

Abstract
Hypertension is a risk factor for cardiovascular disease, the leading cause of death worldwide. Although multiple factors contribute to the pathogenesis of hypertension, studies by Dr. David Barker reporting an inverse relationship between birth weight and blood pressure led to the hypothesis that slow growth during fetal life increases blood pressure and the risk for cardiovascular disease in later life. It is now recognized that growth during infancy and childhood in addition to exposure to adverse influences during fetal life contribute to the developmental programming of increased cardiovascular risk. Numerous epidemiological studies support the link between influences during early life with later cardiovascular health; experimental models provide proof of principle and indicate that numerous mechanisms contribute to the developmental origins of chronic disease. Sex impacts the severity of cardiovascular risk in experimental models of developmental insult. Yet, few studies examine the influence of sex on blood pressure and cardiovascular health in low birth weight men and women. Fewer still assess how aging impacts sex differences in programmed cardiovascular risk. Thus, the aim of this review is to highlight current data regarding sex differences in the developmental programming of blood pressure and cardiovascular disease.
Keywords: Developmental programming, low birth weight, sex differences, blood pressure, aging
Introduction
Over the last 30 years a large body of epidemiological literature supports the inverse relationship between birth weight and blood pressure (BP) reported by Dr. David Barker in 1986. In this study Dr. Barker reported a link between impaired fetal growth and ischemic heart disease in adulthood [1]. Using an atlas of England and Wales, Barker compared the geographical distribution of infant mortality between 1921 and 1925 with death rates in adults from ischemic heart disease between 1968 and 1978 [1]. He noted that the geographical distribution of these deaths was strongly correlated suggesting that factors that heightened the risk of fetal mortality also increased susceptibility to cardiovascular (CV) disease in those individuals that survived their first year of life. Based on this observation, Barker hypothesized that increased susceptibility to CV disease in adulthood resulted from poor fetal nutrition indicative of intrauterine growth restriction (IUGR). Although Dr. Anders Forsdahl initiated the theory that adverse influences in early life such as poor social conditions could increase the risk for CV in later life [2], Dr. Barker was the first to propose that undernutrition in utero resulted in structural and physiological changes that allowed the fetus to survive an adverse nutritional environment but at the expense of an increased risk for coronary heart disease in later life [3]. Since Barker’s original observation, investigation into the fetal origins of CV disease has grown exponentially and expanded to include investigation into the early life origins of metabolic and reproductive consequences as well as expansion of the window for timing of adverse consequences on later chronic health. Thus, early observations by Barker linking birth weight and blood pressure now include the influence of early postnatal events on vulnerability to chronic disease.
It is well established that men have higher blood pressure that age-matched women prior to menopause [4]. Few epidemiological studies report the effect of sex on the developmental origins of increased BP and risk for CV disease. Numerous experimental studies denote a strong sex difference in the developmental programming of BP with male offspring exhibiting a significant increase in BP in young adulthood whereas female offspring remaining normotensive [5–8]. However, females exposed to an insult in early life do not remain protected against the developmental programming of increased BP [9]. Low birth weight (LBW) defined as 5.5 pounds of less, affects approximately 8% of all births in the United States. Therefore, this review will highlight the developmental origins of hypertension and CV disease and current data related to the effect of sex and aging on the relationship between early life and BP.
Birth weight and blood pressure: the effect of childhood growth and age on the developmental programming of cardiovascular disease
Numerous studies support the inverse relationship between birth weight and BP in children and also indicate the contributory effect of postnatal weight gain on BP following slow fetal growth. A cross-sectional study of nearly 16,000 children indicated a significant negative association between birth weight and absolute BP that became stronger when corrected for body weight [10]. Singhal et al. demonstrated that higher mean arterial pressure (MAP) was promoted by faster weight gain in children born small [11]. A prospective study by Belfort et al. suggested that children born small with the most rapid postnatal weight gain had the highest systolic BP [12]. Hindmarsh et al. demonstrated that weight gain in the first 6 months of life was the greater predictive of BP than birth weight at age 3 [13]. Thus, these studies indicate an important role for temporal changes in growth during early life in the prediction of BP during childhood (Figure 1). Law et al. examined the impact of early growth on BP in adulthood. In this study systolic BP was increased in LBW adults but the highest systolic BP was in observed in LBW individuals that exhibited accelerated growth from 1 to 5 years of age [14]. However, unlike the study by Hindmarsh et al, weight gain in infancy in the absence of impaired fetal growth was not linked to higher BP in adult life [14]. Thus, this finding suggested that accelerated weight gain in early life exacerbated increased CV risk initiated in fetal life (Figure 1). Yet, in the absence of slow fetal growth, accelerated growth in early life exerted an adverse effect on BP in childhood that may not be sustained into adulthood.
Figure 1.
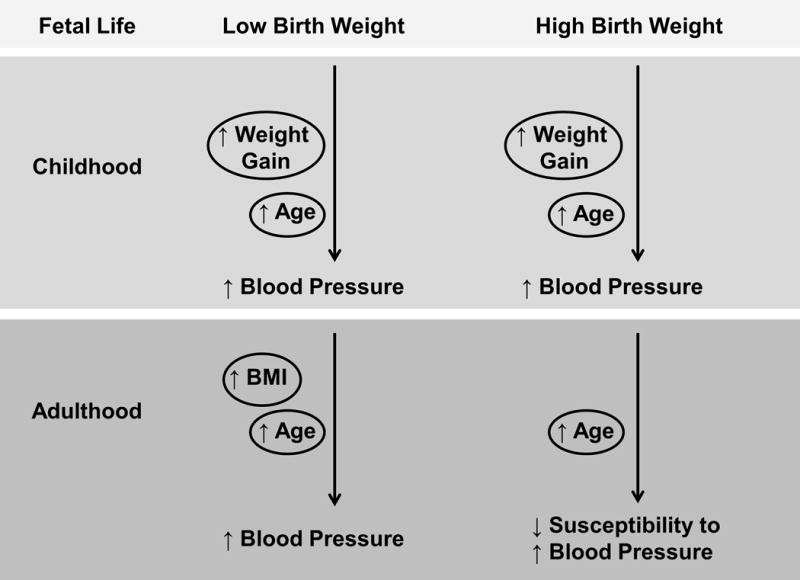
Flowchart of the influences that alter the developmental programming of blood pressure across the lifespan.
BP is known to increase with age within the general population [4] and epidemiological studies are also investigating the effect of age on the relationship between birth weight and BP. Barker et al. reported an inverse relationship between birth weight and systolic BP in childhood and young adulthood; however, the relationship was stronger at age 36 relative to age 10 [15]. Childhood BP is a predictor of adult BP [16]. Thus, this study supported the observation that childhood BP predicted adult BP and also demonstrated that the inverse relationship between birth weight and BP was present in children prior to the contribution of confounding variables such as cigarette smoking. Law et al. demonstrated that the increase in BP in a cohort of low birth weight adults at 22 years of age was amplified from childhood [14]. A similar observation was reported by Moore et al. although this association was greater in those that were heavier in young adulthood [17,18]. Law et al. also reported that the association between birth weight and BP amplified into old age [19]. However, the direct effect of age on BP across the lifespan was not examined within the same cohort in this study [19]. Ulterwal et al. directly tested the effect of age on BP in a longitudinal study followed for 14 years [20]. Although this study noted a strong inverse association between birth weight and systolic BP at all ages, the association was not amplified with increasing age [20]. Thus, conclusions drawn by Ulterwaal et al. were contradictory to the observations reported by Law or Moore. However, using studies published in Medline that involved linear regression analyses of birth weight and BP, Head et al. determined that the association between birth weight and BP became more negative as the age of the cohort increased [21]. Thus, a number of studies indicate that the relationship between birth weight and BP is amplified with age (Figure 1) but additional studies are needed to clarify the effect of age on the relationship between birth weight and BP.
Despite an abundance of evidence supporting the concept that low birth weight babies exhibit an increased predisposition to elevated BP in later life, there is also evidence supporting the theory that high birth weight also increases the risk for elevated BP in childhood (Figure 1). A retrospective longitudinal based study by Bowers et al. demonstrated that higher birth weight was associated with a significantly increased risk for hypertension in both boys and girls with a greater risk of hypertension observed in children with the largest postnatal weight gain [22]. Another longitudinal study by Huang et al. concluded that high birth weight babies exhibited an increased risk for hypertension in childhood but greater weight gain during years 1 to 8 but not the first 12 months of life increased this risk [23]. However, a systematic review and meta-analysis of relevant studies indicated that high birth weight predicted an increase in BP in childhood; yet, the risk of hypertension in adulthood was inversely associated with age. Thus, older high birth weight individuals were less prone to develop hypertension relative to their normal birth weight counterparts [24]. Thus, these studies implicate the importance of growth during fetal life and childhood on the developmental origins of CV disease in later life and emphasize that overnutrition during development can be just as detrimental as undernutrition on CV health.
