

Abstract
Real-time PCR (RT-qPCR) expression analysis is a powerful analytical technique, but reliable results depend on the use of stable reference genes for proper normalization. This study proposed to test the expression stability of 13 candidate reference genes in Setaria viridis, a monocot species recently proposed as a new C4 model plant. Gene expression stability of these genes was assayed across different tissues and developmental stages of Setaria and under drought or aluminum stress. In general, our results showed Protein Kinase, RNA Binding Protein and SDH as the most stable genes. Moreover, pairwise analysis showed that two reference genes were sufficient to normalize the gene expression data under each condition. By contrast, GAPDH and ACT were the least stably expressed genes tested. Validation of suitable reference genes was carried out to profile the expression of P5CS and GolS during abiotic stress. In addition, normalization of gene expression of SuSy, involved in sugar metabolism, was assayed in the developmental dataset. This study provides a list of reliable reference genes for transcript normalization in S. viridis in different tissues and stages of development and under abiotic stresses, which will facilitate genetic studies in this monocot model plant.
Setaria viridis has emerged as a suitable C4 model species for molecular and genetic studies. It is a short, fast-growing, C4 metabolism plant, with its draft genome sequence recently available, making it suitable model plant for genetic and genomics studies1,2. Moreover, S. viridis is highly responsive to Agrobacterium tumefaciens-mediated genetic transformation, with a well-established transformation protocols1,3 and more recently, spike-dipping methods have also been proposed3,4. S. viridis could be used as a model plant for food and bioenergy grasses presenting C4 metabolism such as maize, sorghum, sugarcane and switchgrass. Genetically engineered S. viridis plants can be utilized in a proof-of-concept approach to evaluate phenotypes related to important agricultural traits such as abiotic stress tolerance, resistance to pathogens and improved yield and biomass5,6,7, and the promising genes could be further transferred to a target crop. To achieve this goal, it is important to establish suitable genetic tools, including a reliable gene expression analysis in this species.
Gene expression analysis is an important tool towards the understanding of the complex signaling networks that regulate the different responses observed during the plant life cycle or when they are submitted to different stimulli, and it has been used in many studies for this purpose8,9,10,11. Microarray and RNA-sequencing (RNA-seq) are the most widely used technique to provide a global comprehension of gene expression in plants under a wide range of experimental conditions12. However, reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) is the most sensitive, accurate and reproducible technique compared to microarray and RNA-seq to profile the expression levels of genes. Due to these advantages, RT-qPCR has also been used to validate the expression levels of target genes found as differentially expressed by these two methods. However, to avoid biased results during RT-qPCR analysis, a normalization step of the gene expression data is essential to correct variations between different samples and conditions. Normalization during RT-qPCR analysis is usually performed using a reference gene that must be expressed at stable levels regardless of experimental conditions, cell types, tissue, developmental stage or stress treatment13. Thus, it is necessary to validate the stability of the reference gene under determined experimental condition to ensure proper normalization and a robust RT-qPCR analysis14. Several studies have been published with the aim of identifying suitable reference genes for expression analysis under different stages of the plant life cycle15,16,17,18 or under biotic and abiotic stresses19,20,21,22.
Abiotic stresses in plants cause major losses in agriculture worldwide. Drought, flooding, extreme temperatures conditions (cold, heat and frost), salinity and mineral toxicity are among the main abiotic stresses that negatively affect growth, development and yield of important crops. The knowledge about the physiology, biochemistry and molecular responses involved in abiotic stresses contributes to the discovery of new genes and signaling networks that plants use to cope with these challenges, and it is pivotal for the development of new crop varieties with enhanced tolerance to stress23,24,25,26.
In this study, thirteen genes, actin (ACT), anthranilate phosphoribosyl transferase (APRT), clathrin adaptor complex (CAC), cullin (CUL), elongation factor 1-alpha (EF1α), eukaryotic initiation factor 4-alpha (eIF4α), expressed protein (EXP), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), protein kinase (KIN), RNA-binding protein (BIND), RNA polymerase II (POL), succinate dehydrogenase (SDH) and translation initiation factor SUI1 (SUI) were selected as candidate reference genes. Besides the traditional genes used for transcript normalization in plants, such as GAPDH, actin and elongation factors, we selected other genes based on previous reported studies. Different algorithms and statistical analysis were applied to evaluate the expression stability of the reference genes of S. viridis plants in different tissues (leaves, stems, spikes and roots) and key developmental stages. In addition, the candidate reference genes were tested to normalize the gene expression in plants submitted to drought stress (GolS and P5CS) and in different stages of development (SuSy), in order to validate the results obtained. It is assumed that the genes analyzed will provide robustness to the gene expression analysis during specific experimental conditions in S. viridis plants.
