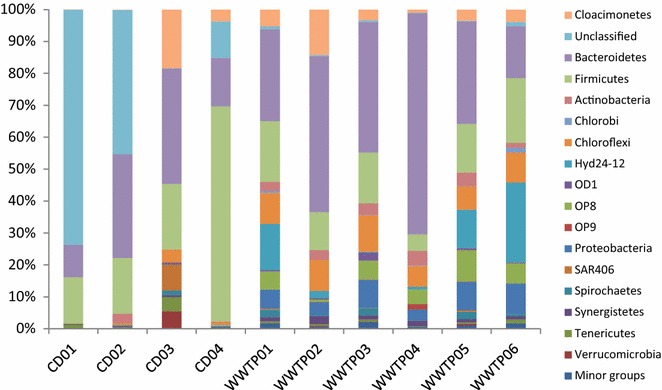
Relative abundance of bacterial 16S rRNA gene at phylum level in 10 industrial-scale biogas plants. CD 01–04 co-digestion plants, WWTP 01–06 wastewater treatment plants
The phylum Firmicutes was detected in all biogas plants, but at different relative abundance, from 5.2 to 67 % (Fig. 3). Within this phylum, the class Clostridia was dominant, with relative abundance of 4.4–64 %. Other classes were also present but at lower levels, such as Bacilli (<2.7 %) and Erysipelotrichi (<0.4 %) (Additional file 1: Figure S1). The order Clostridiales (3.8–56.3 %) was dominant within the clostridia, while Thermoanaerobacterales (<0.6 %) and unclassified orders such as MBA08 and SHA-98 comprised the rest of this class (Additional file 1: Figure S2). In anaerobic environments, Clostridiales has been reported as the main cellulose degrader [10]. This order has frequently been recovered from various digesters operating with mono- and co-digestion of lignocellulosic materials [31, 32, 46, 48–50]. However, the class Clostridia also contains proteolytic members and replacement of cellulolytic clostridia with proteolytic members has been observed when using protein-rich material as substrate with an inoculum originating from a biogas plant processing pig slurry and maize silage [51, 52]. A genus Caldicoprobacter (OTU 481, Additional file 1: Figures S4, S5) within Clostridiales was found at a higher relative abundance (0.68 %) in CD01, 02 and 04 compared to the rest samples (<0.04 %). This genus contains several xylanolytic bacteria [53–55]. The order MBA08 was detected in four samples in the present study CD01 (2.8 %), CD02 (1.5 %), CD04 (6.8 %) and WWTP06 (0.4 %) (Additional file 1: Figures S2, S5). This cluster was first identified in a thermophilic laboratory-scale digester [56] and later also in other thermophilic digesters [31, 57]. The presence of representatives from this cluster in the mesophilic biogas plants included in this study suggests that MBA08 contains organisms growing at wide range of temperatures. The order SHA-98 was observed at comparably higher levels in the high ammonia digesters CD01 and CD02, with relative abundance of 5.5 and 8.8 %, respectively. The levels in the rest of the digesters were 0.3–3.2 %. This order was represented by two dominant OTUs, OTU11 representing the main OTU in the high ammonia digesters CD01/02/04 (up to 7.9 %) and OTU 35, comparably more abundant in all other digesters (up to 2.6 %) (Additional file 1: Figure S5). In line with previous findings by Sundberg and co-workers [45], the family Clostridiaceae had higher relative abundance in WWTPs (2.2–5.4 %) than in CDs (0.2–0.9 %) (Welch’s t test, P < 0.01) (Additional file 1: Figure S3). Notably, the genus Gallicola, classified to the proposed family Tissierellaceae [58], was present at high relative abundance in plant CD04 (46.2 %) (Additional file 1: Figure S5). This family was not detected in any other biogas plant except WWTP06 (0.1 %) (Additional file 1: Figure S4). The first representative of the genus Gallicola was isolated from chicken manure and it has been shown to grow on purines, such as uric acid, xanthine, 6,8-dihydroxypurine, guanine and hypoxanthine [59].
The phylum Bacteroidetes had a relative abundance of 10.1–69.2 % (Fig. 3). Within this phylum, the order Bacteroidales (class Bacteroidia) was dominant in all samples (Additional file 1: Figure S2, S3). Members of the Bacteroidetes are able to degrade various polysaccharides [60, 61]. In a study with batch fermentation of straw and hay, the relative abundance of Bacteroidetes was higher at the end than at the beginning of the batch cultivation, indicating the importance of this phylum for degradation of lignocellulose [36]. An increase in relative abundance of this phylum has also been observed in response to straw addition in a laboratory-scale digester initially operating with cattle manure [31]. However, the Bacteroidetes have been shown to decrease in abundance at high levels of ammonia [41, 42, 51, 62]. In line with this, the lowest relative abundance of this phylum was seen for CD01 and CD04, both with relatively high ammonia levels (Fig. 3). However, CD02 also had a high ammonia level but had a similar relative abundance of Bacteroidetes as the low ammonia digesters. At family level, the Bacteroidaceae (up to 2.3 %), Marinilabiaceae (up to 0.2 %), Porphyromonadaceae (up to 9.2 %), Rikenellaceae (up to 0.7 %) and a few unclassified families were present (Additional file 1: Figure S3). One unclassified family, SB-1, was also observed, with higher abundance in CD03 (12.6 %) and WWTP01-06 (4.0–65.3 %) than in CD01, 02 and 04 (<0.2 %), possibly suggesting sensitivity to high ammonia levels (Table 1; Additional file 1: Figure S5). In addition, the Porphyromonadaceae were observed at relative abundance of 9.8 % in CD02 and 8.0 % in CD03, while other orders represented less than 2.0 %.
The candidate phylum Cloacimonetes (formerly WWE1) was observed at levels of less than 0.1 % in plants CD01 and CD02, but ranged from 1.0 to 14.2 % in the other eight biogas plants (Fig. 3; Additional file 1: Figure S5). Representatives of this phylum were first discovered in AD plants processing sewage sludge [63], but have since been found in a full-scale plant fed energy crop (mainly maize silage) [32] and a laboratory-scale digester fed cattle manure as the sole substrate or co-digested with wheat straw [31]. A study using stable isotopes suggested that members of this phylum are engaged in either cellulose hydrolysis or uptake of cellulose fermentation products [64]. The uncultured cluster at phylum level SAR406 was observed in CD03 with a relative abundance of 8.2 %, while the level in CD01-02 and in all WWTP was less than 1 % (Fig. 3; Additional file 1: Figure S5). The candidate name Marinimicrobia has been proposed for this cluster [65] and preliminary genome analysis suggests that members within this phylum are proteolytic and amino acid degraders [64, 66]. The phylum Proteobacteria was present at a level of 3.4–9.6 % in the WWTP, but less than 0.3 % in the CDs (Fig. 3; Additional file 1: Figure S5). This phylum has previously been found in various digesters processing sewage sludge [45, 67–69], but recently also in mono-digestion of fodder beet silage [46] and in a process co-digesting food waste with Chinese silvergrass [70]. Representatives of the uncultured phylum Hyd24-12 corresponded to 12–25 % in WWTP 01, 05 and 06, but were much less abundant in the other biogas plants (<2.3 %). This uncultured cluster has been found in other methanogenic digesters [71, 72], but its function in the methanogenic environment is still unknown [73]. The phylum Chloroflexi was represented with relative abundance of 6.5–11.5 % and 0.1–3.9 % in the WWTPs and CDs, respectively, and was dominated by an uncultured genus T78 (5.4–9.2 % in WWTPs and 0.1–3.0 % in CDs) (Fig. 3; Additional file 1: Figure S5). This phylum has been previously found in anaerobic digesters processing sewage sludge [74] and in co-digestion of whey permeate and cow manure [75], and members have been suggested to be carbohydrate utilisers [76, 77]. In addition, an uncultured cluster was found at high levels in CD01, 02 and 04, with a relative abundance of 73.7, 45.2 and 11.4 %, respectively (Fig. 3; Additional file 1: Figure S5). This large cluster was represented by one single OTU and had 84 % similarity based on 16S rRNA gene with an uncultured Clostridium (OTU1). The dominance of the community by such a large fraction represented by a single OTU is somewhat surprising and, to our knowledge, has not been reported previously for biogas digesters. Considering the feedstock for these biogas plants, this OTU might represent a protein-fermenting bacterium enriched by the protein-rich feedstock. Alternatively, this bacterium has a selective advantage at the high levels of the free ammonia in these digesters (0.218–0.408 g L−1). Ammonia has in previous studies been shown to have a strong selective pressure on the microbial community [41, 42].
T-RFLP
cel5
The cellulolytic community structures were investigated using T-RFLP combined with clone library analysis (Fig. 4a). Analysis of the inoculum samples revealed that T-RF 275 bp was present in all digesters except CD04, with a higher relative abundance in CD01–02 (51.1–52.1 %) than in the other plants (<21.1 %). T-RF 362 bp was also present at higher levels in CD01–02 (14.0–24.8 %) than in the other samples, with detectable levels only in samples WWTP02–04 (1.3–2.9 %). T-RF 85 bp had the highest relative abundance in CD03 (76.1 %), followed by WWTP01–04 (44.3–71.4 %), WWTP05–06 (14.5–15.4 %) and CD01–02 (2.8–9.2 %), while it was not detected at all in CD04. T-RF 396 bp dominated in CD04 (88.8 %) and WWTP05–06 (66.1–80.8 %), while T-RF 78 bp showed higher relative abundance in WWTP06 (13.1 %), WWTP04 (13.7 %) and WWTP03 (23.9 %), but was present at lower levels (<8 %) in the other plants.
Fig. 4.
