

Abstract
Alzheimer’s disease (AD) is a chronic progressive neurodegenerative condition of the brain, and it is the most common cause of dementia. Several neurobiological etiologies of AD are described in the literature. These include vascular, infectious, toxic, nutritional, metabolic, and inflammatory. However, these heterogeneous etiologies have a common denominator - viz. Inflammation and oxidative stress. Lipopolysaccharide (LPS) elevates the synthesis of proinflammatory cytokines and chemokines; chronically, together they trigger various pathological responses in the periphery and the CNS including dysfunctional memory consolidation and memory decline. Aging - the main risk factor for AD is inherently associated with inflammation. There are several age-related comorbidities that are also associated with inflammation and oxidative stress. Such co-prevailing aggravating factors, therefore, persist against a background of underlying aging-related pathology. They may converge, and their synergistic propagation may modify the disease course. A critical balance exists between homeostasis/repair and inflammatory factors; chronic, unrelenting inflammatory milieu succeeds in promoting a neuroinflammatory and neurodegenerative outcome. Extensive evidence is available that CNS inflammation is associated with neurodegeneration. LPS, proinflammatory cytokines, several mediators secreted by microglia, and oxidative-nitrosative stress in concert play a pivotal role in triggering neuroinflammatory processes and neurodegeneration. The persistent uncontrolled activity of the above factors can potentiate cognitive decline in tandem enhancing vulnerability to AD. Despite significant progress during the past twenty years, the prevention and treatment of AD have been tantalizingly elusive. Current studies strongly suggest that amelioration/prevention of the deleterious effects of inflammation may prove beneficial in preventing AD onset and retarding cognitive dysfunction in aging and AD. A concerted multi-focal therapeutic effort around the inflammation-oxidative-nitrosative stress paradigm may be crucial in preventing and treating AD. This paper informs on such relevant polypharmacy approach.
Keywords: Alzheimer’s disease, aging, LPS, proinflammatory cytokines, neuroinflammation, oxidative stress, nitrosative stress, microglia, amyloid, tau
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder of the aged characterized by the accumulation of amyloid-β (Aβ) and hyperphosphorylated tau (Ptau). These are present aggregated in amyloid plaques, and neurofibrillary tangles respectively - causing synaptic and neuronal loss, and enhancing cognitive dysfunction. AD is a multifactorial neurodegenerative condition and accumulating evidence has shown inflammation to be an integral part of its etiology [1-16]. A host of comorbid conditions are associated with aging; these include - endotoxemia, type 2 diabetes, obesity, metabolic syndrome, obstructive sleep apnea (OSA), and sleep loss. All these conditions upregulate inflammation [17-24]. Indeed, several studies indicate that inflammatory mechanisms are upregulated by the above comorbid conditions. This diverse group of conditions induces primary and secondary mechanisms that upregulate inflammation and increase physiologic dysfunction in older adults. Inflammation is driven by interacting mechanisms including damage-causing proinflammatory mediators, reactive oxygen species (ROS), excitotoxicity, calcium perturbations/increase, and dyshomeostasis, among others. These can be superimposed and synergistic, thus strengthening the deleterious pathophysiological state. There is supportive evidence for inflammation being an initial insult in cognitive decline. Indeed, dysfunctional cognition may also occur in the so-called normal elderly following: a) peripheral infection [25,26], b) delirium due to general anesthesia [27,28], and c) neurotoxic conditions affecting neurons, as in hypoxia/hypoxemia [29,30] - all these are associated with inflammation.
Mild, higher levels of bacterial endotoxin (about ~1-100 pg/mL), persist in circulation in humans with adverse health conditions and unhealthy life styles such as aging, obesity, chronic infections, and chronic alcohol consumption and smoking [31-37]. The causes of mild but persistent increase in plasma endotoxin are several including compromised gut mucosal barriers, dysbiosis (i.e. altered commensal and pathogenic gram-negative microbiota ratio), and vasculature leakage. Lipopolysaccharide (LPS) found in the bacterial wall is present in the intestinal lumen and reaches the circulation causing metabolic endotoxemia. However, inflammation is upregulated when LPS binds to Toll-like receptor 4 (TLR4) [38]. The ongoing smoldering subclinical mild endotoxemia causing low-grade but persistent inflammatory response induces the generation of inflammatory cytokines/mediators [39]. The latter underlie the pathogenesis and propagation of various chronic conditions including obesity, cardiovascular diseases, diabetes, neurodegenerative conditions, and indeed inflammatory diseases [31,40-48]. The pathways involving those mentioned above are being continually investigated and defined [49]. Given the deleterious pathological effects caused by inflammation, engendered by endotoxemia and proinflammatory mediators, therapeutic strategies should target this persistent pathological scenario to prevent and treat the above mentioned debilitating conditions.
Inflammation is meant to be beneficial to clear pathogen and phagocytose apoptotic cells/debris; however, when inflammation remains uncontrolled and becomes chronic, it leads to the production of neurotoxic mediators/proinflammatory cytokine that exacerbates neurodegenerative pathological outcome. Cytokines and Chemokines are proinflammatory proteins that mediate the body’s immune response. Their dysregulation is a cardinal feature in some pathological processes including the neuroinflammation, neurodegeneration, and demyelination. Pathologies in the CNS activate microglia that upregulates the production of these proinflammatory mediators, lead to glial and neuronal injury, and their death (see below). The above mentioned, therefore, orchestrates a scenario of imbalance between homeostasis-repair and inflammatory-neurodegenerative processes.
An excessive inflammatory response is characterized not only by elevated inflammatory cytokines, but by increases in mitochondrial dysfunction, reactive oxygen species (ROS), and nitric oxide (NO). Consequently, there may be damage to the systemic vascular endothelium, redox-glutathione depletion, and mitochondrial respiratory dysfunction causing reduction in ATP and O(2) consumption. Although, ROS are essential as antioxidant defenses in cells; however, an excess of ROS production is harmful to homeostasis. The resulting cellular pathologies, therefore, are a function of mitochondrial dysfunction and an excess of oxidative stress damage. Hence, inflammation and oxidative stress are intertwined [50-54]. LPS-induced ROS signaling from mitochondria has been demonstrated to be critical in macrophage activation [55]. Conversely, antioxidants not only reduce mitochondrial damage, but they may also reduce interleukin-6 levels [54] and decrease LPS-stimulated proinflammatory cytokines [56].
The molecular mechanisms underlying the pathogenesis of sporadic AD are being unraveled on a continuous basis in the literature [1-24]. This enormous amount of data, however, are now available on their involvement in AD pathophysiology. Overall, the purpose of this review is to discuss key concepts of inflammation, immune-reactivity, and oxidative stress in the context of aging, comorbid conditions, neurodegeneration, and cognitive decline. This review describes the salient literature and discusses major studies regarding alterations in proinflammatory mediators and oxidative stress. There is an emphasis on heterogeneous disease conditions promoting neuroinflammation, and inter-relationship between neuroinflammation and neurodegeneration that may lead to cognitive impairment and AD. Additionally, an important pharmacological approach is described that may inhibit both inflammation and oxidative stress, ameliorate cognitive decline, and possibly treat AD.
Aging and inflammation
Aging represents a state of complex multifactorial pathways that involve an ongoing molecular, cellular, and organ damage causing functional loss, disease vulnerability, and eventual death [57]. Dysfunctional aging and an increased risk of death owe their origin to diverse unfavorable factors including genetic, epigenetic, and nongenetic including lifestyle and environmental factors [58].
In aging, there is a 2-4-fold increase in serum levels of inflammatory mediators such as acute phase proteins and cytokines. A variety of factors may maintain this low-grade inflammation; these include - increased adipose tissue, smoking, excess alcohol, indolent infections (asymptomatic microbial), and chronic disorders of gastrointestinal, respiratory, and cardiovascular systems [59-63]. The pathogenetic role of proinflammatory cytokines may conceivably constitute a link between dysfunctional physiology, dysfunctional aging, and co-prevailing persistent age-associated diseases [64].
Interestingly, compared with younger individuals, healthy aged persons may suffer impairment in their memory following deleterious events such as severe bacterial infection, surgery, or psychological trauma. These life events may trigger an increased and prolonged production of proinflammatory cytokines and neuroinflammatory response during aging. This has been shown to be due to sensitization/priming of microglia that is the source of this heightened proinflammatory response. An enhanced neuroinflammatory response may impair synaptic plasticity, and cause a reduction in Arc (important mediator of synaptic plasticity) and BDNF (brain-derived neurotrophic factor) - the important downstream factors. The above-mentioned mechanisms are crucial in impacting/decreasing long-term memory [65,66].
There is an array of proinflammatory factors in addition to cytokines. These, e.g. include homocysteine, C-reactive protein (CRP) and alpha-1-antichymotrypsin (ACT). In a study, the correlation was studied between homocysteine and 6-year cognitive decrease, and the critical role of Interleukin-6 (IL-6), CRP, and ACT. Higher homocysteine was negatively associated with lower information processing speed and a decline in cognitive function. The above negative association was highest in the presence of high level of IL-6. Similarly, higher CRP plus higher homocysteine were associated with decreased memory retention. Further, higher ACT plus higher homocysteine were also associated with lower information processing speed and faster decline. The above data reflect that a combination of inflammatory factors underlies cognitive impairment [67]. Furthermore, high serum CRP levels in association with high IL-6 levels were a significant risk factor for vascular dementia (VaD). Notably, just hyperhomocysteinemia (Hhcy) was correlated with increased risk of AD [68,69].
LPS induces memory impairment in old mice compared to young animals [70-72]. Thus, disruption of cognitive processing [73,74] in the old animals by LPS suggests that in old animals the hippocampus is vulnerable to cytokines, LPS, and an acute infection [75-82]. Importantly, LPS-challenged old animals had difficulty in locating the platform in Morris Maze test. This suggests that cognitive decline can be ameliorated when the neuroinflammatory response is minimized and kept under control [83].
As in neurodegenerative diseases, microglia is primed in aging also [84,85]. Consequently, microglia in the aged brain respond to the inflammatory signals (e.g. infection) and generate more proinflammatory interleukin-1beta (IL-1β) for a longer duration than microglia in younger brains [17]. Indeed, LPS administration also resulted in an elevated proinflammatory cytokine response in the aged brain [84,86,87], as did Escherichia coli administration to older rats [82]. As well as higher proinflammatory cytokine response in the brain, LPS-injected aged mice demonstrated behavioral dysfunction, and a decline in hippocampal-dependent learning and memory - again compared with younger animals [84,86,88]. Similar to rodent data, acute cognitive impairments are common in the elderly humans (aged 65 years or older) suffering from peripheral infection [89,90]. Hence, microglial cell priming, neuroinflammation, and dysfunctional physiological processes activate neuropathology in the aged brain [17,91].
Aging and comorbid age-related diseases might have an impact on synaptic plasticity in the hippocampus, to produce memory failures. For example, the LTP (long-term potentiation) induced in area CA1 (using theta-burst stimulation) is compromised by the combined effects of aging and infection [92]. Likewise, immune challenge e.g. employing intraperitoneal injection of E. coli disrupts hippocampus-dependent memory in aged (24-month-old), but not young (3-month-old), F344xBN rats. Synaptic plasticity and long-term memory strongly depend on BDNF; however, the combined effect of aging plus infection might disrupt BDNF production and its processing in the hippocampus. Indeed, inflammation-related BDNF reduction in synaptosomes may evoke long-term memory disruptions in aging [93]. BNDF is known to function in neuronal protection from dysfunction due to infection or injury; it plays a vital role in hippocampal plasticity processes. However, memory is compromised by deleterious effect of cytokines such as IL-1β. Thus, aging and inflammatory response in the brain depletes BDNF required to maintain memory-related plasticity processes at synapses in the hippocampus [94]. Minocycline, an inhibitor of microglial activation has been shown to restore significant LTP in middle-aged rats administered arthritis adjuvant systemically [95]. (Also, see below).
Heterogeneous comorbid conditions and inflammation: different roads but same destination
Various disease pathologies often co-exist in aging (Figure 1). The predisposition, interaction, and causation of these disease conditions in aging evoke different pathologies. However, since they have an impact on inflammation, it is important to appreciate their role in neuroinflammation in various age-related disease. In keeping with aging being a state of low-grade inflammation, the plasma concentration of LPS and its binding protein are significantly higher in older subjects [96]. This reflects the presence of metabolic endotoxemia, i.e. the presence of increased plasma endotoxin level in old age and metabolic disease [96].
Figure 1.
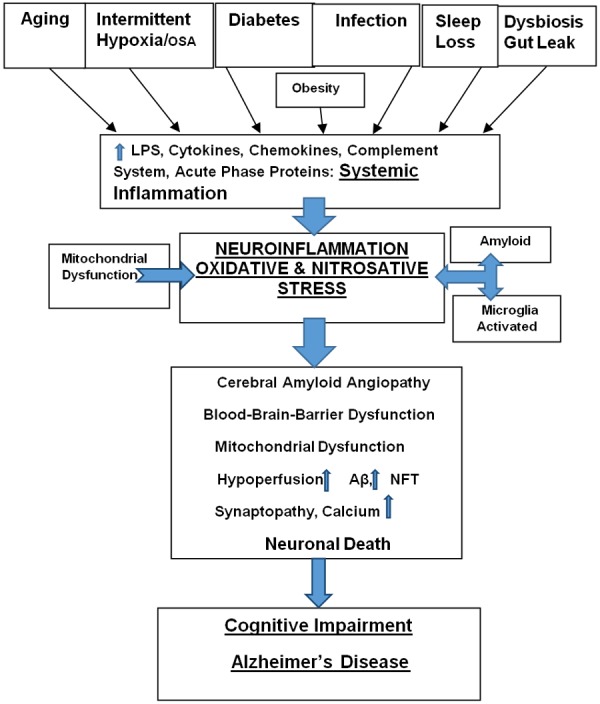
Schematic representation of the pathogenesis of cognitive decline in aging and AD. Aging in conjunction with several comorbid conditions/risk factors including obesity, hypertension, diabetes, hypoxia/OSA, infection, sleep loss and dysbiosis synergistically promote a complex inflammatory milieu. The latter is comprised of LPS, Cytokines, Chemokines, Complement System, and Acute Phase Proteins such as CRP - they are the components of systemic inflammation. The latter triggers neuroinflammation and oxidative-nitrosative stress mediated by activated microglia and amyloid. The above cascade leads to several pathologies including cerebral amyloid angiopathy, blood-brain-barrier dysfunction, mitochondrial dysfunction, and cerebral hypoperfusion. They lead to the accumulation of Aβ, NFT, and neuronal calcium causing neuronal death. An escalating neurodegeneration promotes cognitive/memory decline and AD.
Obesity and inflammation
It is well characterized that negative health outcomes are associated with high levels of fat within the abdominal cavity. Obesity enhances systemic inflammation and proinflammatory cytokines [97-99]. The visceral obesity-induced systemic inflammation is linked to the pathogenesis of insulin resistance [100], type 2 diabetes [101,102], hypertension [103], and cardiovascular disease [104]. The adipose tissue is a rich source of metabolically active adipocytokines including leptin, plasminogen activator inhibitor-1, adiponectin, and resistin [105]. Also, adipose tissue in obese persons secretes high levels of some inflammatory cytokines including TNF-α, IL-6, and IL-8 [106]. Stem cells derived from adipose tissue of obesity showed hyper-responsiveness to hypoxia and strong nuclear factor-κB (NF-κB) activation that may, in turn, sustain inflammation [107]. Hence, NF-κB inhibitors might curtail the obesity-induced inflammation. Systemic inflammation is a precursor of neuroinflammation (see below). The neuroinflammation worsens central pathways of energy regulation/energy balance, perturbs nutrient metabolism, and triggers several disease states such as obesity, diabetes, and cardiovascular problems [108]. These conditions then impact physiological function/homeostasis and perturb neural regulation. Indeed, there is accumulating evidence that high-fat diet and obesity induce gliosis and inflammation in the human and rodent hypothalamus [109,110], and perturb the hypothalamic function [111,112].
