

Summary
Interleukin‐2 (IL‐2) is a critical regulator of immune homeostasis through its non‐redundant role in regulatory T (Treg) cell biology. There is major interest in therapeutic modulation of the IL‐2 pathway to promote immune activation in the context of tumour immunotherapy or to enhance immune suppression in the context of transplantation, autoimmunity and inflammatory diseases. Antibody‐mediated targeting of the high‐affinity IL‐2 receptor α chain (IL‐2Rα or CD25) offers a direct mechanism to target IL‐2 biology and is being actively explored in the clinic. In mouse models, the rat anti‐mouse CD25 clone PC61 has been used extensively to investigate the biology of IL‐2 and Treg cells; however, there has been controversy and conflicting data on the exact in vivo mechanistic function of PC61. Engineering antibodies to alter Fc/Fc receptor interactions can significantly alter their in vivo function. In this study, we re‐engineered the heavy chain constant region of an anti‐CD25 monoclonal antibody to generate variants with highly divergent Fc effector function. Using these anti‐CD25 Fc variants in multiple mouse models, we investigated the in vivo impact of CD25 blockade versus depletion of CD25+ Treg cells on immune homeostasis. We report that immune homeostasis can be maintained during CD25 blockade but aberrant T‐cell activation prevails when CD25+ Treg cells are actively depleted. These results clarify the impact of PC61 on Treg cell biology and reveal an important distinction between CD25 blockade and depletion of CD25+ Treg cells. These findings should inform therapeutic manipulation of the IL‐2 pathway by targeting the high‐affinity IL‐2R.
Keywords: CD25, interleukin‐2, regulatory T cells, therapeutic antibody
Introduction
Interleukin‐2 (IL‐2) is a key nodal regulator of immune homeostasis (reviewed in refs. 1, 2, 3). The importance of IL‐2‐mediated immune regulation in health and well‐being is evidenced by the lethal lymphoid hyperplasia and autoimmune syndrome that develops in mice and humans that are genetically deficient in IL‐2 or components of its receptor.4, 5, 6, 7, 8 Polymorphisms linked to components of the IL‐2 receptor are associated with autoimmune diseases, including multiple sclerosis, type 1 diabetes, coeliac diseases and rheumatoid arthritis.9, 10 Given this central role for IL‐2 in immune control, there is significant interest in therapeutic modulation of the IL‐2 pathway to potentiate cancer immunotherapy, facilitate transplant tolerance and treat autoimmune and inflammatory diseases.3, 11
Interleukin‐2 limits immune activation and maintains immune homeostasis through its non‐redundant role in the development and maintenance of regulatory T (Treg) cells.12, 13, 14, 15 The Treg cells constitutively express the IL‐2 receptor α chain (IL‐2Rα or CD25), the defining component of the high‐affinity IL‐2R complex. Low‐level IL‐2 production by conventional T cells in the steady state is required to maintain Treg cells, which do not produce IL‐2, at the numbers necessary to limit spontaneous T‐cell activation.15, 16, 17, 18 Given this central role for IL‐2 in Treg cell biology, it is critical to determine how a therapeutic agent that targets the IL‐2 pathway will impact Treg cells.
The in vivo impact of a therapeutic monoclonal antibody is determined by both its epitope specificity (e.g. blocking or non‐blocking of ligand interactions) and heavy‐chain constant region (Fc) effector function (e.g. depleting or non‐depleting). Varying the Fc properties of an antibody can significantly affect the biological impact in vivo. In the context of Treg cells and IL‐2 biology, monoclonal antibodies specific for CD25 have been used extensively as research tools in mouse models.19, 20, 21 The monoclonal rat anti‐mouse CD25 clone PC61 is widely used.22 PC61 inhibits IL‐2 binding to CD25 and in vitro it functionally inhibits IL‐2‐mediated T‐cell proliferation.22, 23 Potential in vivo consequences of anti‐CD25 antibodies on Treg cells include blockade of the IL‐2 survival signal, active depletion of CD25‐expressing Treg cells in an Fc‐dependent manner or a combination of the two mechanisms. Determining which mechanism(s) is operative in vivo and the specific impact of PC61 on Treg cells has been controversial.21, 24, 25, 26 Using PC61‐rIgG1, many laboratories have demonstrated a reduction in Treg cells with varying degrees of success (30–50% reduction in Foxp3+ cells in the spleen and lymph node of mice).21, 27 A major caveat in these studies is the assumption that the decline in Treg cell numbers is due to active depletion and not to blockade of the IL‐2 survival signal. It has been suggested that PC61‐rIgG1 treatment resulted in the in vivo ‘functional inactivation’ of Treg cells,25 but this view has been challenged.24, 28 One key aspect underlying this uncertainty is the use of the parental PC61.5 with a rat IgG1 isotype that precludes a direct interpretation of IL‐2 blockade alone. Furthermore, the differential impact of depleting versus non‐depleting anti‐CD25 antibodies on the broader maintenance of immune homeostasis in the steady state is unknown.
In the present study, we engineered the heavy‐chain constant region of PC61 to alter Fc‐mediated effector function without changing antibody specificity. By comparing Fc variants with highly divergent effector function we are able to demonstrate in mouse models the differential effects of actively depleting CD25+ Treg cells through only blockade of CD25 signalling. Our results demonstrate that immune homeostasis can be maintained during CD25 blockade but aberrant immune activation prevails when CD25+ Treg cells are actively depleted. These results should inform the design of monoclonal antibodies that therapeutically target the high‐affinity IL‐2R.
Materials and methods
Mice
Fcer1g −/− (Fcer1gtm1Rav) mice have been previously described29 and were subsequently backcrossed 12 generations on the C57BL/6 background. Fcer1g −/− mice and wild‐type C57BL/6 (B6) control mice were purchased from Taconic Biosciences, Inc. (Germantown, NY). Foxp3eGFP reporter mice (Foxp3tm2Tch) and MOG35‐55‐specific 2D2 T‐cell receptor (TCR) transgenic C57BL/6 mice (Tg(Tcra2D2,Tcrb2D2)1Kuch/J) have been previously described.30, 31 Foxp3eGFP mice and 2D2 mice were purchased from Jackson Laboratories (Bar Harbor, ME). All mice were 10–12 weeks of age at the time of experiments. Animals were housed in conventional rooms in a specific‐pathogen free facility with a 12/12 light–dark cycle. Room temperatures were maintained at 20–23.3°. Animals were socially housed in groups of up to five in Tecniplast caging on BetaChip bedding. All mice were between 20 and 25 g at the beginning of each experiment and all mice had a body conditioning score of 3. The Biogen Institutional Animal Care and Use Committee approved all animal protocols.