Results
Identification of candidate Setaria viridis reference genes
Based on previous systematic studies of reference genes suitable for transcript normalization in monocot27,28,29,30,31 species, thirteen candidate genes were selected to this study. Information about gene names, accession numbers, primer sequences and efficiency and gene description are provided in Supplementary Table S1. Primers were designed to amplify one single PCR product, as confirmed on a 2% agarose gel and melting curve analysis performed in all RT-qPCR assays (see Supplementary Figs S1 and S2). Mean PCR efficiency per gene was estimated using LinRegPCR (version 2016.0) program; the efficiency values ranged from 89 to 96% for reference genes and 90 to 100% for target genes (see Supplementary Table S1). Expression levels of the candidate genes for different developmental stages, drought and aluminum stresses and for all samples combined are presented in Fig. 1. Expression values are inversely proportional to the Cq values, and the mean and range of Cq indicate the most stable genes across all samples and to each experimental set. Cq values of candidate genes from each experimental set presented a high variation, ranging from 18.6 for GAPDH and 32.0 for POL. Expression levels to each subset of the candidate genes for different tissues/organs can be found in Supplementary Fig. S3.
Figure 1. Expression level of reference genes tested in different experimental conditions.

Box plot graphs of Cq values are shown as the first and third quartile. Horizontal lines indicate range of values, black lines indicate median values and circles indicate outliers. (a) All datasets; (b) Developmental stages; (c) Drought stress and (d) Aluminum stress.
Expression stability analysis
Performance of the thirteen genes as potential reference genes for S. viridis was assessed in 31 samples divided into three experimental sets; five developmental stages from whole seedlings, different tissues/organs, and two treatments, including samples submitted to different levels of drought or aluminum stresses. Using geNorm, we estimated two parameters to evaluate the expression stability of these genes; the average expression stability value (M value), based on the pairwise variation between a particular gene compared to all others, and the pairwise variation (Vn/n + 1), which determines the required number of genes to result in a more accurate normalization32.
When considering all dataset, CAC/KIN (M = 0.48) was the best pair to normalize all samples, while GAPDH was the least stable gene (M = 1.6) (Fig. 2a). Comparing with NormFinder, SDH was the most stable gene and CUL/KIN were defined as the best pair for a reliable normalization. In both programs, ACT and GAPDH were ranked as the least stable genes (Tables 1 and 2).
Figure 2. Average expression stability values (M) calculated by geNorm.

A lower value of average expression stability (M) indicates most stable expression. (a) All datasets; (b) Developmental stages; (c) Setaria viridis submitted to drought and (d) aluminum stress.
Table 1. Setaria viridis reference genes ranked according to expression stability as determined by geNorm.
| Ranking |
Total |
Developmental Stages |
Drought Stress |
Aluminum Stress |
||||
|---|---|---|---|---|---|---|---|---|
| geNorm | Stability value | geNorm | Stability value | geNorm | Stability value | geNorm | Stability value | |
| 1 | CAC | 0.48 | EXP | 0.47 | SDH | 0.12 | CAC | 0.32 |
| 2 | KIN | 0.48 | KIN | 0.47 | SUI | 0.12 | KIN | 0.32 |
| 3 | CUL | 0.59 | CAC | 0.48 | CUL | 0.15 | APRT | 0.40 |
| 4 | APRT | 0.69 | CUL | 0.59 | KIN | 0.16 | POL | 0.48 |
| 5 | BIND | 0.87 | BIND | 0.67 | CAC | 0.18 | CUL | 0.54 |
| 6 | EF1α | 0.96 | APRT | 0.73 | eIF4α | 0.20 | eIF4α | 0.59 |
| 7 | SDH | 1.00 | POL | 0.81 | GAPDH | 0.23 | ACT | 0.67 |
| 8 | eIF4α | 1.05 | EF1α | 0.86 | EF1α | 0.26 | SDH | 0.73 |
| 9 | POL | 1.10 | SUI | 0.90 | EXP | 0.29 | BIND | 0.77 |
| 10 | SUI | 1.17 | SDH | 0.94 | POL | 0.33 | EXP | 0.85 |
| 11 | EXP | 1.24 | eIF4α | 0.98 | BIND | 0.37 | SUI | 0.93 |
| 12 | ACT | 1.42 | ACT | 1.22 | APRT | 0.42 | EF1α | 1.02 |
| 13 | GAPDH | 1.59 | GAPDH | 1.42 | ACT | 0.48 | GAPDH | 1.15 |
| Best pair | CAC/KIN | EXP/KIN | SDH/SUI | CAC/KIN | ||||
Table 2. Setaria viridis reference genes ranked according to expression stability as determined by NormFinder.
