

Abstract
Aim
The purpose of our study was to determine whether genes involved in the organization of the hematopoietic niche were dysregulated in patients with primary myelofibrosis (MF) treated with lenalidomide.
Materials and Methods
We used reverse-transcription quantitative polymerase chain reaction to study the expression of a set of genes involved in the organization of the hematopoietic niche in peripheral blood and bone marrow (BM) mononuclear cell (MNC) samples from 32 patients with primary MF who participated in a phase II trial of lenalidomide plus prednisone.
Results
At baseline (before treatment) cyclooxygenase 2 (COX2) was significantly up-regulated, while chemokine (C-X-C motif) receptor 4 (CXCR4), paired box 5 (PAX5) C-terminus, and hypoxia inducible factor 1A(HIF-1α) were significantly down-regulated in BM MNCs from patients with primary MF compared to BM MNCs from healthy individuals. After 9 months of treatment, the expression of suppressor of cytokine signaling 3 (SOCS3) was significantly increased.
Conclusion
Patients with primary MF showed aberrant expression of several genes involved in maintaining BM homeostasis and our findings suggest that treatment with lenalidomide plus prednisone up-regulates SOCS3.
Introduction
Progressive bone marrow (BM) fibrosis is a key feature of primary myelofibrosis (MF), a disease characterized by clonal myeloproliferation. The BM microenvironment comprises of stromal cells, osteoclasts, and endothelial cells, and communication defects between these cells upon expansion of the neoplastic clone results in a functionally-disturbed stromal niche, impaired hematopoiesis and, eventually BM fibrosis. Lenalidomide, an agent which modulates inflammatory cytokine secretion, angiogenesis and the expression of adhesion molecules likely has effects on the BM microenvironment.1 In a phase II trial of lenalidomide plus prednisone in 40 patients with MF conducted at our Institution, 30% of patients achieved an objective response.13 Furthermore, 10 out of 11 patients who responded and had grade 4 reticulin fibrosis at baseline had reduction in fibrosis to grade 2 or less. While newer therapies, such as janus kinase (JAK) inhibitors, have been shown to significantly improve symptoms of MF and quality of life, with the exception of minor reductions in BM fibrosis in some patients after many years of treatment with ruxolitinib,8 lenalidomide is the only therapy shown to significantly reduce BM fibrosis in patients with MF. We hypothesized that lenalidomide may exert its effects, in part, by modulating the expression of genes involved in maintaining the BM stromal niche. To test our hypothesis, we measured the expression of a set of genes involved the organization of the hematopoietic niche in peripheral blood (PB) and BM mononuclear cell (MNC) samples from patients with primary MF who participated in a phase II trial of lenalidomide plus prednisone.13 Genes involved in cell-stroma interactions (secreted protein, acidic, cysteine-rich [SPARC], chemokine [C-X-C motif] receptor 4 [CXCR4]), angiogenesis (cyclo-oxygenase 2 [COX-2]), response to hypoxia (hypoxia inducible factor 1A [HIF-1α], and cell differentiation and signaling (paired box 5 [PAX5] C-terminus, FBJ murine osteosarcoma viral oncogene homolog [FOS], Kristen rat sarcoma viral oncogene homolog [KRAS], suppressor of cytokine signaling 3 [SOCS3] were profiled.
Materials and Methods
All patients gave written informed consent and the study was approved by the Institutional Review Board (PA11-1122) and performed in accordance with the Declaration of Helsinki. BM and PB samples from six hematologically healthy individuals were purchased from Stem Cell Technologies (Vancouver, Canada). BM aspirates and PB samples were available for 13 patients with primary MF. Sequential BM and PB samples were collected at baseline and every 3 months during the course of treatment. However, samples were not available for all 13 patients at all time points due to the fact that some patients discontinued treatment or died, or the samples were of poor quality. For this reason, baseline BM samples available for nine patients and baseline PB samples available for 11 patients (13 patients total) were used for our analysis. Low-density mononuclear cells (MNCs) were isolated from BM aspirates and PB samples using gradient centrifugation with Ficoll Hypaque 1077 (Sigma-Aldrich, St. Louis, MO, USA). Total RNA was isolated from gradient-separated MNCs using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcription was performed with the Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed to quantify the expression levels of SPARC, COX2, CXCR4, Pax5 C-terminus, SOCS3, HIF-1α and β-actin (reference gene) using primer pairs obtained from Applied Biosystems Inc. (Foster City, CA, USA). The primer sequences used are listed in Table 1. qRT-PCR was performed in duplicate for each sample. Gene expression was calculated as ΔCT values, using β-actin as the reference gene. Data are presented as mean ΔCT values with 95% confidence intervals. Student's t-tests were used to compare mean ΔCT values from patient samples at baseline (before treatment) and healthy controls. One-way analysis of variance was used to compare the mean ΔCT values at different time points.
