

Abstract
One of the major challenges in the analysis of human genetic variation is to distinguish mutations that are functionally neutral from those that contribute to disease. BubR1 is a key protein mediating spindle-checkpoint activation that plays a role in the inhibition of the anaphase-promoting complex/cyclosome (APC/C), delaying the onset of anaphase and ensuring proper chromosome segregation. Owing to the importance of BUB1B gene in mitotic checkpoint a functional analysis using different in silico approaches was undertaken to explore the possible associations between genetic mutations and phenotypic variation. In this work we found that 3 nsSNPs I82N, P334L and R814H have a functional effect on protein function and stability. A literature search revealed that R814H was already implicated in human diseases. Additionally, 2 SNPs in the 5′ UTR region was predicted to exhibit a pattern change in the internal ribosome entry site (IRES), and eight MicroRNA binding sites were found to be highly affected due to 3′ UTR SNPs. These in silico predictions will provide useful information in selecting the target SNPs that are likely to have functional impact on the BUB1B gene.
Keywords: Single nucleotide polymorphism (SNP), Pathogenic variants, Spindle assembly checkpoint, BUB1B gene
Highlights
-
•
In this work we found that 3 nsSNPs including I82N, P334L and R814H have a functional effect on BUBR1 protein function and stability.
-
•
2 SNPs in the 5′UTR region of BUB1B were predicted to exhibit a pattern change in the internal ribosome entry site (IRES).
-
•
Eight MicroRNA binding sites were found to be highly affected due to 3′UTR SNPs.
1. Introduction
The spindle assembly checkpoint (SAC) is a cell-cycle surveillance mechanism that prevents premature anaphase entry until all chromosomes have completely aligned at the metaphase plate. SAC is composed of the checkpoint proteins BubR1, Bub3, and Mad2, associated with the APC/C coactivator Cdc20. The checkpoint system acts to inhibit the activity of the large multi-protein E3 ubiquitin ligase known as the anaphase promoting complex/cyclosome (APC/C), by binding to the co-activating subunit Cdc20. BubR1 is a key protein mediating spindle-checkpoint activation That directly binds to Cdc20 and inhibits APC/C activity (Kaisari et al., 2016). The corresponding BUB1B gene is located on chromosome 15q15 and is composed of 23 exons that encodes 1050 amino acids (Davenport et al., 1999, Hanks et al., 2012). BUB1B mutated in several cancers including colorectal, lung, breast, hematopoietic malignancies and in a rare human hereditary condition called premature chromatid separation syndrome (mosaic variegated aneuploidy) (Kapanidou et al., 2015, Kops et al., 2005, Hanks et al., 2004, Hanks et al., 2006, Matsuura et al., 2006, Suijkerbuijk et al., 2010, Ohshima et al., 2000).
Single nucleotide polymorphisms (SNPs) found in any position throughout the genome in exons, introns, intergenic regions, promoters and enhancers (Drazen et al., 1999). Many SNPs are phenotypically neutral. However, others could predispose human to disease or influence their response to a drug. Nonsynonymous SNPs (nsSNPs) that lead to an amino acid substitution in the corresponding protein product are of particular interest as they are responsible for nearly half of the known gene lesions responsible for human inherited disease (Krawczak et al., 2000).
Computational analyses of BUB1B gene for harmful nsSNPs have not been carried out until now; therefore, we applied different publicly available computational tools according to Fig. 1. The value and novelty of this study is to prioritize SNPs with functional significance from an enormous number of neutral non-risk alleles of BUB1B and provides new insights for further genetic association studies.
Fig. 1.
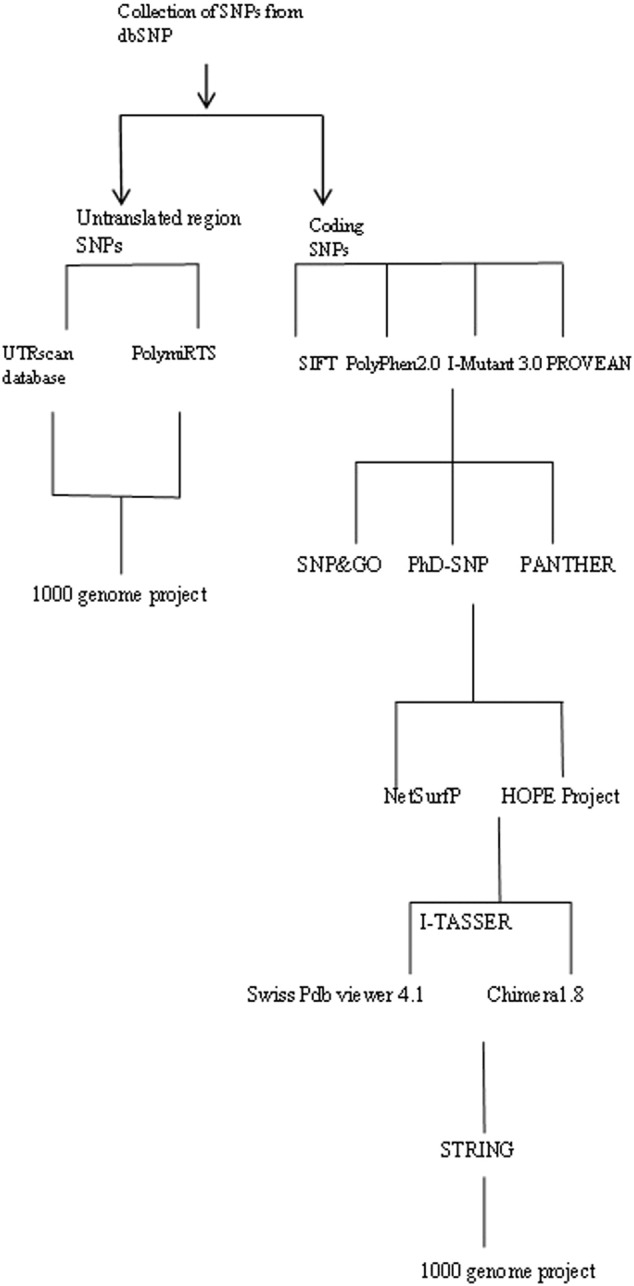
Schematic representation of computational tools for in silico analysis of BUB1B gene.
2. Material and method
2.1. Dataset
The NCBI database of SNPs (Sherry et al., 2001), dbSNP available at http://www.ncbi.nlm.nih.gov/SNP and SWISSProt databases (Bairoch and Apweiler, 1996) were used to obtain the SNP information [SNP ID, amino acid position, mRNA accession number NM_001211, and Protein accession number NP_001202.4] of the human BUB1B gene for our computational analyses.
2.2. Predicting functional context of missense mutation
The functional context of nsSNPs was predicted using SIFT (Sorting Intolerant from Tolerant), PolyPhen 2.0, I-Mutant 3.0 and PROVEAN (Protein Variation Effect Analyzer) (Table 1).
Table 1.
