

Abstract
Exposure to repetitive mild traumatic brain injury (mTBI) is a risk factor for chronic traumatic encephalopathy, which is characterized by patchy deposition of hyperphosphorylated tau aggregates in neurons and astrocytes at the depths of cortical sulci. We developed an mTBI paradigm to explore effects of repetitive concussive-type injury over several months in mice with a human tau genetic background (hTau). Two injuries were induced in the hTau mice weekly over a period of 3 or 4 months and the effects were compared with those in noninjured sham animals. Behavioral and in vivo measures and detailed neuropathological assessments were conducted 6 months after the first injury. Our data confirm impairment in cerebral blood flow and white matter damage. This was accompanied by a 2-fold increase in total tau levels and mild increases in tau oligomers/conformers and pTau (Thr231) species in brain gray matter. There was no evidence of neurofibrillary/astroglial tangles, neuropil threads, or perivascular foci of tau immunoreactivity. There were neurobehavioral deficits (ie, disinhibition and impaired cognitive performance) in the mTBI animals. These data support the relevance of this new mTBI injury model for studying the consequences of chronic repetitive mTBI in humans, and the role of tau in TBI.
Keywords: Axonal injury, Behavior, Cerebral blood flow, Concussion, Glial activation, hTau mice, Tau.
INTRODUCTION
Exposure to traumatic brain injury (TBI) is a risk factor for the later development of chronic neurodegenerative disorders, in particular chronic traumatic encephalopathy (CTE) (1–13). Although it was first described in boxers (14, 15), CTE is now increasingly recognized in autopsy series of nonboxer athletes who participated in a wide range of sports, including American football (11, 13, 16, 17), ice hockey (13), soccer (13), and rugby (18). The neuropathology of CTE is complex and multifaceted (9), but abnormal deposits of hyperphosphorylated tau in perivascular neurons and glia and in a patchy distribution toward the depths of cortical sulci are common to the cases described thus far (13). Furthermore, blows to the head during a single boxing bout are associated with increased total tau in the CSF (19, 20) and athletes (ice hockey) and military personnel with histories of TBI demonstrate an increase in total tau and tau-C-cleaved fragments in plasma (21–23).
The precise incidence and prevalence of CTE, including the threshold of susceptibility to CTE following exposure to TBI, are unknown. Reports of CTE have typically come from individuals exposed to repetitive mild traumatic brain injury (mTBI) through contact-related sports injuries; however, the pathology has also been reported in individuals exposed to single moderate or severe TBI (9, 24). Although data on longitudinal follow-up are lacking, in the cases reported thus far the mean duration of exposure to mTBI in an athlete’s career ranged between 5 and 24 years (11, 25). In addition, a majority of these athletes were exposed to mTBI from their high school years to their late twenties, and some during their mid- to late thirties (13, 25). It has been suggested that the extent of pathology at autopsy correlates not only with the age at death and number of years after retirement but also with the length of exposure to injury (13, 26).
At present, all published autopsy cases have been retrospective studies typically involving small sample sizes and subject to selection bias and incomplete premortem data. These limitations make it difficult to derive a comprehensive understanding of the neuropathological sequelae of exposure to TBI. Furthermore, many current preclinical models fail to recapitulate the clinical and neuropathological consequences of mTBI, the cumulative exposures typical of human sports-associated injuries or the pathologies of CTE. Therefore, there is a critical need to develop an animal model that is relevant to lifetime exposure, sports-associated, repetitive mTBI in humans.