Yet, not all studies indicate relevance for birth weight on BP in later life. A systematic review by Huxley et al. noted that most studies reporting a negative effect of birth weight on blood pressure adjusted for current weight, but smaller sample size studies were more likely to be biased in the results due to failure to account for other confounding variables [25]. Falkner et al. reported no association between birth weight and BP in children [26]. Similarly, no significant correlation between birth weight and systolic BP was observed in an adolescent cohort where blood pressure was determined via 24-hour ambulatory monitoring [27]. Yet, Lurbe et al. reported that 24-hour systolic BP was significantly higher in the lowest birth weight group when measured via the 24 hour ambulatory method but not by conventional office methods [28]. Another study using data collected from US Collaborative Perinatal Project reported that birth weight was positively associated with BP in childhood [29]. Thus, in spite of the ample amount of literature supporting the hypothesis of an inverse relationship between birth weight and future BP, a number of studies fail to demonstrate a negative relationship. Whether accuracy of birth weight information, the use of conventional versus 24 hour ambulatory monitoring of BP or age of the cohort affects these findings requires more extensive investigation.
Birth weight and blood pressure: the effect of sex on the developmental programming of cardiovascular disease
The effect of sex on the association between birth weight and BP varies by age (Figure 2). In a large cohort studied at 10 years of age Barker et al. reported that BP did not differ in LBW boys relative to LBW girls [15]. In a longitudinal study conducted by Moore and colleagues, men and women exhibited an inverse correlation between birth weight and BP at age 20 [18]. However, after adjustment for current weight, the correlation was stronger in LBW women relative to LBW men [18]. Yet, a longitudinal study by Law et al. reported that systolic BP at age 22 was higher in LBW men relative to LBW women, an effect not attenuated by adult body mass index (BMI) [14]. Jarvelin et al. showed that men exhibited an inverse relationship between birth weight and BP at age 31 regardless of current BMI whereas gestational age and BP showed a greater association than birth weight and BP in women [30]. Barker et al. also examined the inverse relationship between birth weight and BP in men and women at 36 years of age in a large cohort born the same week in 1946 [15]. This study demonstrated an inverse relationship between birth weight and BP in both men and women although systolic BP was higher in LBW men relative to age-matched LBW women [15]. A retrospective study by Chen et al. investigating the effects of prenatal and early childhood famine exposure on risk for hypertension in adulthood indicated that women but not men at age 46 born during the Chinese famine had a higher risk for developing hypertension in adulthood [31]. Gamborg and others examined the inverse relationship between birth weight and systolic BP in a meta-regression analysis of 20 Nordic studies that adjusted for confounding factors including BMI, smoking, socioeconomic status and gestational age [32]. The inverse relationship between birth weight and systolic BP was significant for both sexes, the association became stronger with age, but the association was stronger in females at age 50 than men [32]. No sex difference in the birth weight to BP relationship was observed in another meta-analysis that separated studies in children (under 18) or adults [33]. However, the authors found that few performed formal tests for statistical interactions when investigating sex differences suggesting that more rigorous studies are needed to address the importance of sex in programmed CV risk.
Figure 2.
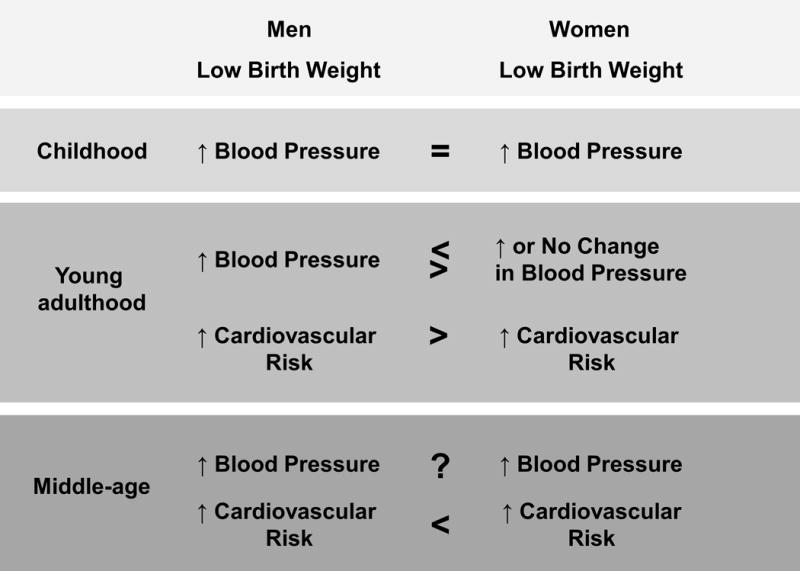
Summary of age-specific sex differences in blood pressure and cardiovascular risk in low birth weight men and women.
Hypertension is a risk factor for CV disease and birth weight also affects an individual’s risk for CV disease. Vos et al. compared birth weight to absolute risk for coronary heart disease using data from the Atherosclerosis Risk in Young Adults (ARYA) study [34]. Overall risk calculated using the Framingham risk score was greater in LBW men relative to LBW women in young adulthood [34] (Figure 2). However, Lawlor et al. reported that by middle age the risk for CV disease was increased in LBW women relative to men [35] (Figure 2). The mechanisms that contribute to the shift in greater risk for CV disease in LBW women relative to LBW men with age are unknown. Early onset menopause increases the risk for heart disease and stroke in women [36]. Several studies suggest that LBW is associated with early onset menopause [37, 38]. Whether reproductive status is linked to CV risk in LBW women is not known but these studies implicate the need for further studies to elucidate the effect of reproductive health on CV risk in LBW women.
Birth weight and blood pressure: Summary of epidemiological studies
Thus, despite a number of studies that dispute the inverse relationship between birth weight and blood pressure, numerous compelling epidemiological studies suggest that birth weight is inversely associated with BP. Whether men and women differ in programmed CV risk across their lifespan is not entirely clear but LBW men (Figure 2). The etiology of influences during early life that alter birth weight and later increased BP and CV risk may contribute to these differential findings. The transition from childhood through puberty and earlier age at time of menopause may also influence whether sex affects the birth weight and blood pressure relationship and contribute to sex difference in programmed risk that occurs with aging. Yet, experimental studies provide proof of principle and allow investigation into the mechanisms that contribute to the developmental origins of increased BP. In addition, these studies are investigating how sex affects later programmed CV risk.
Experimental models of developmental origins of adult disease: Proof of Principle
Models of developmental programming were derived to study chronic diseases such as hypertension, altered endocrine function, and obesity. Experimental models mimic the human conditions that alter fetal growth and development and are associated with increased risk for chronic disease. A consistent finding in many of these models includes sex differences in BP following a developmental insult (Table 1). Therefore, these experimental models are useful to investigate the mechanisms that contribute to the etiology of increased CV risk and to investigate the mechanisms that contribute to the sexual dimorphic programming of increased BP.
Table I.
Sex differences in birth weight and blood pressure: findings from experimental studies
| Model | Age | Blood pressure | Method | Reference |
|---|---|---|---|---|
| Mechanical Reduction in Uterine Perfusion | 16 weeks 16 weeks 12 months |
Male: Increased Female: Normotensive Female: Increased |
Telemetry Telemetry Telemetry |
41 51 9 |
| Bilateral Uterine Ligation | 12 weeks 22 weeks 18 months |
Male: Increased Male: Increased Female: Normotensive |
Telemetry Tail Cuff Tail Cuff |
64 67 68 |
| Prenatal Hypoxia | 12 months 12 months |
Male: Increased Female: Normotensive |
Tail Cuff Tail Cuff |
76 76 |
| Modest Protein Restriction (9% versus 18%) | 21 weeks 21 weeks 12 months |
Male: Increased Female: Normotensive Female: Increased |
Catheter Catheter Telemetry |
6 7 79 |
| Severe Protein Restriction (5–6% versus 18%) | 4 weeks 4 weeks 10 weeks 10 months |
Male: Increased Female: Increased Male: Increased Female: Increased |
Tail Cuff Tail Cuff Tail Cuff Tail Cuff |
80 80 80 80 |
| Prenatal Dexamethasone | 6 months 6 months |
Male: Increased Female: Normotensive |
Tail Cuff Tail Cuff |
8 8 |
| Prenatal Nicotine | 22 months 22 months |
Male: Increased Female: Normotensive |
Catheter Catheter |
107 107 |
Experimental model: Placental insufficiency
Placental insufficiency is a model of fetal programming that mimics the etiology of preeclampsia. Preeclampsia is a disease of high blood pressure during pregnancy [39] and offspring of pregnancies complicated by preeclampsia exhibit a significant increase in BP [40]. Intrauterine growth restriction within the Western world is more likely due to poor placental perfusion implicating that this is a relevant model for investigation into the mechanisms that contribute to the inverse relationship between birth weight and BP. The model of placental insufficiency induced via a reduction in uterine perfusion results in IUGR and a sex difference in blood pressure with male IUGR offspring exhibiting a significant increase in BP in young adulthood whereas female IUGR offspring are normotensive [5] (Figures 3 and 4). Ojeda et al. demonstrated that castration at 10 weeks of age completely abolished hypertension in male IUGR offspring whereas it had no effect on BP in age-matched male control offspring [41] (Figure 3). Castration normalized testosterone values; however, serum testosterone levels were two-fold greater in intact male IUGR relative to intact male control [41] suggesting that hypertension in male IUGR offspring was testosterone dependent. The contribution of sex steroids in the developmental programming of hypertension may involve modulation of neurohormonal systems. It is well established that sex hormones can alter expression of the renin-angiotensin system (RAS) in a sex-specific manner [42]. The RAS is a hormonal cascade that contributes to the regulation of the long-term control of BP and volume homeostasis. Blockade of the RAS with enalapril, an angiotensin converting enzyme (ACE) inhibitor, abolished the significant increase in BP in male IUGR offspring [41] (Figure 3) implicating an important role for the RAS in the etiology of IUGR-induced hypertension in male offspring. Renal expression of renin and angiotensinogen are androgen dependent in the male spontaneously hypertensive rat (SHR) [43]. Grigore et al. reported that mRNA expression of renal renin and angiotensinogen expression were increased in hypertensive male IUGR offspring at 16 weeks of age relative to age-matched normotensive male controls [44]. So despite the increase in BP in male IUGR offspring, renal expression of renin remained inappropriately elevated. Although renal expression of angiotensin II (Ang II) or its receptor (AT1R) was not altered, male IUGR offspring exhibited an enhanced pressor response to acute Ang II that was also abolished by castration [45] providing further support for an important role for testosterone in the programming of increased BP in male IUGR offspring. Similar to activation of RAS, testosterone increases oxidative stress in the male SHR [46–47]. Thus, these findings suggest oxidative stress as another potential mechanism in the developmental programming of hypertension. Tempol, a superoxide dismutase mimetic, normalized BP in male IUGR offspring relative to male control [48] (Figure 3). In addition to the effects on BP, chronic tempol in male IUGR offspring caused a significant reduction in F2-isoprostanes, a marker of renal oxidative stress, and expression of renal markers of oxidative stress that were elevated in untreated male IUGR offspring relative to male control counterparts [48] suggesting that reactive oxygen species (ROS) may also be a contributory factor in the etiology of hypertension in male IUGR rats. Thus, these findings indicate testosterone may contribute to elevated BP in male IUGR via up-regulation of the RAS and increased production of ROS (Figure 3).