In silico approaches available as online tools.
| Server | Feature | URL | Reference |
|---|---|---|---|
| SIFT | Focuses more on the sequence preservation over the evolutionary time in predicting the effect of residue substitutions on function. | http://sift.bii.a-star.edu.sg/index.html | Kumar et al. (2009), Magesh and Doss (2014) |
| PolyPhen 2.0 | Sequence and structure based method that predicts the possible impact of an amino acid substitution on the structure and function of a protein. | http://genetics.bwh.harvard.edu/pph2 | Adzhubei et al. (2010) |
| I-Mutant 3.0 | Support vector machine (SVM) based predictors of protein stability changes upon single amino acid substitution. | http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi | Capriotti et al. (2008) |
| PROVEAN | Sequence based predictor that estimates whether a protein sequence variation affects protein function. | http://provean.jcvi.org | Choi et al. (2012) |
| SNP&GO | Support vector machine (SVM) based web server that combine protein structural/functional parameters and sequence analysis derived information. | http://snps.biofold.org/snps-and-go/snps-and-go.html | Magesh and Doss (2014) |
| PhD-SNP | SVM based on evolutionary information. | http://gpcr.biocomp.unibo.it/cgi/predictors/PhD-SNP/PhD-SNP.cgi | Magesh and Doss (2014) |
| PANTHER | Protein family and subfamily database that predicts the frequency of occurrence of amino acid at a particular position in evolutionary related protein sequences. | http://pantherdb.org/tools/csnpScoreForm.jsp | Mi et al. (2005) |
| UTRscan | Analyzing functional impacts of UTR SNPs. | http://www/.ba.itb.cnr.it/BIG/UTRScan | Pesole et al. (1999) |
| NetSurfP | Analysis of SNP effects on surface and solvent accessibility of protein. | http://www.cbs.dtu.dk/services/NetSurfP | Petersen et al. (2009) |
| I-TASSER | Protein structure prediction server. | http://zhanglab.ccmb.med.umich.edu/I-TASSER | Zhang (2008) |
| HOPE Project | An automatic mutant analysis server for studying the structural features of native protein and the variant models. | http://www.cmbi.ru.nl/hope/home | Venselaar et al. (2010) |
| STRING | Database of known and predicted protein-protein interactions. | http://string-db.org | Von Mering (et al. 2005) |
| PolymiRTS | The polymorphic microRNA target sites are classifies into four classes: ‘D’ (the derived allele disrupts a conserved microRNA site), ‘N’ (the derived allele disrupts a nonconserved microRNA site), ‘C’ (the derived allele creates a new microRNA site) and ‘O’ (other cases when the ancestral allele cannot be determined unambiguously). | http://compbio.uthsc.edu/miRSNP/ | Bhattacharya et al. (2013) |
| 1000 Genomes Project | A resource about human genetic variation that will be used in many studies of particular phenotypes, such as complex diseases or drug response. | http://www.1000genomes.org | Via et al. (2010) |
SIFT predict whether an amino acid substitution in a protein would be tolerated or damaging. The amino acid substitution is predicted damaging when the score is below or equal to 0.05, and tolerated if the score is greater than 0.05 (Ng and Henikoff, 2003). PolyPhen input is the amino acid sequence of protein or SNP identifier with the nsSNP. The output levels of probably damaging and possibly damaging were classified as functionally significant (≤ 0.5) and the benign level being classified as tolerated (≥ 0.5) (Ramensky et al., 2002). I-Mutant 3.0 performed analyses based on the protein sequence combined with mutational position and correlated new residue and the output result of the predicted free energy change (DDG) classifies the prediction into one of three classes: largely unstable (DDG < − 0.5 kcal/mol), largely stable (DDG > − 0.5 kcal/mol), or neutral (− 0.5 ≤ DDG ≥ 0.5 kcal/mol) (Capriotti et al., 2008). PROVEAN is able to provide predictions for any type of protein sequence variations including amino acid substitutions, and in-frame insertions and deletions (Choi et al., 2012). The PROVEAN predict a protein variant to be neutral if the score is above the threshold. The cutoff score − 2.5 indicates a deleterious substitution (Manickam et al., 2014).
Furthermore, we used SNP&GO, PHD-SNP Predictor of human deleterious single nucleotide polymorphisms and PANTHER (Protein Analysis Through Evolutionary Relationships) tools to filter the disease-associated nsSNPs (Table 1).
SNP&GO predict SNPs are or are not disease-associated with including the protein FASTA sequence and Gene Ontology terms. The probability score higher than 0.5 indicates the disease related effect of mutation on the parent protein function (Calabrese et al., 2009). PhD-SNP predicts whether the given amino acid substitution leads to disease associated or neutral along with the reliability index score (Capriotti et al., 2006). PANTHER comprehensive software system predicts the likelihood of a particular nsSNP to cause a functional impact on a protein. The cutoff subPSEC score − 3 indicates a deleterious substitution (Thomas et al., 2003).
2.3. Biophysical validation of nsSNPs
NetSurfP predicts the surface and, solvent accessibility of amino acids, using the amino acid FASTA sequence. The solvent accessibility has been predicted in two classes as either buried or exposed, based on the accessibility of the amino acid residues to the solvent, respectively. The reliability of this prediction method is in the form of Z-score. The Z-score highlights the surface prediction reliability, but is not associated with the secondary structure (Petersen et al., 2009).
Finding 3D structure of proteins is helpful in predicting the impact of SNPs on the structural level and in showing the degrees of alteration. I-TASSER generates a full length model of proteins by excising continuous fragments from threading alignments and then reassembling them using replica-exchanged Monte Carlo simulations. Low temperature replicas (decoys) generated during the simulation are clustered by SPICKER and the top five cluster centroids are selected for generating full atomic models. The quality of prediction models was reflected in the form of c-scores (− 5 to 2) (Roy et al., 2010, Roy et al., 2012). The native structure was mutated with the most deleterious amino acid substitution predicted in this study, using Swiss PDB viewer and Chimera (Kaplan and Littlejohn, 2001, Pettersen et al., 2004).
In addition, we used HOPE Project that provides the 3D structural visualization of mutated proteins, and gives the results by using UniProtKB and predictions from DAS-servers. FASTA sequence of whole protein and selection of mutant variants is considered to be an input option, the output is based on the structural variation between the mutant and the wild-type residues (Venselaar et al., 2010).
2.4. Predictions of protein-protein interactions
STRING (Search Tool for the Retrieval of Interacting proteins) is a database and web resource dedicated to protein–protein interactions, including direct (physical) and indirect (functional) interactions (Jensen et al., 2009); the database contains information from: genomic context, experimental repositories, co-expression and public text collections (Szklarczyk et al., 2011).
2.5. Functional SNPs in UTR found by the UTRscan
The UTRscan program allows one to search the user-submitted sequences for any of the patterns collected in the UTR site (Grillo et al., 2010). If different sequences for each UTR SNP are found to have different functional patterns, that the particular UTR SNP is predicted to have functional significance.
2.6. PolymiRTS database (version 3.0) for polymorphism in microRNA target site
PolymiRTS database was designed specifically for the analysis of non-coding SNPs namely 3′ UTR. The polymorphic microRNA target sites are classified into four classes according to Table 1 (Bhattacharya et al., 2013). PolymiRTS of ‘D’ may cause loss of normal repression; PolymiRTS of class ‘C’ may cause abnormal gene repression control. Therefore, these two classes of PolymiRTS are most likely to have functional impacts.
2.7. 1000 Genomes Project
The 1000 Genomes Project (Consortium, 2010) is sequencing the entire genome of approximately 2500 individuals from different worldwide populations. The aim of the1000 Genomes Project is to determine most of the genetic variation that occurs at a population frequency greater than 1%.
3. Results
3.1. SNP dataset from dbSNP
The BUB1B gene investigated in this work was retrieved from dbSNP database (Table 4). It contained a total of 827 SNPs: 90 were non-synonymous SNPs (nsSNPs), 57 were in non-coding regions, which comprises of 21 SNPs in 5′ UTR region and 36 SNPs in 3′ UTR region. The rest were in the intron region. We selected non-synonymous coding SNPs, 5′ and 3′ UTR region SNPs for our investigation.