Different animal models have been developed to explore the effects of repetitive TBI; most of these experimental models utilize a closed skull mTBI paradigm involving a pneumatic or electromagnetic impactor or weight drop model (27–39). Although these models have described consistent pathologic findings, such as white matter degradation and gliosis, the findings in relation to tau have been less consistent, with some reporting increases in multiple pathogenic tau species following injury (27, 28, 32, 36, 39), whereas others do not (30–31, 33, 38). Moreover, in most of those studies that report evidence of tau-dependent TBI pathology, the authors only examined acute time points after injury, with only 1 study showing evidence of persistent tau pathology at a chronic time point of 6 months after injury (36). Our previous mTBI model in “aged” hTau mice, which express the 6 isoforms of human tau on a null murine background, demonstrated augmentation of tau pathology 3 weeks after injury (39); however, this persistent tau pathology was not observed in younger hTau or wild-type mice exposed to mTBI following examinations up to 12 months postinjury (29, 33; Mouzon B, Bachmeier C, Ferro A, Crynen G, Acker CM, Davies P, Ojo JO, Mullan M, Stewart W, Crawford F. personal communication, 2016). It appears, therefore, that persistent tau pathology is lacking in most TBI models. Additionally, in all the models to date that have depicted increases in intra-axonal or intrasomal phosphorylated tau, other components of CTE-like pathology, such as neurofibrillary or astroglial tangles, neuropil threads, and perivascular phosphorylated tau were absent. These difficulties and relatively few successes in recapitulating tau-associated pathologies after TBI may reflect failure to adequately model the required variables in human injury exposure, such as chronicity of impacts, age at exposure, or duration of exposure to injuries (40).
In this study, we set out to develop a new paradigm for our previously established mouse model of mTBI (29, 33, 35, 39). Our goals were to focus primarily on 2 main aspects: (I) the chronic pathological consequences following cumulative exposure to repeated mTBI over a prolonged exposure period, and (II) the influence of a human tau genetic background on “tau pathology” following this exposure protocol in mTBI. With regard to the latter, we used the hTau mouse model (41) in which we previously demonstrated TBI-dependent tau pathology in “aged” animals exposed to mTBI (39). In addition to examining protein biochemistry profiles in our model, we also present Supplementary Data characterizing brain lipid and peripheral inflammatory profiles and neurobehavioral measures.
In this first report, we confirm a 2-fold increase in total tau levels and mild increases in tau oligomers/conformers and pTau (Thr231) species in the gray matter up to 3 months after cessation of injury, which may be on a continuum of progressive and persistent TBI-dependent tau pathology.
MATERIALS AND METHODS
Animals
Transgenic mice expressing Human Tau on a C57BL/6 and null murine tau background (generated as previously described [41]) were purchased from Jackson Laboratories, Bar Harbor, Maine. All mice were12 weeks old at the start of this study (average weight, 18 g). Animals were housed in standard cages under a 12-hour light/12-hour dark schedule at ambient temperature controlled between 22 °C and 23 °C under specific pathogen-free conditions. Animals were given food and water ad libitum and maintained under veterinary supervision throughout the study. There was no evidence of disease among the colony. Mice of both sexes were randomly assigned to experimental groups (n = 12 each for sham and injured animals). Two animals in the injury group were euthanized due to development of severe dermatitis of unknown reasons. Experiments were performed in accordance with Office of Laboratory Animal Welfare and National Institutes of Health guidelines under a protocol approved by the Roskamp Institute Institutional Animal Care and Use Committee. All analyses were carried out blind to study group assignment.
Experimental mTBI
The experimental TBI methods were performed, as previously described (29). Briefly, mice were anesthetized with 1.5 L per minute of oxygen and 3% isoflurane for 3 minutes. After shaving of the injury site, mice were transferred into a stereotaxic frame (Just For Mice Stereotaxic Instrument, Stoelting, Wood Dale, Illinois) mounted with an electromagnetic controlled impact device (Impact One Stereotaxic Motorized Impactor, Richmond, Illinois). Heads were positioned and fixed in the device, which prevented lateral movements as the impact was delivered. All mice were placed on a heating pad to maintain their body temperature at 37 °C. A 5-mm blunt metal impactor tip attached to the electromagnetic motorized device was centered on the scalp and positioned above the midsagittal suture before each impact using the NeuroLab controller. On satisfactory positioning, the tip was retracted and the depth was adjusted to the desired level. The scalp was gently stretched by hand to restrict lateralization of the impact and to prevent the rod from delivering an inadequate trauma load at an irregular angle. Injury parameters were 5 m per second strike velocity, 1.0 mm strike depth, 200 milliseconds dwell time, and a force of 72N. This sublethal impact does not cause direct tissue damage to the injury site, and there is no development of skull fracture or subdural hemorrhage, even after repetitive injuries. Mice in the repeat mTBI (r-mTBI) group received 2 impacts every week for 3 or 4 months (ie, 24 or 32 impacts), with an interinjury time of 72 to 96 hours. Repetitive sham control mice received anesthesia of the same frequency and duration (∼3 minutes per session) as their r-mTBI counterparts. Animals were grouped as repetitive shams or repetitive injury. This mixed paradigm was chosen to mimic the heterogeneity of cumulative mTBI exposures in the human setting. After each impact, the mice were allowed to recover on a heating pad set at 37 °C to prevent hypothermia. When they became ambulatory, the mice were returned to their cages and carefully monitored for any abnormalities.