Engineering and production of PC61‐mIgG2a and PC61‐mIgG1(N297Q) variants
The PC61 5.3 hybridoma expressing PC61, a rat anti‐mouse CD25 IgG1 monoclonal antibody, was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and grown as recommended by the ATCC.22 Total cellular RNA was extracted (Qiagen RNeasy; Qiagen, Hilden, Germany) from hybridoma cells and cDNA was generated using random hexamer priming (SUPERSCRIPT III; Invitrogen, Carlsbad, CA). Vh and Vl gene segments were PCR amplified using degenerate primers and cloned into pCR4 (Invitrogen TOPO/TA). Inserts from multiple independent sub‐clones were sequenced and BLAST analyses confirmed rat immunoglobulin variable domain identity. Coding sequence was confirmed by comparison to the N‐terminal protein sequence of PC61.5 monoclonal antibody generated by Sanger sequencing. Verified heavy‐chain and light‐chain variable regions were then sub‐cloned into mammalian expression plasmids for expression as the rat–mouse chimeric antibodies PC61‐mIgG2a and PC61‐mIgG1(N297Q). Plasmids were used to generate stably transfected Chinese Hamster Ovary cells for production of the PC61 variants. Monoclonal antibody was purified from conditioned Chinese Hamster Ovary supernatants by protein A chromatography and size exclusion. Peak protein fractions were pooled and analysed for endotoxin (< 0·1 EU/mg) and aggregation level (< 5%). See Supplementary material (Fig. S1) for workflow schematic and for degenerate oligonucleotide and sequencing details (Table S1).
In vivo use of PC61‐mIgG2a and PC61‐mIgG1(N297Q) variants
For all in vivo experiments, antibodies were dosed by intraperitoneal injection at 500 µg/mouse in 200 µl PBS every 7 days. Dosing occurred in the morning while mice were in their home cage. Intraperitoneal injection was chosen to allow for minimal handling of the mice. A PBS vehicle control arm was used in each experiment. For 1‐week dosing experiments, wild‐type and Fcer1 g −/− mice were randomized into three groups with five mice per group. For 4‐week dosing experiments, the groups remained the same but the number of animals was increased to eight mice in the antibody groups and six mice in the PBS group. All mice were measured as a single unit. The dose and frequency were determined by pharmacokinetic studies to maintain complete receptor saturation, which was confirmed at the termination of each experiment by flow cytometry of splenocytes (data not shown). At the end of the dosing regimen, spleen and lymph node were harvested to evaluate cellular populations and cytokines by flow cytometry.
Experimental autoimmune encephalomyelitis
For experimental autoimmune encephalomyelitis (EAE) experiments, animals were weighed daily and monitored for clinical signs of disease using a standard five‐point scale as follows: Clinical Score (CS) 0, no sign of disease; CS 1, complete tail paralysis; CS 2, severe unilateral hind leg paresis or moderate bilateral paresis, hind limb weakness and affected gait; CS 3, partial hind limb paralysis, non‐weight bearing on hind limbs while moving; CS 4, complete bilateral hind limb paralysis; CS 5, moribund. Ameliorative care was given to mice with a CS of 3 or greater as follows: CS 3, hydrogel and food on floor of cage; CS 4, subcutaneous fluid supplementation. An investigator blinded to treatment status scored EAE experiments.
Flow cytometry, binding assay and STAT5 PhosFlow assay
For Foxp3 staining, cells were stained with anti‐CD3 (145‐2C11), anti‐CD4 (RM4‐5), anti‐CD8 (53‐6.7), anti‐CD25 (PC61) and anti‐CD62L (MEL‐14) from BD Biosciences (San Jose, CA), anti‐CD44 (IM7) from eBioscience (San Diego, CA) and anti‐CD25 (7D4) engineered from rat‐IgM isotype to human‐IgG1 isotype at Biogen (Cambridge, MA). Cells were then fixed and permeabilized with a Foxp3‐specific buffer system (eBioscience) and stained with anti‐Foxp3 (FJK‐16s).
For intracellular cytokine staining, cells were activated ex vivo with 50 ng/ml PMA and 500 ng/ml ionomycin in the presence of GolgiStop Protein Transport Inhibitor (BD Biosciences) for 4 hr. Cells were then stained with anti‐CD3 (145‐2C11), anti‐CD4 (RM4‐5) and anti‐CD8 (53‐6.7) from BD Biosciences and anti‐CD44 (IM7) from eBioscience, fixed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences) and stained with anti‐IL‐17A (TC11‐18H10) and anti‐interferon‐γ (IFN‐γ) (XMG1.2) from BD Biosciences.
For analysis of CD25 binding by the PC61 Fc‐variants, splenocytes from FOXP3eGFP mice were incubated with PC61‐mIgG2a, PC61‐mIgG1(N297Q) or PBS for 20 min at 4°. Cells were then stained with PC61‐allophycocyanin (BD Biosciences) for 20 min at 4°.
For analysis of signal transducer and activator of transcription 5 (STAT5) phosphorylation, splenocytes from naive FoxP3eGFP mice were stimulated with IL‐2 in the presence of PC61‐mIgG2a, PC61‐mIgG1(N297Q), anti‐HEL‐mIgG1+ anti‐HEL‐mIgG2a (isotype controls), or PBS. After 15 min, cells were fixed and permeabilized using the BD PhosFlow protocol with Perm III buffer (BD Biosciences). Cells were stained with anti‐CD3 (500A2), anti‐CD4 (RM4‐5), anti‐CD8 (53‐6.7) and phospho‐STAT5 (pY694) from BD Biosciences (San Jose, CA).
In all experiments, at least 50 000 events were collected on an LSR II, LSR Fortessa X‐20 or CALIBUR (BD Biosciences) and analysed using flowjo software (Tree Star, Ashland, OR).
In vitro Treg suppression assay
T responder cells were obtained by FACS sorting CD4+ TCR‐β + CD44low CD62Lhigh GFP− cells from Foxp3eGFP reporter mice, and labelled with CellTrace Violet (Life Technologies, Carlsbad, CA). Splenic CD3− cells were FACS sorted from Foxp3eGFP reporter mice, irradiated at 2500 rads and used as feeder cells. CD25high GFP+ and CD25low/− GFP+ Treg cells were FACS sorted from Foxp3eGFP reporter mice that had been treated for 4 weeks with PC61‐mIgG2a, PC61‐mIgG1(N297Q) or PBS. T responder cells (5 × 104) were cultured with irradiated feeder cells (2·5 × 105) in the absence or presence of Treg cell subpopulations isolated from the indicated mice at the indicated cell numbers. The cultures were stimulated with 0·25 µg/ml of anti‐CD3 and analysed for the dilution of CellTrace Violet at 60–72 hr. The data were acquired on an LSR Fortessa X‐20 (BD Biosciences) and analysed using flowjo. All cell populations were sorted to > 90% purity on a FACS Aria III (BD Biosciences).
Statistical analysis
graphpad prism software (GraphPad, La Jolla, CA) was used to perform all statistical analyses. For comparison of two independent groups, Student independent sample t‐test was used. For analysis of EAE experiments, the Mann–Whitney U‐test was performed. All P‐values < 0·05 were considered significant.