Table 4.
Surface accessibility of wild-type and mutant variants in BUB1B.
| Amino acid | Class assignment | Position | Relative surface accessibility | Absolute surface accessibility | Z-fit score for RSA prediction |
|---|---|---|---|---|---|
| I | Buried | 82 | 0.02 | 3.829 | 1.418 |
| N | Buried | 0.022 | 3.177 | 1.383 | |
| P | Exposed | 334 | 0.546 | 77.520 | − 1.915 |
| L | 0.349 | 63.975 | − 0.779 | ||
| R | Exposed | 814 | 0.436 | 99.798 | − 0.340 |
| H | Exposed | 0.423 | 76.998 | − 0.280 |
3.2. Prediction of functional mutations
Of the 90 nsSNPs used in our analysis, 18 nsSNPs were identified to be deleterious with SIFT and the results were listed in Table 2.
Table 2.
List of nsSNP analysis by SIFT, PolyPhen-2, I-Mutant 3.0 PROVEAN respectively.
| rsID | Amino acid change | SIFT | Score | PolyPhen-2 | Score | I-Mutant 3.0 | Score | PROVEAN | Score |
|---|---|---|---|---|---|---|---|---|---|
| rs38678332 | G37V | Tolerated | 0.17 | Benign | 0.181 | Large decrease | − 0.46 | Neutral | − 1.187 |
| rs52798733 | E641V | Damaging | 0.03 | Probably damaging | 0.981 | Large decrease | − 0.04 | Neutral | − 2.256 |
| rs53178613 | R256K | Tolerated | 0.35 | Probably damaging | 0.992 | Large decrease | − 0.52 | Neutral | − 0.978 |
| rs53231959 | K170E | Damaging | 0.05 | Possibly damaging | 0.897 | Neutral | − 0.53 | Deleterious | − 3.155 |
| rs53396744 | I272N | Damaging | 0.02 | Probably damaging | 0.980 | Large decrease | − 1.75 | Deleterious | − 3.292 |
| rs53429711 | R36Q | Damaging | 0 | Probably damaging | 1.000 | Neutral | − 0.65 | Deleterious | − 3.718 |
| rs54188126 | I625M | Tolerated | 0.11 | Probably damaging | 0.980 | Large decrease | − 1.71 | Neutral | − 0.454 |
| rs54578440 | A348V | Tolerated | 0.25 | Probably damaging | 1.000 | Large decrease | 0.01 | Neutral | − 1.909 |
| rs54653854 | I156V | Tolerated | 0.93 | Probably damaging | 0.966 | Large decrease | − 1.01 | Neutral | − 0.489 |
| rs54660763 | H836Y | Tolerated | 1 | Benign | 0.004 | Neutral | 0.44 | Neutral | − 2.169 |
| rs54865001 | M353T | Tolerated | 0.48 | Probably damaging | 0.992 | Large decrease | − 0.62 | Neutral | − 2.259 |
| rs55238070 | A173V | Tolerated | 0.27 | Benign | 0.034 | Large decrease | − 0.45 | Neutral | − 0.601 |
| rs55342059 | K539Q | Tolerated | 0.24 | Possibly damaging | 0.682 | Large decrease | − 0.79 | Neutral | − 0.111 |
| rs55355571 | I82N | Damaging | 0 | Probably damaging | 1.000 | Large decrease | − 2.12 | Deleterious | − 6.284 |
| rs55478232 | G8A | Tolerated | 0.09 | Benign | 0.018 | Large increase | − 0.11 | Neutral | − 0.595 |
| rs55619315 | R616H | Tolerated | 0.52 | Benign | 0.003 | Large decrease | − 1.29 | Neutral | 1.972 |
| rs55752197 | R677H | Tolerated | 0.12 | Benign | 0.002 | Large decrease | − 1.39 | Neutral | − 0.882 |
| rs55935830 | D576E | Tolerated | 1 | Benign | 0.003 | Neutral | − 0.14 | Neutral | 0.056 |
| rs56791614 | R194Q | Tolerated | 0.2 | Probably damaging | 1.000 | Neutral | − 0. 45 | Neutral | − 0.742 |
| rs57105655 | T100M | Tolerated | 0.06 | Probably damaging | 0.998 | Large increase | − 0.16 | Neutral | − 1.801 |
| rs57153880 | G316D | Tolerated | 0.37 | Benign | 0.024 | Large decrease | − 0.76 | Deleterious | − 3.017 |
| rs57759191 | R727C | Tolerated | 0.09 | Probably damaging | 1.000 | Large decrease | − 0.69 | Neutral | − 2.061 |
| rs28989181 | L844F | Damaging | 0 | Probably damaging | 0.998 | Large decrease | − 0.82 | Neutral | 2.454 |
| rs28989182 | R814H | Damaging | 0 | Probably damaging | 1.000 | Large decrease | − 1.43 | Deleterious | − 2.880 |
| rs28989187 | R550Q | Tolerated | 0.86 | Benign | 0.001 | Large decrease | − 0.78 | Neutral | 0.332 |
| rs56079734 | T40M | Damaging | 0.05 | Probably damaging | 1.000 | Neutral | 0.01 | Neutral | − 1.811 |
| rs1017842 | E390D | Tolerated | 0.46 | Benign | 0.102 | Large decrease | − 0.31 | Neutral | − 0.530 |
| rs1801528 | V618A | Tolerated | 1 | Benign | 0.000 | Large decrease | − 1.43 | Neutral | 1.441 |
| rs17851677 | P378S | Damaging | 0.04 | Possibly damaging | 0.804 | Large decrease | − 1.10 | Deleterious | − 3.228 |
| rs28989188 | E409D | Tolerated | 0.24 | Probably damaging | 1.000 | Large decrease | − 0.49 | Neutral | − 1.355 |
| rs35923791 | N133S | Tolerated | 1 | Benign | 0.248 | Large decrease | − 0.35 | Neutral | − 0.664 |
| rs56158360 | R244H | Damaging | 0.02 | Probably damaging | 1.000 | Large decrease | − 1.08 | Deleterious | − 4.461 |
| rs75763304 | Q460K | Tolerated | 0.72 | Benign | 0.072 | Neutral | 0.13 | Neutral | − 1.236 |
| rs76546181 | F531S | Tolerated | 0.56 | Probably damaging | 1.000 | Large decrease | − 1.68 | Neutral | − 2.074 |
| rs77520855 | Y162H | Damaging | 0.04 | Probably damaging | 0.960 | Large decrease | − 1.09 | Neutral | − 2.414 |
| rs117485407 | T471M | Tolerated | 0.1 | Possibly damaging | 0.579 | Neutral | 0.32 | Neutral | − 0.615 |
| rs138332995 | P544S | Tolerated | 0.74 | Benign | 0.181 | Large decrease | − 1.59 | Neutral | − 1.109 |
| rs139226455 | P800S | Tolerated | 0.07 | Possibly damaging | 0.839 | Large decrease | − 1.85 | Deleterious | − 3.780 |
| rs140368608 | K779R | Tolerated | 0.64 | Benign | 0.073 | Neutral | − 0.30 | Neutral | − 1.176 |
| rs141953425 | P334L | Damaging | 0.01 | Possibly damaging | 0.453 | Large decrease | − 0.80 | Deleterious | − 5.244 |
| rs142705245 | A784V | Tolerated | 0.38 | Benign | 0.019 | Large decrease | − 0.20 | Neutral | 0.604 |
| rs143346774 | H850R | Tolerated | 0.61 | Probably damaging | 0.998 | Neutral | 0.16 | Neutral | − 1.165 |
| rs143559902 | D675E | Tolerated | 1 | Benign | 0.003 | Neutral | − 0.37 | Neutral | − 0.008 |
| rs145026343 | C825F | Tolerated | 0.35 | Probably damaging | 0.966 | Large decrease | − 0.13 | Deleterious | − 4.601 |