Three-Chamber Test for Social Interaction and Novelty Recognition Test
All neurobehavioral tests were conducted 6 months after the first injury. Two social behaviors (social interaction and social memory/novelty recognition) were quantified using a rectangular 3-chamber test that includes a middle chamber with 2 doors leading to 2 separate (left and right) chambers, each containing a steel cage enclosure. After 5 minutes of habituation in the 3-chamber compartment, each mouse (experimental subject) was placed in the middle chamber and allowed to explore for 10 minutes, with the right chamber empty but an unfamiliar congener (Stranger I) held in the steel cage enclosure in the left chamber. Social interaction was determined by measuring the number of entries by the experimental subject into the chamber holding the unfamiliar congener versus the empty chamber. To measure social memory (or novelty recognition), a new novel stimulus mouse (Stranger II) was subsequently placed in the previously empty right chamber. The same parameters as above were measured to determine the preference of the experimental subject for Stranger I or Stranger II.
Elevated Plus Maze
The elevated plus maze consists of a plus-shaped apparatus with 2 open and 2 enclosed arms, each with an open roof, elevated 50 to 70 cm from the floor in a dimly lit room. Each mouse was placed at the junction of the 4 arms of the maze, facing the open arm. The mice were allowed to maneuver within the maze freely for 5 minutes; the number of entries and duration in each arm (open/closed) were recorded with the aid of a video tracking system to evaluate anxiety effects based on the animal’s aversion to open spaces.
Open Field Test
The open field was conducted in a large circular maze (120-cm diameter) setup in a brightly lit room. Animals were placed in the center of the maze and the number of entries/time spent in a predefined center zone and around the walls of the maze was recorded over a 15-minute trial to evaluate anxiety effects and sensorimotor activity.
Cerebral Blood Flow Measurements
Cerebral blood flow (CBF) was measured prior to death at 6 months post-first injury time point. The mice were anesthetized with a gas mixture of 3% isoflurane, 0.9 L min−1 nitrous oxide, and 0.5 L min−1 oxygen. Animals were then immobilized on a mouse stereotaxic table and maintained under anesthesia with a mouse anesthetic mask (Kopf Instruments, Tujunga, California) delivering 3% isoflurane, 0.5 L min−1 nitrous oxide, and 0.3 L min−1 oxygen. Rectal temperature was maintained at 37 °C using a mice homeothermic blanket system (Harvard Apparatus, Holliston, Massachusetts). An incision was made through the scalp and the skin retracted to expose the skull, which was then cleaned with a sterile cotton swab. Animals were maintained on a mixture of 1.5% isoflurane, 0.5 L min−1 nitrous oxide, and 0.3 L min−1 oxygen. Cortical perfusion was measured with the Laser-Doppler Perfusion Imager from Moor Instruments (Wilmington, Delaware), as previously described (42, 43). A computer-controlled optical scanner directed a low-power He-Ne laser beam over the exposed cortex. The scanner head was positioned parallel to the cerebral cortex at a distance of 26 cm. The scanning procedure took 1 minute and 21 seconds for measurements of 5,538 pixels covering an area of 0.64 cm2. At each measuring site, the beam illuminated the tissue to a depth of 0.5 mm. An image color-coded to denote specific relative perfusion levels was displayed on a video monitor. All images were acquired at 2-minute intervals for a period of 30 minutes (15 images for each animal). All images were stored in computer memory for subsequent analysis. For each animal, a square area of 0.05 cm2 (360 pixels) equally distributed between the right and left hemispheres was defined and applied to each image of the series in order to measure the CBF in the frontal and occipital cortex. CBF was also measured in the entire cortex by manually delineating for each mouse the cortex area (0.51–0.54 cm2 corresponding to 3,504–3,714 pixels). Relative perfusion values for each area studied were presented as percentage of CBF values in injured compared with sham controls.