Results
Fc‐engineered anti‐CD25 variants bind CD25 and inhibit IL‐2‐induced STAT5 phosphorylation in Treg cells
Anti‐mCD25 clone PC61 blocks IL‐2 binding to CD25 and inhibits high affinity IL‐2R signalling.22, 23 To distinguish in vivo effects due to Fc‐mediated depletion versus blockade of CD25‐mediated IL‐2 signalling, we engineered a PC61‐mIgG2a variant that was expected to have strong Fc effector function and depletion activity (effector‐competent) and a second antibody variant, PC61‐mIgG1(N297Q), expected to have minimal to no Fc‐mediated effector function and no depletion activity (see Materials and methods for detailed description see Supplementary material, Fig. S1). Mouse IgG1 isotype has poor depletion capability and the N297Q mutation alters a critical N‐glycosylation site to diminish the Fc‐FcR interaction.32
Given that the antibody variants differ only in the Fc portion of the antibody, the ability to bind CD25 and block CD25‐mediated IL‐2 signalling should be identical. We first tested the Fc‐variants in a cell‐binding assay. Splenocytes from Foxp3eGFP mice were incubated with PC61‐mIgG1(N297Q), PC61‐mIgG2a or PBS. PC61‐allophycocyanin was then used to detect unbound CD25 by flow cytometry. Both PC61‐mIgG1(N297Q) and PC61‐mIgG2a had equal binding to CD25+ Treg cells (Fig. 1a). To test for functional inhibition of CD25 signalling, Treg cells were stimulated with IL‐2 in the presence of PC61‐mIgG1(N297Q), PC61‐mIgG2a or isotype control and STAT5 phosphorylation was measured. At the concentrations of IL‐2 tested, both PC61 Fc‐variants inhibited STAT5 phosphorylation equally (Fig. 1b).
Figure 1.
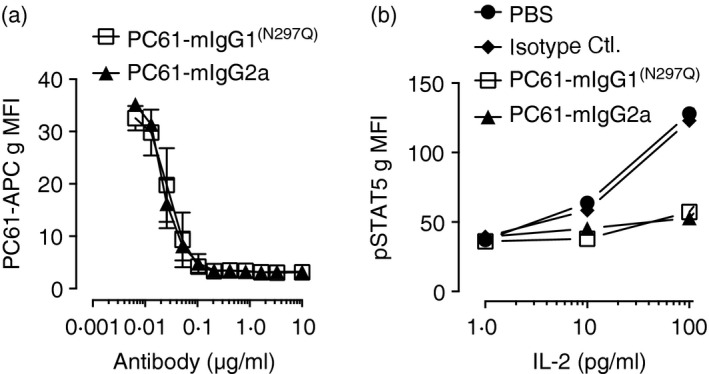
Fc‐engineered anti‐CD25 variants equally bind to CD25 and block interleukin‐2 (IL‐2) mediated signal transducer and activator of transcription 5 (STAT5) phosphorylation. (a) Splenocytes from Foxp3e GFP mice were incubated with PC61‐mIgG1(N297Q), PC61‐mIgG2a or PBS for 20 min. Binding was determined using allophycocyanin‐conjugated PC61 to detect unbound CD25 by flow cytometry. Data are shown as CD25 geometric mean fluorescent intensity (gMFI) from GFP + cells (mean ± SD). (b) Splenocytes from Foxp3eGFP mice were incubated with PC61‐mIgG1(N297Q), PC61‐mIgG2a, isotype control or PBS for 20 min followed by stimulation with IL‐2 for 15 min. Data are shown as pSTAT5 gMFI from GFP + cells. Data are representative of two independent experiments.
Fc‐engineered anti‐CD25 variants differentially impact Treg cells in vivo
In vivo effects of the antibody variants were characterized in wild‐type B6 and Fcer1 g −/− mice. The Fcer1 g −/− mice lack surface expression of activating Fc receptors (FcγRI, FcγRIII, FcεRI, FcγRIV),29, 33 and so were used to evaluate the contribution of antibody Fc/FcR‐dependent effects. Among immune cells, Treg cells express the highest amounts of CD25 and in the steady state constitute roughly 80% of CD25‐expressing cells in lymph node and spleen (data not shown). We analysed the impact of the antibody variants on the percentage and absolute number of Treg cells in spleens as quantified by Foxp3 (Fig. 2a–d). The absolute number of Foxp3+ cells trended down in PC61‐mIgG1(N297Q)‐treated mice (13% decrease) and was significantly decreased in PC61‐mIgG2a‐treated mice (62% decrease) (Fig. 2c). Trends were similar when Foxp3+ cells are measured as a percentage of CD4+ T cells with the decline upon treatment with PC61‐mIgG1(N297Q) reaching significance (17% decrease) but being greater in the PC61‐mIgG2a‐treated mice (61% decrease) (Fig. 2d).
Figure 2.
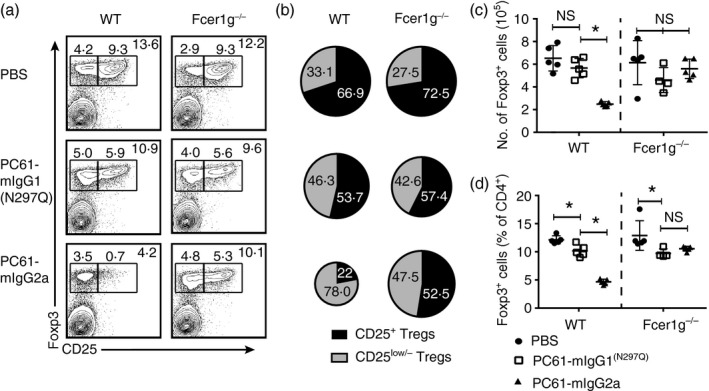
Fc‐engineered anti‐CD25 variants differentially impact regulatory T (Treg) cells in vivo. (a–d) B6 wild‐type and Fcer1g −/− mice were given a single dose of PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control and killed 7 days post‐injection for flow cytometric analysis of splenocytes. (a) Representative dot plots gated on Foxp3+ cells that are either CD25+ or CD25low/− within a CD3+ CD4+ gate. (B) Distribution of CD25+ Treg cells and CD25low/− Treg cells within the Foxp3+ population (mean). (C) Absolute number of Foxp3+ cells (mean ± SD). (D) Percentage of Foxp3+ cells within the CD3+ CD4+ gate (mean ± SD). Data are representative of three (a–d) independent experiments. *P < 0·05; ns, not significant.