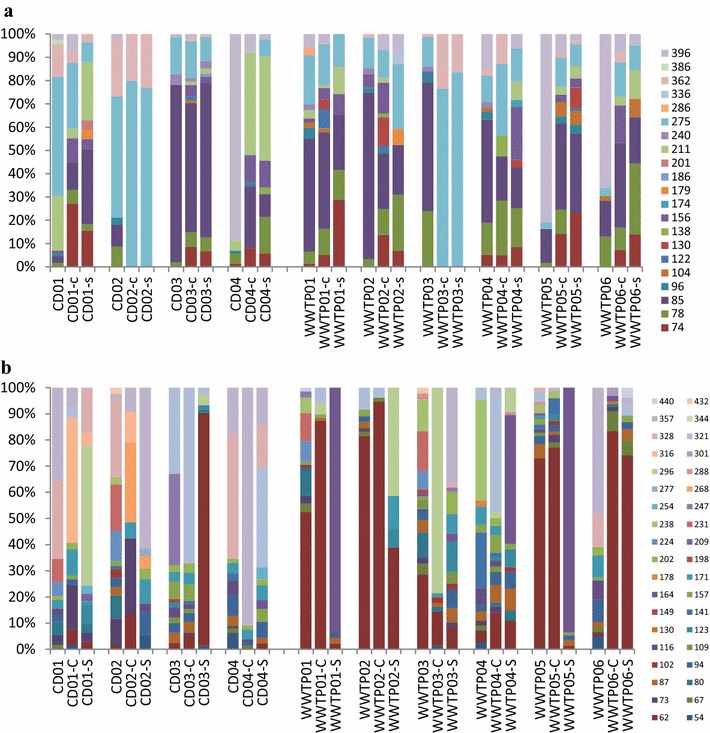
T-RFLP profile representing the community of a glycoside hydrolase gene family 5 (cel5) and b glycoside hydrolase gene family 48 (cel48) in 10 industrial-scale biogas plants: CD01–04/c/s and WWTP 01–06/c/s refer to inoculum analysed at the starting point of a batch cultivation and at the end point using cellulose (-c) and straw (-s) as substrate, respectively
Incubation with cellulose and straw in the batch test clearly changed the cel5 TRFLP profile compared to the one observed at the beginning of the experiment. However, the response varied both with inoculum and with substrate (Fig. 4a). In CD01, the relative abundances of the dominant T-RFs 275 and 362 bp were lower after the incubation with cellulose (28.3, 14.0 %) compared to levels at the beginning of the batch test. Instead the abundance of T-RFs 74, 78, 85, 156 and 396 bp were comparably higher (27.2, 6.0, 11.5, 10.6 and 7.4 %, respectively) than at the starting point (0, 1.8, 2.8, 1.2 and 2.6 %, respectively). For CD02, a similar decrease was seen for T-RF 362 bp, while with this inoculum T-RF 275 bp increased to 79.9 %. The other T-RFs (i.e. 74, 78, 85, 156 and 396 bp) that increased to a smaller extent in CD01 were not detected at the end point for this inoculum. Using straw as substrate resulted in a similar T-RFLP profile change as for cellulose for both CD01 and CD02. For CD03 and WWTP01–04, the relative abundance of the most dominant T-RF 85 bp decreased after incubation with both cellulose and straw. For WWTP03, the level of T-RFs 78 and 85 bp was reduced to non-detectable, while instead T-RFs 275 and 362 bp increased to 76.5 and 23.5 % for cellulose (83.6 and 16.4 % for straw). However, at the end point using both substrates, T-RFs 74, 78 and 156 bp increased their level for CD03, WWTP01–02 and WWTP04, except T-RFs 74 and 156 bp for WWTP04c. For WWTP04c, the level of T-RFs 275 and 362 bp increased. T-RF 122 bp increased in CD03c and WWTP01–02c, while T-RF 130 bp increased in WWTP01–02c. For CD04 and WWTP05–06, the most pronounced change was for the dominant T-RF 396 bp, which decreased to 7.9, 8.1 and 7.8 %, respectively. Instead, for CD04c, T-RFs 211, 85, 156 and 74 bp increased to 44.1, 26.5, 11.9 and 8.2 %, respectively. In addition, for CD04 s T-RF 78 bp increased to 15.8 %. For WWTP05, T-RFs 74, 78, 85 and 275 bp increased to 14.1, 10.5, 37.1 and 12.4 %, respectively, for cellulose, while T-RFs 74, 85 and 275 bp increased to 23.4, 33.7 and 9.5 %, respectively, for straw. The profile of WWTP06 was similar to that of WWTP05 but, in addition, T-RFs 156 and 78 bp increased to 16.1 and 30.5 % for WWTP06c and WWTP06 s, respectively.
cel48
The T-RFLP profile obtained by the cel48 primer set of the inoculum samples showed a high relative abundance of T-RF 62 in the majority of samples from the WWTPs, with WWTP01, 02, 03 and 05 having relative abundance of 28.6–81.5 % (Fig. 4b). The level in WWTP04 and WWTP06 was lower, 4.8 and 1.4 %, respectively. This T-RF was only detected in two CDs (CD02–03) and at low levels (1.5–2.3 %). For WWTP04, the dominant T-RFs were instead 141 bp (21.2 %) and 238 bp (38.6 %), while for WWTP06, the two most dominant T-RFs were 357 bp (48.2 %) and 328 bp (12.8 %). For CD01 and CD04, the T-RFLP profile was dominated by T-RFs 357 bp (35.4 and 17.4 %) and 328 bp, (30.1 and 48.0 %). For CD02, the level of T-RF 328 bp was similar (29.9 %), but T-RF 357 bp represented only 1.9 %. Instead, T-RFs 231, 224 and 73 bp were present at a level of 17.9, 11.5 and 10.2 %, respectively. For CD03, the two most dominant T-RFs were 247 bp (34.9 %) and 321 bp (33.0 %), which were present at a relatively low level in all other biogas plants.
In contrast to the cel5 community, the end point cel48 community in most cases was not the same when using cellulose and straw as substrate (Fig. 4b). For CD01, the dominant T-RFs 328 and 357 bp decreased in both cases, but T-RF 316 bp (47.9 %) increased in the cellulose incubations and T-RF 296 bp (53.6 %) increased after incubation with straw. For CD02, the dominant T-RF after incubation with straw and cellulose was 268 bp (30.5 %) and 357 bp (61.6 %), respectively. For CD03, the starting point T-RF 247 bp disappeared for both end points, while T-RF 321 bp increased in response to cellulose addition (67.0 %), but declined in abundance when straw was used as substrate (2.5 %). In addition, T-RF 62 bp increased to 89.0 % when straw was used. For CD04, T-RF 357 bp dominated after digestion with cellulose (90.9 %), while T-RFs 321 bp (38.4 %), 328 bp (16.2 %) and 357 bp (14.1 %) dominated in the straw cultures. For WWTPs, after digestion with cellulose, the major peak was T-RF 62 bp (13.0–94.6 %). In addition, T-RFs 296 bp (78.7 %) and 321 bp (45.1 %) were high in WWTP03 and 04, respectively. For the end point with straw, T-RF 62 bp remained as the major peak in WWTP02 (38.9) and 06 (74.1 %) and T-RF 296 bp was also high in WWTP02 (41.4 %). For WWTP01 s and 04–05 s, the dominant peak changed to T-RF 164 bp, with a relative abundance of 94.0, 49.3 and 93.6 %, respectively. For WWTP03 s, T-RF 357 bp, with a level of 36.3 %, was the major peak in this plant.
Clone libraries and phylogenetic analysis
Sequencing of 118 and 215 clones from cel5 and cel48 libraries resulted in 10 and 11 OTUs, respectively. All OTUs have low similarity to characterised bacteria, with one exception of OTU10, which partial sequence shared 100 % identity to Clostridium cellulolyticum. For twenty out of 21 OTUs the closest cultivated bacterial phyla belonged to Bacteroidetes or Firmicutes, one OTU was close to Actinobacteria (cel48 OTU06, Table 4). This is in agreement with previous studies investigating the bacterial community in biogas digesters using the same primers that all analysed clones were close to Bacteroidetes and Firmicutes [29]. Although the partial deduced amino acid sequences are as short as 100 and 130 amino acid residues, respectively, both trees are supported by high bootstrap values.
Table 4.
