

Abstract
Background
Neonatal exposure to anaesthetics such as sevoflurane has been reported to result in behavioural deficits in rodents. However, while oxidative injury is thought to play an underlying pathological role, the mechanisms of neurotoxicity remain unclear. In the present study, we investigated whether the NADPH oxidase inhibitor apocynin protects against long-term memory impairment produced by neonatal sevoflurane exposure in mice.
Methods
Postnatal day six mice were divided into four groups; (1) non-anaesthesia, (2) intraperitoneal apocynin (50 mg kg−1) treatment, (3) 3% sevoflurane exposure for 6 h, and (4) apocynin treatment combined with sevoflurane exposure. Superoxide concentrations and NADPH oxidase expression in the brain were determined using dihydroethidium fluorescence and immunoblotting, respectively. Cleaved caspase-3 immunoblotting was used for the detection of apoptosis, and cytochrome c immunoblotting was performed to evaluate mitochondrial function. Long-term cognitive impairment was evaluated using the fear conditioning test in adulthood.
Results
Sevoflurane exposure increased concentrations of superoxide (109%) and the NADPH oxidase subunit p22phox (39%) in the brain, and apocynin abolished these increases. Neonatal sevoflurane exposure caused learning deficits in adulthood. Apocynin also maintained long-term memory function in mice given neonatal sevoflurane exposure, and it reduced apoptosis and decreased cytochrome c concentrations in the brains of these mice.
Conclusions
Apocynin reduces neuronal apoptosis and protects against long-term memory impairment in mice, neonatally exposed to sevoflurane by reducing superoxide concentrations. These findings suggest that NADPH oxidase inhibitors may protect against cognitive dysfunction resulting from neonatal anaesthesia.
Keywords: anaesthesia, anaesthetics, brain, neurotoxicity, paediatric, sevoflurane
Editor's key points.
Exposure to sevoflurane in the neonatal period results in subsequent memory impairment.
Oxidative damage via increased NADPH oxidase activity may be involved.
Neonatal mice were exposed to sevoflurane with and without the NADPH oxidase inhibitor apocynin.
Mice exposed to sevoflurane had memory impairment in adulthood and apocynin reduced this.
Neonatal exposure to anaesthetics, such as sevoflurane, has been reported to induce neuronal apoptosis and produce behavioural deficits in rodents, although the mechanisms of neurotoxicity are still unclear.1–6 Recent studies have demonstrated that the co-administration of a non-selective antioxidant with i.v. or volatile anaesthetics protects rodent neonates against long-term cognitive impairment and neuronal apoptosis.7–9 This suggests that reducing oxidative stress is crucial for preventing memory impairment produced by neonatal anaesthetic exposure.
Superoxide, which is generated from molecular oxygen, is a precursor of several highly reactive oxygen species, and therefore, it is necessary to fully understand its role in oxidative stress and neuronal pathologies.10 Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is one of the most important sources of superoxide.11,12 Indeed, previous studies have shown that superoxide production by NADPH oxidase underlies cognitive impairment after cerebral ischaemia and traumatic brain injury.13–15 However, the role of NADPH oxidase in memory impairment has not been examined in animals exposed to anaesthesia in the neonatal period. Furthermore, it is not known whether targeting NADPH oxidase during anaesthesia protects the neonatal brain from long-term memory deficits. Therefore, in the present study, we investigated whether the NADPH oxidase inhibitor apocynin protects against long-term memory deficits produced by neonatal sevoflurane exposure.
Methods
Ethical approval
The Animal Care and Use Committee of Tokyo Medical and Dental University approved the protocol and the study was performed in accordance with relevant aspects of ARRIVE guidelines.
Experimental mice
Postnatal day five C57BL/6 mice (average body weight: 2.7 g), as a litter with their mother, were purchased from SLC (SLC Japan Inc., Shizuoka, Japan). Mice were housed under a 12-h light-dark cycle (lights on from 08:00 to 20:00), and room temperature was maintained at 21 (1°) C. The same number of pups from each litter was used for the experiments to reduce variability related to the use of different litters (total number of pups used=110). For the behavioural study, male mice were weaned at three or four weeks of age and housed four mice per cage for each experimental group. All mice had ad libitum access to water and food. Behavioural testing was carried out only on male mice to avoid potential variability caused by the oestrous cycle.16
Anaesthesia
We used sevoflurane anaesthesia protocol as previously published.3 On postnatal day six, mice were placed in a humidified chamber (180×180×200 mm) heated to 38 (1)°C, and were exposed to 3% sevoflurane in 40% oxygen for 6 h, which was delivered via a calibrated flowmeter (Shinano, Tokyo, Japan), with a total gas flow of 1 L min−1. During sevoflurane anaesthesia, all mice maintained spontaneous breathing and eventually exhibited the loss of the righting reflex.