| rs145028054 | E184Q | Damaging | 0.01 | Benign | 0.362 | Neutral | 0.07 | Neutral | − 2.086 |
| rs145184714 | A335T | Tolerated | 0.84 | Benign | 0.005 | Large decrease | − 0.78 | Neutral | 0.308 |
| rs145578529 | I567V | Tolerated | 0.3 | Possibly damaging | 0.512 | Large decrease | − 1.10 | Neutral | − 0.081 |
| rs146387899 | L258F | Tolerated | 0.81 | Benign | 0.074 | Large decrease | − 0.64 | Neutral | − 1.786 |
| rs146795655 | T493I | Tolerated | 0.16 | Benign | 0.001 | Neutral | − 0.26 | Neutral | − 0.913 |
| rs146821149 | R886S | Damaging | 0.03 | Benign | 0.002 | Large decrease | − 1.25 | Neutral | − 0.328 |
| rs147150527 | G376V | Tolerated | 0.17 | Benign | 0.181 | Large decrease | − 0.46 | Neutral | − 1.187 |
| rs147549987 | V4M | Tolerated | 0.23 | Benign | 0.000 | Neutral | − 0.68 | Neutral | − 0.282 |
| rs147832586 | S83G | Damaging | 0.01 | Benign | 0.061 | Large decrease | − 0.86 | Neutral | − 1.807 |
| rs148159407 | N26D | Tolerated | 0.52 | Possibly damaging | 0.913 | Neutral | − 0.43 | Deleterious | − 2.827 |
| rs148348158 | T648I | Tolerated | 0.19 | Benign | 0.000 | Large decrease | 0.25 | Neutral | − 1.427 |
| rs149628229 | D579G | Tolerated | 0.1 | Benign | 0.328 | Large decrease | − 1.20 | Neutral | − 1.894 |
| rs149955447 | E813A | Tolerated | 0.07 | Probably damaging | 0.997 | Large decrease | − 0.69 | Neutral | − 2.258 |
| rs150707631 | S797A | Tolerated | 0.13 | Possibly damaging | 0.495 | Large decrease | − 0.79 | Neutral | − 1.043 |
| rs150983783 | R421Q | Tolerated | 0.15 | Benign | 0.178 | Neutral | − 0.79 | Neutral | − 1.197 |
| rs181352808 | H836Q | Tolerated | 0.29 | Probably damaging | 0.959 | Neutral | − 0.22 | Deleterious | − 2.528 |
| rs184449375 | M626V | Tolerated | 0.61 | Benign | 0.000 | Large decrease | − 0.84 | Neutral | 0.072 |
| rs190909040 | Y343F | Tolerated | 0.14 | Possibly damaging | 0.925 | Large decrease | − 0.13 | Neutral | − 1.814 |
| rs199509124 | P222L | Damaging | 0.01 | Possibly damaging | 0.774 | Large increase | − 0.20 | Deleterious | − 4.125 |
| rs199743655 | V274A | Damaging | 0 | Possibly damaging | 0.866 | Large decrease | − 1.00 | Deleterious | − 3.319 |
| rs200060772 | S691L | Tolerated | 0.08 | Probably damaging | 0.999 | Neutral | − 0.32 | Neutral | − 2.173 |
| rs200788206 | Q350K | Tolerated | 0.79 | Probably damaging | 0.984 | Large decrease | 0.10 | Neutral | − 1.593 |
| rs200997833 | K542R | Tolerated | 0.35 | Benign | 0.055 | Neutral | − 0.28 | Neutral | − 0.375 |
| rs201251790 | R421W | Tolerated | 0.09 | Benign | 0.021 | Neutral | − 0.38 | Neutral | − 1.629 |
| rs201360106 | E21K | Tolerated | 0.09 | Probably damaging | 1.000 | Large decrease | − 0.63 | Deleterious | − 2.504 |
| rs202114756 | S384G | Tolerated | 0.31 | Possibly damaging | 0.860 | Large decrease | − 0.57 | Neutral | − 1.503 |
| rs202132335 | A739G | Damaging | 0.0 | Benign | 0.072 | Large decrease | − 1.08 | Neutral | − 1.113 |
| rs367543489 | Q829E | Tolerated | 0.11 | Probably damaging | 0.999 | Neutral | − 0.10 | Neutral | − 1.197 |
| rs368023159 | K488N | Tolerated | 0.15 | Benign | 0.004 | Neutral | − 0.60 | Neutral | − 0.119 |
| rs368079817 | Q42R | Tolerated | 0.25 | Probably damaging | 0.999 | Neutral | − 0.06 | Neutral | − 1.985 |
| rs368996088 | F781L | Tolerated | 0.16 | Benign | 0.075 | Large decrease | − 1.32 | Neutral | − 1.964 |
| rs370388424 | P640L | Tolerated | 0.29 | Benign | 0.020 | Large decrease | − 0.60 | Neutral | − 0.985 |
| rs370655726 | C356S | Tolerated | 0.74 | Probably damaging | 1.000 | Large decrease | − 0.69 | Deleterious | − 3.683 |
| rs371124423 | C700R | Tolerated | 0.38 | Benign | 0.068 | Large decrease | − 0.26 | Neutral | − 1.228 |
| rs371305662 | T291K | Tolerated | 0.84 | Benign | 0.100 | Large decrease | − 0.46 | Neutral | − 1.934 |
| rs372003254 | D846E | Tolerated | 0.1 | Possibly damaging | 0.626 | Neutral | 0.24 | Neutral | − 1.695 |
| rs372569297 | I755T | Tolerated | 0.75 | Benign | 0.000 | Large decrease | − 1.59 | Neutral | − 0.814 |
| rs373256667 | K454R | Tolerated | 0.21 | Probably damaging | 1.000 | Neutral | 0.12 | Neutral | − 0.935 |
| rs373789523 | T658I | Tolerated | 0.16 | Benign | 0.000 | Large decrease | 0.25 | Neutral | − 1.951 |
| rs373830262 | A108T | Tolerated | 0.49 | Probably damaging | 0.982 | Large decrease | − 0.71 | Neutral | − 0.949 |
| rs374682772 | V333I | Tolerated | 0.35 | Benign | 0.002 | Large decrease | − 0.84 | Neutral | − 0.423 |
| rs375105548 | I854V | Tolerated | 0.44 | Benign | 0.007 | Large decrease | − 0.65 | Neutral | − 0.137 |
| rs375388175 | I703T | Tolerated | 0.76 | Probably damaging | 0.985 | Large decrease | − 1.94 | Neutral | − 1.952 |
| rs375798678 | Q181R | Tolerated | 1 | Benign | 0.000 | Neutral | 0.06 | Neutral | 0.416 |
| rs375885859 | C51R | Tolerated | 0.11 | Benign | 0.000 | Neutral | 0.07 | Neutral | − 1.379 |
| rs376072541 | P632L | Tolerated | 0.1 | Benign | 0.001 | Large decrease | − 0.40 | Neutral | − 2.406 |
A total of 46 nsSNPs was predicted to be damaging and the remaining 44 nsSNPs were categorized as benign with Polyphen 2.0 and the results were listed inTable 2. Out of 90 nsSNPs, 27 nsSNP were predicted to be neutral mutation (− 0.5 ≤ DDG ≤ 0.5 kcal/mol), 60 nsSNP were predicted to be “large decrease” (≤−0.5 kcal/mol) and 3 nsSNP were predicted to be “large increase” (> 0.5 kcal/mol). I-Mutant 3.0 predicted 63 of SNPs to affect the stability of protein structure (Table 2). All the nsSNPs submitted to SIFT and PolyPhen 2.0 and I-Mutant 3.0 were submitted as input to PROVEAN. Out of 90 nsSNPs, 17 nsSNP were predicted to be deleterious and 73 were found to be neutral (Table 2).
