

Abstract
Traumatic brain injury (TBI) is one of the major cause of morbidity and mortality and it affects more than 1.7 million people in the USA. A couple of regenerative pathways including activation of hypoxia-inducible transcription factor 1 alpha (HIF-1α) are initiated to reduce cellular damage following TBI; however endogenous activation of these pathways is not enough to provide neuroprotection after TBI. Thus we aimed to see whether sustained activation of HIF-1α can provide neuroprotection and neurorepair following TBI. We found that chronic treatment with dimethyloxaloylglycine (DMOG) markedly increases the expression level of HIF-1α and mRNA levels of its downstream proteins such as Vascular endothelial growth factor (VEGF), Phosphoinositide-dependent kinase-1 and 4 (PDK1, PDK4) and Erythropoietin (EPO). Treatment of DMOG activates a major cell survival protein kinase Akt and reduces both cell death and lesion volume following TBI. Moreover, administration of DMOG augments cluster of differentiation 31 (CD31) staining in pericontusional cortex after TBI, which suggests that DMOG stimulates angiogenesis after TBI. Treatment with DMOG also improves both memory and motor functions after TBI. Taken together our results suggest that sustained activation of HIF-1α provides significant neuroprotection following TBI.
Keywords: DMOG, HIF-1α, angiogenesis, traumatic brain injury, cell death, lesion volume
1. Introduction
Traumatic Brain Injury (TBI) is a major cause of morbidity and mortality and affects more than 1.7 million people throughout the USA annually. TBI is a contributing factor to a third (30.5%) of all injury-related deaths in the USA (Rondina et al., 2005). Direct medical costs and indirect costs of TBI have totaled an estimated $76.5 billion in the USA (Chen et al., 2011; Humphreys et al., 2013; Kayani et al., 2009). TBI is multifactorial in nature and is characterized by several secondary mechanisms including cell death (Farook et al., 2013; Laird et al., 2010; Raghupathi, 2004; Stoica and Faden, 2010), decrease in blood vessels and angiogenesis (Xiong et al., 2010), and impairment in neurobehavioral outcomes (Bachevalier and Meunier, 1996; Brunson et al., 2001).
TBI-induced cell death has been shown with poorer prognosis in TBI patients due to the complexity of the nature of cell death. TBI induces cell death in multiple cell types, however, neuronal cell death has been considered as the predominant focus in TBI-associated cell death. Recently, my laboratory, as well as other groups, have shown that inactivation of a pro-survival kinase, Akt acts as a major mechanism for neuronal cell death following TBI. Akt, a serine/threonine-specific protein kinase, plays a key role in multiple cellular processes such as metabolism, apoptosis, cell proliferation, synaptic plasticity and protein synthesis.
A couple of regenerative pathways such as activation of hypoxia-inducible transcription factor 1 (HIF-1α) are initiated to reduce cellular damage including following TBI (Li et al., 2013; Sheldon et al., 2009). HIF-1α is a heterodimer composed of constitutively expressed HIF-1β and regulatory HIF-1α, regulated by cellular oxygen concentration. Under hypoxic conditions HIF-1α accumulates, allowing nuclear HIF-1α dimerization and transcription (Shenaq et al., 2012; Shi, 2009; Trollmann and Gassmann, 2009), resulting in cell survival by activating factors such as VEGF, PDK1, PDK4 and EPO (Cao et al., 2009; Dery et al., 2005; Hewitson and Schofield, 2004; Ke and Costa, 2006; Tang et al., 2004). Increase in the level of BDNF is important for regulation of the coordination between a neuron and glial cell activity which is essential for neuronal survival following trauma (Del Puerto et al., 2013; Navarro et al., 2007). BDNF can be synthesized by the transcriptional activation of CREB (Merz et al., 2011; Sen and Snyder, 2011; Tao et al., 1998), which is a downstream substrate of cell survival protein kinase Akt (Du and Montminy, 1998). Activation of Akt depends on its phosphorylation at T308 and S473 (Vivanco and Sawyers, 2002) residues and augmentation in the level of phosphorylation of Akt leads to activation of several proteins including CREB by increasing its phosphorylation at residue S133 mediated by Akt (Caravatta et al., 2008; Du and Montminy, 1998).
Dimethyloxaloylglycine (DMOG) is a cell-permeable prolyl-4-hydroxylase inhibitor, which upregulates HIF-1α (Bruick and McKnight, 2001; Jaakkola et al., 2001). It has been discovered that the DMOG mediates neuroprotection against normoxic oxidative death via HIF-1α and CREB-Independent Pathways (Siddiq et al., 2009). DMOG also attenuates myocardial injury in a rabbit ischemia-reperfusion model (Ockaili et al., 2005). Here, we used DMOG to examine whether sustained activation of HIF-1α provides neuroprotection and neuro-repair following TBI.
In the present study, we have provided evidence in support of the fact that sustained activation of HIF-1α via administration of DMOG leads to a sustained increase in VEGF which in turn stimulates angiogenesis through activation of Akt and CREB signaling pathway. Our study also suggests that treatment with DMOG provide neuroprotection and improvement in cognitive functions following TBI.
2. Materials and Methods
2.1 Drugs
DMOG was purchased from Cayman, US and was dissolved in saline. Saline was delivered by intraperitoneal injection to sham and TBI groups of mice. DMOG (20 mg/kg) was delivered by intraperitoneal (i.p.) injection to our DMOG and DMOG + TBI groups for 22 days period after either sham or TBI. Since DMOG was dissolved in saline, only sham and TBI mice received saline for 22 days as controls. The detailed outline of the Experimental design regarding an overview of the various sample points is as follows.
