

Abstract
Calcium, sodium and potassium channel blockers are widely prescribed medications for a variety of health problems, most frequently for cardiac arrhythmias, hypertension, angina pectoris and other disorders. However, chronic application of channel blockers is associated with numerous side effects, including worsening cardiac pathology. For example, nifedipine, a calcium-channel blocker was found to be associated with increased mortality and increased risk for myocardial infarction. In addition to the side effects mentioned above by different channel blockers, these drugs can cause arterial wall damage, thereby contributing to vascular wall structure destabilization and promoting events facilitating rupture of plaques. Collagen synthesis is regulated by ascorbic acid, which is also essential for its optimum structure as a cofactor in lysine and proline hydroxylation, a precondition for optimum crosslinking of collagen and elastin. Therefore, the main objective in this study was to evaluate effects of various types of channel blockers on intracellular accumulation and cellular functions of ascorbate, specifically in relation to formation and extracellular deposition of major collagen types relevant for vascular function. Effects of select Na- and Ca- channel blockers on collagen synthesis and deposition were evaluated in cultured human dermal fibroblasts and aortic smooth muscle cells by immunoassay. All channel blockers tested demonstrated inhibitory effects on collagen type I deposition to the ECM by fibroblasts, each to a different degree. Ascorbic acid significantly increased collagen I ECM deposition. Nifedipine (50 µM), a representative of channel blockers tested, significantly reduced ascorbic acid and ascorbyl palmitate-dependent ECM deposition of collagen type l and collagen type lV by cultured aortic smooth muscle cells. In addition, nifedipine (50 µM) significantly reduced ascorbate-dependent collagen type l and type lV synthesis by cultured aortic smooth muscle cells, assayed by measuring intracellular collagen content. We observed increased intracellular levels of ascorbate under supplementation with elevated doses of ascorbic acid, as well as its lipid soluble derivative ascorbyl palmitate. Nifedipine reduced ascorbic acid intracellular influx in cultured aortic smooth muscle cells with nifedipine (50 µM) compared to control. Adverse effects of nifedipine were neutralized either by an increased level of cell supplementation with ascorbic acid or by substituting it with ascorbyl palmitate. These studies suggest that adverse effects of channel blockers could be caused by their weakening the arterial wall integrity by interfering with proper extracellular matrix formation. In conclusion, these studies confirm the adverse effects of channel blockers on collagen type l and lV deposition, the key ECM components essential for maintaining optimal structural integrity of the arterial walls. Ascorbate supplementation reversed channel blocker inhibition of these collagen types synthesis and deposition. The results of this study imply the benefits of ascorbate and ascorbate palmitate supplementation in medical management of cardiovascular disease in order to compensate for adverse effects of channel blockers.
Keywords: Cardiovascular disease, collagen synthesis, channel blockers, ascorbic acid, ascorbyl palmitate, anti-arrhythmic
Introduction
Calcium, sodium and potassium channel blockers are widely prescribed medications for a variety of health problems, most frequently for cardiac arrhythmias, hypertension, angina pectoris and other disorders, with sales reaching $6 billion worldwide [1]. These agents are associated with problematic side effects. For example, nifedipine, a calcium-channel blocker was found to be associated with increased mortality and increased risk for myocardial infarction [2,3] and breast cancer [4].
Sodium channel blockers, which impair conduction of Na+ through sodium channels, are used in the treatment of cardiac arrhythmias and are classified as “Type I” (membrane stabilizing agents) in the Vaughn Williams classification [5-7]. These drugs bind to and block the fast sodium channels that are responsible for the rapid depolarization (phase 0) of fast-response cardiac action potentials. This type of medication can serve as an important mechanism for suppressing tachycardias that are caused by abnormal conduction [7].
Na blockers, which include such drugs as quinidine and procainamide, are used for treating supraventricular tachycardia, ventricular tachycardia, symptomatic ventricular premature beats, and prevention of ventricular fibrillation. Side effects of quinidine include blurred vision, tinnitus, headache, psychosis, cramping and nausea, and enhancement of digitalis toxicity [7]. Capsaicin is considered a natural Na channel blocker. Most local anesthetics used clinically are relatively hydrophobic molecules that gain access to their blocking site on the sodium channel by diffusing into or through the cell membrane. These anesthetics block sodium channels and thereby the excitability of all neurons, not just sensory neurons [8].
Calcium channel blockers inhibit the movement of calcium through slow calcium channels of cell membranes in the myocardium, cardiac conduction tissues, and vascular smooth muscle [5]. Currently approved calcium channel blockers bind to L-type calcium channels located on the vascular smooth muscle, cardiac myocytes, and cardiac nodal tissue (sinoatrial and atrioventricular nodes) [9]. They are divided into two categories based upon their predominant physiologic effects: dihydropyridines (predominantly vasodilators) such as nifedepine and non-dihydropyridines (reduce vascular permeability affecting cardiac contractility and conduction) such as verapamil and dilitiazem [10].