The accuracy of the in silico techniques for prioritizing deleterious SNPs can be increased by combining different computational methods. Out of 90 nsSNPs, SIFT, PolyPhen, I-Mutant 3.0 and PROVEAN predicted 8 nsSNPs as deleterious (Fig. 2).
Fig. 2.
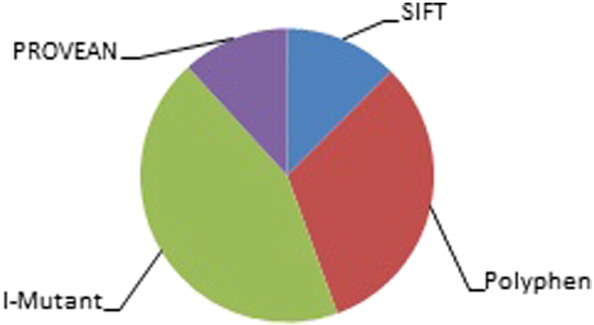
List of functionally significant mutations.
SNPs&GO, PhD-SNP and PANTHER were performed to validate the results obtained from four tools. Out of 8 nsSNPs that predicted to be deleterious with SIFT, Polyphen, I-Mutant and PROVEAN; SNP&GO predicted 3 nsSNP, PhD-SNP predicted 4 nsSNP and PANTHER predicted 5 nsSNP to be associated with disease (Table 3).
Table 3.
List of nsSNP predicted as disease associated byPHD-SNP,SNP&GO and PANTHER server.
| rsID | Amino acid change | PHD-SNP | SNP&GO | PANTHER | subPSEC score |
|---|---|---|---|---|---|
| rs527987333 | I82N | Disease | Disease | Deleterious | − 5.89613 |
| rs53396744 | I272N | Disease | Neutral | Tolerated | − 2.75316 |
| rs199743655 | V274A | Neutral | Neutral | Tolerated | − 2.50206 |
| rs199509124 | P222L | Neutral | Neutral | Deleterious | − 3.07979 |
| rs141953425 | P334L | Disease | Disease | Deleterious | − 3.73233 |
| rs56158360 | R244H | Neutral | Neutral | Deleterious | − 4.34981 |
| rs17851677 | P378S | Neutral | Neutral | Deleterious | − 3.97828 |
| rs28989182 | R814H | Disease | Disease | Deleterious | − 7.86508 |
Finally out of 90 nsSNP, we found 3 nsSNPs namely rs55355571 (I82N), rs141953425 (P334L) and rs28989182 (R814H) that are common in all (SIFT, Polyphen, I-Mutant, PROVEAN, PHD-SNP, SNP&GO, PANTHER) prediction.
3.3. In silico biophysical validation of nsSNPs
Based on the in silico analyses performed, 3 nsSNPs was selected for further analyses. The location and the type of a mutated residue affect the stability of the protein. In particular, as the solvent accessibility of a residue decreases, the stability of the protein due to mutation decreases. NetSurfP Z-score allows the identification of the most reliable/unreliable predictions for both buried and exposed amino acids. A huge drift in the Z-score was not observed for 3 nsSNPs as given in Table 5. For any of 3 nsSNPs, the class assignment does not change.
Table 5.
Top 10 templates used by I-TASSER to create the high quality models for human BUB1B secondary structure.
| Rank | PDB hit | Iden1 | Iden2 | Cov. | Norm. Z-score |
|---|---|---|---|---|---|
| 1 | 3e7eA | 0.22 | 0.08 | 0.31 | 1.71 |
| 2 | 1vw1A | 0.08 | 0.20 | 0.87 | 2.36 |
| 3 | 3e7eA | 0.22 | 0.08 | 0.31 | 1.40 |
| 4 | 4jspB | 0.09 | 0.19 | 0.92 | 1.44 |
| 5 | 3e7eA | 0.23 | 0.08 | 0.30 | 2.68 |
| 6 | 3cm9S | 0.07 | 0.11 | 0.55 | 1.19 |
| 7 | 3e7eA | 0.22 | 0.08 | 0.31 | 2.84 |
| 8 | 4jspB | 0.08 | 0.19 | 0.89 | 2.06 |
| 9 | 3e7eA | 0.22 | 0.08 | 0.31 | 1.51 |
| 10 | 4kf7A | 0.10 | 0.19 | 0.85 | 2.31 |
The I-TASSER tool created the 5 full-length models for BubR1 protein (with C-scores: − 0.24, − 1.01, − 2.83, − 2.95 and − 3.00) by excising top 10 structures with C-scores after targeting the PDB library hits (Table 5).Top 10 proteins in the PDB which are structurally closest to the predicted models. Among the 5 predicted models, model 1 (Fig. 3) carried the high-quality confidence in the form of C-score (− 0.24), TM-score (0.68 ± 0.12), and the RMSD (9.6 ± 4.6 Å) (Table 6). We did not perform any molecular dynamics structure optimization; therefore, our 3D homology model is a preliminary model implicating the disruptive role of the SNPs.
Fig. 3.
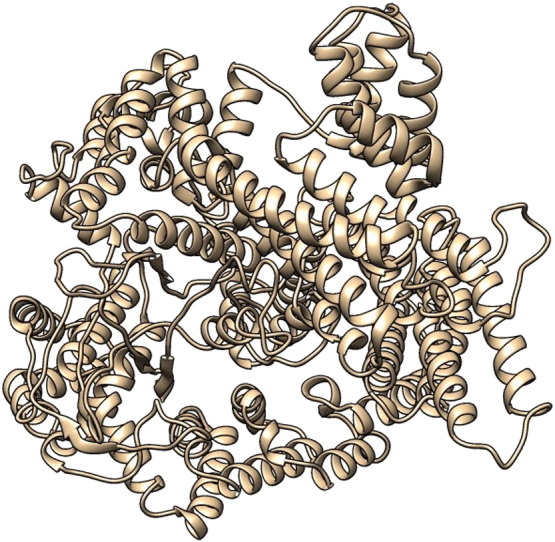
3D structure of BubR1 predicted with I-TASSER.
Table 6.