| Ranking |
Total |
Developmental Stages |
Drought Stress |
Aluminum Stress |
||||
|---|---|---|---|---|---|---|---|---|
| NormFinder | Stability value | NormFinder | Stability value | NormFinder | Stability value | NormFinder | Stability value | |
| 1 | SDH | 0.06 | BIND | 0.06 | SDH | −0.00097 | CUL | 0.02 |
| 2 | BIND | 0.10 | SDH | 0.09 | KIN | −0.00066 | ACT | 0.07 |
| 3 | CAC | 0.29 | EF1α | 0.10 | CUL | 0.00001 | CAC | 0.10 |
| 4 | eIF4α | 0.34 | eIF4α | 0.22 | SUI | 0.01 | SDH | 0.10 |
| 5 | EF1α | 0.37 | CAC | 0.25 | CAC | 0.01 | POL | 0.13 |
| 6 | POL | 0.38 | EXP | 0.29 | EXP | 0.03 | BIND | 0.15 |
| 7 | CUL | 0.44 | SUI | 0.29 | eIF4α | 0.04 | KIN | 0.23 |
| 8 | APRT | 0.45 | POL | 0.33 | GAPDH | 0.07 | eIF4α | 0.32 |
| 9 | KIN | 0.57 | CUL | 0.44 | BIND | 0.07 | APRT | 0.35 |
| 10 | EXP | 0.78 | APRT | 0.46 | EF1α | 0.09 | EXP | 0.47 |
| 11 | SUI | 0.94 | KIN | 0.49 | POL | 0.15 | SUI | 0.57 |
| 12 | ACT | 1.94 | ACT | 2.25 | APRT | 0.17 | EF1α | 0.99 |
| 13 | GAPDH | 2.56 | GAPDH | 2.75 | ACT | 0.28 | GAPDH | 1.43 |
| Best pair | SDH/BIND | BIND/SDH | KIN/CUL | CUL/BIND | ||||
Due to the heterogeneity of these samples and conditions, each experimental set was analyzed individually using both algorithms. While geNorm performs a stepwise exclusion of the least stably expressed gene32, NormFinder uses a model-based approach, which calculates both inter- and intra-group variability to estimate the stability of gene expression33. Estimative of the best reference genes in each experimental set exhibited some particularities. For developmental stages, EXP/KIN pair (M = 0.47) was ranked as the most stable gene pair, while BIND was the most stable gene by NormFinder, followed by SDH (Tables 1 and 2; Fig. 2b). Once again, ACT/GAPDH showed the highest variation and hence, they were not suitable for normalization in different stages of development (Tables 1 and 2). We also analyzed the expression stability of these candidate genes in samples derived from whole seedlings in vegetative phase and in each tissue/organ at the subsequent stages of development. geNorm and NormFinder excluded the same reference genes, but defined different pair of genes as the best reference genes to each particular subset of tissue/organ. In general, SDH and eIF4α were selected as the preferred reference genes when considering both algorithms (see Supplementary Tables S2 and S3; Supplementary Fig. S4).
For drought treatment, SDH/SUI pair and SDH gene were considered the most stable genes according to geNorm and NormFinder, respectively. The best pair according to NormFinder was KIN/CUL, whereas ACT was estimated as the most variable reference gene by both algorithms (Fig. 2c; Tables 1 and 2).
For aluminum treatment, CAC/KIN pair presented the best performance, according to geNorm (Fig. 2d and Table 1). Although CUL was the most stable, according to NormFinder, CAC was ranked in the top-three position (Table 2). In both geNorm and NormFinder, EXP, SUI, EF1α and GAPDH showed the highest variation among all the reference genes tested under Al3+ treatment (Tables 1 and 2).
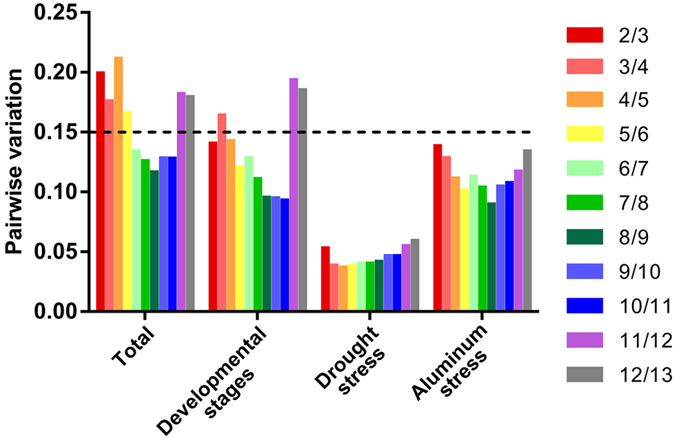
In addition, to define the best pair using geNorm, we also estimated the pairwise variation to determine the minimal number of genes for reliable normalization. Assuming a cut-off of Vn/n + 1 ≤ 0.15, it was determined that the use of only the top two reference genes for each experimental set would be the appropriate number of genes required for normalization (Fig. 3; Supplementary Fig. S5). When the entire dataset were considered, the number of genes increased to six (Fig. 3).
Figure 3. Pairwise variation (V) to define the optimal number of reference genes required to a reliable normalization to each dataset.
Validation of the selected reference genes to different experimental conditions
In order to validate the selection of reference genes in S. viridis under drought treatment, the expression levels of GolS and P5CS, two marker genes for drought stress, were normalized to the best pair (SDH/SUI) or the most variable reference gene (ACT), according to geNorm (Fig. 2c). Normalization of transcripts using SDH/SUI showed that transcript levels increased significantly upon 0 (Ψw-1.50 MPa; permanent wilting point), 25 (Ψw-1.125 MPa; severe stress) and 50% (Ψw-0.75 MPa; moderate stress) of soil water content (SWC) for GolS (Fig. 4a) and under 0 and 25% of SWC for P5CS (Fig. 4b), when compared to 100% of SWC (Ψw-0.03 MPa; field capacity). These results were expected, since P5CS and GolS genes are known to increase their expression levels under drought conditions in plants34,35,36. By contrast, normalization using ACT as reference gene resulted in an overestimated relative expression level of both target genes (Fig. 4a,b). In addition, relative expression of GolS (normalized to ACT) during moderate stress was not significantly different compared to control (Fig. 4a).
Figure 4. Relative expression level of target genes using the most and least stable pair of reference genes to each experimental condition, as determined by geNorm.