Dihydropyridines may cause reflex tachycardia, flushing, headache, and ankle swelling [9]. Ditiazem and verapamil depress cardiac conduction and cause bradycardia [9]. Natural Ca++ channel blockers include magnesium, due to magnesium’s ability to block calcium from entering muscle and heart cells, but with less powerful effects than other Ca++ channel blockers.
In addition to the side effects mentioned above by different channel blockers, these drugs can cause arterial wall damage. For example, Roth et al. reported that Ca++ channel blockers such as verapamil, ditiazem and others significantly decreased the constitutive and platelet-derived growth factor-dependent collagen deposition in the ECM formed by human vascular smooth muscle cells and fibroblasts [11]. The drugs inhibited the expression of fibrillary collagens type I and III and of basement membrane type IV collagen [11]. Furthermore, Ca++ channel blockers specifically increased the proteolytic activity of the 72-kDa type IV collagenase [11], thereby contributing to vascular wall structure destabilization and promoting events facilitating rupture of plaques.
Collagen is a critical component of vascular walls, cartilage and is an essential component of connective tissue in the body. Collagen synthesis is regulated by ascorbic acid, which is also essential for its optimum structure as a cofactor in lysine and proline hydroxylation, a precondition for optimum crosslinking of collagen and elastin. According to the Rath concept, impairment of the vascular wall structure due to inadequate collagen production is a triggering event in excessive deposition of lipoproteins (Lp(a) and LDL) as a biological ‘repair’ process and an underlying factor in atherosclerosis [12]. A recent study by Cha et al. [13] confirmed this concept stressing the importance of vascular collagen synthesis in preventing vascular deposition of lipoproteins and development of atherosclerosis.
The wide use of channel blockers, which have to be taken for decades, or for life, and its negative impact on collagen production, would imply its implication on further cardiovascular events and other components of extracellular matrix.
Therefore, the main objective in this study was to evaluate effects of various types of channel blockers on intracellular accumulation and cellular functions of ascorbate, specifically in relation to formation and extracellular deposition of major collagen types relevant for vascular function.
Materials and methods
Reagents
All reagents were from Sigma-Aldrich (St. Louis, MO) except when indicated differently.
Cell cultures
Normal human dermal fibroblasts (DF) were supplied by ATCC (Manassas, VA). Human aortic smooth muscle cells (AoSMC) were purchased from Cambrix (East Rutherford, NJ). Both cell cultures were maintained in DMEM medium (ATCC) containing antibiotics and 5% fetal bovine serum (FBS, ATCC). All cell cultures were maintained at 37°C and 5% CO2 atmosphere. Cell viability was monitored with MTT assay. None of the used experimental conditions resulted in statistically significant cell death (data not shown).
Collagen production by human cultured cells
For experiments, DF or AoSMC, at 5th to 8th passages, were seeded on collagen type I-covered plastic plates (Becton-Dickinson, collagen I isolated from rat tail tendon) at density of 25,000/cm2 and grown to confluence for 5-7 days. For intracellular immunoassay experiments, plain plastic 96-well plates were used. Tested compounds were added to cells at indicated concentrations for 72 h in DMEM supplemented with 2% FBS and cell-produced extracellular matrix was exposed by sequential treatment with 0.5% Triton X100 and 20 mM ammonium sulfate in phosphate buffered saline (PBS, Life Technologies) for 3 min each at room temperature (RT) as described previously [14]. After four washes with PBS, ECM layers were treated with 1% bovine serum albumin (BSA) in PBS for one hour at RT and immediately used in experiments. Alternatively, cell layers were washed three times with PBS and fixed with 3% formaldehyde in PBS at 4°C for one hour. Fixed cell layers were washed four times with PBS and treated with 1% BSA/PBS for one hour at RT.
Collagen and beta-actin immunoassays
Immunoasssays were done as described previously [15 ] by sequential incubation with primary monoclonal antibodies specific to human collagen type I or IV or human beta-actin in 1% BSA/PBS for 2 hours followed by 1 hour incubation with secondary goat anti-mouse IgG antibodies labeled with horse radish peroxidase (HRP). Retained peroxidase activity was measured after the last washing cycle (three times with 0.1% BSA/PBS) using TMB peroxidase substrate reagent (Rockland). Optical density was read with plate reader (Molecular Devices) at 450 nm and expressed as percentage of control cell samples incubated in unsupplemented 1% BSA/DMEM.