For the experimental treatments, the pups from the same litter were randomly divided into four groups; (1) non-anaesthesia (NA group, control mice), (2) NADPH oxidase inhibitor treatment (apocynin) group where mice received intraperitoneal apocynin, 50 mg kg−1,17,18 in a total injection volume of 10 µl, (3) 3% sevoflurane exposure (SEVO group, where mice received 3% sevoflurane for 6 h), and (4) apocynin in combination with sevoflurane (apocynin+SEVO group, where mice received 3% sevoflurane for 6 h in addition to intraperitoneal apocynin, 50 mg kg−1). Apocynin powder (Abcam, Cambridge, MA, USA) was dissolved in ethanol to make a stock solution, which was then diluted with phosphate buffered saline (PBS) to the appropriate concentration for administration. As pups are very small, injection volumes were limited to 10 µl and so the percentage of alcohol in the solution needed to be sufficient to fully dissolve the apocynin powder. However, during perfusion, the abdominal tissue did not show any sign of damage 24 h after anaesthesia induction, suggesting that local ethanol toxicity was negligible. The preparation of apocynin i.p. injection was done according to previously published studies.17,18 Mice in the NA and apocynin groups received carrier gas. Mice were returned to the original litter after treatment.
Detection of superoxide
Mice were anaesthetized using i.p. pentobarbital (50 mg kg−1) and adequate anaesthesia was ascertained by lack of response to tail pinch. They were then rapidly transcardially perfused with 10 ml cold 0.1 M PBS and the brain was quickly removed and immediately frozen at −80°C. Slices, 10-μm-thick, were cut on a cryostat and mounted onto microscope slides. Concentrations of superoxide in the brain were assessed using dihydroethidium (DHE) (Sigma-Aldrich, St. Louis, MO, USA) fluorescence.19 Each slice was incubated with 10 μM DHE for 30 min at 37°C in the dark. After incubation, the sections were washed with PBS (pH 7.4) three times and analysed by fluorescence microscopy using the Zeiss LSM Image Browser (Carl Zeiss MicroImaging, Jena, Germany). Mean DHE fluorescence values of five regions of interest in the cortex layer (two for each slice) were quantified.
Histopathological evaluation
For histopathology, mice were anaesthetized as before, 18 h after treatment, and perfusion was performed. The brain was removed and immersed in 4% paraformaldehyde in PBS overnight at 4°C. Subsequently, 50-μm-thick coronal sections were cut, and then washed with PBS in 0.3% Triton X-100 for 10 min, and endogenous peroxidase activity was blocked with 1% H2O2 for 30 min. Thereafter, sections were incubated with rabbit anti-cleaved caspase-3 antibody (Cell Signaling Technology; 1:300) in PBS/Tween-20 overnight at 4°C for detecting apoptosis. Sections were incubated with peroxidase-conjugated secondary antibody (EnVision+System, Dako, Tokyo, Japan) for 2 h and washed with PBS. Then, the sections were reacted with 3, 3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's instructions. The sections were mounted on slides and counterstained with 0.5% neutral red solution. Our previous investigation3 showed that the apoptotic response to sevoflurane was robust over the whole brain.
Western blot analysis
For protein analysis using western blotting, mice were anaesthetized as before and 18 h after treatment, whole brains were quickly removed, and immediately homogenized on ice in 100 μl homogenization buffer containing 20 µM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM Na4P2O7, and protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). After centrifugation at 21,000×g for 30 min at 4°C, the supernatant was removed and stored at −80°C until use. The amount of protein in each sample was measured using a protein assay kit (BCA; Pierce, Rockford, IL, USA).