I-TASSER results carrying C-score, TM-score and RMSD regarding selected secondary structure (native protein model 1).
| Model | C-score | Exp. TM-score | Exp. RMSD | No. of decoys | Cluster density |
|---|---|---|---|---|---|
| Model 1 | − 0.24 | 0.68 ± 0.12 | 9.6 ± 4.6 Å | 312 | 0.3039 |
| Model2 | − 1.01 | 258 | 0.1404 | ||
| Model3 | − 2.83 | 60 | 0.0228 | ||
| Model4 | − 2.95 | 57 | 0.0202 | ||
| Model5 | − 3.00 | 57 | 0.0191 |
Project Hope revealed the 3D structure of the proteins with its new residue. Furthermore, it described the reaction and physiochemical properties of these candidates. Here we present the results upon each candidate and discuss the conformational variations and interactions with the neighboring amino acids:
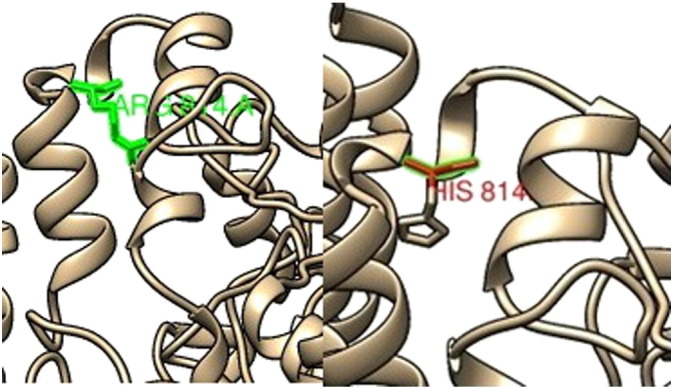
A/G Mutation (rs28989182) caused substitution of the amino acid from Arginine into a Histidine at position 814 (R814H). For this variant the mutated residue is smaller (Fig. 1); this might lead to loss of interactions. The wild-type residue was positively charged, the mutated residue is neutral. Only this residue type was found at this position. Mutation of a 100% conserved residue is usually damaging for the protein. Additionally, the structural analysis of H814 showed some clashes for Phe822 which may contribute to the extra energy in the protein structure, and hence the decrease in stability (Fig. 4).
Fig. 4.
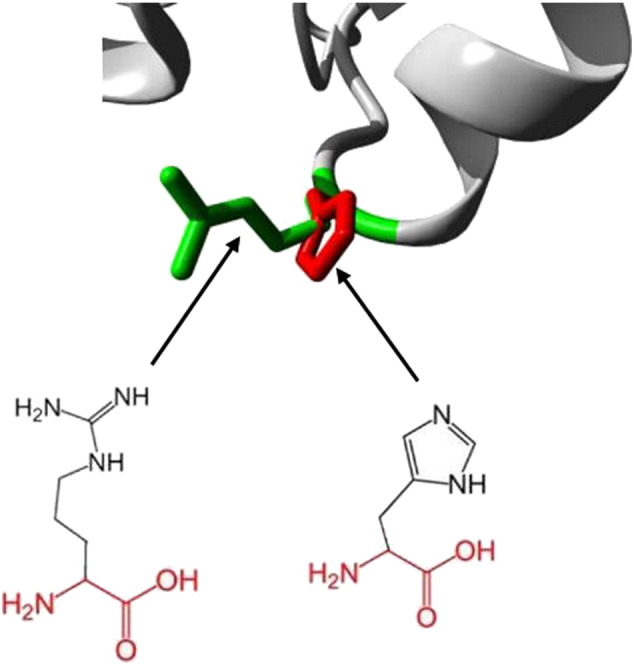
Deep view of superimposed structure of wild and mutant residue at 814 position. The main protein core is shown in gray color while the wild type and mutated residues are shown in green and red colors respectively. SNP ID: rs28989182, protein position 814 changed from Arginine to Histidine.
A/T mutation (rs553555716) resulted in a change of the Isoleucine to Aspargine at position 82 (I82N). The wild type residue is smaller and more hydrophobic than the mutated residue (Fig. 6). The residue is buried in the core of a domain, annotated in UniProt as: “BUB1 N terminal”. The mutation will cause loss of hydrophobic interactions in the core of the protein. This residue is part of an interprotein domain named “Mitotic checkpoint serine/threonine protein kinase Bub1/Mitotic spindle checkpoint component Mad3” (IPR015661).
Fig. 6.
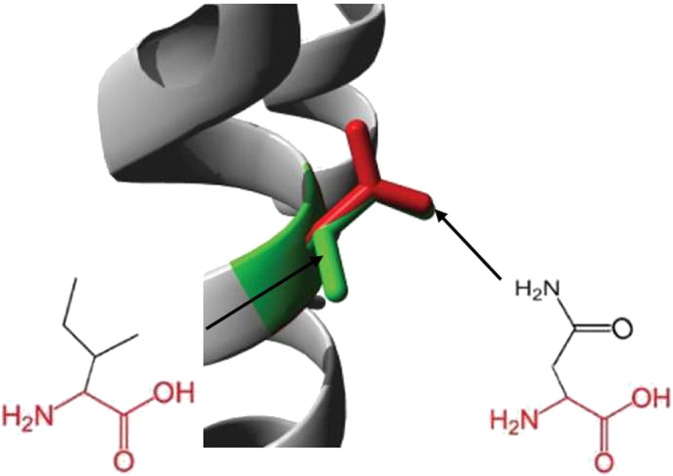
Superimposed structure of (wild type) Ile and (mutant) Asn residues at 82 position.
C/T (rs141953425) leads to conversion of Proline into a Leucine at position 334 (P334L). The wild-type and mutant amino acid differs in sizes; the mutated residue is bigger and this might lead to displace of the mutant residue. Prolines are known to have a very rigid structure, sometimes forcing the backbone in a specific conformation. The mutation can disturb this special conformation. This residue is part of an inter protein domain named “Mitotic checkpoint serine/threonine protein kinase Bub1/Mitotic spindle checkpoint component Mad3”.
Chimera (Fig. 5, Fig. 7, Fig. 8) and Swiss PDB viewer were used to visualize the structural features of amino acids in native and mutant protein chains. During structural visualization for all 3mutations, only mutant residue (Histidine) at 814 position showed a network of clashes with Phe822 (Fig. 9).
Fig. 5.
SNP ID: rs28989182, protein position 814 changed from Arginine (green) to Histidine (red).
Fig. 7.
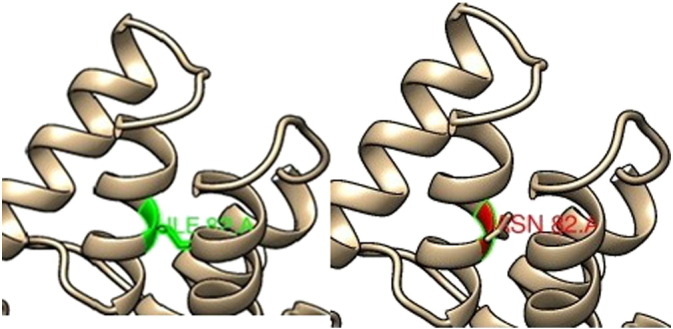
SNP ID: rs553555716, protein position 82 changed from Isoleucine (left image) to Aspargine (right image).
Fig. 8.
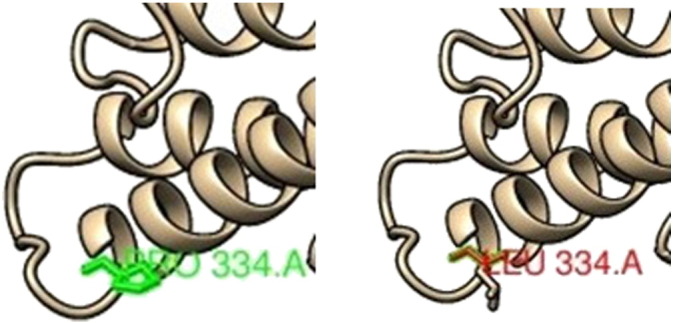
SNP ID: rs141953425, protein position 334 changed from Proline (left image) to Leucine (right image).