Clone sequences of cel5 and cel48 obtained from industrial-scale biogas processes
| Clone | T-RFs (bp) | Most closely related microorganism | Identity (%) | Accession number |
|---|---|---|---|---|
| Cel5 | ||||
| OTU01 | 56 | Ruminococcus callidus | 84.8 | WP_021681794 |
| OTU02a | 85 | Echinicola vietnamensis | 58.0 | WP_015264998 |
| OTU03b | 85 | Echinicola vietnamensis | 61.4 | WP_015264998 |
| OTU04c | 106 | Flavobacterium sp. | 66.3 | WP_007808671 |
| OTU05 | 136 | Niastella koreensis | 73.7 | AEV98714 |
| OTU06d | 211 | Marinilabilia salmonicolor | 66.3 | WP_036163195 |
| OTU07e | 275 | Mahella australiensis | 57.4 | AEE96311 |
| OTU08f | 362 | Mahella australiensis | 58.4 | AEE96311 |
| OTU09 | 375 | Clostridium sp. | 72.1 | WP_033166154 |
| OTU10 | 396 | Clostridium cellulolyticum | 100 | WP_015924614 |
| Cel48 | ||||
| OTU01 | 68 | Clostridium stercorarium DSM 8532 | 57.9 | AGI39871 |
| OTU02 | 68 | Clostridium sp. Iso6-17a | 50.0 | ADM52292 |
| OTU03 | 177 | Ruminococcus sp. CAG:254 | 97.5 | CCZ84184 |
| OTU04g | 205 | Clostridium sp. Iso6-24 | 79.0 | ADM52293 |
| OTU05 | 238 | Ruminococcus sp. HUN007 | 83.2 | WP_049962845 |
| OTU06 | 238 | Streptomyces griseorubens | 61.0 | WP_037642616 |
| OTU07h | 247 | Acetivibrio cellulolyticus | 71.2 | WP_010681059 |
| OTU08 | 290 | Clostridium acetobutylicum | 50.5 | WP_010964229 |
| OTU09i | 321 | Ruminiclostridium thermocellum | 75.0 | ACT46162 |
| OTU10j | 328 | Clostridium termitidis CT1112 | 73.8 | EMS73539 |
| OTU11k | 358 | Clostridium straminisolvens JCM 21531 | 78.8 | GAE90081 |
aMost closely related to uncultured bacterium AGW24153 (identity: 100 %) from laboratory-scale anaerobic reactor
bMost closely related to uncultured bacterium AGW24153 (identity: 89.1 %) from laboratory-scale anaerobic reactor
cMost closely related to uncultured bacterium ACV50344 (identity: 73.3 %) from lignocellulose-based sulphate-reducing bioreactor
dMost closely related to uncultured bacterium AGO64733 (identity: 75.0 %) from anaerobic digester sludge
eMost closely related to uncultured bacterium AEV59723 (identity: 77.0 %) from laboratory biogas digester treating rice straw
fMost closely related to uncultured bacterium AEV59723 (identity: 76.0 %) from laboratory biogas digester treating rice straw
gMost closely related to uncultured bacterium AGO64695 (identity: 99.0 %) from anaerobic digester sludge
hMost closely related to uncultured bacterium AGO64692 (identity: 75.2 %) from anaerobic digester sludge
iMost closely related to uncultured bacterium AGO64682 (identity: 85.6 %) from anaerobic digester sludge
jMost closely related to uncultured bacterium AGO64673 (identity: 99.1 %) from anaerobic digester sludge
kMost closely related to uncultured bacterium AGO64695 (identity: 99.0 %) from anaerobic digester sludge
cel5
T-RF 275 bp, represented by OTU07 and present in all digesters except CD04, and T-RF 362 bp (OTU08), highly abundant in CD01 and CD02, were both most closely related to an uncultured bacterium (AEV59723), with an identity of 77.0 and 76.0 %, respectively. Mahella australiensis (phylum Firmicutes) was the closest cultivated bacterium, with an identity of 57.4 and 58.4 %, respectively (Table 4, Fig. 5). M. australiensis is able to ferment different carbohydrates, including cellobiose [78]. It has previously been detected in thermophilic digesters operating with chicken manure, where it is suggested to be sensitive to ammonia [79, 80]. This species has also been observed in the cel5 community in a CSTR digester fed cow manure and steam-exploded straw operating at 44 °C [29]. T-RF 85 bp, present at high level in most digesters and represented by OTU02 and OTU03, was closely related to an uncultured bacterium (AGW24153), with an identity of 100 and 89.1 %, respectively. This OTU has previously been observed during anaerobic digestion of cow manure and steam-exploded straw at 37 °C [29]. The closest cultivated bacterium was Echinicola vietnamensis, with an identity of 58 and 61.4 %, respectively. E. vietnamensis was first isolated from seawater [81], and is able to hydrolyse starch and grow up to 44 °C with 15 % NaCl. T-RF 396 bp (OTU10), dominating in CD04, WWTP05 and WWTP06, was verified as C. cellulolyticum (identity 100 %). This is a well-studied non-ruminal mesophilic cellulolytic bacterium, initially isolated from decayed grass [82] that has the ability to degrade cellulose and hemicellulose to acetate, ethanol, lactate and H2 [83]. Moreover, it has been shown that bioaugmentation with C. cellulolyticum can increase the cellulose degradation efficiency of wheat straw during batch cultivation [84]. T-RF 78 bp, which increased in most of the digesters in response to cellulose and straw, was unfortunately not found in the clone library in this study. However, in a previous study investigating the cel5 community in digesters operating with manure and steam-exploded straw, a T-RF of the same size was shown to correspond to a clone related to Eubacterium siraeum (49.5 % identity) [29]. The abundance of the organism in that study was shown to increase from 14 to 52 % with increasing temperature from 37 to 52 °C. This bacterium has been isolated from human faeces and belongs to a genus commonly existing in the human gut and involved in cellulose degradation [85] [86]. T-RF 211 bp, which increased in the CDs in response to both straw and cellulose, was represented by OTU06. This OTU was related to an uncultured bacterium (AGO64733; 75.0 % identity) previously identified in a laboratory-scale reactor operating with manure and straw [29] and grouped with P-OTU05 and 07, identified in the same study (Fig. 5). The closest cultured relative, Marinilabiliasalmonicolor (phylum Bacteroidetes, 66.3 % identity) was isolated in seawater and has been shown to have the ability to degrade cellulose, monomeric sugars and starch [87].
Fig. 5.
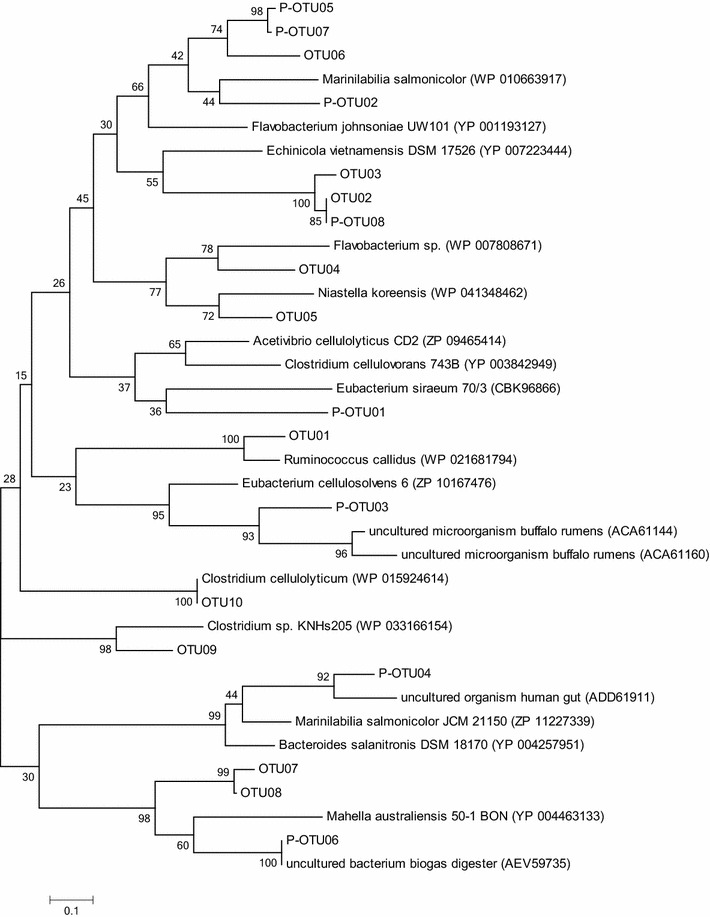
Phylogenetic tree of sequences from glycoside hydrolase gene family 5 retrieved from different industrial-scale biogas processes. OTUs operational taxonomic units identified in this study, P-OTUs operational taxonomic units identified in Sun et al. [29])
cel48
T-RF 238 bp (OTU05), dominating in WWTP04, was most closely related to Ruminococcus sp. (identity 82.3 %). T-RFs 358 bp (OTU11) and 328 bp (OTU10), dominating in WWTP06, were most closely related to uncultured bacterium AGO64695 (99.0 % identity) and AGO64673 (99.1 % identity) as identified previously [29]. The closest cultivated relative was Clostridium straminisolvens JCM 21,531(78.8 % identity) and Clostridium termitidis CT1112 (73.8 % identity) for OTU11 and OTU10, respectively (Fig. 6). Both C. straminisolvens and C. termitidis have previously been shown to have the ability to degrade a variety of monomeric sugars, as well as cellobiose [88, 89]. Clostridium straminisolvens has been detected in various types of biogas digesters, such as an anaerobic thermophilic digester fed municipal waste [24] and an anaerobic mesophilic digester fed pig manure, rice straw [90, 91]. The T-RF with size 238 bp was also found in digester WWTP03 at an abundance of 11.9 %. In this case, the corresponding clone (OTU06) was closely related to Streptomyces griseorubens (61 % identity). This result highlights one drawback of T-RFLP analysis, with several different sequences resulting in the same T-RF size, and the importance of combining this method with a clone library. S. griseorubens has been isolated from soil in both India and China, and the ability to degrade lignocellulose has been demonstrated [92–94]. Clones OTU07 and OTU09 represented T-RFs 247 bp and 321 bp, respectively, which were present at comparatively high levels in CD03. For these, two uncultured bacteria (AGO64692; identity 75.2 % and AGO64682; identity 85.6 %), respectively, both retrieved from anaerobic digester sludge, had the highest similarity. The closest known bacteria were Acetivibrio cellulolyticus (71.2 % identity) and Ruminiclostridium thermocellum (75 % identity), respectively. A. cellulolyticus can utilise cellulose, cellobiose and salicin and has previously been found in a methanogenic enrichment culture from municipal sewage sludge [95]. R. thermocellum, synonym of Clostridium thermocellum, is a well-studied anaerobic cellulose-degrading thermophilic organism which has been suggested as a potential candidate for different biotechnological applications [96]. It has been isolated from plants and from cow and horse manure [97] and has been demonstrated to play a key role in cellulolytic degradation in different types of biomethane production digesters [98–100]. After incubation with straw, T-RFs 296 and 164 bp increased in abundance in CD01 and WWTP01, respectively. These two T-RFs could not be found in the clone libraries. However, in a previous study of laboratory-scale digesters operating with manure and straw at 44 °C [29], both T-RFs were found and suggested to represent species closely related to A. cellulolyticum (74.3 % identity) and Ruminococcus champanellensis (61.8 % identity), respectively. R. champanellensis is able to utilise cellulose, cellobiose and xylan, but not starch and pectin [101]. OTU02 (T-RF 68) and OTU08 (T-RF 290) grouped together in the phylogenetic tree (Fig. 6) and showed similarity to Ruminococcus, but with relatively low identity (50.0 and 50.5 %, respectively). OTU04, 09 and 11 grouped together with P-OTU03 and 08 from a previous study investigating potential cellulose-degrading bacteria in digesters operating with straw and cow manure [29].
Fig. 6.