Intracellular ascorbic acid assay
AoSMC were seeded and grown to confluent layers in plastic 24-well plates (Becton-Dickinson) and treated with nifedipine and ascorbates for 90 min as described above. Intracellular ascorbic acid was extracted as described by Lane and Lawen [16]. Briefly, cells were washed three times with ice-cold PBS on ice and incubated with 300 µl of 0.1% saponin in 1% ethanol in PBS for 10 min on orbit mixer. Samples were collected to Eppendorf tubes and centrifuged at 13,000 x g for 5 min. Supernatants were used in ascorbic acid assay (Ascorbic Acid Colorimetric Assay Kit II FRASC, BioVision) in accordance with manufacturer’s protocol.
Statistical analysis
Results in figures are means ± SD from three or more repetitions from the most representative of at least two independent experiments. Differences between samples were estimated with a two-tailed Student’s t-test using MedCalc software (Mariakerke, Belgium) and accepted as significant at p levels less than 0.05.
Results
Effect of channel blockers and ascorbate on collagen type I ECM deposition by fibroblasts
The results presented in Figure 1A show that all channel blockers tested inhibited collagen type I deposition to the ECM by fibroblasts, however, each one to a different degree. In the presence of ascorbic acid, the ECM deposition of collagen I was significantly increased. Among tested channel blockers, capsaicin demonstrated slight, insignificant inhibition of collagen type I. However, in the presence of 50 µM ascorbate, collagen I deposition significantly increased (164%-219%, p≤0.0005) at each concentration of capsaicin compared to the level of collagen I deposited under capsaicin without ascorbate.
Figure 1.

A. Effects of capsaicin (natural Na+ blocker) on collagen type I ECM deposition by cultured human skin fibroblasts (* indicates significance of p≤0.005 between ascorbate and ascorbate + capsaicin ECM collagen l deposition); B. Effects of ditiazem (Ca++ blocker) on collagen type I ECM deposition by human skin fibroblasts (* indicates significance of p<0.0001 between ascorbate vs ascorbate and ditiazem ECM collagen l deposition); C. Effects of lidocaine (Na+ blocker) on collagen type I ECM deposition by cultured human skin fibroblasts (* indicates significance of p<0.0001 between ascorbate and ascorbate + lidocaine ECM collagen l deposition; ** indicates significance of p≤0.01 between ECM collagen l deposition at increased concentrations of lidocaine compared to control); D. Effects of nifedipine (Ca++ blocker) on collagen type I ECM deposition by cultured human skin fibroblasts (* indicates significance of p<0.0001 between ascorbate and ascorbate + nifedipine ECM collagen l deposition; ** indicates significance of p≤0.05 between ECM collagen l deposition at increased concentrations of nifedipine compared to control); E. Effects of quinidine (Na+ blocker) on collagen type I ECM deposition by cultured human skin fibroblasts (* indicates significance of p≤0.006 between ascorbate and ascorbate + quinidine ECM collagen l deposition); F. Effects of verapamil (Ca++ blocker) on collagen type I ECM deposition by cultured human skin fibroblasts (* indicates significance of p≤0.002 between ascorbate and ascorbate + verapamil ECM collagen l deposition).
As presented in Figure 1B, ditiazem showed up to 13.1% inhibition of collagen type I, but this difference did not reach statistical significance. Trend line R2 = 0.5045. However, in the presence of 50 µM ascorbate, collagen I deposition significantly increased from 206% to 266%, (p<0.0001) at each concentration of ditiazem compared to the level of collagen I deposited under ditiazem without ascorbate.
Figure 1C shows that in the presence of lidocaine there was significant (19.3%-23.7%, p≤0.01) inhibition of collagen type I (trend line R2 = 0.4756). Treatment with 50 µM ascorbate resulted in a significant increase of collagen I, from 237% to 258% (p<0.0001), at each concentration of lidocaine compared to the level of collagen I deposited under lidocaine only.
The effects of nifedipine on collagen I deposition by FB are presented in Figure 1D. Nifedipine shows significant (13%-20.3%, p≤0.05) inhibition of collagen type I, except at a concentration of 100 µM, (trend line R2 = 0.3628). In the presence of 50 µM ascorbate there was a significant increase of collagen I deposition by FB (117%-244%, p<0.0001) at each concentration of nifedipine compared to the level of collagen I deposited under nifedipine without ascorbate.
In the presence of quinidine there was 6% to 12.2% inhibition of collagen type I, but the difference did not reach statistical significance, trend line R2 = 0.2047. See Figure 1E. By including ascorbate at 50 µM concentration, there was a significant increase of collagen I deposition by FB (137%-217%, p≤0.006) at each concentration of quinidine compared to the level of collagen I deposited under quinidine without ascorbate.