The proportion of remaining Treg cells expressing CD25 was altered differently by the antibody variants. Of the Treg cells that remained in wild‐type mice treated with PC61‐mIgG2a, approximately 78% were very low or negative for CD25 expression, compared with 46·3% in the PC61‐mIgG1(N297Q)‐treated wild‐type mice and 33·1% in the PBS‐treated wild‐type mice (Fig. 2a,b). The decline in CD25‐expressing Treg cells observed with PC61‐mIgG2a treatment is consistent with effective Fc‐dependent depletion of the population. As CD25 expression is linked to IL‐2 signalling,34 the reductions in the proportion of CD25‐expressing Treg cells observed with PC61‐mIgG1(N297Q) treatment of wild‐type mice and with both antibodies in the Fcer1 g −/− mice is probably due to blockade of IL‐2 signalling or antibody‐mediated receptor internalization. These results are consistent with previous observations showing that IL‐2 neutralization causes a down‐regulation of CD25 on Foxp3+ cells.12
Aberrant T‐cell activation results from depletion of CD25+ Treg cells but not CD25 blockade
Given the described differences of the PC61 Fc‐variants on Treg cells, the aggregate impact of the antibody variants on immune homeostasis as a consequence of more long‐term dosing was analysed. Wild‐type and Fcer1 g −/− mice were dosed for 4 weeks with antibody variants or vehicle control. Splenocytes were then analysed by flow cytometry for signs of T‐cell activation. Representative dot plots are shown for CD4+ T cells (Fig. 3a) and CD8+ T cells (Fig. 3d). Treatment of wild‐type mice with PC61‐mIgG2a caused an approximately twofold increase in the absolute number and percentage of CD62Llow CD44high CD4+ T cells (Fig. 3b,c). Similarly, the absolute number and percentage of CD62Llow CD44high CD8+ T cells were increased in wild‐type mice treated with PC61‐mIgG2a (Fig. 3d–f). This effect was not seen in Fcer1 g −/− mice. Little to no change in these populations was observed in wild‐type PC61‐mIgG1(N297Q)‐treated mice. At week 4, despite the evidence of T‐cell activation, no significant increase in splenic cellularity was observed in wild‐type mice treated with PC61‐mIgG2a (data not shown).
Figure 3.
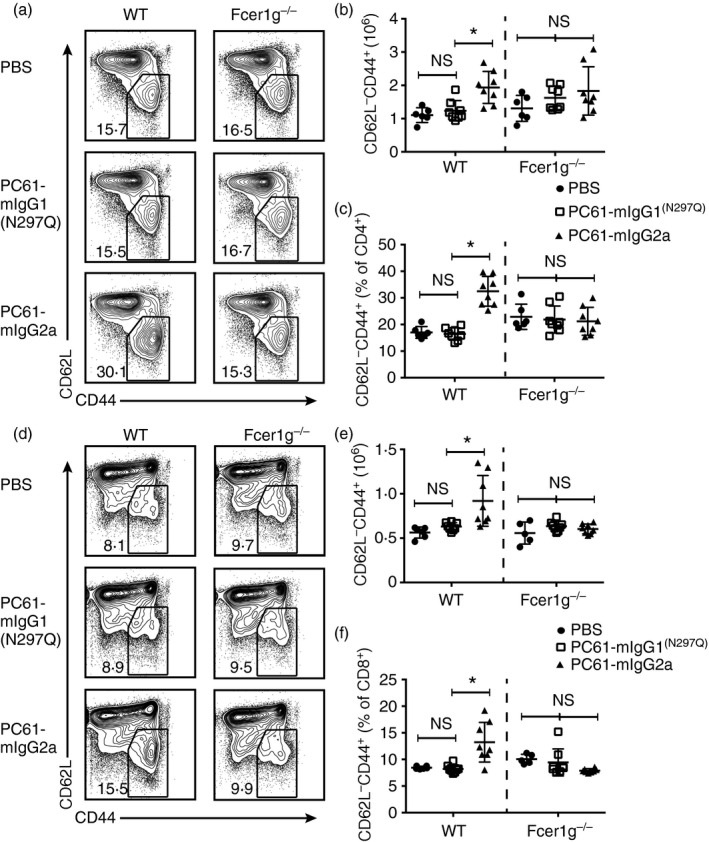
Effector competent PC61‐mIgG2a causes aberrant T‐cell activation whereas PC61‐mIgG1(N297Q) does not. B6 wild‐type and Fcer1g −/− mice were dosed weekly with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control for 4 weeks. Mice were then killed for flow cytometric analysis of splenocytes. (a) Representative dot plots gated on CD62L– CD44high cells within the CD4+ gate. (b) Absolute number of CD4+ CD62L− CD44high cells (mean ± SD). (c) Percentage of CD62L− CD44high cells within the CD4+ gate (mean ± SD). (d) Representative dot plots gated on CD62L− CD44high cells within the CD4− gate. (e) Absolute number of CD4− CD62L− CD44high cells (mean ± SD). (f) Percentage of CD62L− CD44high cells within the CD4− gate (mean ± SD). Data are representative of two independent experiments. *P < 0·05; ns, not significant.
Depletion of CD25+ Treg cells produces aberrant pro‐inflammatory T‐cell responses
Although the dominant role for IL‐2 is on Treg cell homeostasis, IL‐2 also shapes the differentiation of effector T‐cell populations by promoting T helper type 135 and type 236, 37 differentiation yet restraining T helper type 17 differentiation.38, 39 T cells from mice dosed for 4 weeks with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control were analysed for cytokine production upon stimulation ex vivo with PMA/ionomycin followed by intracellular cytokine staining. As seen in representative FACS plots and summarized, the proportion of IFN‐γ + cells within the CD4+ and CD8+ T‐cell populations was significantly increased in PC61‐mIgG2a‐treated mice but not in PC61‐mIgG1(N297Q)‐treated mice (Fig. 4a–c). Although the proportion of IL‐17+ cells within the CD4+ population was slightly increased in both treatment groups, the percentage of cells in any group making IL‐17 was very low (0·1–0·4%) (Fig. 4d).
Figure 4.
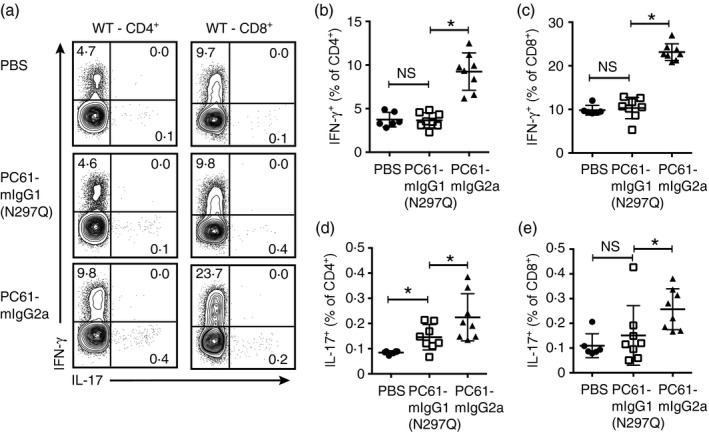
Aberrantly activated T cells in PC61‐mIgG2a‐treated mice have increased pro‐inflammatory cytokine production. B6 wild‐type mice were dosed weekly with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control for 4 weeks. Mice were then killed and splenocytes were activated with PMA (50 ng/ml) and ionomycin (500 ng/ml) for 4 hr and interleukin‐17 (IL‐17) and interferon‐γ (IFN‐γ) were analysed by intracellular cytokine staining. (a) Representative dot plots of IFN‐γ + and IL‐17+ cells within the CD4+ and CD8+ gates. (b) Percentage of IFN‐γ + cells within the CD4+ gate (mean ± SD). (c) Percentage of IFN‐γ + cells within the CD8+ gate (mean ± SD). (d) Percentage of IL‐17+ cells within the CD4+ gate (mean ± SD). Data are representative of three independent experiments. *P < 0·05; ns, not significant.