Figure 3.
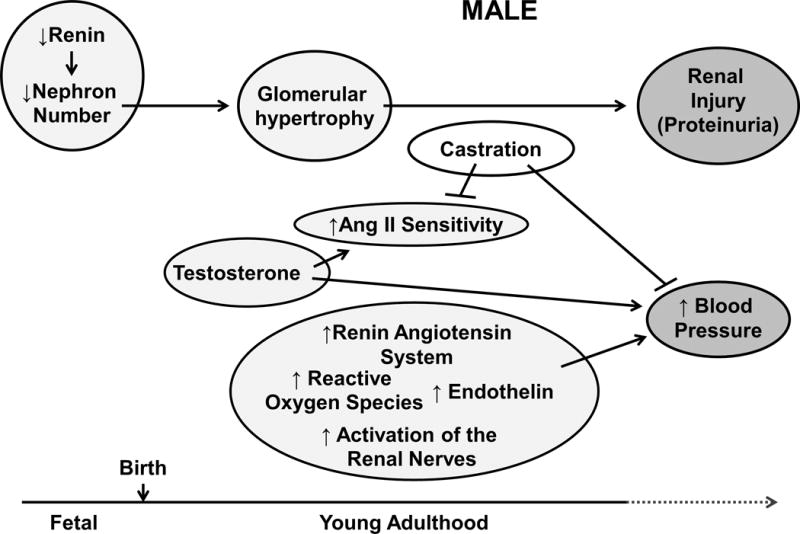
Schematic demonstrating the potential mechanisms that contribute to the developmental programming of increased blood pressure in male offspring.
Figure 4.
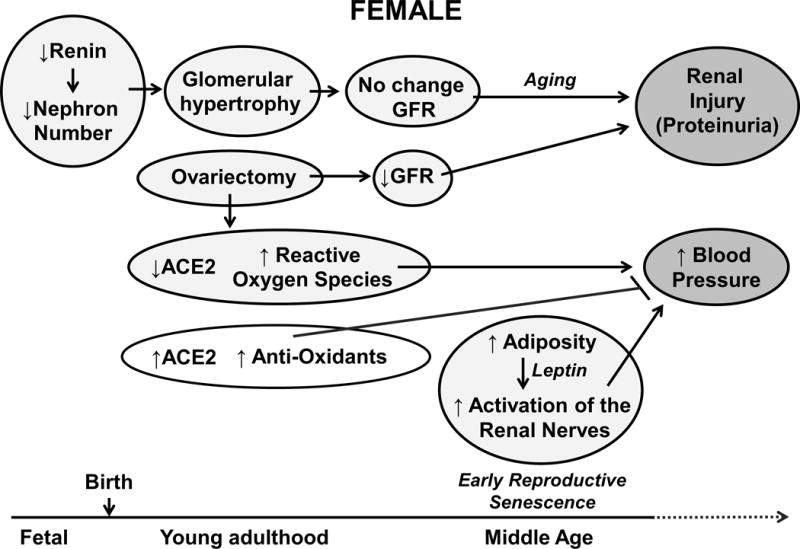
Potential mechanisms for the developmental programming of pro- and anti-hypertensive pathways in female offspring.
In the human population, men exhibit higher BP compared to women through adulthood [49] until women enter menopause [50]. Although the importance of estradiol on CV health in women after menopause is controversial, BP was significantly elevated in female IUGR following ovariectomy whereas ovariectomy had no effect on BP in female control [51] (Figure 4). Restoration of estradiol levels to physiologically relevant levels in ovariectomized offspring reduced BP in control and IUGR to similar levels [51] implicating that estradiol may be protective against the developmental programming of increased BP in female IUGR offspring in young adulthood. In contrast to testosterone’s effects on the RAS, estrogens are important in regulating the vasodepressor arm of the RAS including up-regulation of renal expression of ACE2 and AT2R [52]. The RAS is also implicated in contributing to sex differences in BP control in other models of developmental insults [52] and protection against CV disease in women [53–54]. Chronic blockade of the RAS with the ACE inhibitor, enalapril, abolished hypertension induced via ovariectomy in female IUGR offspring suggesting that the RAS contributes to the development of hypertension following loss of ovarian hormones in female IUGR rats [51]. It is proposed that estrogen modulates alterations in the balance of the vasoconstrictor to vasodilator actions of the RAS by impacting the ACE and ACE2 pathways [53–54]. Renal expression of ACE2 was elevated in intact female IUGR relative to intact female control; ovariectomy abolished this increase [51] (Figure 4) indicating that ovariectomy caused a shift in the vasoconstrictor to vasodilator balance. Renal markers of oxidative stress were not elevated in normotensive female IUGR offspring. However, female IUGR offspring exhibited an increase in expression and activity of renal catalase, an antioxidant [48] (Figure 4). There are numerous studies that indicate that estrogen is protective against hypertension by exhibiting antioxidant effects [55]. Thus, ovarian hormones may contribute to the compensatory increase in renal ACE2 expression in female IUGR offspring to oppose the adverse programming effect of IUGR on BP in female offspring. Up-regulation of anti-oxidant pathways may be another compensatory protective mechanism.
It is well established that BP increases with age in both men and women [56]. Aging is also suggested to amplify the effect of birth weight on BP in LBW individuals [57] suggesting that aging may serve as a secondary influence on BP following a developmental insult. To investigate the impact of aging on BP in female IUGR offspring, female IUGR rats were studied at 12 months of age to avoid the confounding effect of persistent estrus that occurs in the SD rat around 18 months of age [58]. Female IUGR offspring exhibited a significant increase in BP at 12 months of age compared to age-matched controls when measured via radio telemetry [9]. Despite having comparable body weights, female IUGR exhibited a significant increase in total and visceral adiposity and an elevation in circulating leptin compared to age-matched controls [9]. Increased adiposity resulting in increased leptin is also seen in women [59], and is noted to have detrimental outcomes on BP [60]. Leptin is an adipose-derived chemokine that is elevated in obese patients. Experiments from Mark et al., demonstrated that leptin acts as a central mediator between increases in adiposity and elevated BP via activation of the sympathetic nervous system to the kidney [61]. To investigate the role of the renal nerves on the increase in BP that developed by one year of age in female IUGR offspring, female rats underwent either sham or bilateral renal denervation. Hypertension was abolished in the female IUGR offspring following renal denervation whereas no significant change in BP was observed in female age-matched controls [9] (Figure 4). Interestingly, the renal nerves also contributed to the etiology of hypertension that develops in male IUGR offspring in young adulthood [62] (Figure 3) suggesting that IUGR programs a sex specific role for the renal nerves in BP that is age-dependent in male and female IUGR rats. Thus, IUGR female offspring did not remain protected against increased BP indicating the importance of further studies to investigate the impact of age on BP and CV health in low birth weight women.