The results for verapamil, presented on Figure 1F, show 5.6% to 15.5% inhibition of collagen type I by this calcium channel blocker, but the difference did not reach statistical significance (trend line R2 = 0.5383). In the presence of ascorbate (50 µM), collagen I deposition by FB significantly increased (137%-224%, p≤0.002) at each concentration of verapamil compared to the level of collagen I deposited under verapamil without ascorbate.
Effect of nifedipine, ascorbic acid and ascorbyl palmitate on collagen types I and IV deposition by cultured human aortic smooth muscle cells (AoSMC)
Nifedipine, a representative member of the channel blockers class, had similar effects on ascorbic acid dependent collagen I and IV deposition in AoSMC as in FB culture. Comparable results were obtained with verapamil and quinidine (not shown).
As presented in Figure 2A, nifedipine (50 µM) significantly reduced ascorbic acid dependent ECM deposition of collagen type l by cultured aortic smooth muscle cells: 62.3% (p = 0.0001), 41.8% (p = 0.019) and 16.5% (p = 0.177) at 25, 100 and 400 µM ascorbic acid, respectively. Trend line for ascorbic acid with nifedipine R2 = 0.995. Ascorbic acid at 100 and 400 µM increased ECM deposition by 114% and 10.76%, respectively, compared to 25 µM ascorbic acid, trend line R2 = 0.2512.
Figure 2.

A. Effects of 50 μM nifedipine on ascorbic acid dependent ECM deposition of collagen type I by cultured AoSMC (* indicates significance of p≤0.02 between ascorbate and ascorbate + nifedipine ECM collagen l deposition); B. Effects of 50 μM nifedipine on ascorbyl palmitate dependent-ECM deposition of collagen type I by cultured AoSMC (* indicates significance of p≤0.045 between ascorbyl palmitate and ascorbyl palmitate + nifedipine ECM collagen l deposition); C. Effects of 50 μM nifedipine on ascorbic acid dependent ECM deposition of collagen type IV by AoSMC (* indicates significance of p≤0.02 between ascorbate and ascorbate + nifedipine ECM collagen lV deposition); D. Effects of 50 μM nifedipine on ascorbyl palmitate dependent ECM deposition of collagen type IV by AoSMC (* indicates significance of p≤0.02 between ascorbyl palmitate and ascorbyl palmitate + nifedipine ECM collagen lV deposition).
Nifedipine (50 µM) significantly reduced ascorbyl palmitate dependent ECM deposition of collagen type l by cultured aortic smooth muscle cells: 25% (p = 0.005), 38% (p = 0.006), 27.5% (p = 0.045), 11% (p = 0.154) and 26.5% (p = 0.0025) at 1.25, 2.5, 5, 10 and 20 µM ascorbyl palmitate, respectively, as shown in Figure 2B. Trend line for ascorbyl palmitate with nifedipine R2 = 0.7695. Ascorbyl palmitate administered independently increased ECM deposition by 135% (p = 0.013), 189% (p = 0.004), 184% (p = 0.0013) and 212% (p<0.0001) at 2.5, 5, 10 and 20 µM, respectively, compared to 1.25 µM ascorbyl palmitate, trend line R2 = 0.90.
As presented in Figure 2C, in the presence of nifedipine (50 µM), ascorbic acid dependent deposition of collagen type lV by cultured aortic smooth muscle cells was significantly reduced: 36% (p = 0.0029), 54% (p = 0.021) and 43% (p = 0.02) at 25, 100 and 400 µM ascorbic acid, respectively. Trend line for ascorbic acid with nifedipine R2 = 0.9982. Ascorbic acid showed increased collagen IV ECM deposition by 175% (p = 0.053) and 166% (p = 0.0265) at 100 and 400 µM, respectively, compared to 25 µM ascorbic acid, trend line R2 = 0.652.
Figure 2D shows the effects of nifedipine (50 µM) on ascorbyl palmitate dependent ECM deposition of collagen type lV by cultured aortic smooth muscle cells. The results show a significant decrease in collagen IV to 23.5% (p = 0.0459), 22.7% (p = 0.0228), 22% (p = 0.139), 13.5% (p = 0.093) and 31.6% (p = 0.0055) at 1.25, 2.5, 5, 10 and 20 µM ascorbyl palmitate, respectively. Trend line for ascorbyl palmitate with nifedipine R2 = 0.8332. Ascorbyl palmitate alone showed increased collagen IV deposition: 113% (p = 0.228), 144% (p = 0.068), 138% (p = 0.023) and 168% (p = 0.004) at 2.5, 5, 10 and 20 µM, respectively, compared to 1.25 µM ascorbyl palmitate, trend line R2 = 0.8976.