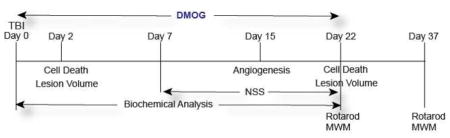
To determine the therapeutic window of the drug, we administered DMOG (20mg/kg) after 1, 4, 6 10 and 12 h after TBI through i.p injections. The neuroprotective effect of DMOG was monitored by TUNEL assay after 2 and 22 days after TBI. DMOG (20mg/kg) was also administered through i.v via tail-vein injections after 30 mins of TBI and continued up to 22 days as described previously(Fenn et al., 2015; Mahmood et al., 2001; Zhang et al., 2015). The neuroprotective effect of DMOG was monitored after 2 and 22 days after TBI by measuring cell death using TUNEL assay.
2.2 TBI procedures
The Committee on Animal Use for Research and Education at Georgia Regents University approved all animal studies, in compliance with NIH guidelines. The procedure was done based on our previously published protocol (Farook et al., 2013; Kapoor et al., 2013; Kimbler et al., 2012). Briefly, adult male C57BL/6 (Jackson Laboratory), mice were anesthetized with xylazine (8 mg/kg)/ketamine (60 mg/kg) and subjected to a sham injury or controlled cortical impact. Briefly, mice were placed in a stereotaxic frame (Ambient Instruments, Richmond, VA, USA) and a 3.5 mm craniotomy was made in the right parietal bone midway between bregma and lambda with the medial edge lateral to the midline, leaving the dura intact. Mice were impacted at 4.5 m/s with a 20-ms dwell time and 1.2-mm depression using a 3-mm-diameter convex tip, mimicking a moderate TBI. Sham-operated mice underwent the identical surgical procedures but were not impacted. The incision was closed with VetBond and mice were allowed to recover. Body temperature was maintained at 37°C using a small animal temperature controller throughout all procedures (Kopf Instruments, Tujunga, CA, USA).
2.3 Neurological function evaluation
Neurological function was evaluated by a modified neurological severity score (NSS) on the day before, and on days 7, 15 and 22 after TBI (Hirjak et al., 2013; Liu et al., 2014). The evaluations consisted of the motor (abnormal movement), sensory (visual, proprioceptive), reflex, and balance tests; and were recorded on a scale of 0 to 10 (normal score, 0; maximal deficit score, 10). They were performed by blinded, trained observers. For the NSS, 1 point was scored for inability to perform the test or the lack of a tested reflex. Thus, a higher score would point to a more severe injury. All mice were given enough time to become familiar with the testing environment before inflicting the brain injury. This was assessed by the mice ability to perform all the tests, and then an NSS was assigned.
2.4 Morris Water Maze (MWM) test
In the Morris water maze, the hidden platform procedure was performed in a circular tank filled with opaque water as described already with minor modifications (Mir et al., 2014; Vorhees and Williams, 2006). For training, either sham or TBI mice treated with or without DMOG at indicated days (8–12 weeks, male) were placed in the tank at four random points and allowed to search and find the platform. In the event, the mouse didn’t find the platform within the 60s time, the mouse was manually put on the platform for extra 30s. During the trials, the mouse was allowed to search for the platform for the 60s. Two trials were given every day and the latency for each trial was recorded. A probe trial was performed on day 22 or 37. The mice were allowed to swim in the tank for the 60s without the platform, and performance was assessed on the basis of the time spent in the quadrant in which the hidden platform was originally located (the right quadrant).
2.5 Rotarod
To examine motor coordination and learning in the mice, a rotor-rod test was performed as Described (Mir et al., 2014). The mice were strictly selected with regard to body weight and eye-opening to reduce data variations. Six trials were continuously performed at 4–40 rpm. After a 30s interval, the time that each mouse stayed on the rod and number of falls was measured (maximum time 120 s for the constant mode and 300 s for the accelerating mode). Trials were performed on three successive days to evaluate motor learning.
2.6 Determination of lesion size
Cortical lesion area was quantified by an investigator blinded to experimental conditions, as described previously (Kabadi et al., 2015)(Kimbler et al., 2012; Laird et al., 2014; Laird et al., 2010). Briefly, out of the total eighty 50-μm sections, every eighth section was analyzed beginning from a random start point (Kabadi et al., 2015). Serial coronal sections were digitized using a Zeiss Axiophot microscope using a 2.5X objective and imported into the OsiriX v2.7.5 32-bit program. Periphery of lesion area was drawn along the perimeter of the injured cortex and lesion volume was calculated as mm3 after 2 and 22 days of TBI with or without treatment of DMOG following previously published protocols (Kabadi et al., 2012; Zhao et al., 2013) The lesion area was outlined using the Stereologer 2000 program (Systems Planning and Analysis, Alexandria, VA, USA) to obtain the final volume measurements.
2.7 Immunohistochemistry
Deeply anesthetized mice were perfused with saline, followed by fixation with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were post-fixed overnight in paraformaldehyde followed by cryoprotection with 30% sucrose (pH 7.4) until brains permeated. Serial coronal sections were prepared using a cryostat microtome (Leica, Wetzlar, Germany) and directly mounted onto glass slides. Sections were incubated at room temperature with 100% normal donkey serum in phosphate-buffered saline containing 0.4% Triton X-100 for 12 h, followed by incubation with the primary antibody against Ki67(a marker of cell division, Cell Signaling)(Acosta et al., 2013), CD31(blood vessel marker, Cell Signaling)(Ge et al., 2014; Tian et al., 2013), NeuN (mature neuron), overnight at 4°C. Sections were then washed and incubated with the appropriate Alexa Fluor-tagged secondary antibody. The omission of primary antibody served as a negative control (Farook et al., 2013; Kapoor et al., 2013).