For sodium dodecyl sulfate polyacrylamide gel electrophoresis, brain protein homogenates were mixed with lane marker sample buffer (Pierce) and denatured for 5 min at 95°C. The samples were cooled, separated using a 10% Mini-PROTEAN TGX™ Gel (Bio-Rad, Hercules, CA, USA), and transferred onto a nitrocellulose membrane (Bio-Rad). Rabbit anti-4-hydroxynonenal (4-HNE) antibody (Abcam; 1:1000) was used to detect concentrations of the oxidative stress marker. Rabbit anti-p22phox and rabbit anti-gp91phox antibodies (Santa Cruz Biotechnology, Dallas, TX, USA; both 1:1000) were used to detect NADPH oxidase subunits. For detecting apoptosis, rabbit anti-cleaved caspase-3 and rabbit anti-cleaved poly [adenosine diphosphate-ribose] polymerase (PARP) antibodies (Cell Signaling Technology, Beverly, MA, USA; both 1:1000) were used. Rabbit anti-cytochrome c antibody (Abcam; 1:5000) was used to evaluate mitochondrial function. After blocking in 5% non-fat milk for 30 min, the membrane was incubated with primary antibody in TBS containing 0.1% Tween-20 overnight at 4°C. The membrane was then incubated with secondary antibody (Abcam; 1:10,000) for 1 h at room temperature after washing with Tris-Buffered Saline and Tween-20. Mouse β-actin antibody (Sigma-Aldrich; 1:5000) was used as the loading control. The protein bands were visualized using a chemiluminescence detection system (SuperSignal West Pico; Pierce). For quantification of the oxidative stress marker 4-HNE, the total intensity of the 4-HNE lane was compared between groups.20,21
Fear conditioning test
To assess long-term cognitive impairment induced by sevoflurane exposure, the fear conditioning test was performed.3,8 Male mice in each experimental group underwent behavioural testing in adulthood (11–13 weeks of age). The fear conditioning test was carried out in the daytime (between 09:00 and 12:00). The movement of mice was monitored using a computer-operated video tracking system. The apparatus used in this study were made by O'Hara & Co., Ltd. (Tokyo, Japan). The conditioning trial for contextual and cued fear conditioning consisted of a 6 min period followed by three conditioned stimulus–unconditioned stimulus pairings, each separated by 1 min. Each pairing was as follows: unconditioned stimulus, 0.5 mA foot shock intensity, 1 s duration; conditioned stimulus, 60 dB white noise, 20 s duration. The unconditioned stimulus was delivered during the last few seconds of the conditioned stimulus presentation. The contextual test was performed in the conditioning chamber for a five min period in the absence of white noise 24 h after conditioning. A cued test (on the same set of mice) was performed by presentation of a cue (60 dB white noise, 3 min duration) in an alternative context with distinct visual and tactile cues 48 h after conditioning. The duration of freezing time was recorded to assess fear memory. The freezing percentage during each test was compared between each group.
Statistical analysis
Data are shown as individual raw data points. Data from behavioural testing are expressed as mean (sd). DHE fluorescence and protein expression are expressed as a percentage of the control (NA group) value. Statistical analysis was performed using SPSS statistical software package (Version 18, SPSS Inc. Chicago, IL). Kruskal-Wallis omnibus test was used for comparisons of non-parametric data and behavioural data were analysed using anova and post hoc Neuman-Keuls multiple comparison test. A P-value<0.05 was considered statistically significant.
Results
Evaluation of oxidative stress
The 6 h neonatal sevoflurane exposure increased concentrations of superoxide in the brain, and the NADPH oxidase inhibitor apocynin abolished this effect of the anaesthetic (Fig. 1, P=0.001). Brain concentrations of the lipid peroxidation marker 4-HNE were also elevated 6 h after sevoflurane exposure, and apocynin inhibited this increase as well (Fig. 1F, P=0.029).
Fig 1.

Concentrations of superoxide and lipid peroxidation in the neonatal mouse brain. Representative images of DHE fluorescence in the brain slice (scale bar=200 μm) showing concentrations of superoxide in the (A) NA, (b) Apocynin, (c) SEVO and (d) Apocynin+SEVO groups. (e) Cumulative data showing concentrations of superoxide in the brain after 6 h sevoflurane exposure (*P<0.05, NA vs SEVO, n=5 each). (f) Representative image and quantitative data showing 4-HNE in the brain after 6 h of sevoflurane exposure (*P<0.05, NA vs SEVO, n=5 each).
NADPH oxidase protein expression
Neonatal sevoflurane exposure increased expression of the NADPH oxidase subunit p22phox in the brain, and apocynin abolished this increase (Fig. 2A, P=0.035). However gp91phox, another NADPH oxidase subunit, remained unchanged in all experimental groups (Fig. 2B)
Fig 2.