Gut microbiota and metabolic endotoxemia-inflammation
High-fat consumption causes changes in gut microbiota, i.e. dysbiosis (an increase in pathogenic bacteria at the expense of commensals), and this generates high plasma concentration of LPS, termed metabolic endotoxemia. The latter has been correlated with a decrease in glucose intolerance, adiposity, oxidative stress, and mRNA expression related to macrophage infiltration in visceral adipose tissue. Of note, high-fat consumption strongly enhanced intestinal permeability via reduced expression of proteins of the tight junctions. This shows a close inter-connection between metabolic endotoxemia, inflammation, and intestinal permeability to LPS. Importantly, metabolic endotoxemia has been shown to upregulate both obesity and insulin resistance [20,113]. Indeed, bacterial LPS is a triggering factor for the above disease states. Hence, LPS may be an important therapeutic target, and lowering plasma LPS concentration could be a viable strategy to ameliorate/retard metabolic diseases [20]. Further, favorable intestinal microbiota e.g. bifidobacteria prevent the deleterious effect of endotoxemia [19,20,31]. Also, therapeutic strategies involving weight loss and probiotic plus prebiotic intake can reduce inflammation, endotoxemia, and associated conditions e.g. insulin resistance [113].
Type 2 diabetes and inflammation
Epidemiological evidence links type 2 diabetes, obesity, hyperinsulinemia, and metabolic syndrome, to AD. There is an inherent association between the development of type 2 diabetes and elevated (but ‘normal range’) levels of circulating inflammatory mediators including CRP, and indices of insulin resistance (IR) [114]. Obesity, (mainly central obesity) is a strong mediator of insulin resistance and type 2 diabetes since it generates proinflammatory cytokines such as IL-6 [115]. The great majority of patients (90%) with diabetes have insulin resistance. Subclinical chronic inflammation has been implicated as an essential pathogenic factor in the development of insulin resistance and type 2 diabetes. Markers for this chronic inflammation include CRP, IL-6 and TNF-α (albeit at low level) [116]. Indeed, inflammation may trigger several mechanisms including insulin resistance and deficiency, impaired insulin receptor, impairment of insulin growth factor (IGF) signaling, glucose toxicity, dysfunctional advanced glycation end products and their receptors, cerebrovascular injury, and vascular inflammation [105,117-119]. Furthermore, the proinflammatory effects of cytokines may involve specific intracellular signaling pathways such as the nuclear factor (NF)-dB, IκB kinase, (IKK), activating Protein-1 (AP-1) and c-Jun NH2-terminal kinase (JNK), and impaired insulin signaling [21]. This provides important perspectives for treating insulin resistance, and glucose intolerance. The antidiabetic drugs that reduce insulin and insulin resistance may, in fact, reduce inflammation as well. Conversely, anti-inflammatory drugs (such as thiazolidinedione) may improve insulin resistance and glucose tolerance.
Obstructive sleep apnea and inflammation
OSA is a common condition whose hallmark feature is intermittent hypoxia (IH). Chronic IH induces inflammation and directly triggers proinflammatory pathways [120-126]. Since obesity is associated with OSA, recent work has shown that IH leads to activation of inflammatory responses in human adipocytes [127]. The latter irrespective of being derived from either visceral or subcutaneous sites showed activated NF-κB DNA-binding activity in IH, compared with normoxic controls. IH upregulated proinflammatory gene expression and mRNA expression in adipocytes that were the source of significantly increased secretion of TNF-α, IL-6, and IL-8 [127]. Recurrence of IH resulted in cellular and systemic inflammation in the rat 3T3-L1 adipocytes. Both mRNA and protein levels of TNF-α, IL-6, and leptin, were significantly high in severe IH group; the level of these proinflammatory markers was proportional to the severity of IH [128]. In OSA patients, TNF-α and TNF-α/IL-10 ratio values were significantly higher compared with the control group; however, IL-10 was much lower. A meta-analysis of 51 studies found that the levels of systemic inflammatory markers were higher in OSA patients compared to control individuals [129]. Importantly, continuous positive airway pressure (C-PAP) therapy for a month decreased TNF-α level, while there was no change in the level of IL-10 [130]. The inflammatory response in IH rodents regarding serum levels of TNF-α, IL-6, and IL-8 showed progressive increment from onset to the 6th week, while the level of anti-inflammatory IL-10 decreased [131]. Further, soluble TNF receptor-1 (sTNF-R1) levels were significantly higher in the OSA patients then the controls [132]. However, three months of CPAP therapy lowered sTNF-R1 [133,134].
Fibrinogen (a coagulation protein) is associated with inflammation, and long-term elevated plasma fibrinogen is linked to an increased risk of cardiovascular diseases (CVD). Compared to controls, fibrinogen is significantly higher in OSA patients [135], as is CVD. Plasminogen activator inhibitor-1 (PAI-1), is the principal inhibitor of tissue plasminogen activator (t-PA); it is higher in both OSA and CVD. OSA adversely affects circadian fibrinolytic balance, and t-PA has a pronounced circadian rhythm. This dysfunctional mechanism may be responsible for increased cardiovascular events in OSA patients [136]. Another important inflammatory marker of systemic inflammation that is elevated in OSA is CRP [137-140]. However, obese OSA patients may contain other elevated proinflammatory cytokines e.g. IL-6, as well as CRP [141]. Six months of CPAP significantly reduced CRP [142].
An interplay has been suggested between OSA, sleep fragmentation, and neuroinflammation that may cause OSA-induced brain injury [29,30]. There is accumulating evidence linking OSA with IR, glucose intolerance, and type2 diabetes [143-145]. Thus, OSA-induced proinflammatory cytokines such as TNF-α and IL-6 are associated with impaired glucose metabolism [143,144]. Hence, chronic OSA could upregulate sustained glucose hypometabolism, inflammatory response, hypertension [146], ischemia due to endothelial dysfunction [147-149], white matter damage [150], and apoptosis in the hippocampus [151,152]; conceivably, these would promote widespread pathology and trigger cognitive dysfunction.
Sleep loss and inflammation
There is insufficient sleep among people in our fast-paced stress-prone society. Sleep disturbance due to conditions such as OSA and short sleep duration increase inflammation. Insufficient sleep enhances blood pressure, glucose dysmetabolism, hormonal dysregulation, and inflammation [153]. Partial sleep deprivation (PSD) (compared with uninterrupted sleep) has been shown to cause significantly higher expression of TNF-α and IL-6 [154]. PSD also activated STAT family proteins; together they enhance the molecular inflammatory signaling pathways [154]. Circadian misalignment due to the wakefulness-sleep schedule perturbation conceivably causes a significant increase in TNF-α, and CRP [155,156]. CRP is a recognized general marker for inflammation [157,158]. Its serum concentration increases immediately after sleep restriction [159]. The CRP level increases, as expected, during both total and PSD conditions [158]. Indeed, long-term sleep restriction may cause increased production of cytokines including IL-β, IL-6, IL-17, as well as CRP [159]. One week of sleep restriction elevated not only mRNA concentrations of TNF-α and IL-1β but reduced endothelial-dependent vasodilatation in healthy individuals [160]. Interestingly, it has been documented recently that decreased sleep may decrease immune function and increase susceptibility to viral infections [161].
Recent data have found that Aβ pathology is associated with non-rapid eye movement (NREM) sleep disruption impacting memory function in older adults. The study found that Aβ burden in medial prefrontal cortex (mPFC) correlated significantly with less NREM sleep, and more memory impairment mediated by impaired hippocampus-dependent memory consolidation [162].
Comments
Disparate aspects of inflammation: LPS, proinflammatory cytokines, and other mediators
LPS (present in the cell wall of Gram-negative bacteria) - a potent endotoxin enhances the levels of proinflammatory cytokines and activates both the neuroimmune and neuroendocrine systems [49]. It also blocks LTP in the hippocampus. Wistar rats administered LPS for three days showed increased level of expression of TNF-α, IL-1β, and IL-6 in the hippocampus (compared with controls). Following seven days of administration, however, Aβ level also increased in the hippocampus. LPS-induced cognitive dysfunction was verified in Morris maze test [163]. Interestingly, even a single injection of LPS impaired hippocampal-dependent learning (spatial learning) [164].
LPS infusion (1.2 mg/kg/day intraperitoneal) for 14 days induced sustained hypertension and a significant increase in plasma level of TNF-α, IL-1β, and CRP. This LPS-induced systemic inflammation was accompanied by activation of microglia, augmentation of TNF-α, IL-1β, or IL-6 protein expression, and O2 production in rostral ventrolateral medulla (RVLM) [165]. The above mentioned were blunted by administration of a cycloxygenase-2 (COX-2) inhibitor, an inhibitor of microglial activation (NS398), a cytokine synthesis inhibitor (pentoxifylline), or minocycline [165]. Neuroinflammation was also associated with a COX-2-dependent downregulation of endothelial NO synthase. The LPS-related pressor response was antagonized by minocycline, pentoxifylline, or tempol (SOD mimetic) [165]. An increase in serum LPS activity is associated with a host of pathologies including higher serum triglyceride, obesity/metabolic syndrome, diabetes, increased blood pressure (mainly diastolic), and cardiometabolic disorders [19,20,31,166-169]. Thus, LPS is linked to the development of metabolic and vascular dysfunctions supporting its role in evoking an immune response in their pathogenesis [166,170,171].
Proinflammatory cytokines are key molecules that modulate immune responses. Their lack of reversibility in persistent inflammation would enhance dyshomeostasis [96]. LPS administration in the aged rats induces prolonged neuroinflammation and astrogliosis in the hippocampus (dentate gyrus) showing higher mRNA expression and protein levels of TNF-α and IL-1β [172]. The secretion of these proinflammatory cytokines increases several fold with daily consumption of alcohol in LPS-treated C57BL/6J mice [173]. Many disparate proinflammatory pathways may underpin aging and age-related diverse comorbid conditions (Figure 1). For example, the proinflammatory molecule eukaryotic translation initiation factor 5A (EIF5A) mediates stress-induced inflammation in diabetes [174]. EIF5A has been shown to upregulate inflammation by stimulating the translation of mRNA encoding inducible NO synthase (iNOS); the latter promotes inflammation-associated cell death. Inflammatory prevalence may be associated with the extracellular release of ATP that activates purinergic P2 receptors and promotes chronic inflammation [175].
Adipose tissue is an important source of inflammation and hypoxia triggers macrophage infiltration in adipose tissue [176-178]. The recurring cessation of breathing in OSA may promote adipose tissue hypoxia and upregulate inflammation in obesity. A major source of cytokine is adipose tissue; its expression of IL-6 is elevated in aging. Upon in vitro treatment of adipose tissue explants with LPS, IL-6 secretion was significantly increased in cultures from 24 months old aged C57BL/6 mice - compared to 4 months old young mice [179]. Further, treatment of these explants with physiological levels of IL-1β induced significant secretion of IL-6; this effect was age-dependent, reflecting a synergistic response of adipose tissues to IL-1β in the aged [179]. However, inflammation in obesity is also associated with elevated cortisol levels [180]. Such enhanced inflammatory response in the obese aged may impact and alter the risk of other age-related comorbid conditions. Healthy obese adults with exacerbated inflammation markers such as CRP and fibrinogen may possess higher fasting glucose (prediabetes). However, these individuals with prediabetes tend to have prehypertension also [181].
Greater levels of serum IL-6 and CRP were associated with faster rates of cognitive decline over nine years among cognitively intact community-dwelling older women. This finding has been correlated with microvascular changes leading to myelin damage, pathological perturbations in neuronal axons, decreased neuron propagation, and impaired processing speed [182].
6,542 middle-aged adults were followed for 19 years prospectively; participants with a history of alcohol abuse, however, showed increased odds of developing severe memory impairment later in life [183]. In the animals, ethanol consumption upregulated basal gene expression of TNF-α, IL-1β, IL-6, and iNOS [184]. Alcohol-induced gut mucosal injury leads to marked increase in the permeability of the gut mucosa to macromolecules such as LPS. Consequently, the release of proinflammatory mediators such as TNF-α, and infiltration of inflammatory cells, e.g., neutrophils, lead to endotoxemia and systemic inflammation [185]. The leaky gut and associated inflammation upregulate depression also [186]. An increase in expression of inflammasome components (i.e. nucleotide-binding domain leucine-rich repeat proteins or NOD-like receptors - NLRP1, NLRP3, and caspase recruitment domain-ASC) and proinflammatory cytokines (such as TNF-α, MCP-1) occurs in the brain of alcohol-fed mice. An increase in IL-1β in alcohol-fed mice further activates inflammasomes [187].
It is well established that cytokines mediate immune and inflammatory responses. TNF-α and IL1-β enhance their mRNA levels as well as affect mRNA levels of other proinflammatory cytokines [188]. Thus, TNF-α and IL1-β induce each other, and indeed their own production [189]. When these cytokines are released into the extracellular space, they stimulate nearby neurons to produce TNF-α and IL1-β. Thus, cytokines render these neurons pathological and dysfunctional. Indeed, pathological neurons produce proinflammatory cytokines and activate microglia [190-196].
The inflammatory process is integral to aging, obesity, IH, hypertension, malnutrition, diabetes, and depression (see above). Also, APOE4 genotype is known to potentiate the induction and regulation of inflammatory processes [197-199]. Furthermore, stress hormones may facilitate inflammation; the induction of TNF-α, IL-1β, IL-6, IL-8, IL-18, and CRP production may occur through the corticotropin-releasing hormone/substance P-histamine axis upregulation. Thus, a dysfunctional neuroendocrine-immune interface may play an important role in the pathogenesis of inflammation-related neuronal pathology and cognitive decline [200].
The effect of mild chronic Hhcy on proinflammatory cytokine levels in the brain, heart, and serum were investigated in rats. Results demonstrated an increase in TNF-α, IL-1β, IL-6, and the chemokine MCP-1 (i.e. CCL(2)) in the hippocampus, as well as an increase in IL-1β and IL-6 levels in the cerebral cortex. Also, an increase in prostaglandin E(2) in the hippocampus and serum of the rats has been documented [201]. These data suggest that homocysteine increase promotes inflammatory status that can contribute to neuronal dysfunctions [202,203] and memory deficit [204]. Recent studies have corroborated these conclusions. Vascular dementia (VaD) is a frequent comorbidity with AD and is estimated to occur in as many as 40% of AD patients. The heterogeneous causes of VaD may include chronic cerebral hypoperfusion, microhemorrhages, hemorrhagic infarcts, or ischemic infarcts. MRI and histopathology revealed the occurrence of significant microhemorrhage, neuroinflammation, and elevated interleukin IL-1β, TNF-α, and IL-6, and the matrix metalloproteinase 2 (MMP2) and MMP9 in the Hhcy mice brain [202]. The Hhcy mice, not surprisingly, showed spatial memory deficit (assessed by the radial-arm water maze), and an increased risk for the neurodegenerative disease.
It is clear from the above mentioned that chronic inflammatory milieu would promote both vascular inflammation and neuroinflammation. The endothelium is the largest receptor-effector end-organ and maintains vascular homeostasis. Inflammation-related endothelial dysfunction is an important perturbation that precedes and accompanies cerebral neuropathology. It leads to cognitive dysfunction as well as sudden cerebrovascular and adverse cardiovascular events. Several cell types infiltrate vasculature in inflammation; these include microglia, astrocytes, perivascular macrophages, and infiltrated peripheral immunocytes (T lymphocyte, B lymphocyte, and dendritic cells) [205,206]. Hypertension also is a major cause of vascular inflammation. Spontaneously hypertensive rats subjected to hypoperfusion (via middle cerebral artery occlusion) showed immune cells of the peripheral blood infiltrated in the ischemic brain. These cells included neutrophils, monocytes, macrophages, and myeloid dendritic cells, lymphatic dendritic cells, microglia, and T cells [207]. This cellular recruitment, therefore, can aggravate damage to the brain tissue by releasing cytotoxic mediators, increasing vascular permeability, and disrupting blood-brain barrier (BBB) [208].