Enzyme-Linked Immunosorbent Assay
To obtain blood specimens to measure glucocorticoid and cytokine levels in plasma, animals were lightly anesthetized with isoflurane prior to euthanasia, and approximately 500 μl of blood was withdrawn into EDTA capillary tubes by cardiac puncture. Samples were centrifuged at 3000g for 3 minutes, and plasma samples (clear supernatant fraction) were flash frozen in liquid nitrogen and stored at −80 °C. Plasma glucocorticoid levels were measured using an enzyme-linked immunosorbent assay (ELISA) kit purchased from Life-sciences-Invitrogen (Grand Island, New York). Cytokine levels (GM-CSF, IL-5, IL-6, IL-1β, TNF, IL-17A, IL-10, IFNγ) were determined using Bio-Plex Pro mouse Th17 panel 8-plex ELISA kit (Bio-Rad, Hercules, California), as instructed by the manufacturer’s guide.
Brain Tissue Preparation and Western Blotting
Six months post-first injury brain tissue was collected following transcardial perfusion by gravity drip with phosphate-buffered saline (PBS). One hemisphere was extracted and dissected into 2 regions (cortex and corpus callosum); these were flash frozen in liquid nitrogen and kept at −80 °C for antibody based/biochemical analyses. The other hemisphere was postfixed in 4% paraformaldehyde for histological and immunohistochemical (IHC) analyses.
For Western blotting analyses, the corpus callosum and cortex from each hemisphere were homogenized in 150 µl and 750 µl (respectively) of PBS (pH 7.4) containing proteinase inhibitor using a probe sonicator. Homogenized samples were spun in a centrifuge at 15000g < 10 minutes and tissue supernatants were collected. Supernatant fractions were either denatured at 95 °C by boiling in Laemmli buffer (Bio-Rad) or prepared under nondenaturing conditions in Laemmli buffer without reducing agent. Samples were then subsequently resolved on 4% to 20% gradient polyacrylamide criterion gels (Bio-Rad) or 4% to 12% gradient NuPAGE Novex Bus-Tris precast polyacrylamide gels (Life Technologies, Grand Island, New York). After electrotransferring, polyvinylidene difluoride membranes were blocked in 5% milk made in Tris-buffered saline and subsequently immunoprobed for different brain-specific primary antibodies overnight (Supplementary Table 1). After a rigorous washing step, membranes were probed with horseradish peroxidase-linked secondary antibodies (Supplementary Table 1). For primary antibodies raised in mice, a 20% superblocking buffer was used in the secondary antibody solution. Immunoblots from cortical tissue samples were analyzed by using a housekeeping gene (β-actin or anti-GAPDH antibody) to quantify the amount of proteins electrotransferred, and signal intensity ratios were quantified by chemiluminescence imaging with the ChemiDocTM XRS (Bio-Rad). Immunoblots from white matter tissue samples for nonreduced tau conformational antibodies were analyzed by using the most abundant bands from a total protein Coomassie stain as a control reference, and signal intensity ratios were quantified by chemiluminescence imaging with the ChemiDoc TM XRS.
Immunohistochemistry
Half brain hemispheres (left side) were fixed in 4% paraformaldehyde for 24–48 hours followed by paraffin embedding. Series of 6-µm-thick sagittal sections were cut throughout the extent of the cortex and hippocampus guided by known bregma coordinates using a microtome (2030 Biocut, Reichert/Leica, Buffalo Grove, Illinois). Cut sections were mounted onto positively charged glass slides (Fisher, Superfrost Plus, Pittsburgh, Pennsylvania).