Table I. Primers used for quantitative reverse transcriptase-polymerase chain reactionq (qRT-PCR).
| Primer | Gene name | Primer sequence |
|---|---|---|
| COX2_F: | Cyclo-oxygenase 2 | 5′-CCT TCCTCCTGTGCCTGATG-3′, |
| COX2_R: | 5′-ACAATCTCATTTGAATCAGGAAGCT-3′ | |
| COX2-6FAM | 5′-TGCCCGACTCCCTTGGGTGTCA-MGBNFQ | |
| SPARC_R: | Secreted protein, acidic, cysteine-rich | 5′-TCTTCCCTGTACACTGGCAGTTC-3′ |
| SPARC_F: | 5′-AGCTCGGTGTGGGAGAGGTA-3′ | |
| SPARC-6FAM | 5′-CAGCTGGACCAGCACCCATTGACA-MGBNFQ | |
| HIF1A_F: | Hypoxia inducible factor 1A | 5′-CTCATCCAAGAAGCCCTAACGTGTT-3′ |
| HIF1A_R: | 5′-GCTTTCTCTGAGCATTCTGCAAAGC-3′ | |
| HIF1A-6FAM | 5′-CCTCAGGAACTGTAGTTCTTTGACTCAAAGCGACA-MGBNFQ | |
| CXCR4: | Chemokine (C-X-C motif) receptor 4 | Hs00607978_s1 (Applied Bioscience) |
| PAX5C | Paired box 5 C-terminus | Hs00277134_m1 (Applied Bioscience) |
| FOS_F: | FBJ murine osteosarcoma viral oncogene homolog | 5′-CGAGCCCTTTGTATGACTTCCT-3′ |
| FOS_R: | 5′-GTCCATGTCTGGCACGGA G-3′ | |
| FOS-6FAM | 5′-CCCAGCATCATCCAGGCCCAGTCA-MGBNFQ | |
| KRAS_F: | Kristen rat sarcoma viral oncogene homolog | 5′-TTCCTACAGGAAGCA AGT AG-3′ |
| KRAS_R: | 5′-CACAAAGAA AGCCCTCCCCA-3′ | |
| KRAS-6FAM | 5′-TTGATGGAGAAACCTGTCTCTTGGCA-MGBNFQ | |
| β-Actin_F: | 5′-GATGGCCACGGCTGCTT-3′ | |
| β-Actin _R: | 5′-ACCGCTCATTGCCAATGG-3′ | |
| β-Actin -6FAM | 5′-ACCACCACGGCCGAGCGGCA-MGMNFQ |
F: Forward primer; R: reverse primer; 6FAM: 6-carboxyfluorescin; MGBNFQ: molecular-groove binding non-fluorescence quencher.
Results and Discussion
Clinical characteristics of the 13 patients whose samples we studied are shown in Table 2. At baseline (before treatment) COX2 was significantly up-regulated, while CXCR4, Pax5 C-terminus, and HIF-1α were significantly down-regulated in BM MNCs from patients compared to healthy BM MNCs (Table 3). Expression of SPARC, KRAS, SOCS3, and FOS and were not significantly different. Although SOCS3 has been shown to be down-regulated in primary MF, in part due to hypermethylation of its promoter, we only detected a significant difference in the expression of SOCS3 in patients with JAK2-negative MF and normal controls (Figure 1B). There were no significant differences in relative gene expression between BM MNC and PB samples; however, when compared with samples from normal controls, significant down-regulation of CXCR4 and HIF-1α in primary MF was only observed in the BM samples. This may be expected since changes in the expression of these genes are likely to be more prominent in the BM.