Bilateral uterine ligation is another model of placental insufficiency that involves a shorter duration of insult but more severe exposure to reduced uterine perfusion. Total ligation of the uterine arteries and veins at day 18 of gestation induces a significant reduction in birth weight and litter size [63] and a significant correlation between birth weight and BP in male IUGR offspring at 12 weeks of age [64]. Schreuder et al. reported that glomerular number was decreased while mean glomerular volume was increased in association with a significant increase in protein excretion in male IUGR rats [65] (Figure 3). A reduction in glomerular number, indicative of a reduction in nephron endowment, is also observed in low birth weight humans [66] implicating the physiological relevance of this model. Moritz et al. reported that male IUGR offspring exhibited a significant increase in BP relative to normal male birth weight offspring [67]; yet, despite a significant reduction in total glomerular number associated with an increase in individual glomerular volume, BP was not increased with in female IUGR although creatinine levels were elevated in later life [68] (Figure 3). Thus, findings using this model suggested that male offspring were more susceptible to an in utero insult than their female counterparts. In addition, these studies demonstrated the complexity of the effect of the fetal environment on later BP and CV risk.
Experimental model: Prenatal hypoxia
Prenatal hypoxia is another experimental model of developmental programming that is linked with increased risk for CVD [69–70]. This model of intrauterine stress induced by long-term hypoxia deprives the fetus of oxygen vital for proper development. Maternal food intake is also reduced potentially causing another developmental programming effect [69, 71–72]. Epidemiological studies indicate that in utero insults influence endothelial function in fetal life [73], children [74], or young adults [75]. Experimental models of prenatal hypoxia support these observations noted in low birth weight individuals via a mechanism that involves impairment in the nitric oxide system [71]. Studies by Williams et al. demonstrated that prenatal hypoxia programmed premature aging of vasculature in IUGR male offspring [69, 71] whereas Morton et al. demonstrated that female IUGR offspring exhibited a loss of NO-induced vasodilation that occurred at a later age than male IUGR [70]. To further examine the sex difference in the developmental programming of CV risk Bourque et al. investigated vascular reactivity to endothelin-1 (ET-1), a potent vasoconstrictor [76]. Male IUGR exhibited a greater vascular response to ET-1 compared to male controls, yet there was no difference in response to ET-1 in female offspring [76] (Figure 3). BP was increased in male IUGR by 12 months of age but female IUGR remained normotensive relative to age-matched female controls [76]. Thus, these results indicated that males were more susceptible to CV risk following the developmental insult of maternal hypoxia indicative of the sex-specific programming of BP observed in other models of placental insufficiency.
Experimental model: Prenatal maternal protein restriction
Maternal protein restriction during gestation was one of the first models developed to investigate the link between birth weight, BP and CV risk [77]. This model which mimics chronic undernutrition in regions that suffer from extreme malnutrition in developing countries is useful for studying the mechanisms by which early growth patterns are related to adult cardiovascular disease. However, the developmental programming effects of a maternal low protein diet vary greatly due to differences in dietary components of the protein restricted diets [77–78] and the method used for measurement of BP [6–7,79–80]. Additionally, the severity of the restriction of protein in the maternal diet also results in sex-specific programming of CV risk [6–7].
This model has been used by numerous investigators and studies by Woods and colleagues reported that moderate maternal protein restriction (9% versus 18% protein) programmed a significant increase in BP in male low-protein offspring [6] that despite a decrease in nephron number was not associated with a change in glomerular filtration rat (GFR) [6]. Glomerular volume was increased [6] providing compensation. However, studies by Woods et al. also demonstrated that a moderate reduction in maternal protein during gestation did not alter BP in female low protein offspring [6]. Joles et al. reported that, fetal exposure to low protein during gestation increased renal injury in the aging male kidney [81] (Figure 3). Yet, an additional study indicated that female offspring exposed to moderate maternal protein restriction during fetal life were protected against the accelerated development of renal injury [79]. Removal of the ovarian hormones accelerated age-related reductions in GFR and worsened albuminuria in female low protein offspring [79] (Figure 4) indicating that similar to the model of placental insufficiency, ovarian hormones played a protective role in the programming of chronic health in female offspring. However, BP was increased in both male and female offspring exposed to a more severe reduction in protein intake within the maternal diet (6% versus 18% reduction) [77]. A similar finding was confirmed by Vehaskari et al. [80]. Mortality rates were greater in the low protein offspring in this study [80] indicating that prenatal protein restriction moderately shortened the lifespan of the low protein offspring. Castration did not attenuate hypertension programmed by prenatal protein restriction in the male offspring [82]. However, unlike male IUGR offspring in the model of placental insufficiency [41], circulating testosterone levels were not elevated by prenatal exposure to low protein [82]. Thus, these studies indicated that females exposed to a developmental insult do not remain protected against increased CV risk in later life; yet, programming of increased CV and renal risk was related to the severity of the developmental insult.
The RAS is also implicated in the etiology of hypertension induced by maternal protein restriction. Blockade of the RAS by enalapril abolished the increase in BP in low protein offspring relative to untreated low protein rats (Figure 3) but was unable to prevent the increase in mortality programmed by this developmental insult [83]. The RAS is important in renal development [52]. Renal expression of renin and angiotensinogen were reduced during nephrogenesis in offspring exposed to maternal protein restriction [6]. Nephron number was also decreased indicating renal development was impaired in association with suppression of the RAS [6] (Figure 3). Suppression of the RAS during nephrogenesis is also reported for models of developmental programming induced by placental insufficiency [84–85]. Thus, these results indicated that developmental programming results in temporal changes in the RAS with the importance of the RAS in the programming of impaired renal development and increased BP implicated despite the different model of developmental insult [6, 41, 44, 83] (Figure 3).
To elucidate the differential effect of exposure to a prenatal versus a postnatal diet of maternal protein restriction, Lozano et al. examined offspring that were cross-fostered from mothers fed a normal or a low protein diet during pregnancy to mothers fed a normal or low protein diet during lactation [86]. Results indicated that cross-fostering of an offspring from a low protein mother (6% protein) to a mother on regular chow (18%) prevented the development of hypertension; whereas cross-fostering of an offspring from a mother on regular chow to a mother on the 6% or protein restricted diet was sufficient to induce an increase in BP [86]. Therefore, these results indicated that the postnatal environment modulated the effect of the prenatal environment on BP and was able to reverse or induce an altered phenotype. These studies mimic findings noted in humans where temporal changes in weight gain alter BP in childhood [10–13]
Experimental model: Global nutrient restriction
A global restriction in nutrient intake is also used to investigate the link between developmental influences and later CV risk. Global nutrient restriction during gestation in the guinea pig reduced birth weight and increased BP in male offspring [87]. Global nutrient restriction in one generation also resulted in transgenerational effect with BP increased in male offspring of the F2 and F3 generations despite the absence of additional insult during their gestational period [88]. The effect of exposure to maternal undernutrition in the F1 also impaired vascular function in the F1 offspring and subsequent generations [88] Nutrient restriction initiated prior to pregnancy also reduced life expectancy in the second-generation male offspring [89]. Thus, these studies suggested that fetal exposure to nutrient restriction in one generation not only increased CV risk in the F1 generation, but also transmitted that risk to the next generations of male offspring.
Experimental model: Early Life Stress
Exposure to socioeconomic stress in early life or prenatal exposure to maternal stress is associated with an increased risk for CV disease [90–91,7]. Loria et al. demonstrated that maternal separation as a model of early life stress (ELS) did not program a significant increase in baseline BP [92]. However, BP was significantly increased to a greater extent in male ELS pups exposed to chronic Ang II compared to male control offspring [92]. The development of hypersensitivity to chronic Ang II was delayed in female ELS rats [92] implicating a sex difference in the BP response to a vasoactive factor. Circulating testosterone levels were increased in male ELS rats exposed to chronic Ang II and castration prevented hypersensitivity to chronic Ang II in male ELS rats [93] (Figure 3). Thus, testosterone also contributes to the etiology of enhanced sensitivity to Ang II that originated from exposure to an adverse environment during early life.
Experimental model: Prenatal Glucocorticoid
Prenatal administration of glucocorticoids are often given to pregnant women to accelerate fetal pulmonary maturation [94]; yet, experimental studies indicate that glucocorticoids may exert an adverse CV effect on the developing fetus with consequences manifesting in later life [95]. Rat offspring born to dams that received a daily dexamethasone injection throughout gestation exhibited a 50% reduction in nephron number in association with a 30% reduction in GFR in young adulthood [96]. BP was elevated by 2 months of age in the dexamethasone exposed offspring [96] indicating that fetal exposure to glucocorticoids was sufficient to increase CV risk in the offspring. Another study resulted in hypertension at 6 months of age in male but not female rat offspring exposed to prenatal glucocorticoids [8]. Male offspring of pregnant ewes exposed to betamethasone during gestation developed hypertension associated with a decrease in nephron number, and a reduction in sodium excretion relative to male controls [97]. However, female dexamethasone exposed ewes did not demonstrate an impaired sodium excretion [97]. Thus, these studies indicated that glucocorticoid exposure during fetal life had sex-specific effects on renal function that were not species specific. Singh et al. demonstrated that a reduction in nephron number in rat pups exposed to glucocorticoids during prenatal life was associated with suppression of the renal RAS [98] (Figure 3). Bilateral renal denervation abolished increased systolic BP in male rat offspring exposed to prenatal dexamethasone [99] (Figure 3). Thus, the mechanisms that contribute to increased BP following fetal exposure to glucocorticoids includes pathways reported to contribute to the etiology of increased programmed CV risk in other models of developmental insult.