Inflammation implicated in the etiopathogenesis of AD
Accumulating evidence supports the major role of inflammation in the etiopathogenesis of AD [209,210]. As described above, proinflammatory mediators activate various signaling pathways that lead to neuronal injury (Figure 1). Neurons, glial cells, and vasculature are all implicated as being different cerebral components of neuroinflammation in AD, as are synapses [211-213]. Aβ-induced microglial cell activation (see below) triggers TNF-α or IL1-β cytokine synthesis and promotes neuroinflammation [214-217]. Systemic inflammation can upregulate neuroinflammation [218,219]. In the preclinical stage of AD, systemic inflammation has been correlated with the development of AD [220,221]. Indeed, early neuroinflammation occurs in the amnestic mild cognitive impairment (aMCI), underscoring that this pathology is a feature of prodromal cognitive decline [212,222]. The expression of proinflammatory cytokines in middle age is an early risk factor and predictor of cognitive decline/AD in old age [223].
The underlying primary event in the etiopathogenesis of AD has been emphasized in the data presented by van Exel et al. [224]. They [224] studied characteristics associated with genetic risk preceding AD development. They compared middle-aged offsprings of AD patients with offsprings of cognitively intact non-AD parents. The offsprings of AD patients showed enhanced proinflammatory responses (and increased hypertension). A greater proinflammatory response to inflammatory challenge found in the offsprings of AD patients, therefore, reflected their elevated genetic risk, and the susceptibility/predisposition to AD in the future. This was in keeping with well-documented alterations in innate immunity/inflammation found by other workers [210]. (Also, see above). Other susceptibility genes for AD include CLU, PICALM, and CRI [225,226]; it is noteworthy that CLU codes for clusterin, and CRI, complement receptor 1, - and both are involved in inflammation/innate immunity.
It has been pointed out in the literature that LPS-induced inflammation promotes AD pathology by altering Aβ transport at the BBB [227] and decreasing the central clearance of Aβ [228]. Alteration of BBB effectively increases brain influx of Aβ but decreases its efflux [227]. It has been discussed above that low-grade systemic inflammation is rampant and accompanies aging and various comorbid diseases in AD [229,230]. Obesity (e.g.) in aging is associated with significant systemic inflammation, that disruption BBB [231]. The resulting neuroinflammation and oxidative stress in the hippocampus likely contribute to the significant cognitive decline observed in obese aged animals. Both peripheral and central increases in proinflammatory cytokine levels result in increased Aβ1-42 in the hippocampus and cognitive deficits [232,233].
The Honolulu-Asia Aging Study found that elevated CRP levels are related to a significantly increased risk for dementia [234]. The Rotterdam Study also reported a relationship between elevated levels of α-1 antichymotrypsin (ACT), CRP, IL-6, Intracellular adhesion molecule-1 (ICAM), and vascular cell adhesion molecule-1 (VCAM) - and an increased risk for AD [235]. Chronic inflammation (in 3xTg-AD mice) - related elevation of TNF signaling (in the hippocampus) [236], and intraneuronal amyloid and Ptau may promote neuronal death [236-238].
Inflammation-activated microglia and abeta exacerbation
Normally, Microglia is innate immune cells of the brain; they constantly scan the tissue, respond to pathological signals, and protect CNS homeostasis. They encapsulate pathogenic foci and remove apoptotic cells/debris thereby defending the CNS tissue integrity. In neurodegenerative conditions, however, activated microglia and macrophages (peripherally-derived) take on strong proinflammatory function and generate proinflammatory mediators - including TNF-α, IL1-β, chemokines, complement factors, and other neurotoxic factors such as NO, ROS, and proteolytic enzymes. They all enhance Aβ production and plaque accumulation. Although microglia should be beneficial by generating anti-Aβ antibodies and stimulating clearance of amyloid plaques; the activated microglia promote Aβ pathology. Indeed, proinflammatory microglia develops in aging, traumatic brain injury, and neurodegenerative disease [239].
AD is characterized by Aβ plaques, neuronal atrophy, and degeneration in the hippocampus and cortex. Chronic inflammation contributes to the onset and progression of the above mentioned AD-related pathologies. Following intraperitoneal administration of LPS, C57BL/6J mice showed significantly higher levels of Aβ1-42 in their hippocampus with cognitive deficits [232]. Similarly, neuroinflammation attenuated memory retrieval (specifically impairing context discrimination memory via disruption of pattern separation processes) in hippocampus-requiring tasks in Sprague-Dawley rats [240]. OSA is a low-grade inflammatory condition associated with neuronal degeneration [29,30] (see above). Exposure to IH induces low-grade neuroinflammation in the dorsal hippocampus of C57BL/6J mice. Chronic IH in conjunction with LPS elevated IL-6 mRNA, caused microglial changes, and enhanced cognitive impairment [241] (Figure 1).
A vicious cycle occurs between neuroinflammation and Aβ accumulation; activated microglia potentiates Aβ deposition and generates inflammatory mediators that in turn enhance Aβ level [242]. LPS administration induces a significant inflammatory response in both cortex and hippocampal formation in AD transgenic mice (see above); the resulting axonal pathology noted in these regions is secondary to increases in β-site of amyloid precursor protein cleaving enzyme (BACE-1) and soluble Aβ [212]. These AD-like data reflect that amyloidogenic axonal pathology and dendritic degeneration arise from LPS-induced neuroinflammation [212]. Other studies have also shown myelin/axonal injury in the cortex following LPS administration and an increase in IL-1, Aβ protein precursor (AβPP), and Aβ [243].
Inflammation, AD pathology, and oxidative stress
Oxidative damage is a common and early feature of AD and other neurodegenerative conditions. Neuroinflammation implicated in cognitive decline enhances ROS and reactive NO species (RNS) that are neurotoxic. Oxidative stress plays an important role in neural injury and cognitive impairments [59,60]. Oxidative stress can arise from several sources including disease state, IH/OSA, sleep restriction, unhealthy lifestyle including excessive caloric intake, malnutrition, and excessive alcohol consumption [29,30,59,60,146-149]. The nocturnal IH may induce the production of ROS and, therefore, cause local and systemic inflammation (Figure 1). ROS contributes to the “age-related cascade of neurodegeneration”; it contributes to accumulating oxidative damage in conjunction with protein aggregation, metabolic dysfunction, and inflammation [50-54,97].
Thiamine deficiency (TD) induces a region-selective neuronal loss in the brain, accompanied by impairment of oxidative metabolism [244]. Free radical injury to the brain was assessed using CSF isoprostane concentrations (with age, sex, race, cigarette smoking, BMI, APOE ε4 allele, and CSF biomarkers of AD as confounders). The results were consistent with an age-related increase in free radical injury in the human brain. The concentration of CSF isoprostane has been shown to increase with age by approximately 10% from age 45 to 71 years in healthy, cognitively normal adults. Also, the CSF isoprostane concentration also increases by approximately >10% for every 5-U increase in BMI, as well as other life style modifications (e.g. smoking) [245]. The above studies highlight the importance of lifestyle modification (reducing high BMI and smoking) in decreasing free radical injury to the brain [245].
There is a potential contribution by ROS to homocysteine-related neurotoxicity and neuronal damage. The latter derives from ROS as well as increased Ca2+ influx. ROS can be generated extracellularly by homocysteine [246] following excessive N-methyl-D-aspartate (NMDA) receptor stimulation [247,248]. Importantly, SOD and catalase may offer protection from neuronal damage [247,248].
Inflammation, AD pathology, and nitrosative stress
The OSA patients demonstrate decreased plasma levels of NO metabolites, and increased production of superoxide (by neutrophils and monocytes), isoprostane (in breath condensate) [249,250], and exhaled NO (eNO), carbon monoxide (eCO), nitrates, and hydrogen peroxide (H2O2) [251,252]. Oxidant stress and inflammation are potential mediators of IH-induced vascular dysfunction [253,254]. There is direct evidence that OSA is a major determinant of endothelial dysfunction, inflammation, and elevated oxidative stress in obese patients [254]. Indeed, both ROS and RNS may lead to a breakdown of endothelial-derived NO and exaggerated lipid peroxidation [255-257].
IH induces inflammation and results in significant oxidative injury in sleep-wake regions of the brain; this is associated with hypersomnolence and increased susceptibility to short-term sleep loss. Compared to mice exposed to sham IH, those exposed to IH develop reduced mean sleep latency. Following two weeks of IH, the oxidative injury was present in regions of the basal forebrain and brainstem reflected by elevated isoprostane (22%), increased protein carbonylation (50%), and increased nitration (200%) [258].
Neuroinflammation enhances both ROS and RNS species [259] (Figure 1); both are neurotoxic and induce mitochondrial damage, upregulate caspases, increase calcium, and promote AD and other neurodegenerative diseases [260-262]. IL-1β and TNF-α in combination caused marked neuronal injury; their synergistic action being required to disrupt memory consolidation [263]. Brain cell cultures treated with TNF-α and IL-1β generated substantial amounts of NO. Blockade of NO production with an NO synthase inhibitor was accompanied by a marked reduction (about 45%) of neuronal injury - suggesting that NO plays a significant role in neurotoxicity. The addition of NMDA receptor antagonists to the brain cell cultures also blocked TNF-α and IL-1β-induced neurotoxicity (by 55%), implicating the involvement of NMDA receptors in neurotoxicity [264]. Both intracellular and extracellular glutamate levels are elevated following TNF-α and IL-1β treatment. Pre-treatment with NMDA receptor antagonist MK-801 blocked cytokine-induced neurotoxicity [265].
Inflammation, AD pathology, and Tau
Tau phosphorylation and NFT are early hallmarks of AD. Neuroinflammation implicated in tau pathology [266] promotes tau, its aggregation and neurodegeneration in humans [267-270], as well as in animals [266,271-275] (Figure 1). The above-mentioned progression of tau pathology following induction of brain inflammation was elegantly exemplified in hTau mice [266,276]. Furthermore, blocking or enhancing IL1-β expression decreases [277] or increases [278] tau pathology, respectively. Importantly, the former ameliorates cognition in 3xTg-AD mice [277]. Several corroborative studies have documented inflammation preceding tau-mediated neuronal loss/neurodegeneration [271,279]. Based on the above studies, it is emphasized that inflammation and activated microglia are essential in driving tau pathology and memory impairment [280]. However, tau pathology in 3xTg-AD transgenic mice may develop independently of Aβ generation [281,282].
There is an age-related enhancement of Ptau species in the cortex and hippocampus of rTg4510 and other transgenic mice [281,282,289,290]. Hypoglycemia enhances the AMPK-Akt-GSK3 pathway and tau hyperphosphorylation [291]. The central angiotensin II elevates Ptau levels via glycogen synthase kinase 3β (GSK 3β) and other tau kinases. However, Ptau and associated cognitive impairment were attenuated by losartan and the GSK 3β inhibitor [292]. These three conditions mentioned above are associated with inflammation (see above). Indeed, neuroinflammation upregulates p38 mitogen-activated protein kinase (MAPK), GSK 3β [278], and cyclin-dependent kinase 5 activity which potentiate tau phosphorylation [297].
The hippocampus in transgenic pR5 mice (expressing the pathogenic mutation P301L in the human tau gene) has a higher turnover of glutamate and glutamine - reflecting a hypermetabolic state [293]. Several data have identified stimulation of inflammation signaling pathway within the brains undergoing tauopathy [294-296]. Consequently, either reducing tau [283-285] or blocking neuroinflammatory pathways may serve as therapeutic targets to attenuate neurotoxicity/neurodegeneration and cognitive decline [286-288].
Neuroprotection-therapeutic approach employing polypharmacy
AD is a multifactorial condition in which a combination of many pathological pathways act synergistically and sequentially, evoking the neurodegenerative milieu. A single drug may not be efficacious - proverbially as “one size does not fit all”. Hence, multicomponent approach (polypharmacy) may be required in potential therapeutic intervention to prevent neurodegeneration and affect AD pathology [298]. There are many therapeutic targets in the pathogenic cascade of inflammation (see above). These may include proinflammatory cytokines, complement system, oxidative stress, nitrosative stress, microglial activation, as well as amyloid and tau accumulation. The following suggested therapeutics may be required in a multicomponent treatment approach; indeed, they may be worthy of clinical trials.
1. It is essential that the immune homeostasis is maintained. High-dose intravenous immunoglobulin G (IgG) antibodies suppress inflammation. Its anti-inflammatory activity is associated with IgG crystallizable fragments [299]. Minocycline has also been shown to attenuate LPS-induced neuroinflammation [300] (see below).
2. MAPKs are implicated in the production of inflammation mediators. LPS treatment activates the MAPKs in both neurons and glia, promoting glia-derived neurotoxic molecules [301]. The p38 MAPK signaling cascade is activated in human AD brain tissue [302]; it is implicated in increasing proinflammatory cytokine levels by glia, following activation with Aβ. An experimental therapeutic “MW01-2-069A-SRM” - an inhibitor of p38 MAPK has been developed. This micromolecule is BBB penetrating, non-toxic, and orally bioavailable; it reversed higher proinflammatory cytokine levels in the hippocampus to normal level in the animal model [303].
3. Fructose-1, 6-bisphosphate (FBP - a glycolytic intermediate) reduced the expression of iNOS and inhibited LPS-induced NO production in a dose-dependent manner [305]. Although, FBP has anti-inflammatory and immunomodulatory properties, the underlying mechanisms of these functions have not been characterized.
4. The peroxisome proliferator activated receptor-gamma (PPAR-γ) agonist pioglitazone inhibits iNOS activity in neurons and NO generation by microglia - following LPS-induced pathology [304]. Pioglitazone inhibits LPS-induced phosphorylation of p38 MAPK [304] and inhibits the expression of proinflammatory genes. It negatively regulates microglial activation and APP processing with a 27% reduction in Aβ1-42 level [306]. This has been corroborated by subsequent studies. Treatment of the triple transgenic 3xTg-AD mouse with pioglitazone (for four months) resulted in reduced serum cholesterol, improved learning, decreased hippocampal Aβ and tau deposits, and improved short- and long-term plasticity [307]. While pioglitazone is ineffective in multiple sclerosis, it has documented effectiveness in suppressing oxidative stress, NFκB signal activation, inflammation, and neuronal degeneration in other inflammatory conditions [308].
There is controversy surrounding common non-steroidal anti-inflammatory (NSAID) regarding its effectiveness in protecting against AD; several anti-inflammatory treatment trials have shown little to no effect on preventing or reversing AD. However, there may be reduced AD incidence after 2 to 3 years of NSAID use in asymptomatic individuals [309]. This, therefore, reflects that in addition to innate inflammatory responses, there may be other concurrent pathological pathways that may require treatment [310].
5. MitoQ is an antioxidant that protects mitochondria from oxidative damage/stress. MitoQ is a derivative of the antioxidant ubiquinone, with antioxidant and anti-apoptotic properties. In inflammatory condition, MitoQ decreased mitochondrial ROS in mice; it suppressed the NLRP3 inflammasome activation that is responsible for the maturation of IL-1β and IL-18 [311,312]. It also decreased neurodegeneration by decreasing ROS production and lipid peroxidation, increasing MnSOD activity and glutathione levels, and reducing protein and DNA oxidation [313]. Similarly, treatment with the mitochondria-targeted antioxidant, MitoTEMPO inhibited gut barrier dysfunction and suppressed colitis, thus enhancing barrier function and inhibiting proinflammatory cytokine generation [314]. Another important compound studied was mitochondria-protecting agent acetyl-L-carnitine (ALC). ALC enhanced SOD and reduced oxidative damage to the BBB [315].