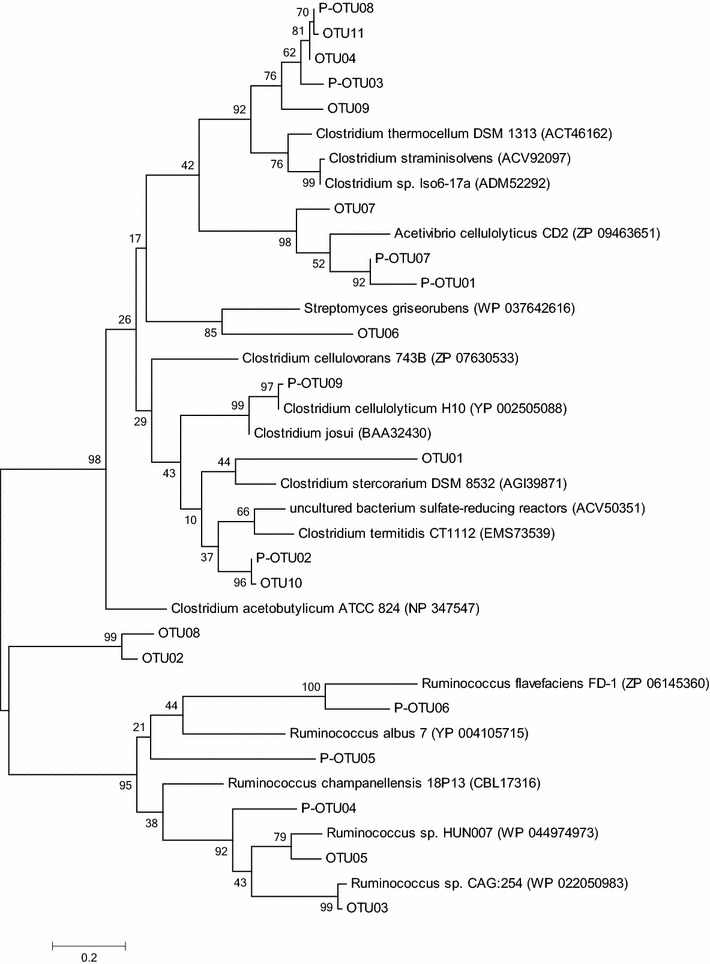
Phylogenetic tree of sequences from glycoside hydrolase gene family 48 retrieved from different industrial-scale biogas processes. OTUs operational taxonomic units identified in this study, P-OTUs operational taxonomic units identified in Sun et al. [29]
Correlation of microbial community structure with process parameters
To identify possible correlations between microbial community composition within the inoculum and biogas process operating parameters (Table 1) and batch digestion performance (Table 2) using cellulose and straw as substrate, CCA was performed. On including the process parameters (free ammonia, OLR, HRT and VFA) and process performance (inverse of days needed to reach 50 and 80 % of the final methane potential for cellulose and straw, C50/80 and S50/80), the first two dimensions of the CCA plot explained around 58.7 % (16S amplicon sequencing; Fig. 7a) and 79.6 % (cel5, Fig. 7b) of the variation in relative abundance of OTUs and T-RFs, respectively. For cel48, no clear clustering of community structure or clear influence of parameter was found (data not shown).
Fig. 7.
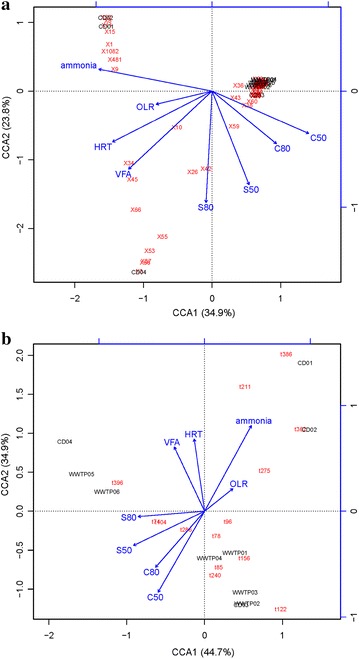
Canonical correspondence analysis (CCA) of a the major OTUs at genus level of the 16S rRNA gene and b the T-RFs of cel5 community, within 10 industrial-scale biogas plants. CD 01–04 co-digestion plants, WWTP 01–06 wastewater treatment plants. Ammonia free ammonia, OLR organic loading rate, HRT hydraulic retention time, VFA volatile fatty acids, C50/80 and S50/80: the inverse of days needed to reach 50/80 % of final methane potential for cellulose and straw, respectively
The CCA plot of amplicon sequencing data (Fig. 7a) showed a similar pattern to the weighted UniFrac PCoA plot (Fig. 2b). The free ammonia concentration was identified as the one important factor influencing the community structure, determined by analysis of both the 16S rRNA and cel5 gene. Batch degradation performance, i.e. the time to reach 50 and 80 % degradation of both cellulose and straw, on the other hand, was negatively associated with the level of free ammonia in the original process in both analyses. At present, not much information is available in the literature regarding the impact of ammonia on the degradation of cellulose and the obtained results so far are contradictory [21]. In the 16S rRNA amplicon dataset, a few OTUs were found to be positively correlated with ammonia level, including the highly dominant OTU in both digester CD01 and CD02. This OTU was also negatively correlated with the batch process performance. For the cel5 community, the dominant T-RFs 362 bp showed a positive correlation with ammonia concentration, while the T-RFs 396 and 85 bp were correlated with process performance. One of these T-RFs (TRF 396) represented a clone with 100 % similarity to C. cellulolyticum, recently shown to increase the cellulose degradation efficiency of wheat straw during bioaugmentation [84]. The batch test results cannot be completely transferred to the industrial-scale process, i.e. the degradation in the batch test might not be the same as in the continuous full-scale system. Still the microbial composition in the inoculums, shaped by the process parameters in the full-scale plant, will most likely impact on the outcome of the BMP test, as has been shown in previous studies [102].
Conclusions
The overall bacterial communities within the investigated co-digestion and WWTP plants were separated from each other, probably owing to differences in substrate and/or operating parameters used by these two groups of biogas plants. Moreover, the diversity was lower in CD compared with WWTP plants. Among the ten plants, two showed clearly lower degradation efficiency of straw and cellulose, measured during batch cultivation. These two plants also had the lowest bacterial diversity (species richness and evenness) and the highest level of ammonia. Ten of 21 OTUs obtained from clone libraries based on the glycosidase hydrolase gene sequence were mainly distantly related to known organisms, while the rest were related to partial sequences of unknown bacteria, although according to the phylogenetic analysis still related to saccharolytic or cellulolytic bacteria. Statistical analysis identified ammonia as a parameter with a strong impact on the cel5 community, while no clear trend could be seen for the cel48 community. This indicates that ammonia not only influences the methanogenic community structure in biogas processes, but also shapes the community of bacteria involved in the hydrolysis step. Interestingly, two dominant T-RFs from the cel5 community were positively correlated with batch process performance, i.e. the degradation efficiency of straw and cellulose. One of these T-RFs represented a clone with 100 % similarity to C. cellulolyticum, recently shown to be of importance for the degradation of wheat straw. The presence of this bacterium was negatively correlated to the ammonia level, supporting the idea that ammonia might have a negative impact on the degradation of lignocellulosic material.
Methods
Biogas plants
In total, the main digesters of 10 different industrial-scale biogas plants were investigated. These digesters were all operating at mesophilic temperature (range 37–38 °C), six processing sludge from wastewater treatment plants (WWTP01–06) and four co-digesting various organic waste fractions (CD01–04), such as agricultural waste, source-separated organic municipal household waste and slaughterhouse waste (Table 1). All investigated biogas plants had been in operation for several years under similar conditions regarding substrate and operating parameters.
Bio-methane potential test
The methane potential of cellulose and straw was determined by biochemical methane potential (BMP) analysis [103]. The substrate used in the BMP test was cellulose (C6663, Sigma-Aldrich, MO, USA) and wheat straw (mechanical chopped into 1–2 cm). Inoculum was collected from the different biogas plants and incubated at 37 °C for 4–6 days prior to the BMP test to decrease gas production from endogenous material. The batch anaerobic digestion was performed in 309 mL serum bottles, to which substrate was added corresponding to 3 g volatile solids (VS) per liter, and the inoculum:substrate ratio was between 2:1 and 4:1. The final liquid volume was set to 193 mL in all bottles by adding tap water. For each inoculum, triplicate bottles were set up for straw and for cellulose. Triplicate bottles filled only with the inoculum, corresponding to the same volume as in the test bottles, were also set up to measure background production of methane (CH4). All bottles were incubated at 37 °C on a rotary shaker at 130 rpm. Total gas production and methane content were continuously monitored over 60 days by pressure measurement combined with gas sampling and gas chromatograph (GC) analysis of gas composition [103]. The accumulated methane production was calculated and the gas production from the control was deducted. The volumetric methane value was normalised to standard temperature (273.15 K) and pressure (1 bar) using the ideal gas law, and finally expressed as N mL CH4 g VS−1 [104]. The days needed to reach 50 and 80 % of final methane potential was used as a measure of degradation capacity.
Analytical methods
Content of total solids (TS) and VS in the inoculum samples was measured according to the international standard methods [105]. Total Kjeldahl nitrogen (TKN) and ammonium-nitrogen (NH4-N) were analysed according to the International Standardization Operation (ISO) methods (ISO 10,694, 1995 and ISO 13,878, 1998). The concentration of free ammonia was calculated from the NH4-N concentration, pH and temperature according to Hansen et al. [106]. The volatile fatty acid (VFA) content in the digester samples was determined by high-performance liquid chromatography (HPLC) analysis as described previously [103].
DNA extraction
Samples for microbial community analysis were taken from the inoculum on the start day of the BMP test and at the end of the anaerobic batch test. A 15 mL sample was withdrawn from each inoculum and each batch test. The latter samples were designated CD 01–04c, WWTP 01–06c and CD 01–04 s, WWTP 01–06 s, for samples from incubation with cellulose and straw, respectively. All samples were stored at −20 °C until the extraction of DNA. Total genomic DNA was extracted in triplicate using the FastDNA Spin kit for soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer’s instructions, with the modifications that aliquots of 200 μL digester sample were used for extraction and 60 μL water was used in the final DNA elution. The concentrations of extracted DNA were measured using a Nano Vue spectrophotometer (GE Healthcare, Buckinghamshire, UK).