(a,b) Transcription levels of GolS and P5CS genes in Setaria viridis submitted to drought stress treatment (0 (Ψw-1.50 MPa; permanent wilting point), 25 (Ψw-1.125 MPa; severe stress), 50 (Ψw-0.75 MPa; moderate stress) and 100% (Ψw-0.03 MPa; field capacity), respectively; (c) Transcription levels of SuSy in tree developmental stages. Bars indicate the standard error (±SE) calculated from three biological replicates. The asterisks indicate statistically significant with respect to control (ANOVA followed by Tukey’s test).
Similarly, top ranked reference genes samples for developmental stages datasets were validated using SuSy gene, responsible for sucrose synthase production and involved in the sucrose metabolism in plants. SuSy expression is known to increase during the plant life cycle in C4 species such as sugarcane and sorghum37,38,39. The increase of SuSy expression levels during the late stages of S. viridis development was confirmed using EXP/KIN, the best pair of reference genes for the developmental stages dataset (Fig. 4c). When GAPDH, the least stable gene for developmental stages dataset, was used to normalize SuSy expression, no significant difference was observed.
Discussion
RT-qPCR has emerged as the standard method for gene expression profiling due to its high sensibility, reproducibility and large dynamic range with a potential increasing sample throughput13,40. However, a reliable RT-qPCR quantification assay depends upon the selection of stable reference genes for a proper normalization of the expression levels measured41. Suitable reference genes should be stably expressed in all samples and experimental conditions under evaluation. Therefore, many studies have been conducted to provide a systematic selection of reliable reference genes candidates to different plant species15,17. In S. viridis, a recent study reported the selection and validation of fifteen reference genes. They found gene that encoded a phosphoglucomutase, folylpolyglutamate synthase and cullin as the most stable reference genes. However, this study was focused on the validation of reference genes only for a leaf gradient dataset and stages of development17. In the present study, we selected thirteen candidate genes in S. viridis to be tested in a total of 112 samples, comprising three experimental sets: developmental stages, different tissues/organs and two abiotic (drought and aluminum) stresses. The selected candidate genes were chosen based on previous studies that identified suitable reference genes in monocot species27,28,29,30,31.
We were able to carry out two simple methods of RNA extraction that yielded a high quality and quantity for all samples. RNA from most tissues/organs of S. viridis was extracted using the TRIzol method (Thermo Scientific), except for RNA from roots, which was extracted using a LiCl method42. The SYBR Green detection dye13 was used in the RT-qPCR for transcript detection and the results obtained were analyzed by different algorithms to verify the stability of the candidate genes expression.
Analysis in both geNorm and NormFinder showed some differences in the top-ranked genes but both programs were more consistent to exclude the least stable genes. These discrepancies reflect differences between the approaches33. The difference was more evident in the developmental stages dataset, possibly due to the higher heterogeneity of samples assessed in each subset (different tissues/organs and developmental stages), when compared to the abiotic stress conditions (treated versus untreated samples). Since NormFinder estimates both inter- and intra-group variation and combines them into a stability value, this model-based approach should provide a more precise and robust estimative of expression variation among subsets composed by different sample types33. Differences in the top-ranked genes selected by both methods were also reported in different tissues, flower stages and fruit development in cotton43, fruit development in apple44 and different organs and flower developmental stages in citrus45. In this study, the gene pairs EXP/KIN and BIND/SDH were considered the most suitable pair of genes to normalize samples in different developmental stages, according to geNorm and NormFinder, respectively. Concerning different tissues/organs and both algorithms, SDH/elF4α was ranked as the best gene pair for a proper normalization.
For drought stress, geNorm and NormFinder identified the same four top-ranked genes with few differences in some positions. SDH/SUI was the best pair of reference genes according to geNorm. SDH was the most stable gene, although CUL/KIN was the best pair, according to NormFinder. SDH gene was stable for normalization of genes in Brachypodium distachyon under different developmental stages30. SUI and KIN showed stable expression in different tissues, development and under biotic and abiotic stresses in rice31. The member of the cullin family CUL has been reported as the most stable gene in leaf gradient dataset to S. viridis17 and one of the three-top genes stably expressed in sugarcane under salinity or drought stress27. APRT and ACT were the most variable reference genes under drought stress in our study, thereby they were considered inappropriate to use as a reference gene under this condition. Accordingly, APRT was also found unstable in sugarcane under abiotic stress conditions28, whereas ACT was selected as a suitable reference gene to switchgrass under drought and salinity treatment46, demonstrating that even phylogenetically similar species show differential responses to gene expression under the same experimental conditions.
Considering Setaria seedlings submitted to aluminum stress, we found by geNorm that CAC and KIN were the most stable reference genes tested, although CAC was only ranked as the third position, according to NormFinder. In contrast, four genes (EXP, SUI, EF1a and GAPDH) changed significantly in their expression levels and then should be carefully evaluated before using them to normalize the expression of target genes in S. viridis under similar stress conditions. Unlike to drought or other abiotic stresses in which several studies reported the selection of reference genes in different plant species29,47,48,49 to the best of our knowledge, this is the first report describing the validation of reference genes for aluminum stress condition in plants.