2.8 Confocal microscopy
Immunofluorescence was determined using an LSM 510 Meta confocal laser microscope (Carl Zeiss, Thornwood, NY, USA), as described previously. Cellular colocalization was determined in Z-stack mode using 63X oil immersion Neofluor objective (NA 12.3) with the image size set at 512 × 512 pixels. The following excitation lasers/emission filters settings were used for various chromophores: argon2 laser was used for Alexa Fluor 488, with excitation maxima at 490 nm and emission in the range 505–530 nm. A HeNe1 laser was used for Alexa Fluor 594 with excitation maxima at 543 nm and emission in the range 568–615 nm. Z-stacks (20 optical slices) were collected at optimal pinhole diameter at 12-bit pixel depth and converted into three-dimensional projection images using LSM 510 Meta imaging software (Carl Zeiss) (Farook et al., 2013; Kapoor et al., 2013).
2.9 TUNEL staining
Mice were subjected to CCI model of TBI followed by treatment with DMOG for 2 days. As a control, mice were treated with saline after TBI. Brain sections were stained with NeuN of both treated and untreated samples as described earlier. Sections were processed for TUNEL assay using a commercial kit that labels DNA strand breaks with fluorescein isothiocyanate (FITC; In Situ Cell Death Detection Kit, Roche Molecular Biochemicals, Mannheim, Germany). Selected sections were pretreated with 20 mg/mL proteinase-K in 10 mM Tris-HCl at 37°C for 15 minutes. These sections were then rinsed in PBS and incubated in 0.3% hydrogen peroxide dissolved in anhydrous methanol for ten minutes. The sections were then incubated in 0.1% sodium citrate and 0.1% Triton X-100 solution for two minutes at 2 to 8°C. After several washes with PBS, sections were incubated with 50 μL of TUNEL reaction mixture with terminal deoxynucleotidyltransferase (TdT) for 60 minutes at 37°C under humidified conditions, and neuronal nuclei were stained with DAPI. Each section was observed and photographed under a confocal microscope. Negative controls were obtained by omitting the TdT enzyme (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014). The number of TUNEL positive cells (Green) and Ki67 positive cells (red) were measured by Image J software.
2.10 Western blotting
Whole tissue lysates were prepared from 3-mm coronal sections centered upon the site of impact. A 1-mm micro-punch was collected from the pericontusional cortex or from the corresponding pericontusional hemisphere as described previously(Farook et al., 2013; Kapoor et al., 2013). Tissue was placed in complete RIPA buffer, sonicated, and centrifuged for 120 min at 124 000 × g at 4°C. Thirty micrograms of protein were resolved on a 4–20% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane. Blots were incubated overnight at 4°C in primary antibody Akt, phosphorylated-Akt (T308), phosphorylated-CREB (S133), HIF-1α, HIF-1β, Actin, VEGF and BDNF followed by a 2 h incubation with an Alexa Fluor-tagged secondary antibody at room temperature, per our laboratory. Blots were visualized using a Li-Cor Odyssey near-infrared imaging system and densitometry analysis was performed using Quantity One software (Bio-Rad, Foster City, CA, USA) (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014). The intensity of each band was determined by Image J software and the changes in the experimental band was represented as fold changes as described previously (Sen et al., 2012) (Sen et al., 2008).
2.11 Quantitative Real-Time PCR
Quantitative real-time PCR was performed as described previously (Farook et al., 2013; Kapoor et al., 2013; Mir et al., 2014). PCR reactions (25 μL) contained 12.5 μL of PCR Sybr Green mix (New England BioLabs) with 0.3 μM primers. At the end of the 35 cycles of amplification, a dissociation curve was performed in which Sybr Green was measured at 1°C intervals between 50°C and 100°C. Results were normalized using total input DNA and expressed as bound/Input (percentage). The primers used to carry out PCR analysis are as follows VEGF: 5′ CTCACCAAAGCCAGCACATA 3′ and 5′ AAATGCTTTCTCCGCTCTGA 3′; EPO: 5′ ACCACCCCACCTGCTCCACTC 3′ and 5′ GTTCGTCGGTCCACCACGGT 3′; eNOS: 5′ GGCTGGGTTTAGGGCTGT 3′ and 5′ GCTGTGGTCTGGTGCTGGT 3′; PDK-1: 5′ TTCACGTCACGCTGGGCGAG 3′ and 5′ GGGCACAGCACGGGACGTTT 3′; PDK-4 5′ GATGAAGGCAGCCCGCTTCG 3′ and 5′ TGCTTCATGGACAGCGGGGA 3′; 18S: 5′ GACTCAACACGGGAAACCTC 3′ and 5′ ATGCCAGAGTCTCGTTCGTT 3′.
2.12 Statistical analysis
All data are presented as the mean ± SEM. The effects of treatments were analyzed using a one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test. Results are expressed as mean±S.E.M. A p-value <0.05 was considered to be statistically significant. Two-way repeated-measures ANOVAs with LSD posthoc tests were used to determine statistical significance between behavioral measures. One-way ANOVAs with the appropriate LSD posthoc tests were used to compare experimental groups. For all analyzes, p < 0.05 was considered significant.