Expression of NADPH oxidase subunits in the neonatal mouse brain. Representative images and relative expression concentrations of (a) p22phox and (b) gp91phox 6 h after sevoflurane exposure (*P<0.05, NA vs SEVO, n=5 each).
Mitochondrial damage and apoptosis
The 6 h neonatal sevoflurane exposure significantly increased concentrations of cytochrome c in the brain (Fig. 3, P=0.038), and simultaneously increased concentrations of cleaved caspase-3 (Fig. 4A-D, P=0.010), and concentrations of another apoptosis marker, PARP (Fig. 4E). Apocynin reduced concentrations of cytochrome c and apoptosis in the brains of these mice (Figs 3 and 4).
Fig 3.
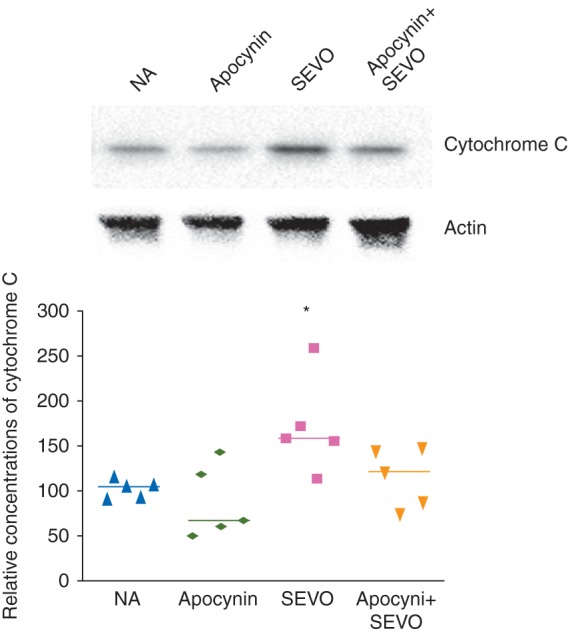
Representative image and relative concentrations of cytochrome C in the brain 6 h after sevoflurane exposure (*P<0.05, NA vs SEVO, n=5 each).
Fig 4.

Representative images showing caspase-3 activation in the retrosplenial cortex of the brain (scale bar=80 μm). Black dots indicate cleaved caspase-3-positive cells in (a) NA, (b) SEVO and (c) Apocynin+SEVO groups. (e) Representative image and relative cumulative concentrations of cleaved caspase-3 in 6 h after sevoflurane exposure (*P<0.05, NA vs SEVO, n=5 each). (e) Representative image showing expression of the apoptosis marker, PARP, in the retrosplenial cortex of the brain after sevoflurane exposure.
Evaluation of long-term memory
Neonatal sevoflurane exposure significantly reduced the freezing response in the contextual fear conditioning test (Fig. 5A, P<0.05) and in the cued fear conditioning test (Fig. 5B, P<0.05). Co-administration of apocynin abrogated this decline in long-term memory function (Fig. 5A, P<0.05; and (Fig. 5B, P<0.05). These results indicated that co-administration of apocynin could suppress impairment of memory subsequent deficits caused by neonatal sevoflurane exposure.
Fig 5.

Long-term memory evaluation using fear conditioning tests in adult mice with or without a 6 h period of neonatal sevoflurane exposure. (a) Freezing response in the contextual test (*P<0.05, NA vs SEVO; NA: n=14; Apocynin: n=12; SEVO: n=12; Apocynin+SEVO: n=11). (b) Freezing response in the cued test (*P<0.05, NA vs SEVO; NA: n=13; Apocynin: n=12; SEVO: n=10; Apocynin+SEVO: n=9). Mean and sd shown.
Discussion
Consistent with previous studies,1–5,7–9 neonatal sevoflurane exposure produced an approximately 40% reduction in subsequent freezing responses in the contextual and cued fear conditioning tests, indicating that neonatal anaesthesia impairs both hippocampus-dependent and independent memory. Neonatal sevoflurane exposure increased concentrations of superoxide and the oxidative stress marker 4-HNE. It also simultaneously increased mitochondrial damage and apoptosis in the brain. These results indicate that sevoflurane anaesthesia induces oxidative stress by increasing superoxide production, which in turn results in neuronal damage. Importantly, the co-administration of apocynin22 prevented mitochondrial damage and apoptosis by abolishing oxidative stress, and further preserved long-term memory function induced by neonatal sevoflurane exposure. These results suggest that inhibiting NADPH oxidase protects against long-term memory impairment induced by neonatal sevoflurane exposure by alleviating oxidative stress-induced neuronal damage and dysfunction.