The suitability of the top ranked genes for the normalization of transcript studies under different stages of development of Setaria was performed by studying the expression of a sucrose synthase gene (SuSy). Our previous results (unpublished data) demonstrated that SuSy (Sevir.4G039300) expression increased in the late stages of S. viridis development, and such expression is accompanied by high levels of sucrose in the culms of the plant in the same developmental stage (data not shown). According to geNorm, the most suitable gene pair for the transcript normalization in different developmental stages was EXP/KIN (Table 1). In fact, using EXP/KIN to normalize SuSy expression in three different stages of development, we were able to demonstrate that the expression of this gene increased as the plant reached the last stage (Figs 4c and 5). In contrast, using the least stable gene GAPDH was not suitable to normalize SuSy expression in the different plant developmental stages. We also performed the normalization of the expression of two drought marker genes, PC5S and GolS, using SDH/SUI as reference genes, suggested as the most stable pair of genes under drought stress according to geNorm. The P5CS gene codifies for Δ1-pyrroline-5-carboxylate synthetase, which catalyzes the rate-limiting step in the biosynthesis of proline, an amino acid that accumulates in plants as the water deficit becomes more severe50,51. GolS is the gene responsible for the transcription of galactinol synthase, a key enzyme involved in raffinose family oligosaccharide biosynthesis, which is highly expressed under abiotic stress conditions35,52,53. Our results demonstrated the reliability of SDH/SUI as reference genes to normalize the transcription of P5CS and GolS, as the expression levels of the marker genes increased as the water deficit becomes more severe (Fig. 4a,b). These results were not observed when ACT, the least stable reference gene for drought stress, was used for normalization.
Figure 5. Developmental stages of Setaria viridis used in this study.

(a) 1 - early vegetative phase (EVP) and 2 - late vegetative phase (LVP), bar = 2 cm; (b) 3 - transition phase (TP), 4 - reproductive phase (RP) and 5 - advanced phase (AP), bar = 10 cm.
In summary, we found that the stability of expression of reference genes varied depending on the experimental dataset tested. In general, KIN, BIND and SDH were the best ranked reference genes considering developmental set and drought stress treatments. In contrast, traditional genes such as GAPDH and ACT varied significantly among our conditions. Different tissues, developmental stages and even different abiotic stresses could influence the expression stability of reference genes. This was more evident when we evaluated the expression stability of reference genes to each tissue or developmental stage individually. In addition, validation of target genes P5CS and GolS normalized to the best pair of reference genes or the most variable gene showed that the arbitrary use of a reference gene without a prior selection could lead to a misinterpretation of data. These results reinforce the need of a systematic evaluation of appropriate reference genes to each particular condition to improve the reliability of gene expression assays and avoid biased results. In this sense, this study provides a list of suitable S. viridis reference genes for validation procedures.
Material and Methods
Plant material and growth conditions
Seeds of S. viridis (accession A10.1) were treated with concentrated sulfuric acid to promote dormancy break3. After the mature seeds were germinated on half strength MS medium54 in growth chamber (Conviron) under 16 h photoperiod, 25 ± 2 °C and light intensity of 150 μmol m−2s−1 for ten days (considered early vegetative phase). After this period, S. viridis seedlings were transferred to pots containing latosoil, substrate (Plantmax) and vermiculite (Agrifloc, Brasil Minérios) mixture (3:1:0.5; w/w/w). Plants were maintained in growth chamber (Fitotron) under 16 h photoperiod of 500 μmol m−2s−1 light intensity, 26 ± 2 °C and 65% relative humidity.
S. viridis tissues, organs and developmental stages
S. viridis seedlings were harvested at the early vegetative phase (EVP) 10 days after in vitro germination and the late vegetative phase (LVP) 7 days after planting (DAP). In the transition (TP; 25 DAP) and reproductive (RP; 32 DAP) phases, the plants were separated into leaf, stem and root tissues. Spikes and other organs were harvested in the advanced phase (AP; 39 DAP) (Fig. 5). Pools of three whole seedlings or tissues were harvested comprising three biological samples. Samples were transferred to liquid nitrogen and stored at −80◦ C until analyses.
Drought treatment
Plants at the reproductive phase (32 DAP) were maintained in growth chamber in the same conditions described above. Four levels of soil water content (SWC) were used, 0 (Ψw-1.50 MPa; permanent wilting point), 25 (Ψw-1.125 MPa; severe stress), 50 (Ψw-0.75 MPa; moderate stress) and 100% (Ψw-0.03 MPa; field capacity). The net photosynthetic rate was assessed to characterize the water stress using an open gas exchange system with a 6 cm2 clamp-on leaf cuvette (LI-6400XT, LICOR). Photosynthetic photon flux density (PPFD) was fixed at 1,500 μmol m−2 s−1, using a red-blue LED light source built into the leaf cuvette. Twenty-four hours after the permanent wilting point, whole +2 leaves (second fully expanded leaf with visible ligule) were harvested from six plants and pooled composing each of the three biological samples. Collected tissues were frozen immediately in liquid nitrogen and stored at −80 °C.
Aluminum treatment
Seedlings in early vegetative phase were submitted to 500 μM CaCl2 solution, in the absence or presence of {20} μM Al3+, pH 4.2, in hydroponic system for 24 hours. Roots of 120 plantlets were harvested, separated in three biological replicates (40 each) and immediately frozen in liquid nitrogen and stored at −80 °C.