3. Results
3.1 Treatment with DMOG increases nuclear accumulation of HIF-1α following TBI
Activation of HIF1α depends on its protein level and the formation of the active transcriptional complex (dimer of HIF1α and HIF1β) inside the nucleus. Thus, we monitored the protein level of HIF1α by Western blot hybridization and the interaction between HIF1α and HIF1β by co-immunoprecipitation after administration of various doses of DMOG ranging from 0–50mg/kg following TBI. We found that after treatment with 20mg/kg of DMOG the protein level of HIF1α and the interaction between HIF1α and HIF1β was increased to the extent of 3.2 fold. However, treatment with 50mg/kg of DMOG did not further increase either the protein level (Fig 1A, B) or the interaction between HIF1α and HIF1β (Fig 1A, C). Thus, we continued to use 20mg/kg of DMOG treatment to assess the neuroprotective properties of DMOG.
Fig. 1. Treatment with DMOG activates HIF-1α.

(A)Western blot analysis of the expression level of HIF-1α and interaction of HIF-1α and HIF-1β (A) in pericontusional cortex after TBI after treatment of DMOG in a dose-dependent manner.(B–C) Quantitation of the protein level of HIF-1α (B) and interaction of HIF-1α and HIF-1β in the pericontusional cortex (C) after treatment with DMOG as dose dependent manner (0–50 mg/kg). (D) Western blot analysis of the expression level of HIF-1α in pericontusional cortex after TBI after treatment of DMOG in a time-dependent manner (7–27 days).(E) Quantitation of the protein level of HIF-1α after treatment of DMOG in a time-dependent manner (7–27 days). (F–G) Western blot analysis of the expression level of HIF-1α (F) and quantitation of its level (G) with or without treatment of DMOG following TBI. (H–I) The interaction of HIF-1α and HIF-1β in the pericontusional cortex (H) and quantitation of its level (I) with or without treatment of DMOG following TBI. (J–K) The cytosolic and nuclear level of HIF-1α (J) and quantitation of its level (K) with or without treatment of DMOG following TBI. *P<0.01, n=5–7, one-way ANOVA, mean ± S.E.M.
We also administered DMOG (20 mg/kg) for 7, 15, 22 and 27 days after TBI and protein level of HIF1α was monitored by Western blot hybridization. We found that protein level of HIF1α was increased to more than 3.5 fold after 22 days of treatment with DMOG, however, it was not further increased after 27 days of treatment (Fig. 1D, E). Thus, we decided to use 1–22 days DMOG treatment after TBI.
To further confirm the specificity of DMOG on the protein level of HIF-1α, we treated mice with DMOG (20 mg/kg; i.p) for 22 days following TBI. We measured the protein level of HIF-1α in pericontusional cortex starting from day 7 to day 22 after DMOG treatment in TBI mice. We found that level of HIF-1α was significantly increased in TBI+DMOG mice compared to TBI groups throughout day 7 to 22 (Fig 1F, G), however, treatment with the vehicle has not effected on the protein level of HIF1α; suggesting that DMOG stabilizes HIF-1α in a specific manner. However in sham mice, there is no alteration in the level of HIF-1α protein level.
We also studied the interaction between HIF-1α and HIF-1β by co-immunoprecipitation (Co-IP) assay in both TBI and TBI+DMOG group of mice along with sham-operated mice. Our data also show that the interaction between HIF-1α and HIF-1β was significantly increased at day 7 of DMOG treatment following TBI and the interaction between HIF-1α and HIF-1β remained unaltered even at day 22 in TBI+DMOG group of mice; although the interaction between HIF-1α and HIF-1β was significantly reduced in TBI group of mice (Fig. 1H, I). Treatment with the vehicle has no effect on the interaction between HIF1α and HIF1β following TBI. Moreover, we did not see any alteration in the interaction between HIF-1α and HIF-1β in sham-operated mice. Thus, our data suggests that DMOG treatment specifically increases the interaction between HIF1α andHIF-1β following TBI.
We have also measured the nuclear and cytosolic fraction of HIF-1α after treatment of DMOG of TBI mice. We found that DMOG treatment leads to an increase in the nuclear level of HIF-1α following TBI compared to TBI + vehicle control. However there is no difference in the nuclear level of HIF-1α between sham and sham + vehicle group of mice (Fig. 1J, K).
3.2 Treatment with DMOG upregulates HIF-1α-regulated genes
To see whether treatment with DMOG has any effect on the mRNA levels of genes regulated by HIF-1α, we have done real-time RT-PCR analysis of VEGF (Fig. 2A), EPO (Fig. 2B), PDK1(Fig. 2C), PDK4 (Fig. 2D) and eNOS (Fig. 2E) in both TBI+DMOG and TBI groups of mice. We found that mRNA levels of all these genes were decreased significantly after 1, 3, 7, 15 and 22 days after TBI, however their levels were increased after treatment with DMOG. In sham-operated mice, the mRNA levels of these genes remained unaltered with or without treatment of DMOG.
Fig. 2. Effect of DMOG treatment on mRNA levels of VEGF, EPO, PDK1, PDK4 and eNOS following TBI.

quantitative RT-PCR analysis of mRNA level of VEGF (A), EPO (B), pdk1 (C), pdk4 (D) and eNOS (E) in the pericontusional cortex with or without treatment of DMOG in TBI mice. *P<0.01, n=5–7, one-way ANOVA, mean±S.E.M.