Experimental model: Prenatal Nicotine
Maternal smoking is associated with an increased risk of BP in postnatal life [100]. Nicotine is one of the major components of cigarette smoke, and it readily crosses the placenta into the fetal circulation [101]. Excess nicotine causes permanent changes in the nicotinic receptors and alterations in activity of the central and peripheral nervous systems [102] implicating nicotine exposure as a mediator of reprogramming of vascular tone and alterations in BP. Prenatal exposure to nicotine during gestation at a dose that is comparable to moderate to heavy smoke inhalation reduced birth weight in offspring of nicotine-treated dams [103]. Male nicotine-treated offspring exhibited vascular dysfunction not observed in their female littermates [103]. Male nicotine-treated offspring also exhibited an enhanced increase in the BP response to acute Ang II relative to Ang II-treated male controls whereas the increase in BP in response to acute Ang II was not elevated in female prenatal nicotine offspring relative to their female control counterparts [104]. Thus, this study demonstrated a sex difference in vascular function and the BP response to acute Ang II following prenatal exposure to nicotine. Xiao et al. also reported that ovariectomy induced a significant increase in the BP response to acute Ang II [105]; ovariectomy also increased vascular AT1R expression in female nicotine-treated offspring [106]. Estrogen replacement reversed these findings [105] implicating that estradiol was protective against the developmental programming of CV risk through a mechanism that involved modulation of the RAS (Figure 4). Ovariectomy also induced a significant increase in vascular production of reactive oxygen species in female offspring exposed to prenatal nicotine relative to ovariectomized controls that was reversed by estradiol replacement [105] (Figure 4).. However, protection against increased CV risk in female offspring following prenatal nicotine exposure was lost with aging. BP responses to acute Ang II were significantly enhanced in female rats exposed to prenatal nicotine when aged to 22 months [107]. Thus, this study demonstrated that aging as a second hit further exacerbated the adverse programming of CV risk in male and female offspring following exposure to nicotine during fetal life.
Conclusion
The etiology of hypertension is not well understood. Epidemiological studies indicate that birth weight is inversely associated with BP suggesting that adverse influences during early life may contribute to the development of hypertension in later life. Whether LBW results in sex-specific programming of higher BP within the human population is unclear (Figure 2). Accuracy of birth weight records, differences in age at the time of study, a lack of consideration for confounding variables such as weight gain in early life, and accuracy of the methodology used for BP measurement may contribute to conflicting results. Clearly additional studies using precise anthropometric measurements at birth, consideration for confounding factors that include adjustment for weight gain during early life and current BMI, 24 hour ambulatory measure of BP conducted at specific time points that coincide with childhood, early adulthood, and age after menopause are needed to effectively address the effect of sex and aging on the developmental programming of blood pressure.
Numerous experimental studies that mimic the causes of LBW are being utilized to investigate the etiology of the developmental origins of high blood pressure. These models demonstrate a distinct sex difference with males affected to a greater extent than females in young adulthood (Table I). Yet, experimental studies also suggest that females do not remain protected against increased BP in later life (Table I). Reporting of sex differences also varies in experimental studies. Accuracy of the methodology used for measure of BP, severity of the developmental insult or the age of the animal at the time of study may be contributing factors. Yet, animal studies are better able to control for confounding variables and thus, experimental studies provide strong proof of principle that a moderate insult during early life programs a sex difference in BP in young adulthood. Experimental studies also implicate a role for sex steroids and involvement of the RAS, the renal nerves, endothelin and oxidative stress in the etiology of BP regulation following a developmental insult (Figures 3 and 4). Recent studies indicate that maternal undernutrition in the rat programs early reproductive senescence [108–109] (Figure 4). Whether this is linked to loss of protection against CV risk is not yet known. However, further studies are needed to clarify the mechanisms that contribute to the development of increased BP in LBW individuals and the effect of sex and aging across the lifespan on CV risk following a developmental insult in order to determine the clinical implications for these findings.
Summary Statement.
Compelling epidemiology studies suggest that birth weight is inversely associated with blood pressure. Experimental studies indicate a sex difference with males more susceptible to developmental insults than females. However, experimental studies suggest that aging abolishes the sex difference exacerbating cardiovascular risk in the female.
Clinical Perspective.
Compelling evidence from experimental models indicate that the in utero environment leads to sex differences in the long-term control of blood pressure with males more adversely affected relative to females in young adulthood. However, epidemiological studies provide inconsistent results.
Experimental models are identifying potential mechanisms that contribute to sex differences in the developmental programming of blood pressure. These studies implicate the renin angiotensin system, the renal nerves, endothelin and oxidative stress with modulation by sex steroids providing a contributory role. Experimental studies also suggest that aging can abolish sex differences in blood pressure implicating a loss of protection against programmed cardiovascular risk in females in later life.
Few epidemiological or population studies are investigating the effect of sex on blood pressure in low birth weight individuals. Therefore, further investigation into whether sex affects blood pressure following low birth weight and whether aging serves as a second insult to exacerbate cardiovascular risk is clearly needed in order to better address therapeutic options and provide better health care for low birth weight individuals as they age.
Acknowledgments
None
Funding Information:
BTA: NIH: HL074927, HL51971, and P20GM104357 and American Heart Association Grant in Aid: GRNT19900004; JHD: NIH: T32HL105324 and American Heart Association Predoctoral Fellowship: 15PRE24700010
Abbreviations List
- Ang II
Angiotensin
- ACE
Angiotensin converting enzyme
- AT1R
Angiotensin type 1 receptor
- BP
Blood pressure
- BMI
Body mass index
- CV
Cardiovascular
- ELS
Early life stress
- IUGR
Intrauterine growth restriction
- MAP
Mean arterial pressure
- RAS
Renin-angiotensin system
- ROS
Reactive oxygen species
- SHR
Spontaneously hypertensive rat
Footnotes
Declarations of Interest:
None
Author Contribution Statement
JHD prepared the initial draft of this manuscript with subsequent editing by BTA.
References
- 1.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- 2.Forsdahl A. Observations throwing light on the high mortality in the county of Finnmark. Is the high mortality today a late effect of very poor living conditions in childhood and adolescence? 1973. Int J Epidemiol. 2002;31:302–308. [PubMed] [Google Scholar]
- 3.Barker DJ, Osmond C. Low birth weight and hypertension. BMJ. 1988;297:134–135. doi: 10.1136/bmj.297.6641.134-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotsis V, Stabouli S, Pitiriga V, Toumanidis S, Papmichael C, Zakopoulos N. Ambulatory blood pressure monitoring and target organ damage: effects of age and sex. Blood Press Monit. 2006;11:9–15. doi: 10.1097/01.mbp.0000189785.59994.20. [DOI] [PubMed] [Google Scholar]
- 5.Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension. 2003;41:457–62. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- 6.Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res. 2001;49:460–467. doi: 10.1203/00006450-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1131–R1136. doi: 10.1152/ajpregu.00037.2003. [DOI] [PubMed] [Google Scholar]