Selection of candidate reference genes and primer design
Thirteen candidate reference genes were selected in the present study using as criterion reference genes previously reported as suitable for transcript normalization in other monocots27,28,29,30,31 subject to different experimental conditions. Initially, we used the Setaria italica genome database (Phytozome 10.1 v) as reference to retrieve the ortholog gene sequences. With the recent release of the S. viridis genome sequence at Phytozome database, we confirmed the identity and specificity of primer sequences used in this study. Using BLASTN algorithm with a default setting and S. italica, Oryza sativa, sugarcane and B. distachyon sequences as queries, Setaria spp. coding sequences with high similarity scores were retrieved (E-value ≤ 1e-90) (see Supplementary Table S1). Primers were designed using Primer Express 3.0 (Applied Biosystems) and PrimerQuest (IDT) tools with the following parameters: Tm around 60 °C and amplicon length of 75 to 150 bp, yielding primer sequences with a length of 19 to 23 nucleotides with an optimum at 20 nucleotides, and a GC content of 45 to 60%. Primers were also designed to span exon-exon junction and allow the amplification of all splicing variants.
RNA isolation and cDNA synthesis
About 200 mg of starting material was used for RNA isolation. Total RNA from all tissues (except roots) was isolated using TRIzol Reagent (Thermo Scientific), according to the manufacturer’s instructions. RNA from roots was extracted using a LiCl method42. Genomic DNA was removed using RQ1 RNase-free DNase (Promega), according to the manufacturer’s instructions. Total RNA was quantified using a NanoDrop ND-1000 Spectrophotometer (Uniscience), and RNA integrity was verified in agarose gel electrophoresis. Reverse transcription reaction was carried out with 1 μg of total RNA and oligo (dT) in a total volume of 20 μL using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific), following the manufacturer’s recommendations. cDNA samples were diluted (1:25) prior to use in RT-qPCR assays.
Quantitative Real-time PCR conditions
RT-qPCR was carried out in a 96-well optical plate with a StepOnePlus Real-Time PCR Systems (Applied Biosystems). Reactions were performed using Platinum SYBR Green PCR SuperMix-UDG with ROX (Invitrogen), 0.2 μM of each primer and 1 μL of diluted cDNA (1:25) in a final volume of 10 μL. The following thermal cycling condition was used for all amplifications: 2 min at 50 °C min, 20 sec at 95 °C, followed by 40 amplification cycles of 95 °C for 3 sec, and 60 °C for 30 sec. After 40 cycles, the specificity of the amplifications was analyzed through the dissociation curve profiles. Quantification cycle threshold (Cq) values per target were manually estimated. Background-corrected raw fluorescence data were imported into LinRegPCR version 2016.0 software for primer efficiency estimation55. The program uses linear regression analysis to fit a straight line and estimate PCR efficiency of each individual sample based on the slope of this line56,57. All assays were performed using three biological replicates with three technical replicates each and a non-template control.
Assessing the expression stability of reference genes
To estimate the expression stability of reference genes, Cq values were converted into non-normalized relative quantities. These values are obtained using the formula Q = EΔCq, where E represents the average efficiency for each gene, and ΔCq represents the difference between the lowest Cq value of a sample of a particular gene and the Cq value of each sample in a dataset40. These data were imported into R/Bioconductor for reference genes selection using geNorm (medgen.ugent.be/∼jvdesomp/geNorm/)32 and NormFinder ( www.mdl.dk/publicationsNormFinder.htm)33 algorithms. Global analysis was performed using all datasets. Subsequently, each experimental set was assessed to define specific reference genes for proper normalization.
Validation of reference genes
Validation of the selected reference genes was carried out in samples of drought treatment and developmental series. In drought treatment, we analyzed the expression pattern of P5CS and GolS, two genes that function as osmoprotectants in drought-stress tolerance in plants34,35. We also quantified the relative expression of SuSy in stem compared to leaf tissues during S. viridis development. Normalization of both target genes was performed using either the two most stable candidate reference genes or the least stable one as determined by geNorm analysis.
Additional Information
How to cite this article: Martins, P. K. et al. Selection of reliable reference genes for RT-qPCR analysis during developmental stages and abiotic stress in Setaria viridis. Sci. Rep. 6, 28348; doi: 10.1038/srep28348 (2016).
Supplementary Material
Acknowledgments
This work was supported by the National Center for Research on Agroenergy (CNPAE), the Brazilian Agricultural Research Corporation (Embrapa) grant n. MP2-02.12.01.008.00.00 and the São Paulo Research Foundation (FAPESP) grant n. 2014/07918-6.
Footnotes
Author Contributions H.B.C.M., A.K.K. and P.K.M. conceived and designed the experiments. P.K.M., V.M. and A.P.R. performed the developmental stage assay. M.F.B, W.R.d.S. and A.P.R. performed the abiotic assays. P.K.M., V.M., F.V., W.R.d.S. and B.A.D.B.d.C. carried out the RT-qPCR assays and analyzed the data. P.K.M., V.M., W.R.d.S. and A.P.R. wrote the manuscript. H.B.C.M. and A.K.K. provided intellectual input and revised the manuscript. All authors read and approved the final manuscript.