3.3 Treatment with DMOG causes sustained activation of both Akt and CREB following TBI
We also measured phosphorylation level of both Akt and CREB proteins in the pericontusional cortex of both TBI and TBI+DMOG group of mice. We found that the phosphorylation level of Akt at residue T308 was decreased significantly in TBI samples compared to sham control. However, treatment with DMOG prevents the decrease in phosphorylation of Akt following TBI (Fig. 3A). Since activated (phosphorylated) Akt is known to activate CREB via increasing its phosphorylation status at S133 residue we also monitored the phosphorylation level of CREB in TBI and TBI+DMOG group along with Sham control. We found that the phosphorylation level of CREB was decreased in TBI samples compared to sham mice. Similar to Akt, treatment with DMOG prevents the reduction of CREB phosphorylation following TBI (Fig. 3B).
Fig. 3. Influence of DMOG on Akt and CREB after TBI.

(A–B) Western blot (A) and quantitative analysis (B) of Akt-phosphorylation at T308 residue (Akt-(P)) in the pericontusional cortex with or without treatment of DMOG in TBI mice. (C–D) Western blot (C) and quantitative analysis (D) of CREB-phosphorylation at S133 residue (CREB-(P)) in the pericontusional cortex with or without treatment of DMOG in TBI mice. *P<0.01, n=5–7, one-way ANOVA, mean ± S.E.M.
3.4 Treatment with DMOG decreases cell death and lesion volume following TBI
To understand whether TBI has any influence the level of cell death, we performed TUNEL assay using pericontusional cortex after administration of DMOG through intraperitoneal (ip) injections. To specify neuronal death, cells were labeled with NeuN; neuronal and total cell death was monitored at 2 and 22 days after TBI. We found that both TUNEL and NeuN positive cells were increased to 70% and 80% after 2 and 22 days of TBI respectively. Treatment with DMOG reduces both neuronal and total cell death to the extent of 80% after 2 and 22 days following TBI (Fig. 4A, B, C).
Fig. 4. Effect of treatment of DMOG on cell death and lesion volume.

(A–C) confocal microscopic analysis of (A) and quantitation (B–C) of TUNEL/NeuN positive cells (B) and total TUNEL positive cells (C) in the pericontusional cortex with or without treatment of DMOG after 2 and 22 days following TBI. (D–E) Measurement of (D) TUNEL/NeuN positive cells (E) and total TUNEL positive cells after administration of DMOG through intravenous route. (F) Mice were administered with DMOG after 1, 4,6,10 and 12 hours after TBI and continued up to 22 days following TBI. Total TUNEL positive cells were quantitated after 2 and 22 days after TBI. (G–H) Nissl staining (G) and quantitation of lesion volume (H) after treatment with DMOG for 2 and 22 days following TBI. *P<0.01, n=5–7, one-way ANOVA, mean ± S.E.M.
To see whether intravenous administration of DMOG (20 mg/kg) provides neuroprotective effects after TBI, we measured the level of cell death after 2 and 22 days after TBI. Consistent with i.p injections, we found that intravenous injection of DMOG significantly reduced neuronal death to the extent of approximately 80% compared to TBI-induced mice (Fig. 4D). Similarly, a total number of TUNEL positive cells were also decreased significantly after 2 and 22 days following TBI (Fig. 4E). Moreover we also measured lesion volume at 2 and 22 days after TBI with or without administration of DMOG. Consistent with cell death data, we found that treatment with DMOG significantly reduced cortical lesion volume to the extent of 52% and 68% after 2 and 22 days of TBI respectively (Fig. 4F, G).
To determine the therapeutic window, DMOG was administered in TBI-mice after 1, 4, 6, 10 and 12 hours after TBI and continued for 22 days. The neuroprotective effects of DMOG were monitored by measuring cell death through TUNEL assay after 2 and 22 days of TBI. We found that administration of DMOG either at 1, 4 or 6h of TBI, the neuroprotective effect of DMOG remains unaltered. However, treatment with DMOG after 10 and 12h of TBI reduced the neuroprotective effect of DMOG to the extent of 53% and 70% compared to the 1–6h data point after 2days of TBI. After 22 days of TBI, DMOG prevented cell death if it was administrated within 6h of TBI; however the neuroprotective effects of DMOG was reduced to the extent of 56 and 74% if DMOG is administered as 10 and 12h after TBI. Thus, we conclude that DMOG can provide maximum neuroprotective effects if it is administered within 1–6h of TBI (Fig 4H).
3.5 Treatment with DMOG upregulates angiogenesis
To examine the effect of DMOG on angiogenesis, we stained newly formed blood vessels with an antibody against the endothelial marker CD31 along with proliferation marker Ki67. We found that TBI leads to a decrease in the level of CD31 and ki67 positive cells compared to sham mice. However, treatment with DMOG augments the level of-of CD31+ki67 positive cells following 15 days of TBI (Fig. 5A, B) along with an increase in total number of CD31 positive cells (Fig. 5C). This data indicates that DMOG may enhance angiogenesis following TBI.
Fig. 5. Effect of DMOG treatment on CD31 and ki67 positive cells.

(A–C) Confocal microscopic analysis (A) and quantitative analysis (B) of CD31/Ki67 positive cells and a total number of CD31 positive cells (C) in the pericontusional cortex with or without treatment of DMOG in TBI mice. *P<0.01, n=5–7, one-way ANOVA, mean ± S.E.M.