Fig. 9.

H-bonding (green discontinuous line) interactions and clashes (pink discontinuous line) of wild type and mutant analogues with the vicinal amino acid residues. (a) At 814 position 2 H-bond is observed with Leu811and Glu817 in both native (Arg) and mutant (His814) structures, but a network of clashes appeared between His814and Phe822. (b) At 82 position, 4 H-bond is observed with Trp78, Asp79, Thr85 and Glu86 in both native (Ile) and mutant (Asn) structures. (c) At 334 position, 2 H-bond is observed with Leu330 and Pro338 in both native (Pro) and mutant (Leu) structures.
3.4. Protein-protein interactions analysis
The interaction analysis revealed that BUB1B is related to Cell Division Cycle 20 homolog (CDC20), Budding Uninhibited By Benzimidazoles 3 (BUB3), Cancer Susceptibility Candidate 5 (CASC5), MAD2 Mitotic Arrest Deficient-Like 1 (MAD2L1), Cell Division Cycle 27 homolog (CDC27), Centromere Protein E (CENPE), BUB1 Mitotic Checkpoint Serine/Threonine Kinase (BUB1), ZW10 Interacting Kinetochore Protein (ZWINT), Anaphase Promoting Complex Subunit 2 (ANAPC2), Cell Division Cycle 16 homolog (CDC16) (Fig. 10). Furthermore, our literature search demonstrated that BubR1 interacts with Bub3, Cdc20, and Mad2 (Kapanidou et al., 2015).
Fig. 10.
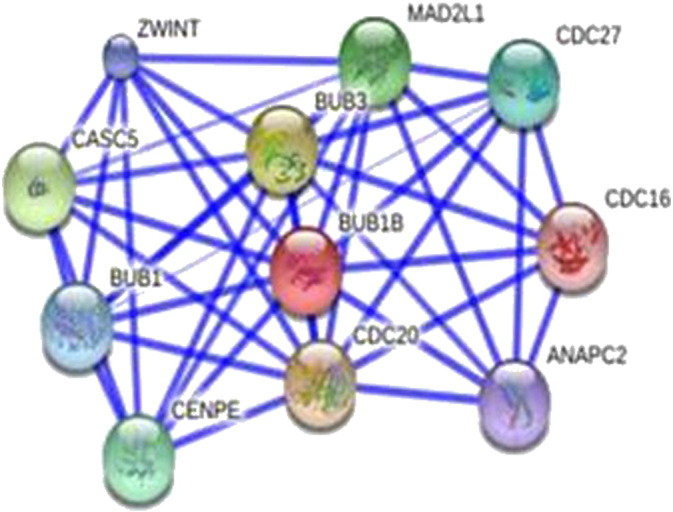
Protein–protein interaction network of BUB1B using STRING 9.0 server.
3.5. Functional SNPs in UTR found by UTRscan server
All of the 57 UTR SNPs was analyzed using UTRscan. After comparing the functional elements for each UTR SNP, we predicted that two SNPs, namely rs375434078 and rs538302864 in 5′ UTR are related to the functional pattern change of IRES (Table 7). Internal ribosome entry site (IRES) is bound by an internal mRNA ribosome that is an alternative mechanism of translation initiation compared to the common 5′-cap dependent ribosome scanning mechanism (Pickering and Willis, 2005).
Table 7.
List of mRNA UTR SNPs that were predicted to be of functional significance by UTRscan server.
| SNP ID | Nucleotide change | UTR position | Functional element change |
|---|---|---|---|
| rs375434078 | C/T | 5′ UTR | IRES → no pattern |
| rs538302864 | A/G | 5′ UTR | IRES → no pattern |
3.6. Functional SNPs in 3′ untranslated regions (UTR) predicted by PolymiRTS database 3.0
Among 36 SNPs in 3′ UTR region of BUB1B gene, 3 functional SNPs was predicted that among them, only one SNP disrupts 8 miRNAs conserved site (ancestral allele with support ≥ 2), while all of them create 8 new miRNA site. The results are listed in Table 8.
Table 8.
Prediction result of PolymiRTS database.
| dbSNP ID | miR ID | Conservation | miRSite | Function class |
|---|---|---|---|---|
| rs149437374 | hsa-miR 130a -3p | 2 | ATGCACTAccatt | D |
| hsa-miR-130b-3p | ||||
| hsa-miR-301a-3p | 2 | ATGCACTAccatt | D | |
| hsa-miR-301b | ||||
| hsa-miR-3666 | 2 | ATGCACTAccatt | D | |
| hsa-miR-4295 | ||||
| hsa-miR-454-3p | 2 | ATGCACTAccatt | D | |
| hsa-miR-4671-3p | 2 | ATGCACTAccatt | D | |
| hsa-miR-323a-5p | 2 | ATGCACTAccatt | D | |
| hsa-miR-876-3p | 2 | ATGCACTAccatt | D | |
| 2 | ATGCACTAccatt | D | ||
| 5 | atgcACCACCAtt | C | ||
| 5 | atgcACCACCAtt | C | ||
| rs143807849 | hsa-miR-539-5p | 4 | CcATTTCTCtcta | C |
| hsa-miR-5680 | ||||
| hsa-miR-6758-5p | 5 | CCATTTCTctcta | C | |
| hsa-miR-6856-5p | 4 | CcatttCTCTCTA | C | |
| 4 | CcatttCTCTCTA | C | ||
| rs1047193 | hsa-miR-4450 | 4 | atgATCCCCAtgt | C |
| hsa-miR-6857-5p | 4 | atgATCCCCAtgt | C |
4. Discussion
The identification of SNPs responsible for specific phenotypes with molecular approaches seems to be expensive and time-consuming (Chen and Sullivan, 2003), hence computational approaches can help in narrowing down the number of missense mutations to be screened in genetic association studies and for a better understanding of the functional and structural aspects of the parent protein.
Previous studies on polymorphisms screening using in silico analysis helped in predicting the functional nsSNPs associated with genes such as G6PD (Rajith, 2011), ATM (Doss and Rajith, 2012), PTEN (Doss and Rajith, 2013), BRAF (Hussain et al., 2012). Our results also revealed that implementations of different algorithms often serve as powerful tools for prioritizing candidate functional nsSNPs. Recent work by Thusberg and Vihinen (2009) compared different in silico tools, out of which SIFT and PolyPhen were reported to have better performance in identifying deleterious nsSNPs. The accuracy of SIFT and PolyPhen 2.0 was further validated by Hicks et al. (2011), which makes these tools more applicable for the prediction. I-Mutant 3.0 was used which evaluate the stability change upon single amino acid mutation that ranked as one of the most reliable predictor based on the work performed by Khan and Vihinen (2010).
Based on these in silico studies, we select SIFT, PolyPhen, I-Mutant, PROVEAN, SNP&GO, PHD-SNP and PANTHER for the screening of functional mutation in BUB1B gene. By comparing the scores of all 7 methods, 3 nsSNPs with positions I82N, P334L and R814H were found to be highly significant.
The 5′ and 3′ UTR SNPs was analyzed using UTRscan. Due to the importance of the translational regulation of microRNAs, we further studied whether the 3′ UTR SNPs changes the profile of microRNA binding to the BUB1B gene using PolymiRTS. Two SNPs in the 5′ UTR was predicted to influence the translation pattern of the BUB1B gene through UTRscan analysis, and three 3′ UTR SNPs may affect microRNA binding sites, as determined through PolymiRTS. Protein-protein interaction analysis showed the interaction of BUB1B with ten different genes. Therefore, any changes in the protein function would have an impact on many pathways involved in disease.