Treg cells maintain suppressive function during CD25 blockade
Given the aberrant T‐cell activation in wild‐type mice treated with PC61‐mIgG2a, we next analysed the impact of chronic dosing on the Treg cell compartment in wild‐type and Fcer1 g −/− mice dosed for 4 weeks with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control. In mice treated with the depleting PC61‐mIgG2a antibody, both the absolute number and percentage of Treg cells was significantly decreased in wild‐type mice but not in Fcer1 g −/− mice (Fig. 5a–c). The percentage of Treg cells, but not absolute number, significantly decreased in mice treated with PC61‐mIgG1(N297Q).
Figure 5.
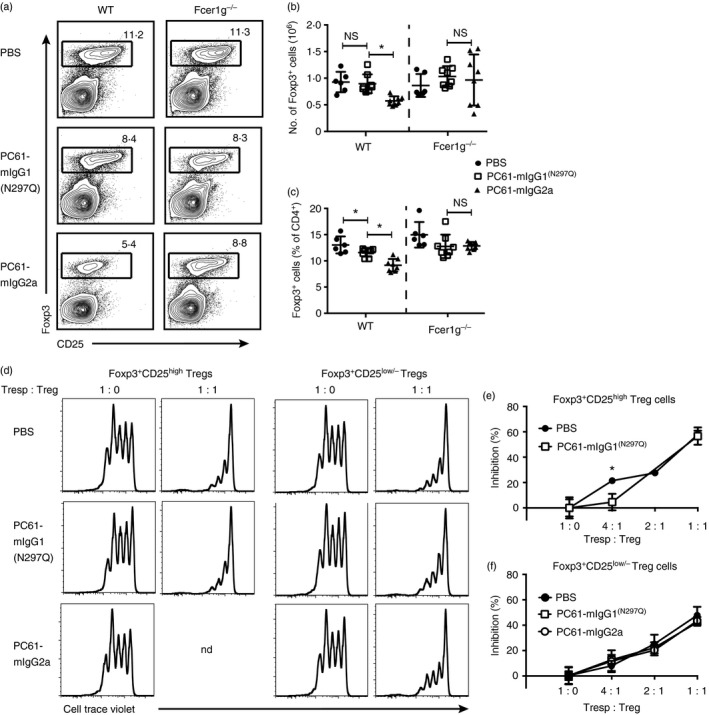
Regulatory T (Treg) cells are differentially impacted by PC61‐mIgG2a versus PC61‐mIgG1(N297Q), but maintain functional suppression. (a–c) C57BL/6 wild‐type and Fcer1 g −/− mice were dosed weekly with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control for 4 weeks. Mice were then killed for flow cytometric analysis of splenocytes. (a) Representative dot plots gated on Foxp3+ cells within the CD4+ gate. (b) Absolute number of Foxp3+ cells (mean ± SD). (c) Percentage of Foxp3+ cells within the CD4+ gate (mean ± SD). (d–f) Foxp3‐eGFP reporter mice were dosed with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control for 4 weeks. Naive CD4+ T cells were FACS sorted from untreated Foxp3e GFP reporter mice and labelled with Cell Trace Violet, then co‐cultured with FACS sorted CD25high or CD25low/− eGFP + cells from indicated mice for 3 days. (d) Representative histograms demonstrating Treg cell suppression of naive CD4+ T‐cell proliferation. (e) Per cent inhibition of naive CD4+ T‐cell proliferation by Foxp3+ CD25high Treg cells (mean ± SD). (f) Per cent inhibition of naive CD4+ T cell proliferation by Foxp3+ CD25low/− Tregs (mean ± SD). Data are representative of two independent experiments. *P < 0·05; ns, not significant; nd, not determined.
A previous study demonstrated that both CD25high and CD25low/− Treg cells are functionally suppressive in vitro.40 Furthermore, Treg cells from CD25−/− mice maintain suppressive function in vitro.12 To determine the impact of the PC61 variants on Treg cell suppressive function, Foxp3eGFP reporter mice were dosed for 4 weeks with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control. Foxp3+ CD25high and Foxp3+ CD25low/− Treg cells were then FACS‐isolated from spleens and in vitro suppression assays were performed. Naive CD4+ T cells from untreated wild‐type mice were used as responder cells in all assays. Figure 5(d) demonstrates responder cell proliferation and Treg cell‐mediated suppression at a 1 : 1 ratio. Foxp3+ CD25high Treg cells from PC61‐mIgG1(N297Q)‐treated and PBS‐treated mice were similarly suppressive, except at the 4 : 1 ratio, where Foxp3+ CD25high Treg cells from PBS‐treated mice demonstrated increased suppression (Fig 5e). Because the PC61‐mIgG2a treatment drastically reduces the number of Foxp3+ CD25high Treg cells, we were unable to sort this population for functional analysis. Foxp3+ CD25low/− Treg cells from all treatment groups were similarly suppressive at all T responder cell to Treg cell ratios tested (Fig. 5f). These data demonstrate that both CD25high and CD25low/− Treg cells maintain suppressive function in vitro during CD25 blockade.
Depletion of CD25+ Treg cells but not CD25 blockade breaks immune tolerance
We next compared the impact of CD25‐blockade with depletion of CD25+ Treg cells in a T‐cell‐driven autoimmune‐prone mouse model. 2D2‐TCR‐tg mice express an MHC class II‐restricted TCR transgene specific for the MOG35‐55 peptide.31 When left un‐manipulated these mice rarely develop clinical signs of EAE (~ 2% at 1‐year of age) but do develop EAE following immunization or treatment with pertussis toxin.31 2D2 mice were dosed weekly with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control and monitored for the development of EAE. In a first experiment, four of ten mice treated with PC61‐mIgG2a broke immune tolerance and developed clinical signs of EAE whereas 2D2 mice treated with PC61‐mIgG1(N297Q) or vehicle control did not (Fig. 6a). Of the mice that developed disease the mean peak score was 3·75. In a repeat of this experiment, five of nine mice treated with PC61‐mIgG2a developed clinical signs of EAE, albeit with slightly delayed kinetics when compared with the first experiment (Fig. 6b). Again, no disease was observed in 2D2 mice treated with PC61‐mIgG1(N297Q) or vehicle control (Fig. 6b). Of the mice that developed disease the mean peak score was 2·6.