- 8.Ortiz LA, Quan A, Zarzar F, Weinberg A, Baum M. Prenatal dexamethasone programs hypertension and renal injury in the rat. Hypertension. 2003;41:328–34. doi: 10.1161/01.hyp.0000049763.51269.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Intapad S, Tull FL, Brown AD, Dasinger JH, Ojeda NB, Fahling JM, Alexander BT. Renal denervation abolishes the age-dependent increase in blood pressure in female intrauterine growth-restricted rats at 12 months of age. Hypertension. 2013;61:828–834. doi: 10.1161/HYPERTENSIONAHA.111.00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Primatesta P, Falaschetti E, Poulter NR. Birth weight and blood pressure in childhood: results from the Health Survey for England. Hypertension. 2005;45:75–79. doi: 10.1161/01.HYP.0000150037.98835.10. [DOI] [PubMed] [Google Scholar]
- 11.Singhal A, Cole TJ, Fewtrell M, Kennedy K, Stephenson T, Elias-Jones A. Promotion of faster weight gain in infants born small for gestational age. Hypertension. 2007;115:213–220. doi: 10.1161/CIRCULATIONAHA.106.617811. [DOI] [PubMed] [Google Scholar]
- 12.Belfort MB, Rifas-Shiman SL, Rich-Edwards J, Kleinman KP, Gillman MW. Size at birth, infant growth, and blood pressure at three years of age. The Journal of Pediatrics. 2007;151:670–674. doi: 10.1016/j.jpeds.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hindmarsh PC, Bryan S, Geary MP, Cole TJ. Effects of current size, postnatal growth, and birth size on blood pressure in early childhood. Pediatrics. 2010;126:e1507–e1513. doi: 10.1542/peds.2010-0358. [DOI] [PubMed] [Google Scholar]
- 14.Law CM, Shiell AW, Newsome CA, Syddall HE, Shinebourne EA, Fayers PM, Martyn CN, de Swiet M. Fetal, infant, and childhood growth and adult blood pressure: a longitudinal study from birth to 22 years of age. Circulation. 2002;105:1088–1092. doi: 10.1161/hc0902.104677. [DOI] [PubMed] [Google Scholar]
- 15.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W, Srinivasan SR, Ruan L, Mei H, Berenson GS. Adult hypertension is associated with blood pressure variability in childhood in blacks and whites: The Bogalusa Heart Study. Am J Hypertens. 2011;24:77–82. doi: 10.1038/ajh.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore VM, Miller AG, Boulton TJC, Cockington RA, Craig IH, Magarey AM, Robinson JS. Placental weight, birth measurements and blood pressure at age 8 years. Arch Dis Child. 1996;74:538–541. doi: 10.1136/adc.74.6.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore VM, Cockington RA, Ryan P, Robinson JS. The relationship between birth weight and blood pressure amplifies from childhood to adulthood. J Hypertens. 1999;17:883–888. doi: 10.1097/00004872-199917070-00003. [DOI] [PubMed] [Google Scholar]
- 19.Law CM, de Swiet M, Osmond C, Fayers PM, Barker DJ, Cruddas AM, Fall CH. Initiation of hypertension in utero and its amplification throughout life. BMJ. 1993;306:24–27. doi: 10.1136/bmj.306.6869.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulterwaal CS, Anthony S, Launer LJ, Witteman JC, Trouwborst AM, Hofman A, Grobbee DE. Birth weight, growth, and blood pressure: an annual follow-up of children aged 5 through 21 years. Hypertension. 1997;30:267–271. doi: 10.1161/01.hyp.30.2.267. [DOI] [PubMed] [Google Scholar]
- 21.Head RF, Tu YK, Gilthorpe MS, Mishra GD, Williams S, Ellison GT. What evidence is there that adjustment for adult height influences the relationship between birth weight and blood pressure? Ann Hum Biol. 2007;34:252–264. doi: 10.1080/03014460701210977. [DOI] [PubMed] [Google Scholar]
- 22.Bowers K, Liu G, Wang P, Ye T, Tian Z, Liu E, Yu Z, Yang X, Klebanoff M, Yeung E, Hu G, Zhang C. Birth weight, postnatal weight change, and risk for high blood pressure among Chinese children. Pediatrics. 2011;127:e1272–e1279. doi: 10.1542/peds.2010-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang RC, Burke V, Newnham JP, Stanley FJ, Kendall GE, Landau LI, Oddy WH, Blake KV, Palmer LJ, Beilin LJ. Perinatal and childhood origins of cardiovascular disease. Int J Obes. 2007;31:236–44. doi: 10.1038/sj.ijo.0803394. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Li H, Liu SJ, Fu GJ, Zhao Y, Xie YJ, Zhang Y, Wang YX. The associations of high birth weight and blood pressure with hypertension in later life: a systematic review and meta-analysis. Hypertens Res. 2013;36:725–733. doi: 10.1038/hr.2013.33. [DOI] [PubMed] [Google Scholar]
- 25.Huxley R, Neil A, Collins R. Unravelling the fetal origins hypothesis: is there really an inverse association between birthweight and subsequent blood pressure? Lancet. 2002;360:659–665. doi: 10.1016/S0140-6736(02)09834-3. [DOI] [PubMed] [Google Scholar]
- 26.Falkner B, Hulman S, Kushner H. Effect of birth weight on blood pressure and body size in early adolescence. Hypertension. 2004;43:203–207. doi: 10.1161/01.HYP.0000109322.72948.24. [DOI] [PubMed] [Google Scholar]
- 27.Salvi P, Meriem C, Temmar M, Marino F, Sari-Ahmed M, Labat C, Alla F, Joly L, Safar ME, Benetos A. Association of current weight and birth weight with blood pressure levels in Saharan and European teenager populations. Am J Hypertens. 2010;23:379–386. doi: 10.1038/ajh.2009.275. [DOI] [PubMed] [Google Scholar]
- 28.Lurbe E, Torro I, Rodriguez C, Alvarez V, Redon J. Birth weight influences blood pressure values and variability in children and adolescents. Hypertension. 2001;38:389–393. doi: 10.1161/01.hyp.38.3.389. [DOI] [PubMed] [Google Scholar]
- 29.Hemachandra AH, Howards PP, Furth SL, Klebanoff MA. Birth weight, postnatal growth, and risk for high blood pressure at 7 years of age: Results from the collaborative perinatal project. Pediatrics. 2007;119:1264–1270. doi: 10.1542/peds.2005-2486. [DOI] [PubMed] [Google Scholar]
- 30.Järvelin MR, Sovio U, King V, Lauren L, Xu B, McCarthy MI, Hartikainen AL, Laitinen J, Zitting P, Rantakallio P, Elliott P. Early life factors and blood pressure at age 31 years in the 1966 northern Finland birth cohort. Hypertension. 2004;44:838–846. doi: 10.1161/01.HYP.0000148304.33869.ee. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Nembhard WN, Stockwell GH. Sex-specific effects of fetal exposure to the 1959–1961 Chinese famine on risk of adult hypertension. Matern Child Health J. 2014;18:527–533. doi: 10.1007/s10995-013-1268-z. [DOI] [PubMed] [Google Scholar]
- 32.Gamborg M, Byberg L, Rasmussen F, Andersen PK, Baker JL, Bengtsson C, Canoy D, Drøyvold W, Eriksson JG, Forsén T, Gunnarsdottir I, Järvelin MR, Koupil I, Lapidus L, Nilsen TI, Olsen SF, Schack-Nielsen L, Thorsdottir I, Tuomainen TP, Sørensen TI, NordNet Study Group Birth weight and systolic blood pressure in adolescence and adulthood: meta-regression analysis of sex- and age-specific results from 20 Nordic studies. Am J Epidemiol. 2007;166:634–645. doi: 10.1093/aje/kwm042. [DOI] [PubMed] [Google Scholar]
- 33.Lawlor DA, Ebrahim S, Davey SG. Is there a sex difference in the association between birth weight and systolic blood pressure in later life? Findings from a meta-regression analysis. Am J Epidemiol. 2002;156:1100–1104. doi: 10.1093/aje/kwf154. [DOI] [PubMed] [Google Scholar]
- 34.Vos LE, Oren A, Bots ML, Gorissen WH, Grobbee DE, Uiterwaal CS. Birth size and coronary heart disease risk score in young adulthood. The Atherosclerosis Risk in Young Adults (ARYA) study. Eur J Epidemiol. 2006;21:33–38. doi: 10.1007/s10654-005-4658-8. [DOI] [PubMed] [Google Scholar]
- 35.Lawlor DA, Ronalds G, Clark H, Smith GD, Leon DA. Birth weight is inversely associated with incident coronary heart disease and stroke among individuals born in the 1950s: findings from the Aberdeen Children of the 1950s prospective cohort study. Circulation. 2005;112:1414–1418. doi: 10.1161/CIRCULATIONAHA.104.528356. [DOI] [PubMed] [Google Scholar]
- 36.Wellons M, Ouyang P, Schreiner PJ, Herrington DM, Vaidya D. Early menopause predicts future coronary heart disease and stroke: the Multi-Ethnic Study of Atherosclerosis. Menopause. 2012;19:1081–1087. doi: 10.1097/gme.0b013e3182517bd0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stein AD, Zybert PA, van der Pal-de Bruin K, Lumey LH. Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: evidence from the Dutch Famine. Eur J Epidemiol. 2006;21:759–765. doi: 10.1007/s10654-006-9065-2. [DOI] [PubMed] [Google Scholar]
- 38.Tom SE, Cooper R, Kuh D, Guralnik JM, Hardy R, Power C. Fetal environment and early age at natural menopause in a British birth cohort study. Hum Reprod. 2010;25:791–798. doi: 10.1093/humrep/dep451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villar J, Carroli G, Wojdyla D, Abalos E, Giordano D, Ba’aqeel H, Farnot U, Bergsjø P, Bakketeig L, Lumbiganon P, Campodónico L, Al-Mazrou Y, Lindheimer M, Kramer M, World Health Organization Antenatal Care Trial Research Group Preeclampsia, gestational hypertension and intrauterine growth restriction, related or independent conditions? Am J Obstet Gynecol. 2006;194:921–931. doi: 10.1016/j.ajog.2005.10.813. [DOI] [PubMed] [Google Scholar]