454-pyrosequencing and 16S rRNA gene sequence analysis
The bacterial communities in inoculum from the 10 industrial biogas plants were investigated by amplification of genomic DNA using polymerase chain reaction (PCR) primers targeting the bacterial 16S rRNA gene and integrated with 454 Life Sciences adaptors 8F (5′-CCT ATC CCC TGT GTG CCT TGG CAG TCT CAG CAA CAG CTA GAG TTT GAT CCT GG-3′) and 515R (5′-CCA TCT CAT CCC TGC GTG TCT CCG ACT CAG NNN NNN NNT TAC CGC GGC TGC T-3′ [107]. Each PCR contained 12.5 µL of Maxima Hot Start PCR Master Mix (Fermentas, Thermo Fisher Scientific, Hudson, NH, USA), 0.5 µM of each primer, 20 ng of DNA template and 9.5 µL of sterile water (25 µL final volume). The PCR protocol was as described in Sun et al. [31]. The size and purity of amplicons were checked by electrophoresis on 2 % agarose gel. Short, non-specific amplification products were removed with AMP beads (AMPure XP, Beckman Coulter Genomics, Danvers, MA, USA) using the manufacturer’s protocol but with a modified bead: DNA volume ratio of 0.7:1. The concentrations of purified products were measured using the Quant-iT dsDNA BR Assay Kit (Invitrogen, Life Technologies Europe, Stockholm, Sweden). All PCR products were pooled in equal molar amounts and sequenced at the Swedish Institute for Infectious Disease Control in Solna (Stockholm, Sweden), using the Roche/454 GS Titanium technology platform. The 16S rRNA sequences were processed as described previously [31] and deposited in the NCBI Sequence Read Archive (SRA) under the accession number PRJNA290173.
T-RFLP
Primers targeting the glycoside hydrolase families 5 (cel5_392F 5′-GAG CAT GGG CTG GAA YHT NGG NAA-3′ and cel5_754R 5′-CAT CAT AAT CTT TGA AGT GGT TTG CAA TYT GDK TCC A-3′) and 48 (cel48_490F 5′ TNA TGG TTG AAG CTC CDG AYT AYG G-3′ and cel48_920R 5′-CCA AAN CCR TAC CAG TTR TCA ACR TC-3′) [86] were used to study the cellulose-degrading bacterial community structures in the different inoculum samples and at the end of the batch test by terminal restriction fragment length polymorphism (T-RFLP) analysis. For the assay, the 5′end of the cel5_754R and cel48_920R primer was labelled with 6-carboxyfluorescein (FAM). PCR amplification of triplicate extractions was conducted using Maxima Hot Start PCR Master Mix (Fermentas, Thermo Fisher Scientific, Hudson, NH, USA). Each PCR contained 12.5 µL of corresponding reaction mix, 1 µL of each primer (0.5 µM final concentration), 1 µL of DNA template (20 times dilution) and 9.5 µL of sterile water. The PCR program used for amplification of cel5 and cel48 included initialisation at 95 °C for 5 min, denaturation at 95 °C for 1 min, annealing for 30 s at 56 °C for cel48 (35 cycles) or at 52 °C for cel5 (45 cycles), elongation at 72 °C for 30 s, followed by a final extension at 72 °C for 10 min. The pooled FAM-labelled amplicons of cel5 and cel48 were purified with QIAquick gel extraction kit (Qiagen, Hilden, Germany) and digested overnight at 37 °C with restriction enzyme MboI (New England Biolabs, Wilbury Way Hitchin, Herts, UK) and AluI (Fermentas, Thermo Fisher Scientific, Hudson, NH, USA). Fluorescently labelled terminal restriction fragments (T-RFs) were separated and detected with ABI3730xl capillary sequencer (Applied Biosystems, Cheshire, UK). GS ROX 500 internal size standard (Applied Biosystems) was included in all assays. The T-RFLP profiles were processed by Peak Scanner software (1.0, Applied Biosystems) and the relative abundance of the individual T-RFs was calculated by dividing the peak area by the total area of all peaks. T-RFs shorter than 70 bp (cel5) and 50 bp (cel48) constituting less than 1 % of the total peak area were excluded as background.
Clone library construction and sequencing analysis
Clone libraries were constructed for the cel5 community with samples retrieved from CD01 and WWTP 02c/03/04/05c and for the cel48 community with samples from CD 01/02/03 and WWTP 03/04/06, as described previously [29]. In brief, triplicate PCRs were conducted for each DNA extraction replicate using the primer (without FAM label) and conditions described above. The resulting nine PCR products per sample were pooled and gel purified with QIAquick gel extraction kit (Qiagen, Hilden, Germany) and ligated into pCR™4-TOPO® vector (Invitrogen, Life Technologies, Grand Island, NY, USA), followed by transformation of the ligation product into TOP10 One Shot® chemically competent Escherichia coli (Invitrogen), according to the manufacturer’s instructions. The sequences obtained were quality checked and edited with the software package Geneious, version 5.6.5 (Biomatters Ltd., Auckland, New Zealand) and subsequently assigned to operational taxonomic units (OTU) at the threshold of 97 % nucleotide identity. The sequences were compared with sequences available in the NCBI GenBank. Alignment of cloned sequences and selected reference sequences, as well as sequences from uncultured bacteria, was conducted using the programme MUSCLE [108]. The phylogenetic trees were constructed with the MEGA programme version 5 using the maximum likelihood method and WAG model [109]. The confidence of the trees was tested by bootstrap resampling analysis for 1000 replicates. All sequences were deposited in the NCBI GenBank database under the accession number KT336110-29 for primer pair cel5 and KT336130-99 for primer cel48.
Statistics
To investigate correlations between the microbial composition of the different biogas plants and the batch test performance using inoculum from the corresponding plants, canonical correspondence analysis (CCA) was performed using the Vegan Community Ecology Package (version 2.3-0, http://CRAN.R-project.org/package=vegan) within R (A language and environment for statistical computing, http://www.R-project.org/). Separate CCA was performed using the amplicon sequencing and T-RFLP data. Only major OTUs, i.e. OTUs representing ≥2 % of total sequences at genus level, were selected for the assay and for T-RFLP the relative abundance of each T-RF was used. The inverse of days needed to reach 50 and 80 % of final methane potential was used as a measure of degradation performance. The process data (Table 1) were included as environmental variables.
Authors’ contributions
LS and TL participated equally in the planning of the study, the laboratory work, the data interpretation and were mainly responsible for writing the manuscript. BM contributed with analytical assistance during the microbiological work, participated in data interpretation and reviewed the manuscript. AS conceived, designed and coordinated the study, participated in supervision and data interpretation and in writing the manuscript. All authors were involved in critical revision of the manuscript, and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Acknowledgements
The authors thank the staff at the large-scale biogas plants for assisting with sampling and for information about operating parameters, Ulf Olsson for assisting with the statistical analysis and Junfeng Liang for help with the BMP test.
Availability of data and material
Sequence data have been deposited in publicly available databases and information about accession numbers is given in the manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This project was funded by the thematic research programme MicroDrive (http://www.slu.se/microdrive), The Swedish Energy Agency (ERA-NET Bioenergy) and the China Scholarship Council (CSC, File No.20 1,307,930,025) for the financial support. This work was also funded in parts by the Research Council of Norway, grant numbers 190877/S60 and 203402/E20.
Abbreviations
- AD
anaerobic digestion
- BMP
biochemical methane potential test
- CCA
canonical correspondence analysis
- CD
co-digestion
- cel5
glycoside hydrolase genes of family 5
- cel48
glycoside hydrolase genes of family 48
- FAM
phosphoramidite fluorochrome 5-carboxy-fluorescein
- GC
gas chromatograph
- HPLC
high pressure liquid chromatography
- HRT
hydraulic retention time
- OLR
organic loading rate
- OTU
operational taxonomic unit
- PCoA
principal coordinates analysis
- PCR
polymeras chain reaction
- TKN
total kjeldahl nitrogen
- TRFLP
terminal restriction fragment length polymorphism
- T-RF
terminal restriction fragment
- TS
total solids
- VFA
volatile fatty acids
- VS
volatile solids
- WWTP
waste water treatment plant
Additional file
10.1186/s13068-016-0543-9 Figure S1 Relative abundance of bacterial 16S rRNA gene at class level in 10 industrial-scale biogas plants. CD 01-04: co-digestion plants; WWTP 01-06: wastewater treatment plants: Figure S2. Relative abundance of bacterial 16S rRNA gene at order level in 10 industrial-scale biogas plants. CD 01-04: co-digestion plants; WWTP 01-06: wastewater treatment plants. Figure S3. Relative abundance of bacterial 16S rRNA gene at family level in 10 industrial-scale biogas plants. CD 01-04: co-digestion plants; WWTP 01-06: wastewater treatment plants. Figure S4. Relative abundance of bacterial 16S rRNA gene at genus level in 10 industrial-scale biogas plants. CD 01-04: co-digestion plants; WWTP 01-06: wastewater treatment plants. Figure S5. OTU heatmap based on bacterial OTUs having relative abundance higher or equal to 0.2% in 10 industrial-scale biogas plants. CD 01-04: co-digestion plants; WWTP 01-06: wastewater treatment plants.
Footnotes
Li Sun and Tong Liu contributed equally to this work
Contributor Information
Li Sun, Email: li.sun@slu.se.
Tong Liu, Email: tong.liu@slu.se.
Bettina Müller, Email: Bettina.muller@slu.se.
Anna Schnürer, Phone: (+46) 18 671000, Email: anna.schnurer@slu.se.