Table II. Baseline characteristics of 13 patients whose samples were analyzed in this study.
| Characteristic | Value |
|---|---|
| Age, median (range), years | 65 (51-83) |
| Gender, N (%) | |
| Male | 7 (54) |
| Female | 6 (46) |
| Cytogenetics, N (%) | |
| Diploid | 6 (46) |
| Abnormal | 7 (54) |
| JAK2V617F mutation, N (%) | |
| Positive | 7 (54) |
| Negative | 4 (31) |
| Not determined | 1 (8) |
| Hemoglobin, mean (range), g/dL | 10.1 (8.1-16.4) |
| Platelet count, mean (range), ×l09/L | 244 (18-704) |
| WBC count, mean (range), ×l09/L | 12 (1.3-28) |
| Spleen size, mean (range), cm | 8.9 (0-22) |
| Performance status, median (range) | 1 (0-2) |
| Prior therapy, N (%) | 8 (69) |
| Number of prior therapies, median (range) | 1 (0-3) |
| Best response (IWG-MRT)*, N (%) | |
| Stable disease | 6 (46) |
| Clinical improvement | 5 (38) |
| Partial response | 1 (8) |
| Complete hematological response | 1 (8) |
| Duration of response, median (range), months | 9 (3-59) |
IWG-MRT: International Working Group for Myelofibrosis Research and Treatment; JAK: janus kinase;
IWG-MRT criteria published in 2006 were used.
Table III. Comparison of mean relative gene expression in patients with primary myelofibrosis and healthy controls at baseline.
| Gene | Gene name | Patients | Controls | p-Value* | ||
|---|---|---|---|---|---|---|
|
| ||||||
| No. of samples analyzed | Mean ΔCT (95% CI) | No. of samples analyzed | Mean ΔCT (95% CI) | |||
| SPARC-BM | Secreted protein, acidic, cysteine-rich | 9 | 4.953 (3.84-6.06) | 6 | 6.207 (5.62-6.79) | 0.0659 |
| SPARC-PB | 11 | 5.852 (4.17-7.54) | 6 | 4.941 (3.93-5.96) | 0.4113 | |
| COX2-BM | Cyclo-oxygenase 2 | 9 | 7.259 (6.27-8.25) | 6 | 8.998 (8.08-9.92) | 0.0132 |
| COX2-PB | 11 | 7.509 (6.32-8.70) | 6 | 11.40 (9.74-13.1) | 0.0004 | |
| KRAS-BM | Kristen rat sarcoma viral oncogene | 9 | 11.60 (7.33-15.87) | 6 | 6.548 (4.526-8.570) | 0.0538 |
| KRAS-PB | 11 | 12.35 (8.24-16.46) | 6 | 7.974 (7.05-8.90) | 0.1064 | |
| FOS-BW | FBJ murine osteosarcoma viral oncogene homolog | 9 | 2.656 (1.952-3.359) | 6 | 2.720 (1.77-3.67) | 0.895 |
| FOS-PB | 11 | 3.349 (2.38-4.32) | 5 | 4.503 (3.53-5.48) | 0.1197 | |
| CXCR4-BM | Chemokine (C-X-C motif) receptor 4 | 9 | 5.072 (3.98-6.17) | 6 | 2.707 (2.30-3.11) | 0.0017 |
| CXCR4-PB | 11 | 4.351 (3.33-5.37) | 4 | 5.284 (4.618-5.949) | 0.2575 | |
| PAX5C-BM | Paired box 5 C-terminus | 6 | 9.539 (7.83-11.25) | 4 | 6.473 (4.80-8.15) | 0.0108 |
| PAX5C-PB | 11 | 9.971 (8.64-11.31) | 3 | 7.482 (5.83-9.13) | 0.0592 | |
| SOCS2-BM | Suppressor of cytokine signaling 2 | 8 | 11.47 (8.46-14.47) | 4 | 8.828 (7.76-9.90) | 0.1634 |
| SOCS2-PB | 11 | 13.19 (10.41-15.97) | 2 | 9.400 (6.80-12.00) | 0.7099 | |
| SOCS3-BM | Suppressor of cytokine signaling 3 | 10 | 5.384 (3.946-6.822) | 4 | 4.724 (1.71-7.74) | 0.5838 |
| SOCS3-PB | 11 | 5.011 (3.50-6.52) | 3 | 5.562 (2.15-8.97) | 0.6984 | |
| HIF1A-BM | Hypoxia inducible factor 1A | 10 | 7.303 (6.34-8.27) | 4 | 5.106 (4.20-6.01) | 0.0095 |
| HIFIA-PB | 11 | 6.259 (5.39-7.13) | 3 | 6.32 (5.90-6.74) | 0.9383 | |
Differences in mean expression between patient sample and control samples were compared using unpaired t-tests. p<0.05 was considered significant. CT: cycle threshold value; CI: confidence interval; BM: bone marrow; PB: peripheral blood.