Effect of nifedipine on ascorbate-dependent stimulation of collagen type I synthesis in AoSMC
In these experiments, the intracellular collagen content was evaluated in relation to the levels of housekeeping intracellular protein, beta-actin.
As shown in Figure 3A, nifedipine (50 µM) significantly reduced ascorbate-dependent intracellular collagen type l synthesis by cultured aortic smooth muscle cells, by 61% (p = 0.0002) and 49.1% (p = 0.0005) at 25 and 100 µM ascorbic acid, respectively. In the presence of 400 µM ascorbic acid and nifedipine, intracellular collagen I increased by 122% (p = 0.0195) compared to control. In the presence of ascorbyl palmitate at 5 and 20 µM, nifedipine significantly reduced ascorbate-dependent collagen type l synthesis by 20.3% (p = 0.0002) and 38% (p = 0.002), respectively. Trend line for ascorbic acid with nifedipine R2 = 0.638 and for ascorbyl palmitate with nifedipine R2 = 0.9056. Trend line for ascorbic acid effect alone on collagen I intracellular content was R2 = 0.874 and that for ascorbyl palmitate alone was R2 = 0.985.
Figure 3.

A. Effects of ascorbic acid on intracellular content of collagen type I in AoSMC in the presence or absence of 50 μM nifedipine (* indicates significance of p≤0.002 between ascorbic acid or ascorbyl palmitate alone vs with nifedipine on intracellular content of collagen type l); B. Effects of ascorbic acid on intracellular content of beta-Actin in AoSMC in the presence or absence of 50 μM nifedipine (* indicates significance of p≤0.03 between ascorbic acid or ascorbyl palmitate alone vs with nifedipine on intracellular content of beta actin); C. Effects of ascorbic acid on intracellular content of collagen type I in AoSMC in the presence or absence of 50 μM nifedipine. Collagen type I content was normalized by beta actin content in corresponding samples (* indicates significance of p≤0.003 between ascorbic acid alone vs with nifedipine on intracellular content of collagen type l).
Nifedipine (50 µM) significantly reduced ascorbate-dependent beta-actin intracellular content in cultured aortic smooth muscle cells compared to control: 29%% (p = 0.0018), 22.6% (p = 0.0061) and 5% (p = 0.54) at 25, 100 and 400 µM ascorbic acid, respectively, as shown in Figure 3B. In the presence of 5 µM and 20 µM of ascorbyl palmitate, nifedipine significantly reduced intracellular beta actin content by 22% (p = 0.013) and 23.6% (p = 0.033), respectively. Trend line for ascorbic acid with nifedipine R2 = 0.5157 and for ascorbyl palmitate with nifedipine R2 = 0.8088. Trend line for ascorbic acid effect alone on collagen I intracellular content was R2 = 0.5157 and that for ascorbyl palmitate alone was R2 = 0.914.
In the presence of nifedipine (50 µM) and ascorbate the intracellular content of collagen type l normalized to beta actin content in aortic smooth muscle cells was reduced compared to control: 46% (p = 0.0005) and 34.4% (p = 0.0033) at 25 and 100 µM ascorbic acid, respectively (Figure 3C). In the presence of 400 µM ascorbic acid, nifedipine increased collagen 1 by 23% (p = 0.017) compared to control. However, nifedipine had no significant effect on collagen I content in the presence of ascorbyl palmitate at 5 µM and 25 µM concentrations. Trend line for ascorbic acid with nifedipine R2 = 0.871 and for ascorbyl palmitate with nifedipine R2 = 0.9918. Trend line for ascorbic acid effect alone on collagen I intracellular content was R2 = 0.6396 and that for ascorbyl palmitate alone was R2 = 0.9442.
Effect of nifedipine on intracellular accumulation of ascorbic acid in AoSMC
Nifedipine (50 µM) had an inhibitory effect on intracellular accumulation of ascorbate in cultured aortic smooth muscle cells compared to control. As shown in Figure 4, in the presence of 100 µM ascorbic acid, nifedipine decreased intracellular ascorbate by 61.5% (p = 0.161) but this difference did not reach statistical significance. However, at 400 µM ascorbic acid, nifedipine significantly reduced (67.7%, p = 0.0337) ascorbate intracellular content. However, nifedipine had no significant effect on intracellular ascorbate levels in the cells incubated with ascorbyl palmitate. We observed that supplementation with 20 µM ascorbyl palmitate resulted in significantly higher (10x, P<0.0001) intracellular levels of ascorbic acid compared to the highest concentrations of ascorbic acid used in experiments, which was 400 µM. Ascorbyl palmitate levels higher than 20 µM were found to be cytotoxic to cultured fibroblasts and smooth muscle cells under our experimental conditions (data not shown).
Figure 4.