References
- Brutnell T. P. et al. Setaria viridis: a model for C4 photosynthesis. Plant Cell 22, 2537–2544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen J. L. et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30, 555–561 (2012). [DOI] [PubMed] [Google Scholar]
- Martins P. K., Ribeiro A. P., da Cunha B. A. D. B., Kobayashi A. K. & Molinari H. B. C. A simple and highly efficient Agrobacterium-mediated transformation protocol for Setaria viridis. Biotechnol. Rep. 6, 41–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha P. & Blumwald E. Spike dip transformation of Setaria viridis. Plant J. 86, 89–101 (2016). [DOI] [PubMed] [Google Scholar]
- Fahlgren N. et al. A versatile phenotyping system and analytics platform reveals diverse temporal responses to water availability in Setaria. Mol. Plant. 8, 1520–1535 (2015). [DOI] [PubMed] [Google Scholar]
- Muthamilarasan M. et al. Global analysis of WRKY transcription factor superfamily in Setaria identifies potential candidates involved in abiotic stress signaling. Front. Plant Sci. 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthamilarasan M. & Prasad M. Advances in Setaria genomics for genetic improvement of cereals and bioenergy grasses. Theor. Appl. Genet. 128, 1–14 (2015). [DOI] [PubMed] [Google Scholar]
- Shu Y., Liu Y., Zhang J., Song L. & Guo C. Genome-wide analysis of the AP2/ERF superfamily genes and their responses to abiotic stress in Medicago truncatula. Front. Plant Sci. 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojta P. et al. Whole transcriptome analysis of transgenic barley with altered cytokinin homeostasis and increased tolerance to drought stress. New Biotechnol . (2016). [DOI] [PubMed] [Google Scholar]
- Miao Z. et al. De novo transcriptome analysis of Medicago falcata reveals novel insights about the mechanisms underlying abiotic stress-responsive pathway. BMC Genomics 16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue R. et al. Genome-wide identification and expression profiling analysis of ZmPIN, ZmPILS, ZmLAX and ZmABCB auxin transporter gene families in maize (Zea mays L.) under various abiotic stresses. PLoS ONE 10, e0118751 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. et al. Expanding frontiers in plant transcriptomics in aid of functional genomics and molecular breeding. Biotechnol. J. 9, 1480–1492 (2014). [DOI] [PubMed] [Google Scholar]
- Wong M. L. & Medrano J. F. Real-time PCR for mRNA quantitation. BioTechniques 39, 75–85 (2005). [DOI] [PubMed] [Google Scholar]
- Kozera B. & Rapacz M. Reference genes in real-time PCR. J. Appl. Genet. 54, 391–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. J., Tian C., Jiang Q., Li X. H. & Zhuang J. Selection of suitable reference genes for qRT-PCR normalization during leaf development and hormonal stimuli in tea plant (Camellia sinensis). Sci. Rep. 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang H., Fu Y., He W., Wang L. & Wei Y. Selection of appropriate reference genes for quantitative real-time PCR in Oxytropis ochrocephala Bunge using transcriptome datasets under abiotic stress treatments. Front. Plant Sci. 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambret-Frotté J. et al. Validating internal control genes for the accurate normalization of qPCR expression analysis of the novel model plant Setaria viridis. PLoS ONE 10, e0135006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay Y., Michaud J. & Dubé M. P. Reference genes for RT-qPCR analysis of environmentally and developmentally regulated gene expression in alfalfa. Am. J. Plant Sci. 6, 132 (2015). [Google Scholar]
- Bansal R. et al. Recommended reference genes for quantitative PCR analysis in soybean have variable stabilities during diverse biotic stresses. PLoS ONE 10, e0134890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. of reference genes for quantitative real-time PCR normalization in creeping bentgrass involved in four abiotic stresses. Plant Cell Rep. 34, 1825–1834 (2015). [DOI] [PubMed] [Google Scholar]
- Li D. et al. Identification and evaluation of reference genes for accurate transcription normalization in safflower under different experimental conditions. PLoS ONE 10, e0140218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C. et al. Selection of suitable reference genes for qPCR normalization under abiotic stresses and hormone stimuli in carrot leaves. PLoS ONE 10, e0117569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathi M. S. & Karaba N. N. Emerging tools, concepts and ideas to track the modulator genes underlying plant drought adaptive traits: An overview. Plant Signal. Behav . (just-accepted) 00-00 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelbart M. V., Hasegawa P. M. & Bailey-Serres J. Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nat. Rev. Genet. 16, 237–251 (2015). [DOI] [PubMed] [Google Scholar]
- Moshelion M. & Altman A. Current challenges and future perspectives of plant and agricultural biotechnology. Trends Biotechnol. 33, 337–342 (2015). [DOI] [PubMed] [Google Scholar]
- Pandey P., Ramegowda V. & Senthil-Kumar M. Shared and unique responses of plants to multiple individual stresses and stress combinations: physiological and molecular mechanisms. Front. Plant Sci. 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Ling H., Wu Q., Xu L. & Que Y. The choice of reference genes for assessing gene expression in sugarcane under salinity and drought stresses. Sci. Rep. 4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H., Wu Q., Guo J., Xu L. & Que Y. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. PLoS ONE 9, e97469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar K., Muthamilarasan M. & Prasad M. Reference genes for quantitative real-time PCR analysis in the model plant foxtail millet (Setaria italica L.) subjected to abiotic stress conditions. Plant Cell Tissue Organ Cult. 115, 13–22 (2013). [Google Scholar]