Interestingly, we found that neonatal sevoflurane exposure increased expression of the NADPH oxidase membrane subunit p22phox in the brain, suggesting that increased NADPH oxidase activity underlies the oxygen free radical overproduction caused by the anaesthetic. Previous studies in animal models of ischaemic and degenerative brain injury have also reported a role for superoxide production resulting from NADPH oxidase activation.13,18,23–27 Another anaesthetic, ketamine, has been reported to induce schizophrenia-like behaviour via NADPH oxidase activation in adult mice.28 These results strongly suggest that activation of NADPH oxidase is a major cause of cerebral malfunction. In previous studied in rodent neonates, 3% sevoflurane exposure did not cause hypoxia and hypoventilation.3,8 The mechanisms through which anaesthetics activate NADPH oxidase in the neonatal brain remain to be clarified.
Our findings are in agreement with previous studies demonstrating that the co-treatment with a non-selective antioxidant, such as EUK-134, along with i.v. or volatile anaesthetics, protects rodent neonates against neuronal apoptosis and long-term cognitive impairments.7,8 Our findings suggest that the inhibition of NADPH oxidase is crucial for alleviating memory impairment in newborn animals subjected to anaesthetic insult, and that such non-selective antioxidants may be neuroprotective via decreased oxidative injury. The present findings, however, do not permit the conclusion that apocynin protects against anaesthetic toxicity in neonatal brain solely through effects on superoxide. The sevoflurane exposure augmented protein expression of p22phox, but not gp91phox (NOX2), in the current study. The NADPH membrane subunit p22phox interacts with the NOX1, NOX2, NOX3 and NOX4 homologues, stabilizing the enzyme complex.12 It is thus possible that several NADPH oxidase homologues, including NOX1, NOX3 and NOX4, play roles in the neuronal toxicity induced by sevoflurane.
Interestingly, Coyoy and colleagues29 found that continuous daily administration of apocynin to rats from postnatal days 2–25 induced a significant change in cerebellar foliation and an alteration in motor behaviour, suggesting that NADPH oxidase is indispensable for proper brain development. In comparison, in the present study, apocynin alone did not affect long-term memory function, although it prevented memory impairment caused by neonatal sevoflurane exposure. These conflicting reports indicate the importance of the timing of apocynin administration.
Conclusions
We demonstrate in this study that apocynin prevents long-term memory impairment in mice neonatally exposed to sevoflurane anaesthesia by decreasing superoxide concentrations through inhibition of NADPH oxidase. These findings suggest that NADPH oxidase inhibitors may have therapeutic potential for protecting against cognitive dysfunction resulting from neonatal anaesthesia in animals and humans. Further studies are needed.
Authors' contributions
Study design/planning: Z.S., M.S.
Study conduct: Z.S., M.S.
Data analysis: Y.A.
Writing paper: Z.S., M.S., Y.A., H.K., K.M.
Revising paper: all authors
Declaration of interest
None declared.
Funding
This work was supported by the Japan Society for the Promotion of Science (Tokyo, Japan; KAKENHI Grant No. 23890054 and 25861361).
Acknowledgement
The authors thank Arimura Koichi, MD, PhD, Department of Neurosurgery, Kobe City Medical Center General Hospital, Kobe, Japan, for the generous advice on RT-PCR analysis and DHE staining.