Figure 1.
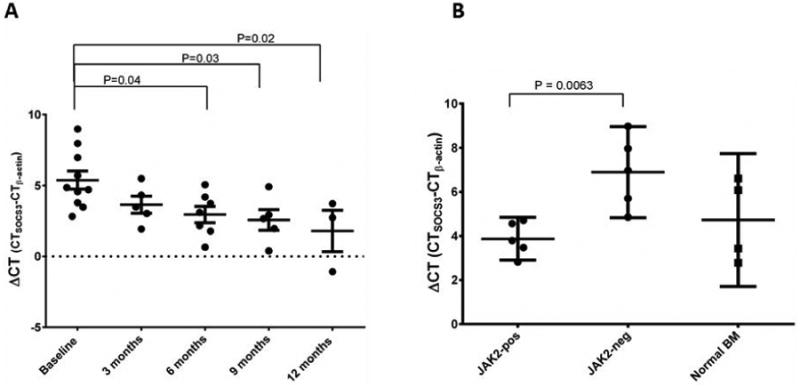
Changes in suppressor of cytokine signaling 3 [SOCS3] gene expression. A: Expression of SOCS3 increased significantly with time on treatment (p=0.02 using one-way analysis of variance). Mean expression at 9, 12 and >14 months was significantly higher than at baseline, as assessed by Dunnett's multiple comparisons test. Horizontal bars represent median±standard deviation. p<0.05 was considered statistically significant. B: SOCS3 expression was significantly higher in patients with the Janus kinase 2 V617F mutation.
Treatment with lenalidomide plus prednisone had no effect on the expression of SPARC, COX-2, CXCR4, Pax5 C-terminus, or HIF-1α. However, the expression of SOCS3 was significantly increased after 9 months of treatment (Figure 1A), suggesting that some of the clinical effects of lenalidomide may be due to a SOCS3-mediated reduction in JAK signaling. Interestingly, at baseline SOCS3 expression was significantly lower in patients without the JAK2V617F mutation than in those with the mutation (p=0.0063; Figure 1B), which is in agreement with a previous study3. By contrast, there was no correlation between JAK2 mutation status and the expression of the other genes. In addition, there was no correlation between expression levels and cytogenetic abnormalities.
Up-regulation of COX2 and down-regulation of CXCR4, PAX5C, and HIF-1α may reflect disruptions in the interactions between cells in the BM microenvironment in primary MF.
For example, down-regulation of CXCR4 in primary MF has been shown in several studies and is thought to contribute to the increased circulation of CD34+ cells in primary MF.1, 6, 15 The observed down-regulation of HIF-1α in the BM from patients with primary MF is consistent with studies showing that a less hypoxic BM microenvironment promotes the aberrant proliferation of hematopoietic progenitor cells in MPNs.2, 9, 14 PAX5 encodes a transcription factor that plays a key role in B-cell development.11 Thus, the reduction in PAX5C expression in primary MF BM compared with normal BM, may be due to the expansion of the myeloid lineage in primary MF. Finally, the up-regulation of COX2, an enzyme involved in the formation of prostaglandins, which are key mediators of inflammation and angiogenesis (among other functions), is consistent with the increased inflammation and angiogenesis seen in BM in primary MF.5, 10, 12
In T-cells from BM in multiple myeloma, lenalidomide has been shown to decrease SOCS1 expression.4 How lenalidomide increases SOCS3 expression in primary MF is therefore not clear. It is intriguing that SOCS3 has been shown to act as part of an E3 ubiquitin ligase complex to promote ubiquitinylation (and hence degradation) of JAK2 and the interleukin 6 receptor common chain (gp130) given the recent finding that lenalidomide exerts it action in part by binding to cereblon as part of an E3 ubiquitin ligase complex.7 In conclusion, we found that patients with primary MF show aberrant expression of several genes involved in maintaining BM homeostasis and our findings suggest that treatment with lenalidomide plus prednisone up-regulates SOCS3. These results should be validated in a larger cohort of patients treated with lenalidomide.