- Molinari H. B. C., Pellny T. L., Freeman J., Shewry P. R. & Mitchell R. A. C. Grass cell wall feruloylation: distribution of bound ferulate and candidate gene expression in Brachypodium distachyon. Front. Plant Sci. 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsai R., Ivanova A., Ng S. & Whelan J. Defining reference genes in Oryza sativa using organ, development, biotic and abiotic transcriptome datasets. BMC Plant Biol. 10, 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, 1–12 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen C. L., Jensen J. L. & Ørntoft T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 (2004). [DOI] [PubMed] [Google Scholar]
- Molinari H. B. C. et al. Evaluation of the stress-inducible production of proline in transgenic sugarcane (Saccharum spp.): osmotic adjustment, chlorophyll fluorescence and oxidative stress. Physiol. Plantarum 130, 218–229 (2007). [Google Scholar]
- Taji T. et al. Important roles of drought and cold inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. Plant J. 29, 417–426 (2002). [DOI] [PubMed] [Google Scholar]
- Nanjo T. et al. Biological functions of proline in morphogenesis and osmotolerance revealed in antisense transgenic Arabidopsis thaliana. Plant J. 18, 185–193 (1999). [DOI] [PubMed] [Google Scholar]
- Chandra A. et al. Expression analysis of genes associated with sucrose accumulation in sugarcane (Saccharum spp. hybrids) varieties differing in content and time of peak sucrose storage. Plant Biol. 17, 608–617 (2015). [DOI] [PubMed] [Google Scholar]
- Ramalashmi K., Prathima P. T., Mohanraj K. & Nair N. V. Expression profiling of sucrose metabolizing genes in Saccharum, Sorghum and their hybrids. Appl. Biochem. Biotechnol. 174, 1510–1519 (2014). [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Miceli F. A. et al. Relationship between sucrose accumulation and activities of sucrose-phosphatase, sucrose synthase, neutral invertase and soluble acid invertase in micropropagated sugarcane plants. Acta Physiol. Plant. 24, 441–446 (2002). [Google Scholar]
- Hellemans J., Mortier G., De Paepe A., Speleman F. & Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruz T. et al. RefGenes: identification of reliable and condition specific reference genes for RT-qPCR data normalization. BMC Genomics 12, 156 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S., Puryear J. & Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116 (1993). [Google Scholar]
- Artico S., Nardeli S. M., Brilhante O., Grossi-de-Sa M. & Alves-Ferreira M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 10, 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perini P., Pasquali G., Margis-Pinheiro M., de Oliveira P. R. D. & Revers L. F. Reference genes for transcriptional analysis of flowering and fruit ripening stages in apple (Malus × domestica Borkh.). Mol. Breed. 34, 829–842 (2014). [Google Scholar]
- Mafra V. et al. Reference genes for accurate transcript normalization in citrus genotypes under different experimental conditions. PLoS ONE 7, e31263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L. et al. Evaluation of candidate reference genes for normalization of quantitative RT-PCR in switchgrass under various abiotic stress conditions. Bioenergy Res. 7, 1201–1211 (2014). [Google Scholar]
- Marcolino-Gomes J. et al. Transcriptome-wide identification of reference genes for expression analysis of soybean responses to drought stress along the day. PLoS ONE 10, e0139051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R. L. D. O. et al. Validation of novel reference genes for reverse transcription quantitative real-time PCR in drought-stressed sugarcane. Sci. World J. ID 35705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulao L. F., Fortunato A. S. & Ramalho J. C. Selection of reference genes for normalizing quantitative real-time PCR gene expression data with multiple variables in Coffea spp. Plant Mol. Biol. Rep. 30, 741–759 (2012). [Google Scholar]
- Su M. et al. Cloning two P5CS genes from bioenergy sorghum and their expression profiles under abiotic stresses and MeJA treatment. Plant Sci. 181, 652–659 (2011). [DOI] [PubMed] [Google Scholar]
- Vendruscolo E. C. G. et al. Stress-induced synthesis of proline confers tolerance to water deficit in transgenic wheat. J. Plant Physiol. 164, 1367–1376 (2007). [DOI] [PubMed] [Google Scholar]
- Santos T. B. D. et al. Galactinol synthase transcriptional profile in two genotypes of Coffea canephora with contrasting tolerance to drought. Genet. Mol. Biol. 38, 182–190 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki K. & Yamaguchi-Shinozaki K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 58, 221–227 (2007). [DOI] [PubMed] [Google Scholar]
- Murashige T. & Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol. Plantarum 15, 473–479 (1962). [Google Scholar]
- Ramakers C., Ruijter J. M., Deprez R. H. & Moorman A. F. M. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339, 62–66 (2003). [DOI] [PubMed] [Google Scholar]
- Ruijter J. M. et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45–e45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle S. E. & Dyer J. M. Cloning and expression of sucrose synthase-1 cDNA from sugarcane. J. Plant Physiol. 158, 129–131 (2001). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.