6. ROS and RNS upregulate glia-mediated inflammation and cause neuronal damage. Minocycline significantly decreases hypoxia-ischemia (HI)-induced brain injury, owing to suppression of microglial activation and inhibition of neurotoxic factors and iNOS. It inhibits oxidative stress evidenced by an 8-isoprostane decrease in the minocycline-treated HI rat brain [316]. It has been shown to suppress pro-inflammatory NO following hypoxic upregulation [317]. Other studies on HI model also found that that doxycycline (a derivative of tetracycline, similar to minocycline) significantly inhibits neuroinflammation in several brain regions including the frontal cortex, striatum, and hippocampus; doxycycline inhibits TNF-α and IL-1β and augments BDNF [318,319]. Interestingly, Aβ1-42 fibrils complexed with C1q (complement factor) upregulated proinflammatory cytokines in human microglia cell cultures, but minocycline decreased this production [320]. Furthermore, minocycline showed long-term neuroprotective property by improving cognitive impairment in the rat by inhibiting astrogliosis [321]. In a detailed analysis, it was shown that minocycline activated BDNF, and increased synaptic plasticity and synaptogenesis. Consequently, minocycline ameliorates cognitive deficits and upregulates neuroplasticity [322]. The above studies indicate that minocycline (and doxycycline) down-regulate microglial toxic factors and provide neuroprotection.
7. Rats subjected to IH (simulating OSA) show significant increases in levels of serum and hippocampal malondialdehyde (MDA, indicators of oxidative stress), mRNA levels of inflammatory mediators, and apoptotic cell death. Melatonin treatment significantly inhibited hippocampal MDA levels, and apoptosis was entirely prevented. It decreased expression of the inflammatory mediators including TNF-α, IL-1β, IL-6, iNOS, and cyclooxygenase-2, but enhanced expression of antioxidant enzymes including glutathione peroxidase, catalase, and copper/zinc SOD in the hippocampus. Thus, melatonin significantly attenuates oxidative stress and the pathogenesis of IH-induced hippocampal pathology [323-325]. Further, melatonin lowered both NO and eNOS and elevated the endothelial function [324]. The neuroprotective effect of melatonin is obvious from its attenuation of Aβ-mediated toxicity, and antioxidant and anti-amyloid effects. Further, it attenuated tau hyperphosphorylation [326]. A recent meta-analysis of randomized clinical trials concluded that “melatonin can be considered as a possible sole or add-on therapy in neurodegenerative disorders” [327]. From the above mentioned it is clear that the antioxidant, pro-mitochondrial (i.e. inhibiting mitochondrial dysfunction), anti-tau, and anti-amyloidogenic impact of melatonin recommends its utilization in aging, MCI, and AD - in conjunction with other selected therapeutic substances described here [328,329].
A polypharmacy therapeutic approach may include melatonin, minocycline, pioglitazone, scavengers of ROS and RNS, as well as those that ameliorate mitochondrial dysfunction. Such a treatment strategy may: (A) reduce the synthesis of inflammation mediators and oxidants at multiple levels, (B) inhibit pathways involved in proinflammatory cytokine, ROS, and RNS signaling, and (C) attenuate mitochondrial and synaptic dysfunction.
Conclusion
The greatest risk factor in the neuropathogenesis of AD is aging. Several age-related comorbid conditions are important additive risk factors in upregulating inflammation and cognitive decline in aging. One of the mechanisms whereby LPS induces brain injury involves activation of TLR-4 on immune cells in neuroinflammation. This initiates activation of inflammatory cells and a generalized inflammatory response; this then results in several pathologies in the CNS including hypoglycemia, white matter injury, and cerebral hypoperfusion [59,330,331].
There is upregulation of proinflammatory cytokines, acute phase reactants, complement molecules, and other inflammatory mediators in AD brains; these are said to contribute the progression of neurodegenerative process. Both astrocytes and microglia are primed to produce several neurotoxic factors in the chronic neurodegenerative state. Consequently, elevated cytokines and chemokines would heighten neuropathology in the vulnerable brain. Synergistic and superimposed insults from a host of sources (described above) would induce neuroinflammatory and neurodegenerative condition and escalate worse functional and memory/cognitive outcomes. Understandably, the mechanisms/pathways of the exaggerated pathological responses due to inflammation and oxidative-nitrosative stress should provide obvious potential targets for therapeutic intervention. The therapeutic neuroprotective strategy pointed out here suggests polypharmacy.
Finally, neuroinflammation exists in very early stages of AD. The neuronal toxicity promoted by chronic inflammation makes it a critical risk factor in the pathogenesis of neurodegenerative diseases in general and AD in particular. An essential goal for research today is to prevent and ameliorate inflammation and to reduce glial activation and neuronal toxicity/degeneration. A targeted therapeutic strategy to abrogate neuroinflammation is of paramount importance and may hold promise to prevent cognitive dysfunction and attenuate (possibly reverse) AD neuropathology.
Disclosure of conflict of interest
None.
References
- 1.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diniz BS, Teixeira AL, Ojopi EB, Talib LL, Mendonça VA, Gattaz WF, Forlenza OV. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J Alzheimers Dis. 2010;22:1305–1311. doi: 10.3233/JAD-2010-100921. [DOI] [PubMed] [Google Scholar]
- 3.Buchhave P, Zetterberg H, Blennow K, Minthon L, Janciauskiene S, Hansson O. Soluble TNF receptors are associated with Aβ metabolism and conversion to dementia in subjects with mild cognitive impairment. Neurobiol Aging. 2010;31:1877–1884. doi: 10.1016/j.neurobiolaging.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol. 2007;82:235–246. doi: 10.1016/S0074-7742(07)82012-5. [DOI] [PubMed] [Google Scholar]
- 5.Forlenza OV, Diniz BS, Talib LL, Mendonça VA, Ojopi EB, Gattaz WF, Teixeira AL. Increased Serum IL-1 Level in Alzheimer’s Disease and Mild Cognitive Impairment. Dement Geriatr Cogn Disord. 2009;28:507–512. doi: 10.1159/000255051. [DOI] [PubMed] [Google Scholar]
- 6.Di Bona D, Plaia A, Vasto S, Cavallone L, Lescai F, Franceschi C, Licastro F, Colonna-Romano G, Lio D, Candore G, Caruso C. Association between the interleukin-1beta polymorphisms and Alzheimer’s disease: a systematic review and meta-analysis. Brain Res Rev. 2008;59:155–163. doi: 10.1016/j.brainresrev.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Déniz-Naranjo MC, Muñoz-Fernandez C, Alemany-Rodríguez MJ, Pérez-Vieitez MC, Aladro-Benito Y, Irurita-Latasa J, Sánchez-García F. Cytokine IL-1 beta but not IL-1 alpha promoter polymorphism is associated with Alzheimer disease in a population from the Canary Islands, Spain. Eur J Neurol. 2008;15:1080–1084. doi: 10.1111/j.1468-1331.2008.02252.x. [DOI] [PubMed] [Google Scholar]
- 8.Jicha GA, Parisi JE, Dickson DW, Johnson K, Cha R, Ivnik RJ, Tangalos EG, Boeve BF, Knopman DS, Braak H, Petersen RC. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674–681. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 9.Petersen RC, Parisi JE, Dickson DW, Johnson KA, Knopman DS, Boeve BF, Jicha GA, Ivnik RJ, Smith GE, Tangalos EG, Braak H, Kokmen E. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 10.Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nature Medicine. 2012;18:1812–189. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- 11.von Bernhardi R, Cornejo F, Parada GE, Eugenín J. Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Front Cell Neurosci. 2015;9:426. doi: 10.3389/fncel.2015.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath I, Jia X, Johansson P, Wang C, Moskalenko R, Steinau A, Forsgren L, Wågberg T, Svensson J, Zetterberg H, Morozova-Roche LA. Pro-inflammatory S100A9 Protein as a Robust Biomarker Differentiating Early Stages of Cognitive Impairment in Alzheimer’s Disease. ACS Chem Neurosci. 2016;7:34–9. doi: 10.1021/acschemneuro.5b00265. [DOI] [PubMed] [Google Scholar]
- 13.Delaby C, Gabelle A, Blum D, Schraen-Maschke S, Moulinier A, Boulanghien J, Séverac D, Buée L, Rème T, Lehmann S. Central Nervous System and Peripheral Inflammatory Processes in Alzheimer’s Disease: Biomarker Profiling Approach. Front Neurol. 2015;6:181. doi: 10.3389/fneur.2015.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dursun E, Gezen-Ak D, Hanağası H, Bilgiç B, Lohmann E, Ertan S, Atasoy İL, Alaylıoğlu M, Araz ÖS, Önal B, Gündüz A, Apaydın H, Kızıltan G, Ulutin T, Gürvit H, Yılmazer S. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J Neuroimmunol. 2015;283:50–57. doi: 10.1016/j.jneuroim.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Cape E, Hall RJ, van Munster BC, de Vries A, Howie SE, Pearson A, Middleton SD, Gillies F, Armstrong IR, White TO, Cunningham C, de Rooij SE, MacLullich AM. Cerebrospinal fluid markers of neuroinflammation in delirium: a role for interleukin-1β in delirium after hip fracture. J Psychosom Res. 2014;77:219–225. doi: 10.1016/j.jpsychores.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P, Li YW, Wang XS, Zou X, Zhang DZ, Wang DX, Li SZ. High serum interleukin-6 level is associated with increased risk of delirium in elderly patients after noncardiac surgery: a prospective cohort study. Chin Med J (Engl) 2013;126:3621–3627. [PubMed] [Google Scholar]
- 17.Hennessy E, Griffin EW, Cunningham C. Astrocytes Are Primed by Chronic Neurodegeneration to Produce Exaggerated Chemokine and Cell Infiltration Responses to Acute Stimulation with the Cytokines IL-1β and TNF-α. J Neuroscience. 2015;35:8411–8422. doi: 10.1523/JNEUROSCI.2745-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fain JN. Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitam Horm. 2006;74:443–447. doi: 10.1016/S0083-6729(06)74018-3. [DOI] [PubMed] [Google Scholar]
- 19.Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50:2374–2383. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- 20.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 21.Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- 22.Daulatzai MA. Memory and Cognitive Dysfunctions in Alzheimer’s disease are Inextricably Intertwined with Neuroinflammation due to Aging, Obesity, Obstructive Sleep Apnea, and other Upstream Risk Factors. In: Costa A, Villalba E, editors. Horizons in Neuroscience Research. New York: Nova Science Publishers Inc; 2012. pp. 69–106. [Google Scholar]
- 23.Daulatzai MA. Neuroinflammation and Dysfunctional Nucleus Tractus Solitarius: Their Role in Neuropathogenesis of Alzheimer’s Dementia. Neurochem Res. 2012;37:846–868. doi: 10.1007/s11064-011-0680-2. [DOI] [PubMed] [Google Scholar]
- 24.Daulatzai MA. Multifactorial Pathologies Promote Inflammation and Enhance vulnerability to Late-Onset Alzheimer’s Disease: Implications for Possible Therapeutic Targets. In: Rahman A, editor. Frontiers in Clinical Drug Research - Alzheimer’s Disorder. Vol. 2. Sharjah: Bentham Publishers; 2014. pp. 103–154. [Google Scholar]
- 25.Lim SL, Rodriguez-Ortiz CJ, Kitazawa M. Infection, systemic inflammation, and Alzheimer’s disease. Microbes Infect. 2015;17:549–556. doi: 10.1016/j.micinf.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;13:e30. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- 27.Cape E, Hall RJ, van Munster BC, de Vries A, Howie SE, Pearson A, Middleton SD, Gillies F, Armstrong IR, White TO, Cunningham C, de Rooij SE, MacLullich AM. Cerebrospinal fluid markers of neuroinflammation in delirium: a role for interleukin-1β in delirium after hip fracture. J Psychosom Res. 2014;77:219–225. doi: 10.1016/j.jpsychores.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasunilashorn SM, Ngo L, Inouye SK, Libermann TA, Jones RN, Alsop DC, Guess J, Jastrzebski S, McElhaney JE, Kuchel GA, Marcantonio ER. Cytokines and Postoperative Delirium in Older Patients Undergoing Major Elective Surgery. J Gerontol A Biol Sci Med Sci. 2015;70:1289–1295. doi: 10.1093/gerona/glv083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daulatzai MA. Death by a Thousand Cuts in Alzheimer’s disease: Hypoxia - The Prodrome. Neurotox Res. 2013;24:216–243. doi: 10.1007/s12640-013-9379-2. [DOI] [PubMed] [Google Scholar]
- 30.Daulatzai MA. Evidence of neurodegeneration in obstructive sleep apnea: Relationship between obstructive sleep apnea and cognitive dysfunction in the elderly. J Neurosci Res. 2015;93:1778–1794. doi: 10.1002/jnr.23634. [DOI] [PubMed] [Google Scholar]
- 31.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 32.Goto T, Eden S, Nordenstam G, Sundh V, Svanborg-Eden C, Mattsby-Baltzer I. Endotoxin levels in sera of elderly individuals. Clin Diagn Lab Immunol. 1994;1:684–688. doi: 10.1128/cdli.1.6.684-688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3:e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szeto CC, Kwan BC, Chow KM, Lai KB, Chung KY, Leung CB, Li PK. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin J Am Soc Nephrol. 2008;3:431–436. doi: 10.2215/CJN.03600807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiedermann CJ, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease prospective results from the Bruneck Study. J Am Coll Cardiol. 1999;34:1975–1981. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 36.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lira FS, Rosa JC, Pimentel GD, Souza HA, Caperuto EC, Carnevali LC Jr, Seelaender M, Damaso AR, Oyama LM, de Mello MT, Santos RV. Endotoxin levels correlate positively with a sedentary lifestyle and negatively with highly trained subjects. Lipids Health Dis. 2010;9:82. doi: 10.1186/1476-511X-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 39.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31:817–844. doi: 10.1210/er.2009-0030. [DOI] [PubMed] [Google Scholar]
- 41.Maachi M, Pieroni L, Bruckert E, Jardel C, Fellahi S, Hainque B, Capeau J, Bastard JP. Systemic low-grade inflammation is related to both circulating and adipose tissue TNFalpha, leptin and IL-6 levels in obese women. Int J Obes Relat Metab Disord. 2004;28:993–997. doi: 10.1038/sj.ijo.0802718. [DOI] [PubMed] [Google Scholar]
- 42.Sun L, Yu Z, Ye X, Zou S, Li H, Yu D, Wu H, Chen Y, Dore J, Clement K, Hu FB, Lin X. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care. 2010;33:1925–1932. doi: 10.2337/dc10-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, Tabita-Martinez J, Sellers KF, Rickels MR, Reilly MP. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59:172–181. doi: 10.2337/db09-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terawaki H, Yokoyama K, Yamada Y, Maruyama Y, Iida R, Hanaoka K, Yamamoto H, Obata T, Hosoya T. Low-grade endotoxemia contributes to chronic inflammation in hemodialysis patients examination with a novel lipopolysaccharide detection method. Ther Apher Dial. 2010;14:477–482. doi: 10.1111/j.1744-9987.2010.00815.x. [DOI] [PubMed] [Google Scholar]
- 45.Laugerette F, Vors C, Geloen A, Chauvin MA, Soulage C, Lambert-Porcheron S, Peretti N, Alligier M, Burcelin R, Laville M, Vidal H, Michalski MC. Emulsified lipids increase endotoxemia possible role in early postprandial low-grade inflammation. J Nutr Biochem. 2011;22:53–59. doi: 10.1016/j.jnutbio.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 46.Moreno-Navarrete JM, Manco M, Ibanez J, Garcia-Fuentes E, Ortega F, Gorostiaga E, Vendrell J, Izquierdo M, Martinez C, Nolfe G, Ricart W, Mingrone G, Tinahones F, Fernandez-Real JM. Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int J Obes (Lond) 2010;34:240–249. doi: 10.1038/ijo.2009.242. [DOI] [PubMed] [Google Scholar]