3.6 Treatment with DMOG improves memory and motor functions after TBI
We found that TBI leads to an increase in neurological score after 7, 15 and 22 days after TBI compared to sham mice. However treatment with DMOG reduces the neurological deficit scores significantly at day 7, 15 and 22 days of post-trauma (Fig. 6A). To monitor whether, treatment with DMOG has any influence on memory impairment following TBI, both TBI and TBI+DMOG group of mice along with sham mice were subjected to Morris water maze tests at day 22 and 37 after TBI. We found that treatment with DMOG, the latency to find the platform was reduced in the TBI+DMOG treated mice compared to TBI mice without treatment with DMOG (Fig. 6B). However, the latency to find the platform is much higher for TBI mice compared to sham mice. Moreover, the time spent in right quadrant (Fig. 6C) in the MWM test was increased in TBI+DMOG group of mice compared to only TBI mice. This data suggests that treatment with DMOG improves neurobehavioral outcomes following TBI.
Fig. 6. Effect on DMOG on motor and memory functions.

(A) The neurological severity score was calculated in TBI mice with or without treatment of DMOG. (B–C) latency to find the platform (B) and time spent in right quadrant (C) were measured in TBI mice with or without treatment of DMOG. A number of falls/min (D) and latency to first fall (E) were measured in both sham and TBI group of mice with or without treatment of DMOG. *P<0.01, n=10–12, mean ± S.E.M.
To monitor the influence of DMOG on motor function, we performed rotarod tests after treatment with DMOG following TBI. We found that TBI leads an increase in falls per min along an increase in a number of falls /min compared to sham + vehicle control. However treatment with DMOG reverses the effect and improve motor functions after TBI (Fig. 6D, E).
4. Discussion
The results of this study show that the administration of DMOG causes sustained increase in transcriptional activation of HIF-1α after TBI that results in neuroprotection following TBI. Enhanced expression of HIF-1α after TBI has been reported by several researchers, however unlike in brain ischemia, the role of HIF-1α in TBI and its outcome is poorly understood. Previously it was shown that TBI leads to an immediate increase in HIF-1α activity; but its level was decreased significantly after 24 h following trauma (Ding et al., 2009). According to our experimental setup, DMOG was administered at a dose of 20 mg/kg once in a day continuously for 22 days following TBI. We found that administration of DMOG provides the desired neuroprotective response such as reduction in cell death and lesion volume after acute treatment for 2 days. Moreover, chronic treatment of DMOG provides a long-term beneficiary response such as stimulation of angiogenesis and reduction in cognitive impairment after 22 days following TBI. To further confirm our proof-of-concept, we also administered DMOG through i.v injections. We found that neuroprotective effects of DMOG remain unaltered irrespective of mode of administration of DMOG in mice model of TBI. Our results further strengthen the fact that DMOG functions as a neuroprotective agent after TBI. Since, the immediate treatment is not feasible in human TBI, we tested the therapeutic window of DMOG after TBI in mice. We found that DMOG can provide maximum neuroprotective effects if it is administered within 1–4h of TBI. It is now well established that the pro-survival effects of Akt after brain injury include anti-apoptotic, pro-angiogenic, and neuroprotective effects (Kilic et al., 2006; Shein et al., 2007). Several studies including ours have shown that TBI leads to an inactivation of Akt within 6h; which triggers cell death, inflammation and increase lesion volume following TBI. Our study shows that the phosphorylation of Akt remained augmented up to 22 days following treatment of DMOG of TBI mice. Activation of Akt by administration of DMOG simultaneously activate CREB by increasing its phosphorylation level and upregulate BDNF level which results in augmentation in cell survival in the injured brain. Consistent with activation of Akt, we found that TBI-induced cell death and lesion volume were also decreased significantly in TBI+DMOG group of mice compared to TBI. Thus, we anticipate that sustained activation of HIF-1α leads to an increase in the phosphorylation level of Akt and CREB that provide neuroprotection after TBI.
Brain angiogenesis may provide the critical neurovascular niches for neuronal remodeling and functional recovery after TBI (Chopp et al., 2008; Li and Chopp, 2009; Lu et al., 2005; Xiong et al., 2008). Thus understanding how neurovascular signals and substrates make the transition from initial injury to angiogenic recovery will be important for developing new therapies for brain injuries (Arai et al., 2009). Our data suggests that treatment with DMOG stimulates angiogenesis after 15 days following TBI which was further supported by an augmentation in the mRNA level of VEGF and EPO following TBI. It was known that VEGF significantly increased angiogenesis by the increase in the number of blood vessels in proximity to the lesion (Thau-Zuchman et al., 2010). Consistent with these studies we found that treatment with DMOG causes an increase in blood vessel marker CD31 level in the pericontusional cortex indicating angiogenesis following TBI. Thus, sustained activation of HIF-1α through treatment of DMOG mimic the effect of administration of exogenous VEGF to TBI-mice (Thau-Zuchman et al., 2010).
We have also shown that treatment with DMOG provides an early improvement in NSS, which proceeded up to the time of reparative events. Moreover, the significant decrease in lesion volume further confirms the early neuroprotective effects of sustained HIF-1α activation via treatment with DMOG. It is important to note that the improvement in outcomes of the motor function directly follows time point associated with angiogenesis and following DMOG treatment in TBI mice. However, during the MWM tests, we found that TBI+DMOG group of mice continued to improve even after 22 days of administration of DMOG in TBI-mice; which suggests that treatment with DMOG could be beneficial against cognitive impairment associated with post-traumatic disorders.
In conclusion, investigating the potential of HIF-1α activation using DMOG as a therapeutic agent in TBI is a novel finding with clinical implications and translational value. In the future, our study will show the feasibility of a new treatment paradigm for TBI, introducing the concept that sustained activation of HIF-1α accelerates neuroprotection, neuro repair and reduces disabilities in TBI survivors.
Highlights.
Chronic administration of DMOG activates HIF1α following TBI.