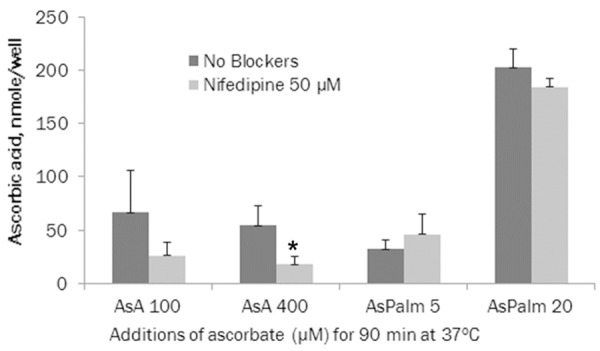
Effects of nifedipine 50 μM on ascorbate accumulation inside human AoSMC (* indicates significance of p≤0.06 between ascorbic acid or ascorbyl palmitate alone vs with nifedipine on intracellular ascorbate content).
Discussion
All channel blockers tested demonstrated inhibitory effects on collagen type I deposition to the ECM by fibroblasts, each to a different degree. Ascorbic acid significantly increased collagen I ECM deposition. Nifedipine, a representative member of the channel blockers class, displayed similar effects on ascorbic acid and on ascorbyl palmitate dependent collagen 1 deposition by AoSMC and by fibroblasts (NHDF). Furthermore, nifedipine demonstrated significant inhibition of collagen lV ECM deposition.
By determining intracellular collagen content in relation to the levels of housekeeping intracellular protein beta-actin, we found that nifedipine counteracted ascorbate-dependent stimulation of collagen type I synthesis in AoSMC. Thus, changes in ECM deposition of collagen seen in previous experiments were caused by its decreased intracellular accumulation (synthesis/protein expression). Nifedipine (50 µM) reduced ascorbate intracellular accumulation in cultured aortic smooth muscle cells compared to control under water-soluble ascorbic acid as well as lipid soluble ascorbyl palmitate. We observed that decreased collagen deposition by this channel blocker can be rescued by supplementation of AoSMC with increased levels of ascorbic acid or with ascorbyl palmitate.
These results confirm the adverse effects of channel blockers on collagen type l and lV ECM deposition. Ascorbate supplementation could reverse the inhibition of these critical ECM components necessary for maintaining optimal structural integrity of the arterial wall. Ascorbic acid is essential for the hydroxylation of lysine and proline, a precondition for optimum crosslinking of collagen and elastin and, thus, for maintaining the integrity of the vascular wall [12,17]. As mentioned earlier, Ca++ channel blockers such as verapamil, ditiazem and others were shown to significantly decrease collagen deposition in the ECM in human vascular smooth muscle cells and fibroblasts. These drugs inhibited the expression of fibrillary collagens type I and II and of basement membrane collagen type IV and increased the proteolytic activity of the 72-kDa type IV collagenase [11].
As in human scurvy, complete dietary ascorbate deprivation has been shown to cause marked alteration in the structural integrity of vascular connective tissue [13,18,19]. In a previous study, we found that hypoascorbemic Gulo-/- mice developed early atherosclerosis accompanied by the deposition of Lp(a) in the intima and deeper layers of the vascular wall, while age-matched mice with high amounts of dietary ascorbate supplementation did not develop atherosclerosis and no Lp(a) detectable in the vascular wall [13]. Disruption of the endothelial layer, impairment of the basement membrane and the structural disintegration of the ECM paralleled the development of scurvy [18]. Since channel blockers have been shown to decrease ascorbate-dependent collagen l and lV, use of channel blockers can lead to increased probability of development of atherosclerosis.
Regular intake of ascorbic acid results in a variable absorption rate, between 70 to 95%, with the degree of absorption decreasing as intake increases [20]. Fractional human absorption of ascorbic acid may be as low as 33% at high intake (1.25 g), but it can reach 98% at low intake (<200 mg) [20]. Ascorbate concentrations over renal re-absorption threshold pass freely into the urine and are excreted. At high dietary doses (corresponding to several hundred mg/day in humans) ascorbate is accumulated in the body until the plasma levels reach the renal resorption threshold, which is about 1.5 mg/dL in men and 1.3 mg/dL in women [21]. Ascorbate at concentrations in the plasma larger than this value (thought to represent body saturation) is rapidly excreted in the urine with a half-life of about 30 minutes. Concentrations less than this threshold value are actively retained by the kidneys, and the excretion half-life for the remainder of the vitamin C store in the body thus increases greatly, with the half-life lengthening as the body stores are depleted. This half-life rises until it is as long as 83 days by the onset of the first symptoms of scurvy [21].