- 47.Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappab and activator protein-1 possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maitra U, Deng H, Glaros T, Baker B, Capelluto DG, Li Z, Li L. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol. 2012;189:1014–1023. doi: 10.4049/jimmunol.1200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Víctor VM, Espulgues JV, Hernández-Mijares A, Rocha M. Oxidative stress and mitochondrial dysfunction in sepsis: a potential therapy with mitochondria-targeted antioxidants. Infect Disord Drug Targets. 2009;9:376–389. doi: 10.2174/187152609788922519. [DOI] [PubMed] [Google Scholar]
- 51.Bivalacqua TJ, Sussan TE, Gebska MA, Strong TD, Berkowitz DE, Biswal S, Burnett AL, Champion HC. Sildenafil inhibits superoxide formation and prevents endothelial dysfunction in a mouse model of secondhand smoke induced erectile dysfunction. J Urol. 2009;181:899–906. doi: 10.1016/j.juro.2008.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 53.Exline MC, Crouser ED. Mitochondrial mechanisms of sepsis induced organ failure. Frontiers Biosc. 2008;13:5031–5041. doi: 10.2741/3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowes DA, Webster NR, Murphy MP, Galley HF. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br J Anaesth. 2013;110:472–480. doi: 10.1093/bja/aes577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Emre Y, Hurtaud C, Nübel T, Criscuolo F, Ricquier D, Cassard-Doulcier AM. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J. 2007;402:271–278. doi: 10.1042/BJ20061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia MZ, Liang YL, Wang H, Chen X, Huang YY, Zhang ZH, Chen YH, Zhang C, Zhao M, Xu DX, Song LH. Melatonin modulates TLR4-mediated inflammatory genes through MyD88- and TRIF-dependent signaling pathways in lipopolysaccharide-stimulated RAW264.7 cells. J Pineal Res. 2012;53:325–334. doi: 10.1111/j.1600-079X.2012.01002.x. [DOI] [PubMed] [Google Scholar]
- 57.Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valdes AM, Glass D, Spector TD. Omics technologies and the study of human ageing. Nat Rev Genet. 2013;14:601–607. doi: 10.1038/nrg3553. [DOI] [PubMed] [Google Scholar]
- 59.Daulatzai MA. Quintessential Risk Factors: Their Role in Promoting Cognitive Dysfunction and Alzheimer’s disease. Neurochem Res. 2012;37:2627–2658. doi: 10.1007/s11064-012-0854-6. [DOI] [PubMed] [Google Scholar]
- 60.Daulatzai MA. Neurotoxic Saboteurs: Straws that Break the Hippo’s (Hippocampus) Back drive Cognitive Impairment and Alzheimer’s disease. Neurotox Res. 2013;24:407–459. doi: 10.1007/s12640-013-9407-2. [DOI] [PubMed] [Google Scholar]
- 61.Daulatzai MA. Chronic Functional Bowel Syndrome Enhances Gut-Brain Axis Dysfunction, Neuroinflammation, Cognitive Impairment and vulnerability to Dementia. Neurochem Res. 2014;39:624–644. doi: 10.1007/s11064-014-1266-6. [DOI] [PubMed] [Google Scholar]
- 62.Daulatzai MA. Non-celiac gluten sensitivity triggers gut dysbiosis, neuroinflammation, gut-brain axis dysfunction, and vulnerability for dementia. CNS Neurol Disord Drug Targets. 2015;14:110–131. doi: 10.2174/1871527314666150202152436. [DOI] [PubMed] [Google Scholar]
- 63.Gregersen I, Holm S, Dahl TB, Halvorsen B, Aukrust P. A focus on inflammation as a major risk factor for atherosclerotic cardiovascular diseases. Expert Rev Cardiovasc Ther. 2016;14:391–403. doi: 10.1586/14779072.2016.1128828. [DOI] [PubMed] [Google Scholar]
- 64.Krabbe KS, Pedersen M, Bruunsgaard H. Inflammatory mediators in the elderly. Exp Gerontol. 2004;39:687–699. doi: 10.1016/j.exger.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 65.Barrientos RM, Frank MG, Watkins LR, Maier SF. Memory impairments in healthy aging: Role of aging-induced microglial sensitization. Aging Dis. 2010;1:212–231. [PMC free article] [PubMed] [Google Scholar]
- 66.Barrientos RM, Frank MG, Watkins LR, Maier SF. Aging-related changes in neuroimmune-endocrine function: implications for hippocampal-dependent cognition. Horm Behav. 2012;62:219–227. doi: 10.1016/j.yhbeh.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van den Kommer TN, Dik MG, Comijs HC, Jonker C, Deeg DJH. Homocysteine and inflammation: Predictors of cognitive decline in older persons? Neurobiol Aging. 2010;31:1700–1709. doi: 10.1016/j.neurobiolaging.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 68.Ravaglia G, Forti P, Maioli F, Montesi F, Rietti E, Pisacane N, Rolfo E, Scali CR, Dalmonte E. Risk factors for dementia: data from the Conselice Study of brain aging. Arch Gerontol Geriatr. 2007;44:311–320. doi: 10.1016/j.archger.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 69.Ravaglia G, Forti P, Maioli F, Chiappelli M, Montesi F, Tumini E, Mariani E, Licastro F, Patterson C. Blood inflammatory markers and risk of dementia: the Conselice Study of Brain Aging. Neurobiol Aging. 2007;28:1810–1820. doi: 10.1016/j.neurobiolaging.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 70.Gibertini M. IL1 beta impairs relational but not procedural rodent learning in a water maze task. Adv Exp Med Biol. 1996;402:207–217. doi: 10.1007/978-1-4613-0407-4_27. [DOI] [PubMed] [Google Scholar]
- 71.Sparkman NL, Kohman RA, Garcia AK, Boehm GW. Peripheral lipopolysaccharide administration impairs two-way active avoidance conditioning in C57BL/6J mice. Physiol Behav. 2005;85:278–288. doi: 10.1016/j.physbeh.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 72.Sparkman NL, Buchanan JB, Heyen JR, Chen J, Beverly JL, Johnson RW. Interleukin-6 facilitates lipopolysaccharide-induced disruption in working memory and expression of other proinflammatory cytokines in hippocampal neuronal cell layers. J Neurosci. 2006;26:10709–107016. doi: 10.1523/JNEUROSCI.3376-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev. 1992;99:195–231. doi: 10.1037/0033-295x.99.2.195. [DOI] [PubMed] [Google Scholar]
- 74.Squire LR, Ojemann JG, Miezin FM, Petersen SE, Videen TO, Raichle ME. Activation of the hippocampus in normal humans: a functional anatomical study of memory. Proc Natl Acad Sci U S A. 1992;89:1837–1841. doi: 10.1073/pnas.89.5.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Sachdev PS, Wen W, Crawford JD, Brodaty H, Baune BT, Kochan NA, Slavin MJ, Reppermund S, Kang K, Trollor JN. The relationship between inflammatory markers and voxel-based gray matter volumes in nondemented older adults. Neurobiol Aging. 2016;37:138–46. doi: 10.1016/j.neurobiolaging.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 76.François A, Rioux Bilan A, Quellard N, Fernandez B, Janet T, Chassaing D, Paccalin M, Terro F, Page G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J Neuroinflammation. 2014;11:139. doi: 10.1186/s12974-014-0139-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bardou I, Brothers HM, Kaercher RM, Hopp SC, Wenk GL. Differential effects of duration and age on the consequences of neuroinflammation in the hippocampus. Neurobiol Aging. 2013;34:2293–2301. doi: 10.1016/j.neurobiolaging.2013.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu Z, Dong Y, Wang H, Culley DJ, Marcantonio ER, Crosby G, Tanzi RE, Zhang Y, Xie Z. Peripheral surgical wounding and age-dependent neuroinflammation in mice. PLoS One. 2014;9:e96752. doi: 10.1371/journal.pone.0096752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hopp SC, Royer S, Brothers HM, Kaercher RM, D’Angelo H, Bardou I, Wenk GL. Age-associated alterations in the time-dependent profile of pro- and anti-inflammatory proteins within the hippocampus in response to acute exposure to interleukin-1β. J Neuroimmunol. 2014;267:86–91. doi: 10.1016/j.jneuroim.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barrientos RM, Kitt MM, Watkins LR, Maier SF. Neuroinflammation in the normal aging hippocampus. Neuroscience. 2015;309:84–99. doi: 10.1016/j.neuroscience.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aubert A, Vega C, Dantzer R, Goodall G. Pyrogens specifically disrupt the acquisition of a task involving cognitive processing in the rat. Brain Behav Immun. 1995;9:129–148. doi: 10.1006/brbi.1995.1013. [DOI] [PubMed] [Google Scholar]
- 82.Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF. Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging. 2006;27:723–732. doi: 10.1016/j.neurobiolaging.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 83.Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, Takata M, Lever IJ, Nanchahal J, Fanselow MS, Maze M. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol. 2010;68:360–368. doi: 10.1002/ana.22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 85.Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- 86.Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2007;22:301–311. doi: 10.1016/j.bbi.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang Y, Henry CJ, Dantzer R, Johnson RW, Godbout JP. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol Aging. 2008;29:1744–1753. doi: 10.1016/j.neurobiolaging.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor J, Castanon N, Kelley KW, Dantzer R, Johnson RW. Aging Exacerbates Depressive-like Behavior in Mice in Response to Activation of the Peripheral Innate Immune System. Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wofford JL, Loehr LR, Schwartz E. Acute cognitive impairment in elderly ED patients: etiologies and outcomes. Am J Emerg Med. 1996;14:649–653. doi: 10.1016/S0735-6757(96)90080-7. [DOI] [PubMed] [Google Scholar]
- 90.Chiovenda P, Vincentelli GM, Alegiani F. Cognitive impairment in elderly ED patients: need for multidimensional assessment for better management after discharge. Am J Emerg Med. 2002;20:332–335. doi: 10.1053/ajem.2002.33785. [DOI] [PubMed] [Google Scholar]
- 91.Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–939. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chapman TR, Barrientos RM, Ahrendsen JT, Maier SF, Patterson SL. Synaptic correlates of increased cognitive vulnerability with aging: peripheral immune challenge and aging interact to disrupt theta-burst late-phase long-term potentiation in hippocampal area CA1. J Neurosci. 2010;30:7598–7603. doi: 10.1523/JNEUROSCI.5172-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cortese GP, Barrientos RM, Maier SF, Patterson SL. Aging and a peripheral immune challenge interact to reduce mature brain-derived neurotrophic factor and activation of TrkB, PLCgamma1, and ERK in hippocampal synaptoneurosomes. J Neurosci. 2011;31:4274–4279. doi: 10.1523/JNEUROSCI.5818-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Patterson SL. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology. 2015;96:11–8. doi: 10.1016/j.neuropharm.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu X, Wu Z, Hayashi Y, Nakanishi H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience. 2012;216:133–142. doi: 10.1016/j.neuroscience.2012.04.050. [DOI] [PubMed] [Google Scholar]
- 96.Ghosh S, Lertwattanarak R, Garduño Jde J, Galeana JJ, Li J, Zamarripa F, Lancaster JL, Mohan S, Hussey S, Musi N. Elevated muscle TLR4 expression and metabolic endotoxemia in human aging. J Gerontol A Biol Sci Med Sci. 2015;70:232–246. doi: 10.1093/gerona/glu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pou KM, Massaro JM, Hoffmann U, Vasan RS, Maurovich-Horvat P, Larson MG, Keaney JF Jr, Meigs JB, Lipinska I, Kathiresan S, Murabito JM, O’Donnell CJ, Benjamin EJ, Fox CS. Visceral and subcutaneous adipose tissue volumes are cross-sectionally related to markers of inflammation and oxidative stress: the Framingham Heart Study. Circulation. 2007;116:1234–1241. doi: 10.1161/CIRCULATIONAHA.107.710509. [DOI] [PubMed] [Google Scholar]
- 98.Speaker KJ, Fleshner M. Interleukin-1 beta: A potential link between stress and the development of visceral obesity. BMC Physiol. 2012;12:8. doi: 10.1186/1472-6793-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lubrano C, Valacchi G, Specchia P, Gnessi L, Rubanenko EP, Shuginina EA, Trukhanov AI, Korkina LG, De Luca C. Integrated Haematological Profiles of Redox Status, Lipid, and Inflammatory Protein Biomarkers in Benign Obesity and Unhealthy Obesity with Metabolic Syndrome. Oxid Med Cell Longev. 2015;2015:490613. doi: 10.1155/2015/490613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Despres JP. The insulin resistance-dyslipidemic syndrome of visceral obesity: effect on patients’ risk. Obes Res. 1998;6(Suppl 1):8S–17S. doi: 10.1002/j.1550-8528.1998.tb00683.x. [DOI] [PubMed] [Google Scholar]
- 101.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 102.Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS. Prevalence of obesity, diabetes, and obesity-related health risk factors. JAMA. 2003;289:76–79. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 103.Harris MM, Stevens J, Thomas N, Schreiner P, Folsom AR. Associations of fat distribution and obesity with hypertension in a bi-ethnic population: the ARIC study: Atherosclerosis Risk in Communities Study. Obes Res. 2000;8:516–524. doi: 10.1038/oby.2000.64. [DOI] [PubMed] [Google Scholar]
- 104.Wilson PW, D’Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med. 2002;162:1867–1872. doi: 10.1001/archinte.162.16.1867. [DOI] [PubMed] [Google Scholar]
- 105.Schernthaner GH, Schernthaner G. Insulin resistance and inflammation in the early phase of type 2 diabetes: potential for therapeutic intervention. Scand J Clin Lab Invest Suppl. 2005;240:30–40. doi: 10.1080/00365510500236119. [DOI] [PubMed] [Google Scholar]
- 106.Coppack SW. Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc. 2001;60:349–356. doi: 10.1079/pns2001110. [DOI] [PubMed] [Google Scholar]
- 107.Petrangeli E, Coroniti G, Brini AT, de Girolamo L, Stanco D, Niada S, Silecchia G, Morgante E, Lubrano C, Russo MA, Salvatori L. Hypoxia Promotes the Inflammatory Response and Stemness Features in Visceral Fat Stem Cells from Obese Subjects. J Cell Physiol. 2016;231:668–679. doi: 10.1002/jcp.25113. [DOI] [PubMed] [Google Scholar]
- 108.Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab. 2013;24:40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dorfman MD, Thaler JP. Hypothalamic inflammation and gliosis in obesity. Curr Opin Endocrinol Diabetes Obes. 2015;22:325–330. doi: 10.1097/MED.0000000000000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Waise TM, Toshinai K, Naznin F, NamKoong C, Md Moin AS, Sakoda H, Nakazato M. One-day high-fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem Biophys Res Commun. 2015;464:1157–1162. doi: 10.1016/j.bbrc.2015.07.097. [DOI] [PubMed] [Google Scholar]
- 111.Sasaki T. Age-Associated Weight Gain, Leptin, and SIRT1: A Possible Role for Hypothalamic SIRT1 in the Prevention of Weight Gain and Aging through Modulation of Leptin Sensitivity. Front Endocrinol (Lausanne) 2015;6:109. doi: 10.3389/fendo.2015.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ornellas F, Souza-Mello V, Mandarim-de-Lacerda CA, Aguila MB. Combined parental obesity augments single-parent obesity effects on hypothalamus inflammation, leptin signaling (JAK/STAT), hyperphagia, and obesity in the adult mice offspring. Physiol Behav. 2016;153:47–55. doi: 10.1016/j.physbeh.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 113.Jialal I, Rajamani U. Endotoxemia of metabolic syndrome: a pivotal mediator of meta-inflammation. Metab Syndr Relat Disord. 2014;12:454–456. doi: 10.1089/met.2014.1504. [DOI] [PubMed] [Google Scholar]