- 40.Davis EF, Lewandowski AJ, Aye C, Williamson W, Boardman H, Huang RC, Mori TA, Newnham J, Beilin LJ, Leeson P. Clinical cardiovascular risk during young adulthood in offspring of hypertensive pregnancies: insights from a 20-year prospective follow-up birth cohort. BMJ Open. 2015;5:e008136. doi: 10.1136/bmjopen-2015-008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ojeda NB, Grigore D, Yanes LL, Iliescu R, Robertson EB, Zhang H, Alexander BT. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2007;292:R758–R763. doi: 10.1152/ajpregu.00311.2006. [DOI] [PubMed] [Google Scholar]
- 42.Fisher M, Baessler A, Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res. 2002;15:672–677. doi: 10.1016/s0008-6363(01)00479-5. [DOI] [PubMed] [Google Scholar]
- 43.Chen YF, Naftilan AJ, Oparil S. Androgen-dependent angiotensinogen and renin messenger RNA expression in hypertensive rats. Hypertension. 1992;19:456–463. doi: 10.1161/01.hyp.19.5.456. [DOI] [PubMed] [Google Scholar]
- 44.Grigore D, Ojeda NB, Robertson EB, Dawson AS, Huffman CA, Bourassa EA, Speth RC, Brosnihan KB, Alexander BT. Placental insufficiency results in temporal alterations in the renin angiotensin system in male hypertensive growth restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2007;293:R804–R8011. doi: 10.1152/ajpregu.00725.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ojeda NB, Royals TP, Black JT, Dasinger JH, Johnson JM, Alexander BT. Enhanced sensitivity to acute angiotensin II is testosterone dependent in adult male growth-restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1421–R1427. doi: 10.1152/ajpregu.00096.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iliescu R, Cucchiarelli VE, Yanes LL, Iles JW, Reckelhoff JF. Impact of androgen-induced oxidative stress on hypertension in male SHR. Am J Physiol Regul Integr Comp Physiol. 2007;292:R731–R735. doi: 10.1152/ajpregu.00353.2006. [DOI] [PubMed] [Google Scholar]
- 47.Alonso-Alvarez C, Bertrand S, Faivre B, Chastel O, Sorci G. Testosterone and oxidative stress: the oxidation handicap hypothesis. Proceedings. 2007;274:819–825. doi: 10.1098/rspb.2006.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ojeda NB, Hennington BS, Williamson DT, Hill ML, Betson NE, Sartori-Valinotti JC, Reckelhoff JF, Royals TP, Alexander BT. Oxidative stress contributes to sex differences in blood pressure in adult growth-restricted offspring. Hypertension. 2012;60:114–122. doi: 10.1161/HYPERTENSIONAHA.112.192955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y, American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 50.Preston RA. Effects of blood pressure reduction on cardiovascular risk estimates in hypertensive postmenopausal women. Climacteric. 2007;10(Suppl):132–141. doi: 10.1080/13697130601114909. [DOI] [PubMed] [Google Scholar]
- 51.Ojeda NB, Grigore D, Robertson EB, Alexander BT. Estrogen protects against increased blood pressure in postpubertal female growth restricted offspring. Hypertension. 2007;50:679–685. doi: 10.1161/HYPERTENSIONAHA.107.091785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moritz KM, Cuffe JS, Wilson LB, Dickinson H, Wlodek ME, Simmons DG, Denton KM. Review: Sex specific programming: a critical role for the renal renin-angiotensin system. Placenta. 2010;31:S40–46. doi: 10.1016/j.placenta.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 53.Baumer AT, Nickenig G, Bohm M. Protection of the cardiovascular system by estrogens. Role of the renin-angiotensin system, Dtsch Med Wochenschr. 2000;125:1444–9. doi: 10.1055/s-2000-8494. [DOI] [PubMed] [Google Scholar]
- 54.Wassmann K, Ghiassi A, Wassmann S, Bohm M, Nickenig G. AT1 receptor antagonism improves endothelial dysfunction in postmenopausal women. Maturitas. 2006;53:176–183. doi: 10.1016/j.maturitas.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 55.Bureau I, Gueux E, Mazur A, Rock E, Roussel AM, Rayssiguier Y. Female rats are protected against oxidative stress during copper deficiency. J Am Coll Nutr. 2003;22:239–246. doi: 10.1080/07315724.2003.10719299. [DOI] [PubMed] [Google Scholar]
- 56.Wiinberg N, Høegholm A, Christensen HR, Bang LE, Mikkelsen KL, Nielsen PE, et al. 24-h ambulatory blood pressure in 352 normal Danish subjects, related to age and gender. Am J Hypertens. 1995;8:978–986. doi: 10.1016/0895-7061(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 57.Davies AA, Smith GD, May MT, Ben-Shlomo Y. Association between birth weight and blood pressure is robust, amplifies with age, and may be underestimated. Hypertension. 2006;48:431–436. doi: 10.1161/01.HYP.0000236551.00191.61. [DOI] [PubMed] [Google Scholar]
- 58.Sokol RZ, Okuda H, Stanczyk FZ, Wolfe GW, Delaney JC, Chapin RE. Normative reproductive indices for male and female adult Sprague-Dawley rats. Contraception. 1999;59:203–207. doi: 10.1016/s0010-7824(99)00017-7. [DOI] [PubMed] [Google Scholar]
- 59.Shah NR, Braverman ER. Measuring adiposity in patients: the utility of body mass index (BMI), percent body fat, and leptin. PLoS One. 2012;7:e33308. doi: 10.1371/journal.pone.0033308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mark AL, Agassandian K, Morgan DA, Liu X, Cassell MD, Rahmouni K. Leptin signaling in the nucleus tractussolitarii increases sympathetic nerve activity to the kidney. Hypertension. 2009;53:375–380. doi: 10.1161/HYPERTENSIONAHA.108.124255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alexander BT, Hendon AE, Ferril G, Dwyer TM. Renal denrvation abolishes hypertension in low birth weight offspring from regnant rats with reduced uterine perfusion. Hypertenison. 2005;45:754–758. doi: 10.1161/01.HYP.0000153319.20340.2a. [DOI] [PubMed] [Google Scholar]
- 63.Lane RH, Chandorkar AK, Flozak AS, Simmons RA. Intrauterine growth retardation alters mitochondrial gene expression and function in fetal and juvenile rat skeletal muscle. Pediatr Res. 1998;43:563–570. doi: 10.1203/00006450-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 64.Schreuder MF, Fodor M, van Wijk JA, Delemarre-van de Waal HA. Association of birth weight with cardiovascular parameters in adult rats during baseline and stressed conditions. Pediatr Res. 2006;59:126–130. doi: 10.1203/01.pdr.0000190576.08501.df. [DOI] [PubMed] [Google Scholar]
- 65.Schreuder MF, Nyengaard JR, Fodor M, van Wijk JA, Delemarre-van de Waal HA. Glomerular number and function are influenced by spontaneous and induced low birth weight in rats. J Am Soc Nephrol. 2005;16:2913–2919. doi: 10.1681/ASN.2004100875. [DOI] [PubMed] [Google Scholar]
- 66.Hughson M, Farris AB, 3rd, Douglas-Denton R, Hoy WE, Bertram JF. Glomerular number and size in autopsy kidneys: the relationship to birth weight. Kidney Int. 2003;63:2113–212. doi: 10.1046/j.1523-1755.2003.00018.x. [DOI] [PubMed] [Google Scholar]
- 67.Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int. 2008;74:187–195. doi: 10.1038/ki.2008.153. [DOI] [PubMed] [Google Scholar]
- 68.Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, Owens JA, Wlodek ME. Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol. 2009;587:2635–2646. doi: 10.1113/jphysiol.2009.170407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williams SJ, Campbell ME, McMillen IC, Davidge ST. Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R360–R367. doi: 10.1152/ajpregu.00178.2004. [DOI] [PubMed] [Google Scholar]
- 70.Morton JS, Rueda-Clausen CF, Davidge ST. Mechanisms of endothelium-dependent vasodilation in male and female, young and aged offspring born growth restricted. Am J Physiol Regul Integr Comp Physiol. 2010;298:R930–R938. doi: 10.1152/ajpregu.00641.2009. [DOI] [PubMed] [Google Scholar]
- 71.Williams SJ, Hemmings DG, Mitchell JM, McMillen IC, Davidge ST. Effects of maternal hypoxia or nutrient restriction during pregnancy on endothelial function in adult male rat offspring. J Physiol. 2005;565:125–135. doi: 10.1113/jphysiol.2005.084889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Geijn HP, Kaylor WM, Jr, Nicola KR, Zuspan FP. Induction of severe intrauterine growth retardation in the Sprague–Dawley rat. Am J Obstet Gynecol. 1980;137:43–47. doi: 10.1016/0002-9378(80)90384-1. [DOI] [PubMed] [Google Scholar]
- 73.Martin H, Hu J, Gennser G, Norman M. Impaired endothelial function and increased carotid stiffness in 9-year-old children with low birthweight. Circulation. 2000;102:2739–2744. doi: 10.1161/01.cir.102.22.2739. [DOI] [PubMed] [Google Scholar]
- 74.Martin H, Gazelius B, Norman M. Impaired acetylcholine-induced vascular relaxation in low birth weight infants: implications for adult hypertension? Pediatr Res. 2000;47:457–462. doi: 10.1203/00006450-200004000-00008. [DOI] [PubMed] [Google Scholar]
- 75.Goodfellow J, Bellamy MF, Gorman ST, Brownlee M, Ramsey MW, Lewis MJ, Davies DP, Henderson AH. Endothelial function is impaired in fit young adults of low birth weight. Cardiovasc Res. 1998;40:600–606. doi: 10.1016/s0008-6363(98)00197-7. [DOI] [PubMed] [Google Scholar]