References
- 1.Börjesson P, Mattiasson B. Biogas as a resource-efficient vehicle fuel. Trends Biotechnol. 2008;26:7–13. doi: 10.1016/j.tibtech.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Möller K, Müller T. Effects of anaerobic digestion on digestate nutrient availability and crop growth: a review. Eng Life Sci. 2012;12:242–257. doi: 10.1002/elsc.201100085. [DOI] [Google Scholar]
- 3.Lozanovski A, Lindner J, Bos U. Environmental evaluation and comparison of selected industrial scale biomethane production facilities across Europe. Int J Life Cycle Assess. 2014;19:1823–1832. doi: 10.1007/s11367-014-0791-5. [DOI] [Google Scholar]
- 4.Börjesson P, Tufvesson LM. Agricultural crop-based biofuels—resource efficiency and environmental performance including direct land use changes. J Clean Prod. 2011;19:108–120. doi: 10.1016/j.jclepro.2010.01.001. [DOI] [Google Scholar]
- 5.Appels L, Lauwers J, Degrève J, Helsen L, Lievens B, Willems K, Van Impe J, Dewil R. Anaerobic digestion in global bio-energy production: potential and research challenges. Renew Sust Energ Rev. 2011;15:4295–4301. doi: 10.1016/j.rser.2011.07.121. [DOI] [Google Scholar]
- 6.Weiland P. Biogas production: current state and perspectives. Appl Microbiol Biotechnol. 2010;85:849–860. doi: 10.1007/s00253-009-2246-7. [DOI] [PubMed] [Google Scholar]
- 7.Sawatdeenarunat C, Surendra KC, Takara D, Oechsner H, Khanal SK. Anaerobic digestion of lignocellulosic biomass: challenges and opportunities. Bioresour Technol. 2015;178:178–186. doi: 10.1016/j.biortech.2014.09.103. [DOI] [PubMed] [Google Scholar]
- 8.Ioelovich M. Recent findings and the energetic potential of plant biomass as a renewable source of biofuels—a review. Bioresources. 2015;10:1879–1914. [Google Scholar]
- 9.Noike T, Endo G, Chang JE, Yaguchi JI, Matsumoto JI. Characteristics of carbohydrate degradation and the rate-limiting step in anaerobic digestion. Biotechnol Bioeng. 1985;27:1482–1489. doi: 10.1002/bit.260271013. [DOI] [PubMed] [Google Scholar]
- 10.Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev. 2002;66:506–577. doi: 10.1128/MMBR.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mata-Alvarez J, Macé S, Llabrés P. Anaerobic digestion of organic solid wastes. An overview of research achievements and perspectives. Bioresour Technol. 2000;74:3–16. doi: 10.1016/S0960-8524(00)00023-7. [DOI] [Google Scholar]
- 12.Yang L, Xu F, Ge X, Li Y. Challenges and strategies for solid-state anaerobic digestion of lignocellulosic biomass. Renew Sust Energ Rev. 2015;44:824–834. doi: 10.1016/j.rser.2015.01.002. [DOI] [Google Scholar]
- 13.Zheng Y, Zhao J, Xu F, Li Y. Pretreatment of lignocellulosic biomass for enhanced biogas production. Prog Energy Combust Sci. 2014;42:35–53. doi: 10.1016/j.pecs.2014.01.001. [DOI] [Google Scholar]
- 14.Čater M, Fanedl L, Malovrh Š, Marinšek Logar R. Biogas production from brewery spent grain enhanced by bioaugmentation with hydrolytic anaerobic bacteria. Bioresour Technol. 2015;186:261–269. doi: 10.1016/j.biortech.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 15.MerlinChristy P, Gopinath LR, Divya D. A review on anaerobic decomposition and enhancement of biogas production through enzymes and microorganisms. Renew Sust Energ Rev. 2014;34:167–173. doi: 10.1016/j.rser.2014.03.010. [DOI] [Google Scholar]
- 16.Zinder SH, Koch M. Non-aceticlastic methanogenesis from acetate: acetate oxidation by a thermophilic syntrophic coculture. Arch Microbiol. 1984;138:263–272. doi: 10.1007/BF00402133. [DOI] [Google Scholar]
- 17.Angelidaki I, Karakashev D, Batstone DJ, Plugge CM, Stams AJM. Chapter sixteen—biomethanation and its potential. In: Amy CR, Stephen WR, editors. Methods enzymol. New York: Academic Press; 2011. pp. 327–351. [DOI] [PubMed] [Google Scholar]
- 18.Himmel ME, Xu Q, Luo Y, Ding S-Y, Lamed R, Bayer EA. Microbial enzyme systems for biomass conversion: emerging paradigms. Biofuels. 2010;1:323–341. doi: 10.4155/bfs.09.25. [DOI] [Google Scholar]
- 19.Medie FM, Davies GJ, Drancourt M, Henrissat B. Genome analyses highlight the different biological roles of cellulases. Nat Rev Micro. 2012;10:227–234. doi: 10.1038/nrmicro2729. [DOI] [PubMed] [Google Scholar]
- 20.Koeck DE, Pechtl A, Zverlov VV, Schwarz WH. Genomics of cellulolytic bacteria. Curr Opin Biotechnol. 2014;29:171–183. doi: 10.1016/j.copbio.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Azman S, Khadem AF, Van Lier JB, Zeeman G, Plugge CM. Presence and role of anaerobic hydrolytic microbes in conversion of lignocellulosic biomass for biogas production. Crit Rev Environ Sci Technol. 2015;45:2523–2564. doi: 10.1080/10643389.2015.1053727. [DOI] [Google Scholar]
- 22.Yang JC, Chynoweth DP, Williams DS, Li A. Clostridium aldrichii sp. nov., a cellulolytic mesophile inhabiting a wood-fermenting anaerobic digester. Int J Syst Bacteriol. 1990;40:268–272. doi: 10.1099/00207713-40-3-268. [DOI] [PubMed] [Google Scholar]
- 23.Palop ML, Valles S, Pinaga F, Flors A. Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium celerecrescens sp. nov. Int J Syst Bacteriol. 1989;39:68–71. doi: 10.1099/00207713-39-1-68. [DOI] [Google Scholar]
- 24.Shiratori H, Ikeno H, Ayame S, Kataoka N, Miya A, Hosono K, Beppu T, Ueda K. Isolation and characterization of a New Clostridium sp. That performs effective cellulosic waste digestion in a thermophilic methanogenic bioreactor. Appl Environ Microbiol. 2006;72:3702–3709. doi: 10.1128/AEM.72.5.3702-3709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiratori H, Sasaya K, Ohiwa H, Ikeno H, Ayame S, Kataoka N, Miya A, Beppu T, Ueda K. Clostridium clariflavum sp. nov. and Clostridium caenicola sp. nov., moderately thermophilic, cellulose-/cellobiose-digesting bacteria isolated from methanogenic sludge. Int J Syst Evol Microbiol. 2009;59:1764–1770. doi: 10.1099/ijs.0.003483-0. [DOI] [PubMed] [Google Scholar]
- 26.Sleat R, Mah RA, Robinson R. Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium cellulovorans sp. nov. Appl Environ Microbiol. 1984;48:88–93. doi: 10.1128/aem.48.1.88-93.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sleat R, Mah RA. Clostridium populeti sp. nov., a cellulolytic species from a woody-biomass digestor. Int J Syst Bacteriol. 1985;35:160–163. doi: 10.1099/00207713-35-2-160. [DOI] [Google Scholar]
- 28.Lebuhn M, Hanreich A, Klocke M, Schluter A, Bauer C, Perez CM. Towards molecular biomarkers for biogas production from lignocellulose-rich substrates. Anaerobe. 2014;29:10–21. doi: 10.1016/j.anaerobe.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Sun L, Müller B, Schnürer A. Biogas production from wheat straw: community structure of cellulose-degrading bacteria. Energy Sustain Soc. 2013;3:15. doi: 10.1186/2192-0567-3-15. [DOI] [Google Scholar]
- 30.Krause L, Diaz NN, Edwards RA, Gartemann KH, Krömeke H, Neuweger H, Pühler A, Runte KJ, Schlüter A, Stoye J, et al. Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J Biotechnol. 2008;136:91–101. doi: 10.1016/j.jbiotec.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Pope PB, Eijsink VGH, Schnürer A. Characterization of microbial community structure during continuous anaerobic digestion of straw and cow manure. Microb Biotechnol. 2015;8:815–827. doi: 10.1111/1751-7915.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lucas R, Kuchenbuch A, Fetzer I, Harms H, Kleinsteuber S. Long-term monitoring reveals stable and remarkably similar microbial communities in parallel full-scale biogas reactors digesting energy crops. FEMS Microbiol Ecol. 2015;91(3). doi:10.1093/femsec/fiv004. [DOI] [PubMed]
- 33.Theuerl S, Kohrs F, Benndorf D, Maus I, Wibberg D, Schlüter A, Kausmann R, Heiermann M, Rapp E, Reichl U, et al. Community shifts in a well-operating agricultural biogas plant: how process variations are handled by the microbiome. Appl Microbiol Biotechnol. 2015;99:7791–7803. doi: 10.1007/s00253-015-6627-9. [DOI] [PubMed] [Google Scholar]
- 34.Guo Z, Zhou A, Yang C, Liang B, Sangeetha T, He Z, Wang L, Cai W, Wang A, Liu W. Enhanced short chain fatty acids production from waste activated sludge conditioning with typical agricultural residues: carbon source composition regulates community functions. Biotechnol Biofuels. 2015;8:192. doi: 10.1186/s13068-015-0369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlüter A, Bekel T, Diaz NN, Dondrup M, Eichenlaub R, Gartemann KH, Krahn I, Krause L, Krömeke H, Kruse O, et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J Biotechnol. 2008;136:77–90. doi: 10.1016/j.jbiotec.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Hanreich A, Schimpf U, Zakrzewski M, Schlüter A, Benndorf D, Heyer R, Rapp E, Pühler A, Reichl U, Klocke M. Metagenome and metaproteome analyses of microbial communities in mesophilic biogas-producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Syst Appl Microbiol. 2013;36:330–338. doi: 10.1016/j.syapm.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Theuretzbacher F, Lizasoain J, Lefever C, Saylor MK, Enguidanos R, Weran N, Gronauer A, Bauer A. Steam explosion pretreatment of wheat straw to improve methane yields: investigation of the degradation kinetics of structural compounds during anaerobic digestion. Bioresour Technol. 2015;179:299–305. doi: 10.1016/j.biortech.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 38.Risberg K, Sun L, Levén L, Horn SJ, Schnürer A. Biogas production from wheat straw and manure—impact of pretreatment and process operating parameters. Bioresour Technol. 2013;149:232–237. doi: 10.1016/j.biortech.2013.09.054. [DOI] [PubMed] [Google Scholar]