- 114.Greenfield JR, Campbell LV. Relationship between inflammation, insulin resistance and type 2 diabetes: ‘cause or effect’? Curr Diabetes Rev. 2006;2:195–211. doi: 10.2174/157339906776818532. [DOI] [PubMed] [Google Scholar]
- 115.Siemińska L, Wojciechowska C, Walczak K, Borowski A, Marek B, Nowak M, Kajdaniuk D, Foltyn W, Kos-Kudła B. Associations between metabolic syndrome, serum thyrotropin, and thyroid antibodies status in postmenopausal women, and the role of interleukin-6. Endokrynol Pol. 2015;66:394–403. doi: 10.5603/EP.2015.0049. [DOI] [PubMed] [Google Scholar]
- 116.Sjoholm A, Nystrom T. Inflammation and the etiology of type 2 diabetes. Diabetes Metab Res Rev. 2006;22:4–10. doi: 10.1002/dmrr.568. [DOI] [PubMed] [Google Scholar]
- 117.Luchsinger JA. Type 2 diabetes and cognitive impairment: linking mechanisms. J Alzheimers Dis. 2012;30(Suppl 2):S185–198. doi: 10.3233/JAD-2012-111433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72:49–66. doi: 10.2165/11597760-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li X, Song D, Leng SX. Link between type 2 diabetes and Alzheimer’s disease: from epidemiology to mechanism and treatment. Clin Interven Aging. 2015;10:1789–1791. doi: 10.2147/CIA.S74042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Arnaud C, Dematteis M, Pepin JL, Baguet JP, Lévy P. Obstructive sleep apnea, immuno-inflammation, and atherosclerosis. Semin Immunopathol. 2009;31:113–125. doi: 10.1007/s00281-009-0148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arnaud C, Poulain L, Lévy P, Dematteis M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis. 2011;219:425–431. doi: 10.1016/j.atherosclerosis.2011.07.122. [DOI] [PubMed] [Google Scholar]
- 122.May AM, Mehra R. Obstructive sleep apnea: role of intermittent hypoxia and inflammation. Semin Respir Crit Care Med. 2014;35:531–544. doi: 10.1055/s-0034-1390023. [DOI] [PubMed] [Google Scholar]
- 123.Iturriaga R, Moya EA, Del Rio R. Inflammation and oxidative stress during intermittent hypoxia: the impact on chemoreception. Exp Physiol. 2015;100:149–155. doi: 10.1113/expphysiol.2014.079525. [DOI] [PubMed] [Google Scholar]
- 124.Thunström E, Glantz H, Fu M, Yucel-Lindberg T, Petzold M, Lindberg K, Peker Y. Increased inflammatory activity in nonobese patients with coronary artery disease and obstructive sleep apnea. Sleep. 2015;38:463–471. doi: 10.5665/sleep.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Unnikrishnan D, Jun J, Polotsky V. Inflammation in sleep apnea: an update. Rev Endocr Metab Disord. 2015;16:25–34. doi: 10.1007/s11154-014-9304-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wu J, Stefaniak J, Hafner C, Schramel JP, Kaun C, Wojta J, Ullrich R, Tretter VE, Markstaller K, Klein KU. Intermittent Hypoxia Causes Inflammation and Injury to Human Adult Cardiac Myocytes. Anesth Analg. 2016;122:373–80. doi: 10.1213/ANE.0000000000001048. [DOI] [PubMed] [Google Scholar]
- 127.Taylor CT, Kent BD, Crinion SJ, McNicholas WT, Ryan S. Human adipocytes are highly sensitive to intermittent hypoxia induced NF-kappaB activity and subsequent inflammatory gene expression. Biochem Biophys Res Commun. 2014;447:660–665. doi: 10.1016/j.bbrc.2014.04.062. [DOI] [PubMed] [Google Scholar]
- 128.He Q, Yang QC, Zhou Q, Zhu H, Niu WY, Feng J, Wang Y, Cao J, Chen BY. Effects of varying degrees of intermittent hypoxia on proinflammatory cytokines and adipokines in rats and 3T3-L1 adipocytes. PLoS One. 2014;9:e86326. doi: 10.1371/journal.pone.0086326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nadeem R, Molnar J, Madbouly EM, Nida M, Aggarwal S, Sajid H, Naseem J, Loomba R. Serum inflammatory markers in obstructive sleep apnea: a meta-analysis. J Clin Sleep Med. 2013;9:1003–1012. doi: 10.5664/jcsm.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang H, Cao H, Wang P, Liu W, Cao F, Chen J. Tumour necrosis factor-α/interleukin-10 ratio in patients with obstructive sleep apnoea hypopnoea syndrome. J Laryngol Otol. 2015;129:73–78. doi: 10.1017/S0022215114002990. [DOI] [PubMed] [Google Scholar]
- 131.Li S, Qian XH, Zhou W, Zhang Y, Feng J, Wan NS, Zhang Z, Guo R, Chen BY. Time-dependent inflammatory factor production and NFκB activation in a rodent model of intermittent hypoxia. Swiss Med Wkly. 2011;141:w13309. doi: 10.4414/smw.2011.13309. [DOI] [PubMed] [Google Scholar]
- 132.Hanikoglu F, Huseyinoglu N, Ozben S, Cort A, Ozdem S, Ozben T. Increased plasma soluble tumor necrosis factor receptor-1 and myeloperoxidase activity in patients with obstructive sleep apnea syndrome. Int J Neurosci. 2015;125:655–662. doi: 10.3109/00207454.2014.960521. [DOI] [PubMed] [Google Scholar]
- 133.Arias MA, García-Río F, Alonso-Fernández A, Hernanz A, Hidalgo R, Martínez-Mateo V, Bartolomé S, Rodríguez-Padial L. CPAP decreases plasma levels of soluble tumour necrosis factor-alpha receptor 1 in obstructive sleep apnoea. Eur Respir J. 2008;32:1009–1015. doi: 10.1183/09031936.00007008. [DOI] [PubMed] [Google Scholar]
- 134.Yue HJ, Mills PJ, Ancoli-Israel S, Loredo JS, Ziegler MG, Dimsdale JE. The roles of TNF-alpha and the soluble TNF receptor I on sleep architecture in OSA. Sleep Breath. 2009;13:263–269. doi: 10.1007/s11325-008-0242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shamsuzzaman A, Amin RS, Calvin AD, Davison D, Somers VK. Severity of obstructive sleep apnea is associated with elevated plasma fibrinogen in otherwise healthy patients. Sleep Breath. 2014;18:761–766. doi: 10.1007/s11325-014-0938-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bagai K, Muldowney JA 3rd, Song Y, Wang L, Bagai J, Artibee KJ, Vaughan DE, Malow BA. Circadian variability of fibrinolytic markers and endothelial function in patients with obstructive sleep apnea. Sleep. 2014;37:359–367. doi: 10.5665/sleep.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lui MM, Lam JC, Mak HK, Xu A, Ooi C, Lam DC, Mak JC, Khong PL, Ip MS. C-reactive protein is associated with obstructive sleep apnea independent of visceral obesity. Chest. 2009;135:950–956. doi: 10.1378/chest.08-1798. [DOI] [PubMed] [Google Scholar]
- 138.Chien MY, Lee P, Tsai YF, Yang PC, Wu YT. C-reactive protein and heart rate recovery in middle-aged men with severe obstructive sleep apnea. Sleep Breath. 2012;16:629–637. doi: 10.1007/s11325-011-0549-2. [DOI] [PubMed] [Google Scholar]
- 139.Guven SF, Turkkani MH, Ciftci B, Ciftci TU, Erdogan Y. The relationship between high-sensitivity C-reactive protein levels and the severity of obstructive sleep apnea. Sleep Breath. 2012;16:217–221. doi: 10.1007/s11325-011-0492-2. [DOI] [PubMed] [Google Scholar]
- 140.Guo Y, Pan L, Ren D, Xie X. Impact of continuous positive airway pressure on C-reactive protein in patients with obstructive sleep apnea: a meta-analysis. Sleep Breath. 2013;17:495–503. doi: 10.1007/s11325-012-0722-2. [DOI] [PubMed] [Google Scholar]
- 141.Arnardottir ES, Maislin G, Schwab RJ, Staley B, Benediktsdottir B, Olafsson I, Juliusson S, Romer M, Gislason T, Pack AI. The interaction of obstructive sleep apnea and obesity on the inflammatory markers C-reactive protein and interleukin-6: the Icelandic Sleep Apnea Cohort. Sleep. 2012;35:921–932. doi: 10.5665/sleep.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schiza SE, Mermigkis C, Panagiotis P, Bouloukaki I, Kallergis E, Tzanakis N, Tzortzaki E, Vlachaki E, Siafakas NM. C-reactive protein evolution in obstructive sleep apnoea patients under CPAP therapy. Eur J Clin Invest. 2010;40:968–975. doi: 10.1111/j.1365-2362.2010.02348.x. [DOI] [PubMed] [Google Scholar]
- 143.Pallayova M, Steele KE, Magnuson TH, Schweitzer MA, Hill NR, Bevans-Fonti S, Schwartz AR. Sleep apnea predicts distinct alterations in glucose homeostasis and biomarkers in obese adults with normal and impaired glucose metabolism. Cardiovasc Diabetol. 2010;9:83. doi: 10.1186/1475-2840-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Appleton SL, Vakulin A, McEvoy RD, Wittert GA, Martin SA, Grant JF, Taylor AW, Antic NA, Catcheside PG, Adams RJ. Nocturnal Hypoxemia and Severe Obstructive Sleep Apnea are Associated with Incident Type 2 Diabetes in a Population Cohort of Men. J Clin Sleep Med. 2015;11:609–614. doi: 10.5664/jcsm.4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kent BD, McNicholas WT, Ryan S. Insulin resistance, glucose intolerance and diabetes mellitus in obstructive sleep apnoea. J Thorac Dis. 2015;7:1343–1357. doi: 10.3978/j.issn.2072-1439.2015.08.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Parati G, Ochoa JE, Bilo G, Mattaliano P, Salvi P, Kario K, Lombardi C. Obstructive sleep apnea syndrome as a cause of resistant hypertension. Hypertens Res. 2014;37:601–613. doi: 10.1038/hr.2014.80. [DOI] [PubMed] [Google Scholar]
- 147.Badran M, Golbidi S, Devlin A, Ayas N, Laher I. Chronic intermittent hypoxia causes endothelial dysfunction in a mouse model of diet-induced obesity. Sleep Med. 2014;15:596–602. doi: 10.1016/j.sleep.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 148.Andaku DK, D’Almeida V, Carneiro G, Hix S, Tufik S, Togeiro SM. Sleepiness, inflammation and oxidative stress markers in middle-aged males with obstructive sleep apnea without metabolic syndrome: a cross-sectional study. Respir Res. 2015;16:3. doi: 10.1186/s12931-015-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang J, Yu W, Gao M, Zhang F, Gu C, Yu Y, Wei Y. Impact of Obstructive Sleep Apnea Syndrome on Endothelial Function, Arterial Stiffening, and Serum Inflammatory Markers: An Updated Meta-analysis and Metaregression of 18 Studies. J Am Heart Assoc. 2015;4:e002454. doi: 10.1161/JAHA.115.002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen HL, Lu CH, Lin HC, Chen PC, Chou K, Lin WM, Tsai NW, Su YJ, Friedman M, Lin CP, Lin WC. White matter damage and systemic inflammation in obstructive sleep apnea. Sleep. 2015;38:361–370. doi: 10.5665/sleep.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sapin E, Peyron C, Roche F, Gay N, Carcenac C, Savasta M, Levy P, Dematteis M. Chronic Intermittent Hypoxia Induces Chronic Low-Grade Neuroinflammation in the Dorsal Hippocampus of Mice. Sleep. 2015;38:1537–1546. doi: 10.5665/sleep.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yuan X, Guo X, Deng Y, Zhu D, Shang J, Liu H. Chronic intermittent hypoxia-induced neuronal apoptosis in the hippocampus is attenuated by telmisartan through suppression of iNOS/NO and inhibition of lipid peroxidation and inflammatory responses. Brain Res. 2015;1596:48–57. doi: 10.1016/j.brainres.2014.11.035. [DOI] [PubMed] [Google Scholar]
- 153.Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. 2009;51:294–302. doi: 10.1016/j.pcad.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Irwin MR, Witarama T, Caudill M, Olmstead R, Breen EC. Sleep loss activates cellular inflammation and signal transducer and activator of transcription (STAT) family proteins in humans. Brain Behav Immun. 2015;47:86–92. doi: 10.1016/j.bbi.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Carroll JE, Carrillo C, Olmstead R, Witarama T, Breen EC, Yokomizo M, Seeman T, Irwin MR. Sleep deprivation and divergent toll-like receptor-4 activation of cellular inflammation in aging. Sleep. 2015;38:205–211. doi: 10.5665/sleep.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wright KP Jr, Drake AL, Frey DJ, Fleshner M, Desouza CA, Gronfier C, Czeisler CA. Influence of sleep deprivation and circadian misalignment on cortisol, inflammatory markers, and cytokine balance. Brain Behav Immun. 2015;47:24–34. doi: 10.1016/j.bbi.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 158.Meier-Ewert HK, Ridker PM, Rifai N, Regan MM, Price NJ, Dinges DF, Mullington JM. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004;43:678–683. doi: 10.1016/j.jacc.2003.07.050. [DOI] [PubMed] [Google Scholar]
- 159.van Leeuwen WM, Lehto M, Karisola P, Lindholm H, Luukkonen R, Sallinen M, Härmä M, Porkka-Heiskanen T, Alenius H. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLoS One. 2009;4:e4589. doi: 10.1371/journal.pone.0004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sauvet F, Drogou C, Bougard C, Arnal PJ, Dispersyn G, Bourrilhon C, Rabat A, Van Beers P, Gomez-Merino D, Faraut B, Leger D, Chennaoui M. Vascular response to 1 week of sleep restriction in healthy subjects. A metabolic response? Int J Cardiol. 2015;190:246–255. doi: 10.1016/j.ijcard.2015.04.119. [DOI] [PubMed] [Google Scholar]
- 161.Prather AA, Janicki-Deverts D, Hall MH, Cohen S. Behaviorally assessed sleep and susceptibility to the common cold. Sleep. 2015;38:1353–1359. doi: 10.5665/sleep.4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18:1051–1057. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhu B, Wang ZG, Ding J, Liu N, Wang DM, Ding LC, Yang C. Chronic lipopolysaccharide exposure induces cognitive dysfunction without affecting BDNF expression in the rat hippocampus. Exp Ther Med. 2014;7:750–754. doi: 10.3892/etm.2014.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shaw KN, Commins S, O’Mara SM. Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behav Brain Res. 2001;124:47–54. doi: 10.1016/s0166-4328(01)00232-7. [DOI] [PubMed] [Google Scholar]
- 165.Wu KL, Chan SH, Chan JY. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation. 2012;9:212. doi: 10.1186/1742-2094-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lassenius MI, Pietiläinen KH, Kaartinen K, Pussinen PJ, Syrjänen J, Forsblom C, Pörsti I, Rissanen A, Kaprio J, Mustonen J, Groop PH, Lehto M. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011;34:1809–1815. doi: 10.2337/dc10-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Boroni Moreira AP, de Cássia Gonçalves Alfenas R. The influence of endotoxemia on the molecular mechanisms of insulin resistance. Nutr Hosp. 2012;27:382–390. doi: 10.1590/S0212-16112012000200007. [DOI] [PubMed] [Google Scholar]
- 168.Boutagy NE, McMillan RP, Frisard MI, Hulver MW. Metabolic endotoxemia with obesity: Is it real and is it relevant? Biochimie. 2016;124:11–20. doi: 10.1016/j.biochi.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kallio KA, Hätönen KA, Lehto M, Salomaa V, Männistö S, Pussinen PJ. Endotoxemia, nutrition, and cardiometabolic disorders. Acta Diabetol. 2015;52:395–304. doi: 10.1007/s00592-014-0662-3. [DOI] [PubMed] [Google Scholar]
- 170.Bradlow HL. Obesity and the gut microbiome: pathophysiological aspects. Horm Mol Biol Clin Investig. 2014;17:53–61. doi: 10.1515/hmbci-2013-0063. [DOI] [PubMed] [Google Scholar]
- 171.Zhou X, Han D, Xu R, Li S, Wu H, Qu C, Wang F, Wang X, Zhao Y. A model of metabolic syndrome and related diseases with intestinal endotoxemia in rats fed a high fat and high sucrose diet. PLoS One. 2014;9:e115148. doi: 10.1371/journal.pone.0115148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fu HQ, Yang T, Xiao W, Fan L, Wu Y, Terrando N, Wang TL. Prolonged neuroinflammation after lipopolysaccharide exposure in aged rats. PLoS One. 2014;9:e106331. doi: 10.1371/journal.pone.0106331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Maier B, Ogihara T, Trace AP, Tersey SA, Robbins RD, Chakrabarti SK, Nunemaker CS, Stull ND, Taylor CA, Thompson JE, Dondero RS, Lewis EC, Dinarello CA, Nadler JL, Mirmira RG. The unique hypusine modification of eIF5A promotes islet beta cell inflammation and dysfunction in mice. J Clin Invest. 2010;120:2156–2170. doi: 10.1172/JCI38924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509:310–317. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH, Smith SR. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. 2009;58:718–725. doi: 10.2337/db08-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Netzer N, Gatterer H, Faulhaber M, Burtscher M, Pramsohler S, Pesta D. Hypoxia, Oxidative Stress and Fat. Biomolecules. 2015;5:1143–1150. doi: 10.3390/biom5021143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wood IS, de Heredia FP, Wang B, Trayhurn P. Cellular hypoxia and adipose tissue dysfunction in obesity. Proc Nutr Soc. 2009;68:370–377. doi: 10.1017/S0029665109990206. [DOI] [PubMed] [Google Scholar]