References
- 1.Jevtovic-Todorovic V, Hartman RE, Izumi Y et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 2003; 23: 876–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fredriksson A, Paontén E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-D-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology 2007; 107: 427–36 [DOI] [PubMed] [Google Scholar]
- 3.Satomoto M, Satoh Y, Terui K et al. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 2009; 110: 628–37 [DOI] [PubMed] [Google Scholar]
- 4.Zheng H, Dong Y, Xu Z et al. Sevoflurane anesthesia in pregnant mice induces neurotoxicity in fetal and offspring mice. Anesthesiology 2013; 118: 516–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen X, Dong Y, Xu Z et al. Selective anesthesia-induced neuroinflammation in developing mouse brain and cognitive impairment. Anesthesiology 2013; 118: 502–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takaenoki Y, Satoh Y, Araki Y et al. Neonatal exposure to sevoflurane in mice causes deficits in maternal behavior later in adulthood. Anesthesiology 2014; 120: 403–15 [DOI] [PubMed] [Google Scholar]
- 7.Boscolo A, Starr JA, Sanchez V et al. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: the importance of free oxygen radicals and mitochondrial integrity. Neurobiol Dis 2012; 45: 1031–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yonamine R, Satoh Y, Kodama M, Araki Y, Kazama T. Coadministration of hydrogen gas as part of the carrier gas mixture suppresses neuronal apoptosis and subsequent behavioral deficits caused by neonatal exposure to sevoflurane in mice. Anesthesiology 2013; 118: 105–13 [DOI] [PubMed] [Google Scholar]
- 9.Yon JH, Carter LB, Reiter RJ, Jevtovic-Todorovic V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis 2006; 21: 522–30 [DOI] [PubMed] [Google Scholar]
- 10.Kinoshita H. Effects of oxidative stress on vascular function, and the role of anesthetics. J Anesth 2012; 26: 141–2 [DOI] [PubMed] [Google Scholar]
- 11.Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004; 3: 205–14 [DOI] [PubMed] [Google Scholar]
- 12.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007; 87: 245–313 [DOI] [PubMed] [Google Scholar]
- 13.Raz L, Zhang QG, Zhou CF et al. Role of rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS One 2010; 5: e12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi DH, Lee KH, Kim JH et al. NADPH oxidase 1, a novel source of ROS in hippocampal neuronal death in vascular dementia. Antioxid Redox Signal 2014; 21: 533–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferreira AP, Rodrigues FS, Della-Pace ID et al. The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: role of inflammatory and oxidative brain damage. Neurochem Int 2013; 63: 583–93 [DOI] [PubMed] [Google Scholar]
- 16.Milad MR, Igoe SA, Lebron-Milad K, Novales JE. Estrous cycle phase and gonadal hormones influence conditioned fear extinction. Neuroscience 2009; 164: 887–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang LL, Ye K, Yang XF, Zheng JS. Apocynin attenuates cerebral infarction after transient focal ischaemia in rats. J Int Med Res 2007; 35: 517–22 [DOI] [PubMed] [Google Scholar]
- 18.Lu XY, Wang HD, Xu JG, Ding K, Li T. NADPH oxidase inhibition improves neurological outcome in experimental traumatic brain injury. Neurochem Int 2014; 69: 14–9 [DOI] [PubMed] [Google Scholar]
- 19.Cheng PW, Ho WY, Su YT et al. Resveratrol decreases fructose-induced oxidative stress, mediated by NADPH oxidase via an AMPK-dependent mechanism. Br J Pharmacol 2014; 171: 2739–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruells CS, Maes K, Rossaint R et al. Sedation using propofol induces similar diaphragm dysfunction and atrophy during spontaneous breathing and mechanical ventilation in rats. Anesthesiology 2014; 120: 665–72 [DOI] [PubMed] [Google Scholar]
- 21.Sango K, Yanagisawa H, Kato K, Kato N, Hirooka H, Watabe K. Differential Effects of High Glucose and Methylglyoxal on Viability and Polyol Metabolism in Immortalized Adult Mouse Schwann Cells. The Open Diabetes Journal 2008; 1: 1–11 [Google Scholar]
- 22.Stefanska J, Pawliczak R. Apocynin: molecular aptitudes. Mediators Inflamm 2008; 2008: 106507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song SX, Gao JL, Wang KJ et al. Attenuation of brain edema and spatial learning deficits by the inhibition of NADPH oxidase activity using apocynin following diffuse traumatic brain injury in rats. Mol Med Rep 2013; 7: 327–31 [DOI] [PubMed] [Google Scholar]
- 24.Hernandes MS, D'Avila JC, Trevelin SC et al. The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J Neuroinflammation 2014; 11: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SH, Lee DY, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci 2005; 25: 4082–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fischer MT, Sharma R, Lim JL et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012; 135: 886–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walder CE, Green SP, Darbonne WC et al. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 1997; 28: 2252–8 [DOI] [PubMed] [Google Scholar]
- 28.Behrens MM, Ali SS, Dao DN et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007; 318: 1645–7 [DOI] [PubMed] [Google Scholar]
- 29.Coyoy A, Olguín-Albuerne M, Martínez-Briseño P, Morán J. Role of reactive oxygen species and NADPH-oxidase in the development of rat cerebellum. Neurochem Int 2013; 62: 998–1011 [DOI] [PubMed] [Google Scholar]