- 76.Bourque SL, Gragasin FS, Quon AL, Mansour Y, Morton JS, Davidge ST. Prenatal hypoxia causes long-term alterations in vascular endothelin-1 function in aged male, but not female, offspring. Hypertension. 2013;62:753–758. doi: 10.1161/HYPERTENSIONAHA.113.01516. [DOI] [PubMed] [Google Scholar]
- 77.Langley SC, Jackson AA. Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clin Sci. 1994;86:217–222. doi: 10.1042/cs0860217. [DOI] [PubMed] [Google Scholar]
- 78.Langley-Evans SC. Critical differences between two low protein diet protocols in th programming of hypertension in the rat. Int J Food Sci Nutr. 2000;51:11–1. doi: 10.1080/096374800100859. [DOI] [PubMed] [Google Scholar]
- 79.Pijacka W, Clifford B, Tilburgs C, Joles JA, Langley-Evans S, McMullen S. Protective role of female gender in programmed accelerated renal aging in the rat. Physiol Rep. 2015;3:e12342. doi: 10.14814/phy2.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vehaskari VM, Aviles DH, Manning J. Prenatal programming of adult hypertension in the rat. Kidney Int. 2001;59:238–245. doi: 10.1046/j.1523-1755.2001.00484.x. [DOI] [PubMed] [Google Scholar]
- 81.Joles JA, Sculley DV, Langley-Evans SC. Proteinuria in aging rats due to low-protein diet during mid-gestation. J Dev Orig Health Dis. 2010;1:75–83. doi: 10.1017/S2040174409990183. [DOI] [PubMed] [Google Scholar]
- 82.Woods LL, Morgan TK, Resko JA. Castration fails to prevent prenatally programmed hypertension in male rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1111–R1116. doi: 10.1152/ajpregu.00803.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manning J, Vehaskari VM. Low birth weight-associated adult hypertension in the rat. Pediatr Nephrol. 2001;16:417–422. doi: 10.1007/s004670000560. [DOI] [PubMed] [Google Scholar]
- 84.Zhang DY, Lumbers ER, Simonetta G, Wu JJ, Owens JA, Robinson JS, McMillen IC. Effects of placental insufficiency on the ovine fetal renin-angiotensin system. Exp Physiol. 2000;85:79–84. [PubMed] [Google Scholar]
- 85.Grigore D, Ojeda NB, Alexander BT. Sex differences in the fetal programming of hypertension. Gend Med. 2008;5:S121–132. doi: 10.1016/j.genm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lozano G, Elmaghrabi A, Salley J, Siddique K, Gattineni J, Baum M. Effect of prenatal programming and postnatal rearing on glomerular filtration rate in adult rats. Am J Physiol Renal Physiol. 2015;308:F411–F419. doi: 10.1152/ajprenal.00593.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kind KL, Simonetta G, Clifton PM, Robinson JS, Owens JA. Effect of maternal feed restriction on blood pressure in the adult guinea pig. Exp Physiol. 2002;87:469–477. doi: 10.1111/j.1469-445x.2002.tb00060.x. [DOI] [PubMed] [Google Scholar]
- 88.Ponzio BF, Carvalho MH, Fortes ZB, do Carmo Franco M. Implications of maternal nutrient restriction in transgenerational programming of hypertension and endothelial dysfunction across F1–F3 offspring. Life Sci. 2012;90:571–577. doi: 10.1016/j.lfs.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 89.Araminaite V, Zalgeviciene V, Simkunaite-Rizgeliene R, Stukas R, Kaminskas A, Tutkuviene J. Maternal caloric restriction prior to pregnancy increases the body weight of the second-generation male offspring and shortens their longevity in rats. Tohoku J Exp Med. 2014;234:41–50. doi: 10.1620/tjem.234.41. [DOI] [PubMed] [Google Scholar]
- 90.Camelo LV, Giatti L, Chor D, Griep RH, Benseñor IM, Santos IS, Kawachi I, Barreto SM. Associations of life course socioeconomic position and job stress with carotid intima-media thickness. The Brazilian Longitudinal Study of Adult Health (ELSA-Brasil) Soc Sci Med. 2015;141:91–99. doi: 10.1016/j.socscimed.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 91.Plana-Ripoll O, Liu X, Momen NC, Parner E, Olsen J, Li J. Prenatal exposure to maternal stress following bereavement and cardiovascular disease: A nationwide population-based and sibling-matched cohort study. Eur J Prev Cardiol. 2015 doi: 10.1177/2047487315585294. pii: 2047487315585294. [DOI] [PubMed] [Google Scholar]
- 92.Loria AS, Pollock DM, Pollock JS. Early life stress sensitizes rats to angiotensin II-induced hypertension and vascular inflammation in adult life. Hypertension. 2010;55:494–499. doi: 10.1161/HYPERTENSIONAHA.109.145391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Loria AS, Yamamoto T, Pollock DM, Pollock JS. Early life stress induces renal dysfunction in adult male rats but not female rats. Am J Physiol Regul Integr Comp Physiol. 2013;304:R121–R129. doi: 10.1152/ajpregu.00364.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crowley P, Chalmers I, Keirse MJ. The effects of corticosteroid administration before preterm delivery: an overview of the evidence from controlled trials. Brit J Obstet Gynaecol. 1990;97:11–25. doi: 10.1111/j.1471-0528.1990.tb01711.x. [DOI] [PubMed] [Google Scholar]
- 95.Reinisch JM, Simon NG, Karow WG, Gandelman R. Prenatal exposure to prednisone in humans and animals retards intrauterine growth. Science. 1978;202:436–438. doi: 10.1126/science.705336. [DOI] [PubMed] [Google Scholar]
- 96.Celsi G, Kistner A, Aizman R, Eklof AC, Ceccatelli S, de Santiago A, Jacobson SH. Prenatal dexamethasone causes oligonephronia, sodium retention, and higher blood pressure in the offspring. Pediatr Res. 1998;44:317–322. doi: 10.1203/00006450-199809000-00009. [DOI] [PubMed] [Google Scholar]
- 97.Tang L, Carey LC, Bi J, Valego N, Sun X, Deibel P, Perrott J, Figueroa JP, Chappell MC, Rose JC. Gender differences in the effects of antenatal betamethasone exposure on renal function in adult sheep. Am J Physiol Regul Integr Comp Physiol. 2009;296:R309–R317. doi: 10.1152/ajpregu.90645.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Singh RR, Cullen-McEwen LA, Kett MM, Boon WM, Dowling J, Bertram JF, Moritz KM. Prenatal corticosterone exposure results in altered AT1/AT2, nephron deficit and hypertension in the rat offspring. J Physiol. 2007;579:503–513. doi: 10.1113/jphysiol.2006.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dagan A, Kwon HM, Dwarakanath V, Baum M. Effect of renal denervation on prenatal programming of hypertension and renal tubular transporter abundance. Am J Physiol Renal Physiol. 2008;295:F29–F34. doi: 10.1152/ajprenal.00123.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blake KV, Gurrin LC, Evans SF, Beilin LJ, Landau LI, Stanley FJ, Newnham JP. Maternal cigarette smoking during pregnancy, low birth weight and subsequent blood pressure in early childhood. Early Hum Dev. 2000;57:137–147. doi: 10.1016/s0378-3782(99)00064-x. [DOI] [PubMed] [Google Scholar]
- 101.Lambers DS, Clark KE. The maternal and fetal physiologic effects of nicotine. Semin Perinatol. 1996;20:115–126. doi: 10.1016/s0146-0005(96)80079-6. [DOI] [PubMed] [Google Scholar]
- 102.Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther. 1998;285:931–945. [PubMed] [Google Scholar]
- 103.Xiao D, Huang X, Lawrence J, Yang S, Zhang L. Fetal and neonatal nicotine exposure differentially regulates vascular contractility in adult male and female offspring. J Pharmacol Exp Ther. 2007;320:654–461. doi: 10.1124/jpet.106.113332. [DOI] [PubMed] [Google Scholar]
- 104.Xiao D, Xu Z, Huang X, Longo LD, Yang S, Zhang L. Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension. 2008;51:1239–1247. doi: 10.1161/HYPERTENSIONAHA.107.106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xiao D, Huang X, Yang S, Zhang L. Estrogen normalizes perinatal nicotine-induced hypertensive responses in adult female rat offspring. Hypertension. 2013;61:1246–54. doi: 10.1161/HYPERTENSIONAHA.113.01152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mao C, Wu J, Xiao D, Lv J, Ding Y, Xu Z, Zhang L. The effect of fetal and neonatal nicotine exposure on renal development of AT(1) and AT(2) receptors. Reprod Toxicol. 2009;27:149–154. doi: 10.1016/j.reprotox.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tao H, Rui C, Zheng J, Tang J, Wu L, Shi A, Chen N, He R, Wu C, Li J, Yin X, Zhang P, Zhu Z, Tao J, Xiao J, Mao C, Xu Z. Angiotensin II-mediated vascular changes in aged offspring rats exposed to perinatal nicotine. Peptides. 2013;44:111–119. doi: 10.1016/j.peptides.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 108.Khorram O, Keen-Rinehart E, Chuang TD, Ross MG, Desai M. Maternal undernutrition induces premature reproductive senescence in adult female rat offspring. Fertil Steril. 2015;103:291–8.e2. doi: 10.1016/j.fertnstert.2014.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bernal AB, Vickers MH, Hampton MB, Poynton RA, Sloboda DM. Maternal undernutrition significantly impacts ovarian follicle number and increases ovarian oxidative stress in adult rat offspring. Plos One. 2010;5:e15558. doi: 10.1371/journal.pone.0015558. [DOI] [PMC free article] [PubMed] [Google Scholar]