Figure 6.
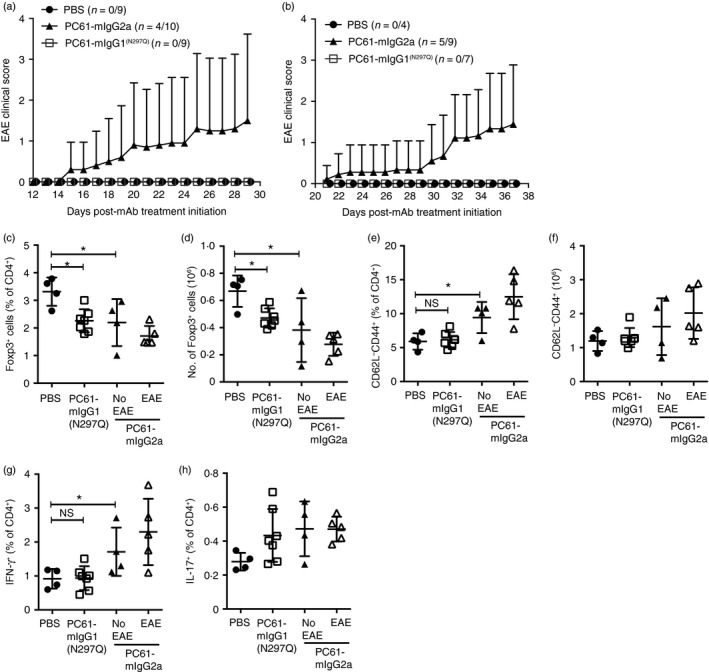
PC61‐mIgG2a‐mediated aberrant T‐cell activation induces experimental autoimmune encephalomyelitis (EAE) in transgenic T‐cell receptor (TCR‐tg) 2D2 mice. TCR‐tg 2D2 mice were dosed weekly with PC61‐mIgG1(N297Q), PC61‐mIgG2a or vehicle control. (a, b) EAE clinical score was assessed in two independent experiments (mean ± SD). Incidence is shown as a ratio of number of mice in each experiment. (c–h) Splenocytes were isolated and analysed by flow cytometry. (c) Absolute number of Foxp3+ cells (mean ± SD). (d) Percentage of Foxp3+ cells within the CD4+ gate (mean ± SD). (e) Absolute number of CD4+ CD62Lhigh cells (mean ± SD). (f) Percentage of CD62L– CD44high cells within the CD4+ gate (mean ± SD). (g) Percentage of interferon‐γ + cells within the CD4+ gate (mean ± SD). (h) Percentage of interleukin‐17+ cells within the CD4+ gate (mean ± SD). *P < 0·05; ns, not significant.
At the conclusion of EAE monitoring, splenic Treg cell and effector T‐cell compartments were characterized by flow cytometry. Additional analysis of the PC61‐mIgG2a‐treated mice that developed EAE was also conducted. As seen in wild‐type mice treated with the antibody variants, Treg cell percentages and numbers declined with treatment and the decline was more pronounced in the PC61‐mIgG2a‐treated group (Fig. 6c,d). Similarly, the numbers and percentages of effector memory CD4+ T cells were only increased in the PC61‐mIgG2a‐treated group (Fig. 6e,f). These cells were predominantly IFN‐γ‐producing (Fig. 6g). PC61‐mIgG2a‐treated mice that developed EAE did not demonstrate statistically significant differences in T‐cell populations from PC61‐mIgG2a‐treated mice that did not develop EAE but there was a trend towards fewer Treg cells and increased effector memory CD4+ T cells in the mice that developed EAE.
Discussion
Given the central role for IL‐2 in Treg cell biology and the interest in therapeutic targeting of the IL‐2 pathway in multiple human diseases, understanding the in vivo consequences of CD25 blockade versus depletion of CD25+ Treg cells on the maintenance of immune homeostasis may inform the design of therapeutic antibodies against the high‐affinity IL‐2R. To this end, we generated and characterized anti‐CD25 Fc variants with full Fc effector function or crippled Fc effector function (mIgG2a versus mIgG1(N297Q), respectively) of the rat anti‐mouse CD25 antibody, clone PC61.
Foxp3‐expressing Treg cells make up roughly 80% of CD25+ lymphocytes and so the effects of the CD25 monoclonal antibodies were predominantly directed against and studied in Treg cells. Seventy per cent of Treg cells were depleted by PC61‐mIgG2a but a fraction of CD25low/− Foxp3‐expressing Treg cells escape depletion. PC61‐mIgG1(N297Q) had a reduced impact on Treg cells, reaching statistical significance when quantified as a percentage of CD4+ T cells (Fig. 2d) but not when based on absolute number of Foxp3‐expressing cells (Fig. 2c). The effect of both antibody variants was equivalent in mice deficient in the FcRγ chain, clearly differentiating the impact of blocking the IL‐2 survival signal versus active depletion of Treg cells. The reduction in the proportion of CD25‐expressing Treg cells and Treg cell CD25 mean fluorescent intensity in the presence of PC61‐mIgG1(N297Q) is consistent with inhibition of IL‐2 signalling and hence IL‐2‐dependent CD25 expression. This is also supported by results showing that genetic deficiency in IL‐2 leads to a reduction in CD25+ Treg cells and an increase in the proportion of CD25low/− Treg cells.12 It is also worth noting that the complete loss of Treg cells in Foxp3‐deficient mice causes a much more aggressive autoimmunity than that seen in CD25 and IL‐2‐deficient mice that lack only 50% of Treg cells.12 Taken together, blockade of CD25‐dependent IL‐2 signalling alone can reduce Treg cell numbers but active depletion with an effector‐competent monoclonal antibody achieved a greater reduction.
Our findings are notable with regard to previous literature that contains conflicting data and debates the ability of anti‐CD25 monoclonal antibodies to deplete Treg cells.21, 24, 25, 26 Numerous laboratories used PC61‐rIgG1 and demonstrated a reduction in Treg cells with varying degrees of success (30–50% reduction in Foxp3+ cells in the spleen and lymph node). One major caveat in these studies is the assumption that the decline in Treg cell numbers is to the result of active depletion and not blockade of the IL‐2 survival signal. In contrast, Kohm et al.25 reported that PC61‐rIgG1 injection resulted in ‘functional inactivation’ of Treg cells rather than active depletion. Setiady et al.24 were the first to demonstrate a role for Fc–FcR interactions in the mechanistic impact of PC61‐rIgG1 on Treg cells. In our study, using PC61 Fc‐variants in both wild‐type and fcerg1 –/– mice, we extend these findings and definitively demonstrate the impact of blocking the CD25‐dependent IL‐2 survival signal versus Fc‐mediated active depletion of CD25+ Treg cells.