Acknowledgments
Grant support: This work was supported in part by a Cancer Center Support Grant from the United States National Cancer Institute (CA016672) to The University of MD Anderson Cancer Center.
Footnotes
S.V. and R.K. designed the study, assisted with data analysis and reviewed the manuscript. A.L. and T.M. designed the study, conducted the experiments, analyzed the data and reviewed the final manuscript. K.N. analyzed the data and wrote the manuscript. The authors have no conflicts of interest to declare.
References
- 1.Bogani C, Ponziani V, Guglielmelli P, et al. Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells. 2008;26:1920–30. doi: 10.1634/stemcells.2008-0377. [DOI] [PubMed] [Google Scholar]
- 2.Desplat V, Faucher JL, Mahon FX, Sbarba PD, Praloran V, Ivanovic Z. Hypoxia Modifies Proliferation and Differentiation of CD34+ CML Cells. STEM CELLS. 2002;20:347–54. doi: 10.1634/stemcells.20-4-347. [DOI] [PubMed] [Google Scholar]
- 3.Fourouclas N, Li J, Gilby DC, et al. Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. 2008 doi: 10.3324/haematol.13043. [DOI] [PubMed] [Google Scholar]
- 4.Gorgun G, Calabrese E, Soydan E, et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood. 2010;116:3227–37. doi: 10.1182/blood-2010-04-279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine & growth factor reviews. 2013;24:133–45. doi: 10.1016/j.cytogfr.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Hasselbalch HC, Skov V, Stauffer Larsen T, et al. Transcriptional profiling of whole blood identifies a unique 5-gene signature for myelofibrosis and imminent myelofibrosis transformation. PLoS One. 2014;9:e85567. doi: 10.1371/journal.pone.0085567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kershaw NJ, Laktyushin A, Nicola NA, Babon JJ. Reconstruction of an active SOCS3-based E3 ubiquitin ligase complex in vitro: identification of the active components and JAK2 and gp130 as substrates. Growth factors (Chur, Switzerland) 2014;32:1–10. doi: 10.3109/08977194.2013.877005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kvasnicka HM, Thiele J, Bueso-Ramos CE, et al. Effects Of Five-Years Of Ruxolitinib Therapy On Bone Marrow Morphology In Patients With Myelofibrosis and Comparison With Best Available Therapy. Blood. 2013;122:4055. [Google Scholar]
- 9.Mitsumori T, Nozaki Y, Kawashima I, et al. Hypoxia inhibits JAK2V617F activation via suppression of SHP-2 function in myeloproliferative neoplasm cells. Experimental hematology. 2014;42:783–92.e1. doi: 10.1016/j.exphem.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Ni H, Barosi G, Hoffman R. Quantitative evaluation of bone marrow angiogenesis in idiopathic myelofibrosis. American journal of clinical pathology. 2006;126:241–7. doi: 10.1309/4YGK-ED5L-WFW4-AVDV. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien P, Morin P, Ouellette RJ, Robichaud GA. The Pax-5 Gene: A Pluripotent Regulator of B-cell Differentiation and Cancer Disease. Cancer Research. 2011;71:7345–50. doi: 10.1158/0008-5472.CAN-11-1874. [DOI] [PubMed] [Google Scholar]
- 12.Ponce CC, Chauffaille MLLF, Ihara SSM, Silva MRR. Increased angiogenesis in primary myelofibrosis: Latent transforming growth factor-β as a possible angiogenic factor. Revista Brasileira de Hematologia e Hemoterapia. 2014;36:322–8. doi: 10.1016/j.bjhh.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quintas-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:4760–6. doi: 10.1200/JCO.2009.22.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers HM, Yu X, Wen J, Smith R, Fibach E, Noguchi CT. Hypoxia alters progression of the erythroid program. Experimental hematology. 2008;36:17–27. doi: 10.1016/j.exphem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosti V, Massa M, Vannucchi AM, et al. The expression of CXCR4 is down-regulated on the CD34+ cells of patients with myelofibrosis with myeloid metaplasia. Blood cells, molecules & diseases. 2007;38:280–6. doi: 10.1016/j.bcmd.2007.01.003. [DOI] [PubMed] [Google Scholar]