In conclusion, we surveyed and compared available databases such as NCBI, dbSNP, 1000 genome project along with in silico prediction programs to assess the effects of deleterious functional variants on the protein functions. Analyzing deleterious nsSNPs by both sequence and structure level has the added advantage of being able to assess the reliability of the generated prediction results by cross-referencing the results from both approaches. One striking observation was the identification of rs28989182 (R814H), that associated with Mosaic Variegated Aneuploidy Syndrome (Bairoch and Apweiler, 1996), lie within a serine/threonine kinase domain of BubR1 protein. Only this residue type was found at this position. Mutation of a 100% conserved residue is usually damaging for the protein. Both I82N and P334L mutations occurred in the N terminal region of BubR1; Therefore, these mutations may compromise its binding to Bub1, Mad2 and cdc20 resulting plausible failure of the corresponding checkpoint. In addition rs149437374 and rs143807849 in 3′ UTR that disrupts a conserved of 8 miRNAs site are genotyped by 1000 genome project; Based on the data obtained through determining the allele frequency in 1000 genome populations, it is observed that the frequency of normal allele is more than the mutant allele. Therefore, it is concluded that rs149437374 and rs143807849 in 3′ UTR are deleterious, so that in different 1000 genome population, it has a low frequency; hence allele frequency reported in 1000 genome project confirmed our results. These results indicate that our approach successfully allowed us in selecting the deleterious SNPs that are likely to have functional impact on the BUB1B gene and contribute to an individual's susceptibility to the disease.
References
- Adzhubei I.A. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A., Apweiler R. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res. 1996;24(1):21–25. doi: 10.1093/nar/24.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A., Ziebarth J.D., Cui Y. PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2013:gkt1028. doi: 10.1093/nar/gkt1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009;30(8):1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- Capriotti E., Calabrese R., Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22(22):2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Capriotti E. A three-state prediction of single point mutations on protein stability changes. BMC Bioinf. 2008;9(Suppl. 2):S6. doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Sullivan P. Single nucleotide polymorphism genotyping: biochemistry, protocol, cost and throughput. Pharmacogenomics J. 2003;3(2):77–96. doi: 10.1038/sj.tpj.6500167. [DOI] [PubMed] [Google Scholar]
- Choi Y. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, G.P. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport J.W. The mouse mitotic checkpoint gene bub1b, a novel bub1 family member, is expressed in a cell cycle-dependent manner. Genomics. 1999;55(1):113–117. doi: 10.1006/geno.1998.5629. [DOI] [PubMed] [Google Scholar]
- Doss C.G.P., Rajith B. Computational refinement of functional single nucleotide polymorphisms associated with ATM gene. PLoS One. 2012;7(4):e34573. doi: 10.1371/journal.pone.0034573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doss C.G.P., Rajith B. A new insight into structural and functional impact of single-nucleotide polymorphisms in PTEN gene. Cell Biochem. Biophys. 2013;66(2):249–263. doi: 10.1007/s12013-012-9472-9. [DOI] [PubMed] [Google Scholar]
- Drazen J.M. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat. Genet. 1999;22(2):168–170. doi: 10.1038/9680. [DOI] [PubMed] [Google Scholar]
- Grillo G. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010;38(Suppl. 1):D75–D80. doi: 10.1093/nar/gkp902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S., Snape K., Rahman N. 2012. BUB1B (Budding Uninhibited by Benzimidazoles 1 Homolog Beta (Yeast)) [Google Scholar]
- Hanks S. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 2004;36(11):1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- Hanks S. Comparative genomic hybridization and BUB1B mutation analyses in childhood cancers associated with mosaic variegated aneuploidy syndrome. Cancer Lett. 2006;239(2):234–238. doi: 10.1016/j.canlet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hicks S. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011;32(6):661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M.R.M. In silico analysis of single nucleotide polymorphisms (SNPs) in human BRAF gene. Gene. 2012;508(2):188–196. doi: 10.1016/j.gene.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Jensen L.J. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Suppl. 1):D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisari S. Intermediates in the assembly of mitotic checkpoint complexes and their role in the regulation of the anaphase-promoting complex. Proc. Natl. Acad. Sci. 2016:201524551. doi: 10.1073/pnas.1524551113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidou M., Lee S., Bolanos-Garcia V.M. BubR1 kinase: protection against aneuploidy and premature aging. Trends Mol. Med. 2015 doi: 10.1016/j.molmed.2015.04.003. [DOI] [PubMed] [Google Scholar]
- Kaplan W., Littlejohn T.G. Swiss-PDB viewer (deep view) Brief. Bioinform. 2001;2(2):195–197. doi: 10.1093/bib/2.2.195. [DOI] [PubMed] [Google Scholar]
- Khan S., Vihinen M. Performance of protein stability predictors. Hum. Mutat. 2010;31(6):675–684. doi: 10.1002/humu.21242. [DOI] [PubMed] [Google Scholar]
- Kops G.J., Weaver B.A., Cleveland D.W. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 2005;5(10):773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Krawczak M. Human gene mutation database—a biomedical information and research resource. Hum. Mutat. 2000;15(1):45–51. doi: 10.1002/(SICI)1098-1004(200001)15:1<45::AID-HUMU10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Magesh R., Doss C.G.P. Computational pipeline to identify and characterize functional mutations in ornithine transcarbamylase deficiency. 3 Biotech. 2014;4(6):621–634. doi: 10.1007/s13205-014-0216-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam M. In silico identification of genetic variants in glucocerebrosidase (GBA) gene involved in Gaucher's disease using multiple software tools. Front. Genet. 2014;5 doi: 10.3389/fgene.2014.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am. J. Med. Genet. A. 2006;140(4):358–367. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- Mi H. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33(Suppl. 1):D284–D288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K. Mutation analysis of mitotic checkpoint genes (hBUB1 and hBUBR1) and microsatellite instability in adult T-cell leukemia/lymphoma. Cancer Lett. 2000;158(2):141–150. doi: 10.1016/s0304-3835(00)00512-7. [DOI] [PubMed] [Google Scholar]
- Pesole G. UTRdb: a specialized database of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 1999;27(1):188–191. doi: 10.1093/nar/27.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009;9(1):51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pickering B.M., Willis A.E. Seminars in Cell & Developmental Biology. Elsevier; 2005. The implications of structured 5′ untranslated regions on translation and disease. [DOI] [PubMed] [Google Scholar]
- Rajith B. Path to facilitate the prediction of functional amino acid substitutions in red blood cell disorders–a computational approach. PLoS One. 2011;6(9):e24607. doi: 10.1371/journal.pone.0024607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30(17):3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Yang J., Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012:gks372. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suijkerbuijk S.J. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 2010;70(12):4891–4900. doi: 10.1158/0008-5472.CAN-09-4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Suppl. 1):D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.D. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13(9):2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J., Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum. Mutat. 2009;30(5):703–714. doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
- Venselaar H. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinf. 2010;11(1):548. doi: 10.1186/1471-2105-11-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via M., Gignoux C., Burchard E.G. The 1000 Genomes Project: new opportunities for research and social challenges. Genome Med. 2010;2(3) doi: 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Mering C. STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33(Suppl. 1):D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008;9(1):40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]