Beyond the impact on Treg cells, anti‐CD25 antibodies may also directly impact conventional T cells that transiently express CD25 upon activation or indirectly by inhibiting Treg cell‐mediated suppression. It is therefore difficult to extrapolate known cell‐type specific effects of CD25‐signalling to an aggregate biological or therapeutic impact in vivo. We evaluated the aggregate biological effect of the antibody variants by long‐term (4 weeks) treatment of otherwise unmanipulated mice. The emergence of sizeable populations of IFN‐γ + CD4+ and CD8+ T cells with an activated phenotype after 4 weeks of treatment with PC61‐mIgG2a is consistent with a breakdown in Treg‐mediated immune tolerance and T‐cell‐driven autoimmune activation. No overt autoimmune sequelae were observed in wild‐type mice at the 4‐week time‐point. In contrast, the effector T‐cell compartment in PC61‐mIgG1(N297Q)‐treated mice remained largely unchanged. Hence the homeostatic balance is tipped toward immune activation by the more significant PC61‐mIgG2a Treg cell depletion whereas a more modest reduction in Treg cells may be balanced by blockade of IL‐2 signalling in effector T cells by PC61‐mIgG1(N297Q). The induction of autoimmune pathology in 2D2 TCR transgenic mice extends these observations by demonstrating that immune activation generated by PC61‐mIgG2a is capable of overcoming central nervous system immune privilege and generating a pathological immune response whereas PC61‐mIgG1(N297Q)‐treated mice remain healthy.
The effects of PC61‐mIgG1(N297Q) are notable with regard to ongoing evaluation of an anti‐hCD25 monoclonal antibody (daclizumab) for the treatment of multiple sclerosis. Daclizumab, a humanized monoclonal antibody that binds CD25 and inhibits high‐affinity IL‐2R signalling, has demonstrated clinical efficacy in relapsing–remitting multiple sclerosis, a T‐cell‐mediated neuroinflammatory disease.41, 42, 43 Despite a human IgG1 isotype, daclizumab has poor antibody‐dependent cellular cytotoxicity.44 Daclizumab causes an ~ 50% reduction in circulating Treg cells;45, 46 however, the mechanism by which daclizumab reduces Treg cell numbers in vivo is unclear and probably involves a combination of inhibiting the IL‐2 survival signal and active depletion. When balanced with impacts on effector T cells and cells of the innate immune system, the net outcome of CD25 blockade is therapeutic benefit. We hypothesize that outside the context of a strong pro‐inflammatory environment, IL‐2 signalling may be critical to drive autoimmune T‐cell pathology. In the context of CD25 blockade but not depletion, as with PC61‐mIgG1(N297Q), the reduction in Treg cells may be outweighed by limiting IL‐2‐dependent effector T‐cell activation. This hypothesis will require further experimentation with anti‐CD25 Fc‐variants in the contexts of active immune responses to self or foreign antigens.
Beyond balancing effector and regulatory T‐cell homeostasis, IL‐2 also plays a role in natural killer (NK) cell biology. Published reports have demonstrated an important role for Treg cell consumption of IL‐2 in limiting NK cell expansion.47, 48 Treg cell depletion in the BDC2.5/NOD mouse model of autoimmune diabetes led to rapid aggressive pancreatitis, mediated in part by the expansion of IFN‐γ‐producing NK cells.49 In our study, we saw only mild increases in NK cell numbers after treatment of naive mice with the PC61 variants (data not shown). This is in contrast to the clinical experience with daclizumab, which causes dramatic increases in the CD56bright subset of human NK cells, which are thought to contribute to the therapeutic efficacy of the drug.50, 51, 52
In conclusion, our results clarify the impact of PC61 on Treg cell biology and reveal an important distinction between CD25 blockade and depletion of CD25+ Treg cells. These findings should help inform therapeutic manipulation of the IL‐2 pathway by targeting the high‐affinity IL‐2R.
Disclosures
All authors were paid employees of Biogen Inc. when this work was conducted. Biogen and AbbVie are jointly developing Daclizumab HYP, a humanized monoclonal antibody against CD25, for the treatment of multiple sclerosis.
Supporting information
Table S1. (a) Degenerate oligonucleotides used to amplify the heavy‐chain variable region. (b) Degenerate oligonucleotides used to amplify the light‐chain variable region.
Table S2. Heavy‐ and light‐chain variable region amino acid sequence obtained from PC61.5.3.
Table S3. Amino acid sequence of the chimeric antibody constructs.
Figure S1. PC61 re‐engineering schematic.
Acknowledgements
The authors would like to thank Ankur Thomas and Davide Gianni for insight regarding the EAE model. This study was supported by Biogen.
Author contributions
All authors designed, performed experiments and/or analysed data. DJH, AFP and JDF conceptualized and wrote the manuscript.
References
- 1. Malek TR, Castro I. Interleukin‐2 receptor signaling: at the interface between tolerance and immunity. Immunity 2010; 33:153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boyman O, Sprent J. The role of interleukin‐2 during homeostasis and activation of the immune system. Nat Rev Immunol 2012; 12:180–90. [DOI] [PubMed] [Google Scholar]
- 3. Liao W, Lin J‐X, Leonard WJ. Interleukin‐2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013; 38:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roifman CM. Human IL‐2 receptor α chain deficiency. Pediatr Res 2000; 48:6–11. [DOI] [PubMed] [Google Scholar]
- 5. Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin‐2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity 1995; 3:521–30. [DOI] [PubMed] [Google Scholar]
- 6. Sharfe N, Dadi HK, Shahar M, Roifman CM. Human immune disorder arising from mutation of the α chain of the interleukin‐2 receptor. Proc Natl Acad Sci USA 1997; 94:3168–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis‐like disease in mice with a disrupted interleukin‐2 gene. Cell 1993; 75:253–61. [DOI] [PubMed] [Google Scholar]
- 8. Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T et al Deregulated T cell activation and autoimmunity in mice lacking interleukin‐2 receptor β . Science 1995; 268:1472–6. [DOI] [PubMed] [Google Scholar]
- 9. Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol 2009; 27:363–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Todd JA. Etiology of type 1 diabetes. Immunity 2010; 32:457–67. [DOI] [PubMed] [Google Scholar]
- 11. Arenas‐Ramirez N, Woytschak J, Boyman O. Interleukin‐2: biology, design and application. Trends Immunol Elsevier Ltd; 2015; 36:736–777. [DOI] [PubMed] [Google Scholar]
- 12. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3‐expressing regulatory T cells. Nat Immunol 2005; 6:1142–51. [DOI] [PubMed] [Google Scholar]
- 13. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL‐2Rβ‐deficient mice. Implications for the nonredundant function of IL‐2. Immunity 2002; 17:167–78. [DOI] [PubMed] [Google Scholar]
- 14. Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med 2002; 196:851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)‐2 and induction of autoimmune disease by IL‐2 neutralization. J Exp Med 2005; 201:723–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amado IF, Berges J, Luther RJ, Mailhe MP, Garcia S, Bandeira A et al IL‐2 coordinates IL‐2‐producing and regulatory T cell interplay. J Exp Med 2013; 210:2707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T et al Foxp3 controls regulatory T‐cell function by interacting with AML1/Runx1. Nature 2007; 446:685–9. [DOI] [PubMed] [Google Scholar]