Ascorbic acid is absorbed in the body by both active transport and simple diffusion. Sodium-dependent active transport-sodium-ascorbate co-transporters (SVCTs) and hexose transporters (GLUTs)-are the two transporters required for absorption. SVCT1 and SVCT2 import the reduced form of ascorbate across plasma membrane [22]. GLUT1 and GLUT3 are the two glucose transporters, and transfer only the dehydroascorbic acid form of ascorbic acid [23]. Although dehydroascorbic acid is absorbed at a higher rate than ascorbate, the amount of dehydroascorbic acid found in plasma and tissues under normal conditions is low, as cells rapidly reduce dehydroascorbic acid to ascorbate [24,25]. Thus, SVCTs appear to be the predominant system for vitamin C transport in the body.
A possible mechanism of ascorbate’s rescuing effect on channel blocker induced inhibition could be competitive inhibition of channel blockers. These conclusions are supported by the rescuing action of increased levels of ascorbate supplementation in channel blocker-dependent inhibition of collagen deposition. Kuo et al. reported that hydrophobic 1.4-dihydropyridine compounds (nifedipine and nicardipine) inhibited Na+ - dependent and Na+ - independent (K+ substituting Na+) accumulation of ascorbic acid in human intestinal Caco-2 cells [26]. Ebersole and Molinoff report that ascorbate inhibits binding of dihydropyridine calcium channel blockers [27]. A study by Grossmann et al. demonstrated that co-infusion of ascorbate and quinidine (K+ blocker) abolished ascorbate-dependent venodilation in human veins [28].
The absence of inhibitory effects of channel blockers on ascorbyl palmitate dependent synthesis, ECM deposition of collagen and on accumulation of intracellular ascorbate from channel blockers apparently could be explained by different pathways of intracellular uptake independent from ion-channel-associated transporters, i.e. through hydrophobic portions of the cellular membrane.
Conclusion
In conclusion, these studies confirm the adverse effects of channel blockers on collagen type l and lV deposition, the key ECM components essential for maintaining optimal structural integrity of the arterial walls. Ascorbate supplementation reversed channel blocker inhibition of synthesis and deposition of these collagen types The results of this study imply the benefits of ascorbate and ascorbate palmitate supplementation in medical management of cardiovascular disease in order to compensate for adverse effects of channel blockers.
Acknowledgements
The research study was funded by Dr. Rath Health Foundation (Santa Clara, CA, USA), a non-profit organization.
Disclosure of conflict of interest
None.
References
- 1.Cox B. Chapter 1: Ion channel drug discovery: a historical perspective, in Ion Channel Drug Discovery, Royal Society of Chemistry. 2014. pp. 1–15. [Google Scholar]
- 2.Psaty BM, Heckbert SR, Koepsell TD, Siscovick DS, Raghunathan TE, Weiss NS, Rosendaal FR, Lemaitre RN, Smith NL, Wahl PW. The risk of myocardial infarction associated with antihypertensive drug therapies. JAMA. 1995;274:620–625. [PubMed] [Google Scholar]
- 3.Furberg CD, Psaty BM, Meyer JV. Nifedipine: Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326–1331. doi: 10.1161/01.cir.92.5.1326. [DOI] [PubMed] [Google Scholar]
- 4.Li CI, Daling JR, Tang MT, Haugen KL, Porter PL, Malone KE. Use of antihypertensive medications and breast cancer risk among women aged 55 to 74 years. JAMA Intern Med. 2013;173:1629–37. doi: 10.1001/jamainternmed.2013.9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sampson KJ, Kass RS. Anti-arrhythmic drugs, in Goodman & Gillman. In: Brunton LL, editor. The Pharmacological Basis of Therapeutics. 12th Edition. McGraw Medical; 2011. [Google Scholar]
- 6.National Library of Medicine: Sodium channel blockers. Updated 2011. Accessed 6/9/2015 http://www.nlm.nih.gov/cgi/mesh/2011/MB_cgi?mode=&term=Sodium+Channel+Blockers.
- 7.Klabunde RE. Cardiovascular pharmacology concepts: Sodium-Channel Blockers (Class I Antiarrhythmics) Revised 12/01/11, Accessed 6/9/15. http://www.cvpharmacology.com/antiarrhy/sodium-blockers.
- 8.Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449:607–610. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- 9.Klabunde RE. Cardiovascular pharmacology concepts: Calcium-Channel Blockers (CCBs) Revised 03/22/15, Accessed 6/9/15. http://www.cvpharmacology.com/vasodilator/CCB.