- 179.Starr ME, Saito M, Evers BM, Saito H. Age-Associated Increase in Cytokine Production During Systemic Inflammation-II: The Role of IL-1β in Age-Dependent IL-6 Upregulation in Adipose Tissue. J Gerontol A Biol Sci Med Sci. 2015;70:1508–1515. doi: 10.1093/gerona/glu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Laudisio A, Bandinelli S, Gemma A, Ferrucci L, Incalzi RA. Associations of Heart Rate with Inflammatory Markers Are Modulated by Gender and Obesity in Older Adults. J Gerontol A Biol Sci Med Sci. 2015;70:899–904. doi: 10.1093/gerona/glu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gupta AK, Johnson WD. Prediabetes and prehypertension in disease free obese adults correlate with an exacerbated systemic proinflammatory milieu. J Inflamm (Lond) 2010;7:36. doi: 10.1186/1476-9255-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Palta P, Xue QL, Deal JA, Fried LP, Walston JD, Carlson MC. Interleukin-6 and C-Reactive Protein Levels and 9-Year Cognitive Decline in Community-Dwelling Older Women: The Women’s Health and Aging Study II. J Gerontol A Biol Sci Med Sci. 2015;70:873–878. doi: 10.1093/gerona/glu132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kuźma E, Llewellyn DJ, Langa KM, Wallace RB, Lang IA. History of alcohol use disorders and risk of severe cognitive impairment: a 19-year prospective cohort study. Am J Geriatr Psychiatry. 2014;22:1047–1054. doi: 10.1016/j.jagp.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fleming S, Toratani S, Shea-Donohue T, Kashiwabara Y, Vogel SN, Metcalf ES. Pro- and anti-inflammatory gene expression in the murine small intestine and liver after chronic exposure to alcohol. Alcohol Clin Exp Res. 2001;25:579–589. [PubMed] [Google Scholar]
- 185.Wang HJ, Zakhari S, Jung MK. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J Gastroenterol. 2010;16:1304–1313. doi: 10.3748/wjg.v16.i11.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Leclercq S, Cani PD, Neyrinck AM, Stärkel P, Jamar F, Mikolajczak M, Delzenne NM, de Timary P. Role of intestinal permeability and inflammation in the biological and ignalling control of alcohol-dependent subjects. Brain Behav Immun. 2012;26:911–918. doi: 10.1016/j.bbi.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 187.Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, Szabo G. Alcohol-induced IL-1β in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leukoc Biol. 2013;94:171–182. doi: 10.1189/jlb.1212659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Churchill L, Taishi P, Wang M, Brandt J, Cearley C, Rehman A, Krueger JM. Brain distribution of cytokine mRNA induced by systemic administration of interleukin-1beta or tumor necrosis factor alpha. Brain Res. 2006;1120:64–73. doi: 10.1016/j.brainres.2006.08.083. [DOI] [PubMed] [Google Scholar]
- 189.Vitkovic L, Konsman JP, Bockaert J, Dantzer R, Homburger V, Jacque C. Cytokine signals propagate through the brain. Mol Psychiatry. 2000;5:604–615. doi: 10.1038/sj.mp.4000813. [DOI] [PubMed] [Google Scholar]
- 190.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 191.Friedman WJ. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp Neurol. 2001;168:23–31. doi: 10.1006/exnr.2000.7595. [DOI] [PubMed] [Google Scholar]
- 192.Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine. 2004;29:1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- 193.Ignatowski TA, Noble BK, Wright JR, Gorfien JL, Heffner RR, Spengler RN. Neuronal-associated tumor necrosis factor (TNF alpha): its role in noradrenergic functioning and modification of its expression following antidepressant drug administration. J Neuroimmunol. 1997;79:84–90. doi: 10.1016/s0165-5728(97)00107-0. [DOI] [PubMed] [Google Scholar]
- 194.Ji JF, Dheen ST, Kumar SD, He BP, Tay SS. Expressions of cytokines and chemokines in the dorsal motor nucleus of the vagus nerve after right vagotomy. Brain Res Mol Brain Res. 2005;142:47–57. doi: 10.1016/j.molbrainres.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 195.Figiel I, Dzwonek K. TNFalpha and TNF receptor 1 expression in the mixed neuronal-glial cultures of hippocampal dentate gyrus exposed to glutamate or trimethyltin. Brain Res. 2007;1131:17–28. doi: 10.1016/j.brainres.2006.10.095. [DOI] [PubMed] [Google Scholar]
- 196.Leyva-Grado VH, Churchill L, Wu M, Williams TJ, Taishi P, Majde JA, Krueger JM. Influenza virus- and cytokine-immunoreactive cells in the murine olfactory and central autonomic nervous systems before and after illness onset. J Neuroimmunol. 2009;211:73–83. doi: 10.1016/j.jneuroim.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS. Association of interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann Neurol. 2000;47:365–368. [PMC free article] [PubMed] [Google Scholar]
- 198.Culpan D, Cram D, Chalmers K, Cornish A, Palmer L, Palmer J, Hughes A, Passmore P, Craig D, Wilcock GK, Kehoe PG, Love S. TNFR-associated factor-2 (TRAF-2) in Alzheimer’s disease. Neurobiol Aging. 2009;30:1052–1060. doi: 10.1016/j.neurobiolaging.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 199.Horsburgh K, McCulloch J, Nilsen M, Roses AD, Nicoll JA. Increased neuronal damage and apoE immunoreactivity in human apolipoprotein E, E4 isoform-specific, transgenic mice after global cerebral ischaemia. Eur J Neurosci. 2000;12:4309–4317. [PubMed] [Google Scholar]
- 200.Elenkov IJ, Iezzoni DG, Daly A, Harris AG, Chrousos GP. Cytokine dysregulation, inflammation and well-being. Neuroimmunomodulation. 2005;12:255–269. doi: 10.1159/000087104. [DOI] [PubMed] [Google Scholar]
- 201.da Cunha AA, Ferreira AG, Loureiro SO, da Cunha MJ, Schmitz F, Netto CA, Wyse AT. Chronic hyperhomocysteinemia increases inflammatory markers in hippocampus and serum of rats. Neurochem Res. 2012;37:1660–1669. doi: 10.1007/s11064-012-0769-2. [DOI] [PubMed] [Google Scholar]
- 202.Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab. 2013;33:708–715. doi: 10.1038/jcbfm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Scherer EB, Loureiro SO, Vuaden FC, da Cunha AA, Schmitz F, Kolling J, Savio LE, Bogo MR, Bonan CD, Netto CA, Wyse AT. Mild Hyperhomocysteinemia Increases Brain Acetylcholinesterase and Proinflammatory Cytokine Levels in Different Tissues. Mol Neurobiol. 2014;50:589–596. doi: 10.1007/s12035-014-8660-6. [DOI] [PubMed] [Google Scholar]
- 204.Streck EL, Bavaresco CS, Netto CA, Wyse AT. Chronic hyperhomocysteinemia provokes a memory deficit in rats in the Morris water maze task. Behav Brain Res. 2004;153:377–381. doi: 10.1016/j.bbr.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 205.Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Blasko I, Grubeck-Loebenstein B. [Vaccination against Alzheimer disease?] . Wien Klin Wochenschr. 2003;115:279–280. doi: 10.1007/BF03040332. [DOI] [PubMed] [Google Scholar]
- 207.Möller K, Boltze J, Pösel C, Seeger J, Stahland T, Wagner D. Sterile inflammation after permanent distal MCA occlusion in hypertensive rats. J Cereb Blood Flow Metab. 2014;34:307–315. doi: 10.1038/jcbfm.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Choi S, Aid S, Choi U, Bosetti F. Cyclooxygenases-1 and -2 differentially modulate leukocyte recruitment into the inflamed brain. Pharmacogenomics J. 2010;10:448–457. doi: 10.1038/tpj.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis. 2010;19:355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 210.Zotova E, Nicoll JA, Kalaria R, Holmes C, Boche D. Inflammation in Alzheimer’s disease: relevance to pathogenesis and therapy. Alzheimers Res Ther. 2010;2:1. doi: 10.1186/alzrt24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhang XM, Cai Y, Xiong K, Cai H, Luo XG, Feng JC, Clough RW, Struble RG, Patrylo PR, Yan XX. Beta-secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur J Neurosci. 2009;30:2271–2283. doi: 10.1111/j.1460-9568.2009.07017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Deng X, Li M, Ai W, He L, Lu D, Patrylo PR, Cai H, Luo X, Li Z, Yan X. Lipolysaccharide-Induced Neuroinflammation Is Associated with Alzheimer-Like Amyloidogenic Axonal Pathology and Dendritic Degeneration in Rats. Adv Alzheimer Dis. 2014;3:78–93. doi: 10.4236/aad.2014.32009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG. Soluble amyloid-β oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci. 2015;9:191. doi: 10.3389/fncel.2015.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 215.Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16. doi: 10.1016/j.arcmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 216.Morales I, Farías G, Maccioni RB. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation. 2010;17:202–204. doi: 10.1159/000258724. [DOI] [PubMed] [Google Scholar]
- 217.Khandelwal PJ, Herman AM, Moussa CE. Inflammation in the early stages of neurodegenerative pathology. J Neuroimmunol. 2011;238:1–11. doi: 10.1016/j.jneuroim.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lampa J, Westman M, Kadetoff D, Agréus AN, Le Maître E, Gillis-Haegerstrand C, Andersson M, Khademi M, Corr M, Christianson CA, Delaney A, Yaksh TL, Kosek E, Svensson CI. Peripheral inflammatory disease associated with centrally activated IL-1 system in humans and mice. Proc Natl Acad Sci U S A. 2012;109:12728–12733. doi: 10.1073/pnas.1118748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schedlowski M, Engler H, Grigoleit JS. Endotoxin-induced experimental systemic inflammation in humans: a model to disentangle immune-to-brain communication. Brain Behav Immun. 2014;35:1–8. doi: 10.1016/j.bbi.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 220.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77:212–218. doi: 10.1212/WNL.0b013e318225ae07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Monson NL, Ireland SJ, Ligocki AJ, Chen D, Rounds WH, Li M, Huebinger RM, Munro Cullum C, Greenberg BM, Stowe AM, Zhang R. Elevated CNS inflammation in patients with preclinical Alzheimer’s disease. J Cereb Blood Flow Metab. 2014;34:30–33. doi: 10.1038/jcbfm.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Singh-Manoux A, Dugravot A, Brunner E, Kumari M, Shipley M, Elbaz A, Kivimaki M. Interleukin-6 and C-reactive protein as predictors of cognitive decline in late midlife. Neurology. 2014;83:486–493. doi: 10.1212/WNL.0000000000000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.van Exel E, Eikelenboom P, Comijs H, Frölich M, Smit JH, Stek ML, Scheltens P, Eefsting JE, Westendorp RG. Vascular factors and markers of inflammation in offspring with a parental history of late-onset Alzheimer disease. Arch Gen Psychiatry. 2009;66:1263–1270. doi: 10.1001/archgenpsychiatry.2009.146. [DOI] [PubMed] [Google Scholar]
- 225.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 227.Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, Banks WA. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun. 2009;23:507–517. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid β efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Rink L, Cakman I, Kirchner H. Altered cytokine production in the elderly. Mech Ageing Dev. 1998;102:199–209. doi: 10.1016/s0047-6374(97)00153-x. [DOI] [PubMed] [Google Scholar]
- 230.Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, Szalai G, Sonntag WE, Ungvari Z, Csiszar A. Obesity in Aging Exacerbates Blood-Brain Barrier Disruption, Neuroinflammation, and Oxidative Stress in the Mouse Hippocampus: Effects on Expression of Genes Involved in Beta-Amyloid Generation and Alzheimer’s Disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–1226. doi: 10.1093/gerona/glt177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kahn MS, Kranjac D, Alonzo CA, Haase JH, Cedillos RO, McLinden KA, Boehm GW, Chumley MJ. Prolonged elevation in hippocampal Aβ and cognitive deficits following repeated endotoxin exposure in the mouse. Behav Brain Res. 2012;229:176–184. doi: 10.1016/j.bbr.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 233.Joshi YB, Giannopoulos PF, Chu J, Praticò D. Modulation of lipopolysaccharide-induced memory insult, γ-secretase, and neuroinflammation in triple transgenic mice by 5-lipoxygenase. Neurobiol Aging. 2014;35:1024–1031. doi: 10.1016/j.neurobiolaging.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Schmidt R, Schmidt H, Curb JD, Masaki K, White LR, Launer LJ. Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Ann Neurol. 2002;52:168–174. doi: 10.1002/ana.10265. [DOI] [PubMed] [Google Scholar]
- 235.Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, Stijnen T, Hofman A, Witteman JC, Breteler MM. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61:668–672. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- 236.Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, LaFerla FM, Callahan LM, Federoff HJ, Bowers WJ. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol. 2008;173:1768–1782. doi: 10.2353/ajpath.2008.080528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Norden DM, Muccigrosso MM, Godbout JP. Microglial Priming and Enhanced Reactivity to Secondary Insult in Aging, and Traumatic CNS injury, and Neurodegenerative Disease. Neuropharmacology. 2015;96:29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Czerniawski J, Miyashita T, Lewandowski G, Guzowski JF. Systemic lipopolysaccharide administration impairs retrieval of context-object discrimination, but not spatial, memory: Evidence for selective disruption of specific hippocampus-dependent memory functions during acute neuroinflammation. Brain Behav Immun. 2015;44:159–166. doi: 10.1016/j.bbi.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sapin E, Peyron C, Roche F, Gay N, Carcenac C, Savasta M, Levy P, Dematteis M. Chronic Intermittent Hypoxia Induces Chronic Low-Grade Neuroinflammation in the Dorsal Hippocampus of Mice. Sleep. 2015;38:1537–46. doi: 10.5665/sleep.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124:307–321. doi: 10.3109/00207454.2013.833510. [DOI] [PubMed] [Google Scholar]
- 243.Zhan X, Cox C, Ander BP, Liu D, Stamova B, Jin LW, Jickling GC, Sharp FR. Inflammation Combined with Ischemia Produces Myelin Injury and Plaque-Like Aggregates of Myelin, Amyloid-β and AβPP in Adult Rat Brain. J Alzheimers Dis. 2015;46:507–523. doi: 10.3233/JAD-143072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zhang Q, Ding H, Li W, Fan Z, Sun A, Luo J, Ke ZJ. Senescence accelerated mouse strain is sensitive to neurodegeneration induced by mild impairment of oxidative metabolism. Brain Res. 2009;1264:111–118. doi: 10.1016/j.brainres.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 245.Peskind ER, Li G, Shofer JB, Millard SP, Leverenz JB, Yu CE, Raskind MA, Quinn JF, Galasko DR, Montine TJ. Influence of Lifestyle Modifications on Age-Related Free Radical Injury to Brain. JAMA Neurol. 2014;71:1150–1154. doi: 10.1001/jamaneurol.2014.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Stamler JS, Osborne JA, Jaraki O, Rabbani LE, Mullins M, Singel D, Loscalzo J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest. 1993;91:308–318. doi: 10.1172/JCI116187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature (London) 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 248.Lipton SA, Kim WK, Choi YB, Kumar S, D’Emilia DM, Rayudu PV, Arnelle DR, Stamler JS. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med. 2000;162:566–570. doi: 10.1164/ajrccm.162.2.9908091. [DOI] [PubMed] [Google Scholar]