- 39.Ferreira LC, Nilsen PJ, Fdz-Polanco F, Pérez-Elvira SI. Biomethane potential of wheat straw: influence of particle size, water impregnation and thermal hydrolysis. Chem Eng J. 2014;242:254–259. doi: 10.1016/j.cej.2013.08.041. [DOI] [Google Scholar]
- 40.Werner JJ, Garcia ML, Perkins SD, Yarasheski KE, Smith SR, Muegge BD, Stadermann FJ, DeRito CM, Floss C, Madsen EL, et al. Microbial community dynamics and stability during an ammonia-induced shift to syntrophic acetate oxidation. Appl Environ Microbiol. 2014;80:3375–3383. doi: 10.1128/AEM.00166-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Vrieze J, Saunders AM, He Y, Fang J, Nielsen PH, Verstraete W, Boon N. Ammonia and temperature determine potential clustering in the anaerobic digestion microbiome. Water Res. 2015;75:312–323. doi: 10.1016/j.watres.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 42.Müller B, Sun L, Westerholm M, Schnürer A. Bacterial community composition and fhs profiles of low- and high-ammonia biogas digesters reveal novel syntrophic acetate-oxidising bacteria. Biotechnol Biofuels. 2016;9:48. doi: 10.1186/s13068-016-0454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carballa M, Smits M, Etchebehere C, Boon N, Verstraete W. Correlations between molecular and operational parameters in continuous lab-scale anaerobic reactors. Appl Microbiol Biotechnol. 2011;89:303–314. doi: 10.1007/s00253-010-2858-y. [DOI] [PubMed] [Google Scholar]
- 44.Wittebolle L, Marzorati M, Clement L, Balloi A, Daffonchio D, Heylen K, De Vos P, Verstraete W, Boon N. Initial community evenness favours functionality under selective stress. Nature. 2009;458:623–626. doi: 10.1038/nature07840. [DOI] [PubMed] [Google Scholar]
- 45.Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH, Sørensen SJ, Karlsson A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol. 2013;85:612–626. doi: 10.1111/1574-6941.12148. [DOI] [PubMed] [Google Scholar]
- 46.Klocke M, Mähnert P, Mundt K, Souidi K, Linke B. Microbial community analysis of a biogas-producing completely stirred tank reactor fed continuously with fodder beet silage as mono-substrate. Syst Appl Microbiol. 2007;30:139–151. doi: 10.1016/j.syapm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 47.Lu X, Rao S, Shen Z, Lee PKH. Substrate induced emergence of different active bacterial and archaeal assemblages during biomethane production. Bioresour Technol. 2013;148:517–524. doi: 10.1016/j.biortech.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 48.Cirne DG, Lehtomäki A, Björnsson L, Blackall LL. Hydrolysis and microbial community analyses in two-stage anaerobic digestion of energy crops. J Appl Microbiol. 2007;103:516–527. doi: 10.1111/j.1365-2672.2006.03270.x. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Vuorela M, Keränen AL, Lehtinen TM, Lensu A, Lehtomäki A, Rintala J. Development of microbial populations in the anaerobic hydrolysis of grass silage for methane production. FEMS Microbiol Ecol. 2010;72:496–506. doi: 10.1111/j.1574-6941.2010.00850.x. [DOI] [PubMed] [Google Scholar]
- 50.Ziganshin AM, Liebetrau J, Proter J, Kleinsteuber S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl Microbiol Biotechnol. 2013;97:5161–5174. doi: 10.1007/s00253-013-4867-0. [DOI] [PubMed] [Google Scholar]
- 51.Kovács E, Wirth R, Maróti G, Bagi Z, Nagy K, Minárovits J, Rákhely G, Kovács KL. Augmented biogas production from protein-rich substrates and associated metagenomic changes. Bioresour Technol. 2015;178:254–261. doi: 10.1016/j.biortech.2014.08.111. [DOI] [PubMed] [Google Scholar]
- 52.Kovács E, Wirth R, Maróti G, Bagi Z, Rákhely G, Kovács KL. Biogas production from protein-rich biomass: fed-batch anaerobic fermentation of casein and of pig blood and associated changes in microbial community composition. PLoS ONE. 2013;8:e77265. doi: 10.1371/journal.pone.0077265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouanane-Darenfed A, Fardeau ML, Gregoire P, Joseph M, Kebbouche-Gana S, Benayad T, Hacene H, Cayol JL, Ollivier B. Caldicoprobacter algeriensis sp. nov. a new thermophilic anaerobic, xylanolytic bacterium isolated from an Algerian hot spring. Curr Microbiol. 2011;62:826–832. doi: 10.1007/s00284-010-9789-9. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama H, Wagner ID, Wiegel J. Caldicoprobacter oshimai gen. nov., sp. nov., an anaerobic, xylanolytic, extremely thermophilic bacterium isolated from sheep faeces, and proposal of Caldicoprobacteraceae fam. nov. Int J Syst Evol Microbiol. 2010;60:67–71. doi: 10.1099/ijs.0.011379-0. [DOI] [PubMed] [Google Scholar]
- 55.Bouanane-Darenfed A, Ben Hania W, Hacene H, Cayol JL, Ollivier B, Fardeau ML. Caldicoprobacter guelmensis sp. nov., a thermophilic, anaerobic, xylanolytic bacterium isolated from a hot spring. Int J Syst Evol Microbiol. 2013;63:2049–2053. doi: 10.1099/ijs.0.043497-0. [DOI] [PubMed] [Google Scholar]
- 56.Tang Y, Shigematsu T, Morimura S, Kida K. The effects of micro-aeration on the phylogenetic diversity of microorganisms in a thermophilic anaerobic municipal solid-waste digester. Water Res. 2004;38:2537–2550. doi: 10.1016/j.watres.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 57.Cheon J, Hong F, Hidaka T, Koshikawa H, Tsuno H. Microbial population dynamics in a thermophilic methane digester fed with garbage. Water Sci Technol. 2007;55:175–182. doi: 10.2166/wst.2007.320. [DOI] [PubMed] [Google Scholar]
- 58.Alauzet C, Marchandin H, Courtin P, Mory F, Lemée L, Pons JL, Chapot-Chartier MP, Lozniewski A, Jumas-Bilak E. Multilocus analysis reveals diversity in the genus Tissierella: description of Tissierella carlieri sp. nov. in the new class Tissierellia classis nov. Syst Appl Microbiol. 2014;37:23–34. doi: 10.1016/j.syapm.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 59.Schiefer-Ullrich H, Andreesen J. Peptostreptococcus barnesae sp. nov., a Gram-positive, anaerobic, obligately purine utilizing coccus from chicken feces. Arch Microbiol. 1985;143:26–31. doi: 10.1007/BF00414763. [DOI] [Google Scholar]
- 60.Hatamoto M, Kaneshige M, Nakamura A, Yamaguchi T. Bacteroides luti sp. nov., an anaerobic, cellulolytic and xylanolytic bacterium isolated from methanogenic sludge. Int J Syst Evol Microbiol. 2014;64:1770–1774. doi: 10.1099/ijs.0.056630-0. [DOI] [PubMed] [Google Scholar]
- 61.Robert C, Chassard C, Lawson PA, Bernalier-Donadille A. Bacteroides cellulosilyticus sp. nov., a cellulolytic bacterium from the human gut microbial community. Int J Syst Evol Microbiol. 2007;57:1516–1520. doi: 10.1099/ijs.0.64998-0. [DOI] [PubMed] [Google Scholar]
- 62.Rui J, Li J, Zhang S, Yan X, Wang Y, Li X. The core populations and co-occurrence patterns of prokaryotic communities in household biogas digesters. Biotechnol Biofuels. 2015;8:1–15. doi: 10.1186/s13068-015-0339-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chouari R, Le Paslier D, Dauga C, Daegelen P, Weissenbach J, Sghir A. Novel major bacterial candidate division within a municipal anaerobic sludge digester. Appl Environ Microbiol. 2005;71:2145–2153. doi: 10.1128/AEM.71.4.2145-2153.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Limam RD, Chouari R, Mazéas L, Wu TD, Li T, Grossin-Debattista J, Guerquin-Kern JL, Saidi M, Landoulsi A, Sghir A, Bouchez T. Members of the uncultured bacterial candidate division WWE1 are implicated in anaerobic digestion of cellulose. MicrobiologyOpen. 2014;3:157–167. doi: 10.1002/mbo3.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng J-F, Darling A, Malfatti S, Swan BK, Gies EA, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–437. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 66.Nobu MK, Narihiro T, Rinke C, Kamagata Y, Tringe SG, Woyke T, Liu W-T. Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J. 2015;9:1710–1722. doi: 10.1038/ismej.2014.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Narihiro T, Sekiguchi Y. Microbial communities in anaerobic digestion processes for waste and wastewater treatment: a microbiological update. Curr Opin Biotechnol. 2007;18:273–278. doi: 10.1016/j.copbio.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 68.Chouari R, Le Paslier D, Daegelen P, Ginestet P, Weissenbach J, Sghir A. Novel predominant archaeal and bacterial groups revealed by molecular analysis of an anaerobic sludge digester. Environ Microbiol. 2005;7:1104–1115. doi: 10.1111/j.1462-2920.2005.00795.x. [DOI] [PubMed] [Google Scholar]
- 69.Riviere D, Desvignes V, Pelletier E, Chaussonnerie S, Guermazi S, Weissenbach J, Li T, Camacho P, Sghir A. Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J. 2009;3:700–714. doi: 10.1038/ismej.2009.2. [DOI] [PubMed] [Google Scholar]
- 70.Wan S, Sun L, Sun J, Luo W. Biogas production and microbial community change during the Co-digestion of food waste with chinese silver grass in a single-stage anaerobic reactor. Biotechnol Bioprocess Eng. 2013;18:1022–1030. doi: 10.1007/s12257-013-0128-4. [DOI] [Google Scholar]
- 71.Zhang Y, Hu M, Li P, Wang X, Meng Q. Trichloroethylene removal and bacterial variations in the up-flow anaerobic sludge blanket reactor in response to temperature shifts. Appl Microbiol Biotechnol. 2015;99:6091–6102. doi: 10.1007/s00253-015-6480-x. [DOI] [PubMed] [Google Scholar]
- 72.Shu D, He Y, Yue H, Wang Q. Microbial structures and community functions of anaerobic sludge in six full-scale wastewater treatment plants as revealed by 454 high-throughput pyrosequencing. Bioresour Technol. 2015;186:163–172. doi: 10.1016/j.biortech.2015.03.072. [DOI] [PubMed] [Google Scholar]