Treatment with DMOG provide neuroprotective functions by reducing cell death and lesion volume after TBI.
Treatment with DMOG augments angiogenesis following TBI.
Treatment with DMOG improves memory impairment and motor functions after TBI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Grimmig B, Diamond DM, Sanberg PR, Bickford PC, Kaneko Y, Borlongan CV. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PloS one. 2013;8:e53376. doi: 10.1371/journal.pone.0053376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Jin G, Navaratna D, Lo EH. Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. The FEBS journal. 2009;276:4644–4652. doi: 10.1111/j.1742-4658.2009.07176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachevalier J, Meunier M. Cerebral ischemia: are the memory deficits associated with hippocampal cell loss? Hippocampus. 1996;6:553–560. doi: 10.1002/(SICI)1098-1063(1996)6:5<553::AID-HIPO8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, Bender R, Chen Y, Baram TZ. Long-term, progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin-releasing hormone reproduce the effects of early-life stress. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8856–8861. doi: 10.1073/pnas.151224898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao D, Hou M, Guan YS, Jiang M, Yang Y, Gou HF. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. BMC cancer. 2009;9:432. doi: 10.1186/1471-2407-9-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravatta L, Sancilio S, di Giacomo V, Rana R, Cataldi A, Di Pietro R. PI3-K/Akt-dependent activation of cAMP-response element-binding (CREB) protein in Jurkat T leukemia cells treated with TRAIL. Journal of cellular physiology. 2008;214:192–200. doi: 10.1002/jcp.21186. [DOI] [PubMed] [Google Scholar]
- Chen YH, Kang JH, Lin HC. Patients with traumatic brain injury: population-based study suggests increased risk of stroke. Stroke; a journal of cerebral circulation. 2011;42:2733–2739. doi: 10.1161/STROKEAHA.111.620112. [DOI] [PubMed] [Google Scholar]
- Chopp M, Li Y, Zhang J. Plasticity and remodeling of brain. Journal of the neurological sciences. 2008;265:97–101. doi: 10.1016/j.jns.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Del Puerto A, Wandosell F, Garrido JJ. Neuronal and glial purinergic receptors functions in neuron development and brain disease. Frontiers in cellular neuroscience. 2013;7:197. doi: 10.3389/fncel.2013.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dery MA, Michaud MD, Richard DE. Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. The international journal of biochemistry & cell biology. 2005;37:535–540. doi: 10.1016/j.biocel.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neuroscience letters. 2009;453:68–72. doi: 10.1016/j.neulet.2009.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. The Journal of biological chemistry. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N. GADD34 induces cell death through inactivation of Akt following traumatic brain injury. Cell death & disease. 2013;4:e754. doi: 10.1038/cddis.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn AM, Skendelas JP, Moussa DN, Muccigrosso MM, Popovich PG, Lifshitz J, Eiferman DS, Godbout JP. Methylene blue attenuates traumatic brain injury-associated neuroinflammation and acute depressive-like behavior in mice. Journal of neurotrauma. 2015;32:127–138. doi: 10.1089/neu.2014.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XT, Lei P, Wang HC, Zhang AL, Han ZL, Chen X, Li SH, Jiang RC, Kang CS, Zhang JN. miR-21 improves the neurological outcome after traumatic brain injury in rats. Scientific reports. 2014;4:6718. doi: 10.1038/srep06718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitson KS, Schofield CJ. The HIF pathway as a therapeutic target. Drug discovery today. 2004;9:704–711. doi: 10.1016/S1359-6446(04)03202-7. [DOI] [PubMed] [Google Scholar]
- Hirjak D, Wolf RC, Stieltjes B, Hauser T, Seidl U, Thiemann U, Schroder J, Thomann PA. Neurological soft signs and brainstem morphology in first-episode schizophrenia. Neuropsychobiology. 2013;68:91–99. doi: 10.1159/000350999. [DOI] [PubMed] [Google Scholar]
- Humphreys I, Wood RL, Phillips CJ, Macey S. The costs of traumatic brain injury: a literature review. ClinicoEconomics and outcomes research : CEOR. 2013;5:281–287. doi: 10.2147/CEOR.S44625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Byrnes KR, Hanscom M, Loane DJ, Faden AI. Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32:137–149. doi: 10.1038/jcbfm.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi SV, Stoica BA, Zimmer DB, Afanador L, Duffy KB, Loane DJ, Faden AI. S100B inhibition reduces behavioral and pathologic changes in experimental traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2015;35:2010–2020. doi: 10.1038/jcbfm.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S, Kim SM, Farook JM, Mir S, Saha R, Sen N. Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:17398–17403. doi: 10.1523/JNEUROSCI.2756-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayani NA, Homan S, Yun S, Zhu BP. Health and economic burden of traumatic brain injury: Missouri, 2001–2005. Public health reports. 2009;124:551–560. doi: 10.1177/003335490912400412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Molecular pharmacology. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- Kilic E, Kilic U, Wang Y, Bassetti CL, Marti HH, Hermann DM. The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:1185–1187. doi: 10.1096/fj.05-4829fje. [DOI] [PubMed] [Google Scholar]