- 10.Triggle DJ. Drug targets in the voltage-gated calcium channel family: why some are and some are not. Assay Drug Dev Technol. 2003;1:719–733. doi: 10.1089/154065803770381075. [DOI] [PubMed] [Google Scholar]
- 11.Roth M, Eickelberg O, Köhler E, Erne P, Block LH. Ca2+ channel blockers modulate metabolism of collagens within the extracellular matrix. Proc Natl Acad Sci U S A. 1996;93:5478–5482. doi: 10.1073/pnas.93.11.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rath M, Pauling L. Hypothesis: Lipoprotein(a) is a surrogate for ascorbate. Proc Natl Acad Sci U S A. 1990;87:6204–6207. doi: 10.1073/pnas.87.16.6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cha J, Niedzwiecki A, Rath M. Hypoascorbemia induces atherosclerosis and vascular deposition of lipoprotein(a) in transgenic mice. Am J Cardiovasc Dis. 2015;5:53–62. [PMC free article] [PubMed] [Google Scholar]
- 14.Ivanov V, Ivanova S, Roomi MW, Kalinovsky T, Niedzwiecki A, Rath M. Extracellular matrix-mediated control of aortic smooth muscle cell growth and migration by a combination of ascorbic acid, lysine, proline, and catechins. J Cardiovasc Pharmacol. 2007;50:541–7. doi: 10.1097/FJC.0b013e318145148e. [DOI] [PubMed] [Google Scholar]
- 15.Ivanov V, Ivanova S, Kalinovsky T, Niedzwiecki A, Rath M. Plant-derived micronutrients suppress monocyte adhesion to cultured human aortic endothelial cell layer by modulating its extracellular matrix composition. J Cardiovasc Pharmacol. 2008;52:55–65. doi: 10.1097/FJC.0b013e31817e692f. [DOI] [PubMed] [Google Scholar]
- 16.Lane DJ, Lawen A. Non-transferrin iron reduction and uptake are regulated by transmembrane ascorbate cycling in K562 cells. J Biol Chem. 2008;283:12701–12708. doi: 10.1074/jbc.M800713200. [DOI] [PubMed] [Google Scholar]
- 17.Pinnell SR. Regulation of collagen biosynthesis by ascorbic acid: a review. Yale J Biol Med. 1985;58:553–9. [PMC free article] [PubMed] [Google Scholar]
- 18.Gore I, Fujinami T, Shirahama T. Endothelial changes produced by ascorbic acid deficiency in guinea pigs. Arch Pathol. 1965;80:371–6. [PubMed] [Google Scholar]
- 19.Maeda N, Hagihara H, Nakata Y, Hiller S, Wilder J, Reddick R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc Natl Acad Sci U S A. 2000;97:841–618. doi: 10.1073/pnas.97.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, Park JB, Lazarev A, Graumlich JF, King J, Cantilena LR. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93:3704–9. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oreopoulos DG, Lindeman RD, VanderJagt DJ, Tzamaloukas AH, Bhagavan HN, Garry PJ. Renal excretion of ascorbic acid: effect of age and sex. J Am Coll Nutr. 1993;12:537–42. doi: 10.1080/07315724.1993.10718349. [DOI] [PubMed] [Google Scholar]
- 22.Savini I, Rossi A, Pierro C, Avigliano L, Catani MV. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids. 2008;34:347–55. doi: 10.1007/s00726-007-0555-7. [DOI] [PubMed] [Google Scholar]
- 23.Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem. 1997;272:18982–9. doi: 10.1074/jbc.272.30.18982. [DOI] [PubMed] [Google Scholar]
- 24.May JM, Qu ZC, Neel DR, Li X. Recycling of vitamin C from its oxidized forms by human endothelial cells. Biochim Biophys Acta. 2003;1640:153–61. doi: 10.1016/s0167-4889(03)00043-0. [DOI] [PubMed] [Google Scholar]
- 25.Packer L. Vitamin C and redox cycling antioxidants. In: Fuchs J, Packer L, editors. Vitamin C in health and disease. New York: M. Dekker; 1997. [Google Scholar]
- 26.Kuo SM, Lin CP, Morehouse HF. Dihydropyridine calcium channel blockers inhibit ascorbic acid accumulation in human intestinal Caco-2 cells. Life Sci. 2001;68:1751–60. doi: 10.1016/s0024-3205(01)00970-5. [DOI] [PubMed] [Google Scholar]
- 27.Ebersole B, Molinoff PB. Identification of ascorbate as an endogenous substance that irreversibly inhibits binding of dihydropyridine calcium channel blockers. J Neurochem. 1992;58:1300–7. doi: 10.1111/j.1471-4159.1992.tb11342.x. [DOI] [PubMed] [Google Scholar]
- 28.Grossmann M, Dobrev D, Himmel HM, Ravens U, Kirch W. Ascorbic acid-induced modulation of venous tone in humans. Hypertension. 2001;37:949–954. doi: 10.1161/01.hyp.37.3.949. [DOI] [PubMed] [Google Scholar]