- 73.Narihiro T, Nobu MK, Kim N-K, Kamagata Y, Liu W-T. The nexus of syntrophy-associated microbiota in anaerobic digestion revealed by long-term enrichment and community survey. Environ Microbiol. 2015;17:1707–1720. doi: 10.1111/1462-2920.12616. [DOI] [PubMed] [Google Scholar]
- 74.Ariesyady HD, Ito T, Okabe S. Functional bacterial and archaeal community structures of major trophic groups in a full-scale anaerobic sludge digester. Water Res. 2007;41:1554–1568. doi: 10.1016/j.watres.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 75.Hagen LH, Vivekanand V, Linjordet R, Pope PB, Eijsink VGH, Horn SJ. Microbial community structure and dynamics during co-digestion of whey permeate and cow manure in continuous stirred tank reactor systems. Bioresour Technol. 2014;171:350–359. doi: 10.1016/j.biortech.2014.08.095. [DOI] [PubMed] [Google Scholar]
- 76.Yamada T, Sekiguchi Y. Cultivation of uncultured Chloroflexi Subphyla: significance and ecophysiology of formerly uncultured Chloroflexi ‘Subphylum I’ with natural and biotechnological relevance. Microbes Environ. 2009;24:205–216. doi: 10.1264/jsme2.ME09151S. [DOI] [PubMed] [Google Scholar]
- 77.Miura Y, Okabe S. Quantification of cell specific uptake activity of microbial products by uncultured chloroflexi by microautoradiography combined with fluorescence in situ hybridization. Environ Sci Technol. 2008;42:7380–7386. doi: 10.1021/es800566e. [DOI] [PubMed] [Google Scholar]
- 78.BonillaSalinas M, Fardeau ML, Thomas P, Cayol JL, Patel BKC, Ollivier B. Mahella australiensis gen. nov., sp. nov., a moderately thermophilic anaerobic bacterium isolated from an Australian oil well. Int J Syst Evol Microbiol. 2004;54:2169–2173. doi: 10.1099/ijs.0.02926-0. [DOI] [PubMed] [Google Scholar]
- 79.Niu Q, Qiao W, Qiang H, Li YY. Microbial community shifts and biogas conversion computation during steady, inhibited and recovered stages of thermophilic methane fermentation on chicken manure with a wide variation of ammonia. Bioresour Technol. 2013;146:223–233. doi: 10.1016/j.biortech.2013.07.038. [DOI] [PubMed] [Google Scholar]
- 80.Niu Q, Takemura Y, Kubota K, Li Y-Y. Comparing mesophilic and thermophilic anaerobic digestion of chicken manure: microbial community dynamics and process resilience. Waste Manage. 2015;43:114–122. doi: 10.1016/j.wasman.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 81.Nedashkovskaya OI, Kim SB, Hoste B, Shin DS, Beleneva IA, Vancanneyt M, Mikhailov VV. Echinicola vietnamensis sp. nov., a member of the phylum Bacteroidetes isolated from seawater. Int J Syst Evol Microbiol. 2007;57:761–763. doi: 10.1099/ijs.0.64546-0. [DOI] [PubMed] [Google Scholar]
- 82.Petitdemange E, Caillet F, Giallo J, Gaudin C. Clostridium cellulolyticum sp. nov., a cellulolytic, mesophilic: species from decayed grass. Int J Syst Bacteriol. 1984;34:155–159. doi: 10.1099/00207713-34-2-155. [DOI] [Google Scholar]
- 83.Giallo J, Gaudin C, Belaich JP, Petitdemange E, Caillet-Mangin F. Metabolism of glucose and cellobiose by cellulolytic mesophilic Clostridium sp. strain H10. Appl Environ Microbiol. 1983;45:843–849. doi: 10.1128/aem.45.3.843-849.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng X, Börner RA, Nges IA, Liu J. Impact of bioaugmentation on biochemical methane potential for wheat straw with addition of Clostridium cellulolyticum. Bioresour Technol. 2014;152:567–571. doi: 10.1016/j.biortech.2013.11.067. [DOI] [PubMed] [Google Scholar]
- 85.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pereyra LP, Hiibel SR, Prieto Riquelme MV, Reardon KF, Pruden A. Detection and quantification of functional genes of cellulose-degrading, fermentative, and sulfate-reducing bacteria and methanogenic Archaea. Appl Environ Microbiol. 2010;76:2192–2202. doi: 10.1128/AEM.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suzuki M, Nakagawa Y, Harayama S, Yamamoto S. Phylogenetic analysis of genus Marinilabilia and related bacteria based on the amino acid sequences of GyrB and emended description of Marinilabilia salmonicolor with Marinilabilia agarovorans as its subjective synonym. Int J Syst Evol Microbiol. 1999;49:1551–1557. doi: 10.1099/00207713-49-4-1551. [DOI] [PubMed] [Google Scholar]
- 88.Hethener P, Brauman A, Garcia JL. Clostridium termitidis sp. nov., a cellulolytic Bacterium from the gut of the wood-feeding termite, Nasutitermes lujae. Syst Appl Microbiol. 1992;15:52–58. doi: 10.1016/S0723-2020(11)80138-4. [DOI] [Google Scholar]
- 89.Kato S, Haruta S, Cui ZJ, Ishii M, Yokota A, Igarashi Y. Clostridium straminisolvens sp. nov., a moderately thermophilic, aerotolerant and cellulolytic bacterium isolated from a cellulose-degrading bacterial community. Int J Syst Evol Microbiol. 2004;54:2043–2047. doi: 10.1099/ijs.0.63148-0. [DOI] [PubMed] [Google Scholar]
- 90.Wei Y, Zhou H, Zhang J, Zhang L, Geng A, Liu F, Zhao G, Wang S, Zhou Z, Yan X. Insight into dominant cellulolytic bacteria from two biogas digesters and their glycoside hydrolase genes. PLoS ONE. 2015;10:e0129921. doi: 10.1371/journal.pone.0129921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park SK, Jang HM, Ha JH, Park JM. Sequential sludge digestion after diverse pre-treatment conditions: sludge removal, methane production and microbial community changes. Bioresour Technol. 2014;162:331–340. doi: 10.1016/j.biortech.2014.03.152. [DOI] [PubMed] [Google Scholar]
- 92.Prasad P, Singh T, Bedi S. Characterization of the cellulolytic enzyme produced by Streptomyces griseorubens (Accession No. AB184139) isolated from Indian soil. J King Saud Univ Sci. 2013;25:245–250. doi: 10.1016/j.jksus.2013.03.003. [DOI] [Google Scholar]
- 93.Feng HW, Zhi YE, Shi WW, Mao L, Zhou P. Isolation, identification and characterization of a straw degrading Streptomyces griseorubens JSD-1. Afr J Microbiol Res. 2013;7:2730–2735. [Google Scholar]
- 94.Feng H, Zhi Y, Sun Y, Wei X, Luo Y, Zhou P. Draft genome sequence of a novel Streptomyces griseorubens strain, JSD-1, active in carbon and nitrogen recycling. Genome Announc. 2014;2:e00650. doi: 10.1128/genomeA.00650-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel GB, Khan AW, Agnew BJ, Colvin JR. Isolation and characterization of an anaerobic, cellulolytic microorganism, Acetivibrio cellulolyticus gen. nov., sp. nov. Int J Syst Bacteriol. 1980;30:179–185. doi: 10.1099/00207713-30-1-179. [DOI] [Google Scholar]
- 96.Akinosho H, Yee K, Close D, Ragauskas A. The emergence of Clostridium thermocellum as a high utility candidate for consolidated bioprocessing applications. Front Chem. 2014;2:66. doi: 10.3389/fchem.2014.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.MahalingeswaraBhat K, Wood TM. The cellulase of the anaerobic bacterium Clostridium thermocellum isolation, dissociation, and reassociation of the cellulosome. Carbohydr Res. 1992;227:293–300. doi: 10.1016/0008-6215(92)85079-F. [DOI] [Google Scholar]
- 98.Lu F, Bize A, Guillot A, Monnet V, Madigou C, Chapleur O, Mazeas L, He P, Bouchez T. Metaproteomics of cellulose methanisation under thermophilic conditions reveals a surprisingly high proteolytic activity. ISME J. 2014;8:88–102. doi: 10.1038/ismej.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lamed R, Setter E, Bayer EA. Characterization of a cellulose-binding, cellulase-containing complex in Clostridium thermocellum. J Bacteriol. 1983;156:828–836. doi: 10.1128/jb.156.2.828-836.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dumitrache A, Wolfaardt G, Allen G, Liss SN, Lynd LR. Form and function of Clostridium thermocellum biofilms. Appl Environ Microbiol. 2013;79:231–239. doi: 10.1128/AEM.02563-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chassard C, Delmas E, Robert C, Lawson PA, Bernalier-Donadille A. Ruminococcus champanellensis sp. nov., a cellulose-degrading bacterium from human gut microbiota. Int J Syst Evol Microbiol. 2012;62:138–143. doi: 10.1099/ijs.0.027375-0. [DOI] [PubMed] [Google Scholar]
- 102.De Vrieze J, Raport L, Willems B, Verbrugge S, Volcke E, Meers E, Angenent LT, Boon N. Inoculum selection influences the biochemical methane potential of agro-industrial substrates. Microb Biotechnol. 2015;8:776–786. doi: 10.1111/1751-7915.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Westerholm M, Hansson M, Schnürer A. Improved biogas production from whole stillage by co-digestion with cattle manure. Bioresour Technol. 2012;114:314–319. doi: 10.1016/j.biortech.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 104.Stromberg S, Nistor M, Liu J. Towards eliminating systematic errors caused by the experimental conditions in Biochemical Methane Potential (BMP) tests. Waste Manag. 2014;34:1939–1948. doi: 10.1016/j.wasman.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 105.APHA. Standard methods for the examination of water and wastewater, 20th edn. American Public Health Association, American Water Works Association, Water Environment Federation; 1998.
- 106.Hansen KH, Angelidaki I, Ahring BK. Anaerobic digestion of swine manure: inhibition by ammonia. Water Res. 1998;32:5–12. doi: 10.1016/S0043-1354(97)00201-7. [DOI] [Google Scholar]
- 107.Stanley D, Geier MS, Hughes RJ, Denman SE, Moore RJ. Highly variable microbiota development in the chicken gastrointestinal tract. PLoS ONE. 2013;8:e84290. doi: 10.1371/journal.pone.0084290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]