- Kimbler DE, Shields J, Yanasak N, Vender JR, Dhandapani KM. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PloS one. 2012;7:e41229. doi: 10.1371/journal.pone.0041229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, Youssef P, Yanasak N, Vender JR, Dhandapani KM. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Sukumari-Ramesh S, Swift AE, Meiler SE, Vender JR, Dhandapani KM. Curcumin attenuates cerebral edema following traumatic brain injury in mice: a possible role for aquaporin-4? Journal of neurochemistry. 2010;113:637–648. doi: 10.1111/j.1471-4159.2010.06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Sun X, Ni Y, Chen X, Guo A. HIF-1alpha involves in neuronal apoptosis after traumatic brain injury in adult rats. Journal of molecular neuroscience : MN. 2013;51:1052–1062. doi: 10.1007/s12031-013-0084-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M. Marrow stromal cell transplantation in stroke and traumatic brain injury. Neuroscience letters. 2009;456:120–123. doi: 10.1016/j.neulet.2008.03.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zou Y, Belegu V, Lv LY, Lin N, Wang TY, McDonald JW, Zhou X, Xia QJ, Wang TH. Co-grafting of neural stem cells with olfactory en sheathing cells promotes neuronal restoration in traumatic brain injury with an anti-inflammatory mechanism. Journal of neuroinflammation. 2014;11:66. doi: 10.1186/1742-2094-11-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Mahmood A, Qu C, Goussev A, Schallert T, Chopp M. Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. Journal of neurotrauma. 2005;22:1011–1017. doi: 10.1089/neu.2005.22.1011. [DOI] [PubMed] [Google Scholar]
- Mahmood A, Lu D, Wang L, Li Y, Lu M, Chopp M. Treatment of traumatic brain injury in female rats with intravenous administration of bone marrow stromal cells. Neurosurgery. 2001;49:1196–1203. discussion 1203–1194. [PubMed] [Google Scholar]
- Merz K, Herold S, Lie DC. CREB in adult neurogenesis--master and partner in the development of adult-born neurons? The European journal of neuroscience. 2011;33:1078–1086. doi: 10.1111/j.1460-9568.2011.07606.x. [DOI] [PubMed] [Google Scholar]
- Mir S, Sen T, Sen N. Cytokine-Induced GAPDH Sulfhydration Affects PSD95 Degradation and Memory. Molecular cell. 2014;56:786–795. doi: 10.1016/j.molcel.2014.10.019. [DOI] [PubMed] [Google Scholar]
- Navarro X, Vivo M, Valero-Cabre A. Neural plasticity after peripheral nerve injury and regeneration. Progress in neurobiology. 2007;82:163–201. doi: 10.1016/j.pneurobio.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Ockaili R, Natarajan R, Salloum F, Fisher BJ, Jones D, Fowler AA, 3rd, Kukreja RC. HIF-1 activation attenuates postischemic myocardial injury: role for heme oxygenase-1 in modulating microvascular chemokine generation. American journal of physiology Heart and circulatory physiology. 2005;289:H542–548. doi: 10.1152/ajpheart.00089.2005. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain pathology. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondina C, Videtta W, Petroni G, Lujan S, Schoon P, Mori LB, Matkovich J, Carney N, Chesnut R. Mortality and morbidity from moderate to severe traumatic brain injury in Argentina. The Journal of head trauma rehabilitation. 2005;20:368–376. doi: 10.1097/00001199-200507000-00008. [DOI] [PubMed] [Google Scholar]
- Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nature cell biology. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Molecular cell. 2012;45:13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Snyder SH. Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20178–20183. doi: 10.1073/pnas.1117820108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein NA, Tsenter J, Alexandrovich AG, Horowitz M, Shohami E. Akt phosphorylation is required for heat acclimation-induced neuroprotection. Journal of neurochemistry. 2007;103:1523–1529. doi: 10.1111/j.1471-4159.2007.04862.x. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Osredkar D, Lee CL, Jiang X, Mu D, Ferriero DM. HIF-1 alpha-deficient mice have increased brain injury after neonatal hypoxia-ischemia. Developmental neuroscience. 2009;31:452–458. doi: 10.1159/000232563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenaq M, Kassem H, Peng C, Schafer S, Ding JY, Fredrickson V, Guthikonda M, Kreipke CW, Rafols JA, Ding Y. Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1alpha after traumatic brain injury (TBI) Journal of the neurological sciences. 2012;323:134–140. doi: 10.1016/j.jns.2012.08.036. [DOI] [PubMed] [Google Scholar]
- Shi H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Current medicinal chemistry. 2009;16:4593–4600. doi: 10.2174/092986709789760779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Thau-Zuchman O, Shohami E, Alexandrovich AG, Leker RR. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2010;30:1008–1016. doi: 10.1038/jcbfm.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian SF, Yang HH, Xiao DP, Huang YJ, He GY, Ma HR, Xia F, Shi XC. Mechanisms of neuroprotection from hypoxia-ischemia (HI) brain injury by up-regulation of cytoglobin (CYGB) in a neonatal rat model. The Journal of biological chemistry. 2013;288:15988–16003. doi: 10.1074/jbc.M112.428789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trollmann R, Gassmann M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain & development. 2009;31:503–509. doi: 10.1016/j.braindev.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature reviews Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature protocols. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Lu D, Qu C, Goussev A, Schallert T, Mahmood A, Chopp M. Effects of erythropoietin on reducing brain damage and improving functional outcome after traumatic brain injury in mice. Journal of neurosurgery. 2008;109:510–521. doi: 10.3171/JNS/2008/109/9/0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Current opinion in investigational drugs. 2010;11:298–308. [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chopp M, Meng Y, Katakowski M, Xin H, Mahmood A, Xiong Y. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. Journal of neurosurgery. 2015;122:856–867. doi: 10.3171/2014.11.JNS14770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Faden AI, Loane DJ, Lipinski MM, Sabirzhanov B, Stoica BA. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:1897–1908. doi: 10.1038/jcbfm.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]