- 250.Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002;165:934–939. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- 251.Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ. 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest. 2003;124:1386–1392. doi: 10.1378/chest.124.4.1386. [DOI] [PubMed] [Google Scholar]
- 252.Petrosyan M, Perraki E, Simoes D, Koutsourelakis I, Vagiakis E, Roussos C, Gratziou C. Exhaled breath markers in patients with obstructive sleep apnoea. Sleep Breath. 2008;12:207–215. doi: 10.1007/s11325-007-0160-8. [DOI] [PubMed] [Google Scholar]
- 253.Morgan BJ. Vascular consequences of intermittent hypoxia. Adv Exp Med Biol. 2007;618:69–84. doi: 10.1007/978-0-387-75434-5_6. [DOI] [PubMed] [Google Scholar]
- 254.Jelic S, Lederer DJ, Adams T, Padeletti M, Colombo PC, Factor PH, Le Jemtel TH. Vascular inflammation in obesity and sleep apnea. Circulation. 2010;121:1014–1021. doi: 10.1161/CIRCULATIONAHA.109.900357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Schulz R, Schmidt D, Blum A, Lopes-Ribeiro X, Lücke C, Mayer K, Olschewski H, Seeger W, Grimminger F. Decreased plasma levels of nitric oxide derivatives in obstructive sleep apnoea - response to CPAP therapy. Thorax. 2000;55:1046–1051. doi: 10.1136/thorax.55.12.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004;27:123–128. [PubMed] [Google Scholar]
- 257.Zhang J, Veasey S. Making sense of oxidative stress in obstructive sleep apnea: mediator or distracter? Front Neurol. 2012;3:179. doi: 10.3389/fneur.2012.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D, Gow A. Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep. 2004;27:194–201. doi: 10.1093/sleep/27.2.194. [DOI] [PubMed] [Google Scholar]
- 259.Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev. 2015;2015:610813. doi: 10.1155/2015/610813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zou J, Crews FT. Inflammasome-IL-1β Signaling Mediates Ethanol Inhibition of Hippocampal Neurogenesis. Front Neurosci. 2012;6:77. doi: 10.3389/fnins.2012.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 262.Maracchioni A, Totaro A, Angelini DF, Di Penta A, Bernardi G, Carri MT, Achsel T. Mitochondrial damage modulates alternative splicing in neuronal cells: implications for neurodegeneration. J Neurochem. 2007;100:142–153. doi: 10.1111/j.1471-4159.2006.04204.x. [DOI] [PubMed] [Google Scholar]
- 263.Thomson LM, Sutherland RJ. Systemic administration of lipopolysaccharide and interleukin-1beta have different effects on memory consolidation. Brain Res Bull. 2005;67:24–29. doi: 10.1016/j.brainresbull.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 264.Chao CC, Hu S, Ehrlich L, Peterson PK. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355–365. doi: 10.1006/brbi.1995.1033. [DOI] [PubMed] [Google Scholar]
- 265.Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, Qian G, Zheng JC. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013;125:897–908. doi: 10.1111/jnc.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138:1738–1755. doi: 10.1093/brain/awv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microsc Res Tech. 2001;54:47–58. doi: 10.1002/jemt.1120. [DOI] [PubMed] [Google Scholar]
- 268.Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. 2001;60:647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- 269.Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C] (R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21:89–93. doi: 10.1002/mds.20668. [DOI] [PubMed] [Google Scholar]
- 270.Bellucci A, Bugiani O, Ghetti B, Spillantini MG. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis. 2011;8:221–229. doi: 10.1159/000322228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol. 2004;165:1643–1652. doi: 10.1016/S0002-9440(10)63421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Ikeda M, Shoji M, Kawarai T, Kawarabayashi T, Matsubara E, Murakami T, Sasaki A, Tomidokoro Y, Ikarashi Y, Kuribara H, Ishiguro K, Hasegawa M, Yen SH, Chishti MA, Harigaya Y, Abe K, Okamoto K, St George-Hyslop P, Westaway D. Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am J Pathol. 2005;166:521–531. doi: 10.1016/S0002-9440(10)62274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 274.Sasaki A, Kawarabayashi T, Murakami T, Matsubara E, Ikeda M, Hagiwara H, Westaway D, George-Hyslop PS, Shoji M, Nakazato Y. Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain Res. 2008;1214:159–168. doi: 10.1016/j.brainres.2008.02.084. [DOI] [PubMed] [Google Scholar]
- 275.Zilka N, Stozicka Z, Kovac A, Pilipcinec E, Bugos O, Novak M. Human misfolded truncated tau protein promotes activation of microglia and leukocyte infiltration in the transgenic rat model of tauopathy. J Neuroimmunol. 2009;209:16–25. doi: 10.1016/j.jneuroim.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 276.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33:5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Jaworski T, Lechat B, Demedts D, Gielis L, Devijver H, Borghgraef P, Duimel H, Verheyen F, Kügler S, Van Leuven F. Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol. 2011;179:2001–2015. doi: 10.1016/j.ajpath.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, Weeber EJ, Bickford PC, Gemma C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci. 2011;31:16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Sanjay W. Pimplikara SW. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106:18367–18372. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for tau versus Aβ-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 283.Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis. 2008;15:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, Hutton ML, Citron M. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011;118:658–667. doi: 10.1111/j.1471-4159.2011.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Zubair AC, Dickson D, Golde TE, Das P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24:548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gabbita SP, Srivastava MK, Eslami P, Johnson MF, Kobritz NK, Tweedie D, Greig NH, Zemlan FP, Sharma SP, Harris-White ME. Early intervention with a small molecule inhibitor for tumor necrosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2012;9:99. doi: 10.1186/1742-2094-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Maphis N, Xu G, Kokiko-Cochran ON, Cardona AE, Ransohoff RM, Lamb BT, Bhaskar K. Loss of tau rescues inflammation-mediated neurodegeneration. Front Neurosci. 2015;9:196. doi: 10.3389/fnins.2015.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Dickey C, Kraft C, Jinwal U, Koren J, Johnson A, Anderson L, Lebson L, Lee D, Dickson D, de Silva R. Binder LI, Morgan D, Lewis J. Aging analysis reveals slowed tau turnover and enhanced stress response in a mouse model of tauopathy. Am J Pathol. 2009;174:228–238. doi: 10.2353/ajpath.2009.080764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A, Blair L, Gordon MN, Dickey CA, Morgan D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56. doi: 10.1186/1742-2094-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Lee CW, Shih YH, Wu SY, Yang T, Lin C, Kuo YM. Hypoglycemia induces tau hyperphosphorylation. Curr Alzheimer Res. 2013;10:298–208. doi: 10.2174/1567205011310030009. [DOI] [PubMed] [Google Scholar]
- 292.Tian M, Zhu D, Xie W, Shi J. Central angiotensin II-induced Alzheimer-like tau phosphorylation in normal rat brains. FEBS Lett. 2012;586:3737–3745. doi: 10.1016/j.febslet.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 293.Nilsen LH, Rae C, Ittner LM, Götz J, Sonnewald U. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J Cereb Blood Flow Metab. 2013;33:684–691. doi: 10.1038/jcbfm.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 295.Lukiw WJ. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem Res. 2004;29:1287–1297. doi: 10.1023/b:nere.0000023615.89699.63. [DOI] [PubMed] [Google Scholar]
- 296.Cui JG, Hill JM, Zhao Y, Lukiw WJ. Expression of inflammatory genes in the primary visual cortex of late-stage Alzheimer’s disease. Neuroreport. 2007;18:115–119. doi: 10.1097/WNR.0b013e32801198bc. [DOI] [PubMed] [Google Scholar]
- 297.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Olanrewaju O, Clare L, Barnes L, Brayne C. A multimodal approach to dementia prevention: A report from the Cambridge Institute of Public Health. Alzheimers Dement (N Y) 2015;1:151–156. doi: 10.1016/j.trci.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel TH2 pathway. Nature. 2011;475:110–113. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Xie Z, Smith CJ, Van Eldik LJ. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004;45:170–179. doi: 10.1002/glia.10314. [DOI] [PubMed] [Google Scholar]
- 302.Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G. p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- 303.Munoz L, Ralay Ranaivo H, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Xing B, Xin T, Hunter RL, Bing G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J Neuroinflammation. 2008;5:4. doi: 10.1186/1742-2094-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kim YC, Park TY, Baik E, Lee SH. Fructose-1,6-bisphosphate attenuates induction of nitric oxide synthase in microglia stimulated with lipopolysaccharide. Life Sci. 2012;90:365–372. doi: 10.1016/j.lfs.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 306.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 307.Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL, Beckett TL, Murphy MP, Chen KC, Blalock EM, Landfield PW, Porter NM, Thibault O. Long-Term Pioglitazone Treatment Improves Learning and Attenuates Pathological Markers in a Mouse Model of Alzheimer’s Disease. J Alzheimers Dis. 2012;30:943–961. doi: 10.3233/JAD-2012-111661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Iida M, Katsuno M, Nakatsuji H, Adachi H, Kondo N, Miyazaki Y, Tohnai G, Ikenaka K, Watanabe H, Yamamoto M, Kishida K, Sobue G. Pioglitazone suppresses neuronal and muscular degeneration caused by polyglutamine-expanded androgen receptors. Hum Mol Genet. 2015;24:314–329. doi: 10.1093/hmg/ddu445. [DOI] [PubMed] [Google Scholar]
- 309.Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, Ismail MS, Martin BK, Mullan MJ, Sabbagh M, Tariot PN. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7:402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Carta AR, Pisanu A. Modulating microglia activity with PPAR-γ agonists: a promising therapy for Parkinson’s disease? Neurotox Res. 2013;23:112–123. doi: 10.1007/s12640-012-9342-7. [DOI] [PubMed] [Google Scholar]
- 311.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 312.Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, Yoon KS, Kim HJ, Park JH, Murphy MP, Kim SS. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11:178. doi: 10.1186/1741-7015-11-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Wani WY, Gudup S, Sunkaria A, Bal A, Singh PP, Kandimalla RJ, Sharma DR, Gill KD. Protective efficacy of mitochondrial targeted antioxidant MitoQ against dichlorvos induced oxidative stress and cell death in rat brain. Neuropharmacology. 2011;61:1193–1201. doi: 10.1016/j.neuropharm.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 314.Wang A, Keita AV, Phan V, McKay CM, Schoultz I, Lee J, Murphy MP, Fernando M, Ronaghan N, Balce D, Yates R, Dicay M, Beck PL, MacNaughton WK, Söderholm JD, McKay DM. Targeting Mitochondria-Derived Reactive Oxygen Species to Reduce Epithelial Barrier Dysfunction and Colitis. Am J Pathol. 2014;184:2516–2527. doi: 10.1016/j.ajpath.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Haorah J, Floreani NA, Knipe B, Persidsky Y. Stabilization of superoxide dismutase by acetyl-l-carnitine in human brain endothelium during alcohol exposure: novel protective approach. Free Radic Biol Med. 2011;51:1601–1609. doi: 10.1016/j.freeradbiomed.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137:425–435. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 317.Lai AY, Todd KG. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia. 2006;53:809–816. doi: 10.1002/glia.20335. [DOI] [PubMed] [Google Scholar]
- 318.Jantzie LL, Cheung PY, Todd KG. Doxycycline reduces cleaved caspase-3 and microglial activation in an animal model of neonatal hypoxia-ischemia. J Cereb Blood Flow Metab. 2005;25:314–324. doi: 10.1038/sj.jcbfm.9600025. [DOI] [PubMed] [Google Scholar]
- 319.Jantzie LL, Todd KG. Doxycycline inhibits proinflammatory cytokines but not acute cerebral cytogenesis after hypoxia-ischemia in neonatal rats. J Psychiatry Neurosci. 2010;35:20–32. doi: 10.1503/jpn.090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Eikelenboom P, Veerhuis R, Familian A, Hoozemans JJ, van Gool WA, Rozemuller AJ. Neuroinflammation in plaque and vascular beta-amyloid disorders: clinical and therapeutic implications. Neurodegener Dis. 2008;5:190–193. doi: 10.1159/000113699. [DOI] [PubMed] [Google Scholar]
- 321.Jin WJ, Feng SW, Feng Z, Lu SM, Qi T, Qian YN. Minocycline improves postoperative cognitive impairment in aged mice by inhibiting astrocytic activation. Neuroreport. 2014;25:1–6. doi: 10.1097/WNR.0000000000000082. [DOI] [PubMed] [Google Scholar]
- 322.Jiang Y, Liu Y, Zhu C, Ma X, Ma L, Zhou L, Huang Q, Cen L, Pi R, Chen X. Minocycline enhances hippocampal memory, neuroplasticity and synapse-associated proteins in aged C57 BL/6 mice. Neurobiol Learn Mem. 2015;121:20–29. doi: 10.1016/j.nlm.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 323.Hung MW, Tipoe GL, Poon AM, Reiter RJ, Fung ML. Protective effect of melatonin against hippocampal injury of rats with intermittent hypoxia. J Pineal Res. 2008;44:214–221. doi: 10.1111/j.1600-079X.2007.00514.x. [DOI] [PubMed] [Google Scholar]
- 324.Hung MW, Kravtsov GM, Lau CF, Poon AM, Tipoe GL, Fung ML. Melatonin ameliorates endothelial dysfunction, vascular inflammation, and systemic hypertension in rats with chronic intermittent hypoxia. J Pineal Res. 2013;55:247–256. doi: 10.1111/jpi.12067. [DOI] [PubMed] [Google Scholar]
- 325.Liu Y, Tipoe GL, Fung ML. Melatonin attenuates intermittent hypoxia-induced lipid peroxidation and local inflammation in rat adrenal medulla. Int J Mol Sci. 2014;15:18437–18452. doi: 10.3390/ijms151018437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Wang JZ, Wang ZF. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol Sin. 2006;27:41–49. doi: 10.1111/j.1745-7254.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- 327.Zhang W, Chen XY, Su SW, Jia QZ, Ding T, Zhu ZN, Zhang T. Exogenous melatonin for sleep disorders in neurodegenerative diseases: a meta-analysis of randomized clinical trials. Neurol Sci. 2016;37:57–65. doi: 10.1007/s10072-015-2357-0. [DOI] [PubMed] [Google Scholar]
- 328.Cardinali DP, Furio AM, Brusco LI. Clinical aspects of melatonin intervention in Alzheimer’s disease progression. Curr Neuropharmacol. 2010;8:218–227. doi: 10.2174/157015910792246209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Cardinali DP, Furio AM, Brusco LI. The use of chronobiotics in the resynchronization of the sleep/wake cycle. Therapeutical application in the early phases of Alzheimer’s disease. Recent Pat Endocr Metab Immune Drug Discov. 2011;5:80–90. doi: 10.2174/187221411799015354. [DOI] [PubMed] [Google Scholar]
- 330.Hagberg H, Peebles D, Mallard C. Models of white matter injury: Comparison of infectious, hypoxic-Ischemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev. 2002;8:30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- 331.Mallard C, Welin AK, Peebles D, Hagberg H, Kjellmer I. White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochem Res. 2003;28:215–223. doi: 10.1023/a:1022368915400. [DOI] [PubMed] [Google Scholar]