- 18. Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC et al FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell 2006; 126:375–87. [DOI] [PubMed] [Google Scholar]
- 19. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self‐tolerance maintained by activated T cells expressing IL‐2 receptor α‐chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol 1995; 155:1151–64. [PubMed] [Google Scholar]
- 20. Taguchi O, Takahashi T. Administration of anti‐interleukin‐2 receptor α antibody in vivo induces localized autoimmune disease. Eur J Immunol 1996; 26:1608–12. [DOI] [PubMed] [Google Scholar]
- 21. McHugh RS, Shevach EM. Cutting edge: depletion of CD4+ CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ‐specific autoimmune disease. J Immunol 2002; 168:5979–83. [DOI] [PubMed] [Google Scholar]
- 22. Lowenthal JW, Corthésy P, Tougne C, Lees R, MacDonald HR, Nabholz M. High and low affinity IL 2 receptors: analysis by IL 2 dissociation rate and reactivity with monoclonal anti‐receptor antibody PC61. J Immunol 1985; 135:3988–94. [PubMed] [Google Scholar]
- 23. Lowenthal JW, Zubler RH, Nabholz M, MacDonald HR. Similarities between interleukin‐2 receptor number and affinity on activated B and T lymphocytes. Nature 1985; 315:669–72. [DOI] [PubMed] [Google Scholar]
- 24. Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+ FOXP3+ Treg cells by the PC61 anti‐CD25 monoclonal antibody is mediated by FcγRIII+ phagocytes. Eur J Immunol 2010; 40:780–6. [DOI] [PubMed] [Google Scholar]
- 25. Kohm AP, McMahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ et al Cutting Edge: anti‐CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+ CD25+ T regulatory cells. J Immunol 2006; 176:3301–5. [DOI] [PubMed] [Google Scholar]
- 26. Couper KN, Blount DG, de Souza JB, Suffia I, Belkaid Y, Riley EM. Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti‐CD25 treatment in malaria‐infected mice. J Immunol 2007; 178:4136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Couper KN, Lanthier PA, Perona‐Wright G, Kummer LW, Chen W, Smiley ST et al Anti‐CD25 antibody‐mediated depletion of effector T cell populations enhances susceptibility of mice to acute but not chronic Toxoplasma gondii infection. J Immunol 2009; 182:3985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stephens LA, Anderton SM. Comment on “Cutting edge: anti‐CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+ CD25+ T regulatory cells”. J Immunol 2006;177:2036 author reply 2037–8. [DOI] [PubMed] [Google Scholar]
- 29. Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell 1994; 76:519–29. [DOI] [PubMed] [Google Scholar]
- 30. Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol 2007; 178:2961–72. [DOI] [PubMed] [Google Scholar]
- 31. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein‐specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 2003; 197:1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse‐human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol 1989; 143:2595–601. [PubMed] [Google Scholar]
- 33. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcγRIV: a novel FcR with distinct IgG subclass specificity. Immunity 2005; 23:41–51. [DOI] [PubMed] [Google Scholar]
- 34. Depper JM, Leonard WJ, Drogula C, Krönke M, Waldmann TA, Greene WC. Interleukin 2 (IL‐2) augments transcription of the IL‐2 receptor gene. Proc Natl Acad Sci USA 1985; 82:4230–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shi M, Lin TH, Appell KC, Berg LJ. Janus‐kinase‐3‐dependent signals induce chromatin remodeling at the Ifng locus during T helper 1 cell differentiation. Immunity 2008; 28:763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh T‐Y et al Priming for T helper type 2 differentiation by interleukin 2‐mediated induction of interleukin 4 receptor α‐chain expression. Nat Immunol 2008; 9:1288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cote‐Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu‐Li J et al Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci USA 2004; 101:3880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z et al Interleukin‐2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26:371–81. [DOI] [PubMed] [Google Scholar]
- 39. Liao W, Lin J‐X, Wang L, Li P, Leonard WJ. Modulation of cytokine receptors by IL‐2 broadly regulates differentiation into helper T cell lineages. Nat Immunol 2011; 12:551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005; 22:329–41. [DOI] [PubMed] [Google Scholar]
- 41. Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue E‐W et al Daclizumab high‐yield process in relapsing‐remitting multiple sclerosis (SELECT): a randomised, double‐blind, placebo‐controlled trial. Lancet 2013; 381:2167–2175. [DOI] [PubMed] [Google Scholar]
- 42. Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J et al Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double‐blind, placebo‐controlled, add‐on trial with interferon β . Lancet Neurol 2010; 9:381–90. [DOI] [PubMed] [Google Scholar]
- 43. Kappos L, Wiendl H, Selmaj K, Arnold DL, Havrdova E, Boyko A et al Daclizumab HYP versus interferon β‐1a in relapsing multiple sclerosis. N Engl J Med 2015; 373:1418–28. [DOI] [PubMed] [Google Scholar]
- 44. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ et al CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med 2012; 4:134ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huss DJ, Mehta DS, Sharma A, You X, Riester KA, Sheridan JP et al In vivo maintenance of human regulatory T cells during CD25 blockade. J Immunol 2014; 194:84–92. [DOI] [PubMed] [Google Scholar]
- 46. Bielekova B, Howard T, Packer AN, Richert N, Blevins G, Ohayon J et al Effect of anti‐CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol 2009; 66:483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gasteiger G, Hemmers S, Bos PD, Sun JC, Rudensky AY. IL‐2‐dependent adaptive control of NK cell homeostasis. J Exp Med 2013; 210:1179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gasteiger G, Hemmers S, Firth MA, Le Floc'h A, Huse M, Sun JC et al IL‐2‐dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med 2013; 210:1167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity 2009; 31:654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bielekova B, Catalfamo M, Reichert‐Scrivner S, Packer A, Cerna M, Waldmann TA et al Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL‐2Rα‐targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci USA 2006; 103:5941–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin JF, Perry JSA, Jakhete NR, Wang X, Bielekova B. An IL‐2 paradox: blocking CD25 on T cells induces IL‐2‐driven activation of CD56bright NK cells. J Immunol 2010; 185:1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Elkins J, Sheridan J, Amaravadi L, Riester K, Selmaj K, Bielekova B et al CD56bright natural killer cells and response to daclizumab HYP in relapsing–remitting MS. Neurol Neuroimmunol Neuroinflamm 2015; 2:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. (a) Degenerate oligonucleotides used to amplify the heavy‐chain variable region. (b) Degenerate oligonucleotides used to amplify the light‐chain variable region.
Table S2. Heavy‐ and light‐chain variable region amino acid sequence obtained from PC61.5.3.
Table S3. Amino acid sequence of the chimeric antibody constructs.
Figure S1. PC61 re‐engineering schematic.