Sections were deparaffinized in xylene and rehydrated in a decreasing gradient of ethanol before the IHC procedure. Sections were rinsed in distilled water and subsequently incubated at room temperature in a solution of endogenous peroxidase blocking solution containing 3% hydrogen peroxide diluted in PBS for 15 minutes. For primary antibodies requiring antigen retrieval, sections were treated with either (I) DAKO target retrieval citrate buffer solution (pH 6) (Dako) for 8 minutes in the microwave, (II) Tris-ethylene-diamine-tetraacetic acid—Tris-EDTA buffer (pH 8), or (III) Proteinase K (IHC World, Woodstock, Maryland). Following antigen retrieval, sections were blocked for 30 minutes to 1 hour in either (I) Dako protein serum-free protein block (Dako), (II) normal blocking serum to which secondary antibody was raised, or (III) mouse immunoglobulin G blocking reagent mouse on mouse (MOM) Kit (Vector Laboratories, Burlingame, California). Sections were immunostained in batches with primary antibodies made up in supersensitive wash buffer or antibody diluent background-reducing agent. Primary antibodies used are listed in Supplementary Table 1. After overnight incubation, sections were rinsed with PBS and transferred to a solution containing the appropriate secondary antibody (Vectastain Elite ABC Kit, Vector Laboratories) for 30 minutes to 1 hour, depending on the specific requirement of the antibody protocol. For antibodies raised in a mouse host, secondary antibodies were diluted in 20% superblock blocking buffer (Pierce, Thermo Fisher Scientific, Rockford, Illinois). After rinsing in water, sections were incubated with avidin-biotin horseradish peroxidase or alkaline phosphatase enzyme solution (Vector Laboratories) solution for 30 minutes.
Immunoreactivity was visualized with 3,3′-diaminobenzidine (DAB) or Vector red alkaline phosphatase substrate. Development with the chromogen was timed and applied as a constant across batches to limit technical variability before progressing to quantitative image analysis. Reactions were terminated by rinsing sections in copious amounts of distilled water. Mounted sections were progressed through a graded series of alcohols (dehydrated), cleared in xylene, and coverslipped with permanent mounting medium. The IHC procedure was previously validated for most antibodies used by including negative control sections in which the primary mouse monoclonal antibodies were omitted and replaced with either blocking agent or biotinylated secondary antibodies alone. Immunoreacted sections were viewed using a motorized Olympus (BX63) upright microscope and photographs were taken using the high-resolution DP72 color digital camera.
Histology
Series of sections were deparaffinized and processed for different histological stains. The Gallyas silver impregnation method was performed to reveal neurofibrillary tangles using a standardized protocol (39). Congo red staining for amyloid plaque was performed using manufacturer’s instructions (Sigma, St. Louis, Missouri). Luxol fast blue (LFB) staining was performed as previously described (29). To detect microhemorrhages in the brain tissue, Prussian blue staining was also performed according to the manufacturer’s guidelines (ENG Scientific Inc., Clifton, New Jersey). Gallyas- and Prussian blue-stained sections were counterstained with Nuclear Fast Red; LFB-stained sections were counterstained with Cresyl violet. Counterstained sections were subsequently processed through an alcohol gradient, xylene, and coverslipped with Permount mounting medium.
Image Analysis and Amyloid Precursor Protein Cell Counts
Immunoreactivity for cell markers (glial fibrillary acidic protein [GFAP], Iba1) was measured by quantitative image analysis (optical segmentation). Analyses were performed in a blinded manner to experimental conditions using coded slides. Multiple regions of interest were analyzed in standardized fashion for each cell marker/antibody. A survey of immunostained tissue sections was first performed independently to verify specific immunoreactivity that was subsequently progressed to quantitative image analysis. Briefly, nonoverlapping red, green, blue (RGB) images were digitally captured randomly within the defined areas from each section (comprising an average of 5 sections per animal for each marker), providing a systematic survey of each region of interest for each animal within a group. A minimum of 15 microscopic fields (×60 magnification) was analyzed per region per animal. The total microscopic field yielding a total area of 3.5 mm2 analyzed for each region per animal. Immunoreacted profiles that were optically segmented were analyzed using CellSens morphometric image analysis software (Olympus, Center Valley, Pennsylvania). A semiautomated RGB histogram-based protocol (specified in the image analysis program) was used to determine the optimal segmentation (threshold setting) for immunoreactivity for each antibody. Immunoreactive profiles discriminated in this manner were used to determine the specific immunoreactive percentage area. Data were separately plotted as the mean percentage area of immunoreactivity per field (% area) ± SE for each region and grouping.
To assess changes in amyloid precursor protein (APP)-positive profiles in the corpus callosum, axonal profiles were determined from a series of 5 sections. Fifteen separate images spanning a total area of 3.52 mm2 were examined. Only clearly distinguishable profiles with distinct morphological characteristics of axonal bulbs or swellings were included in the counts. Data were plotted separately as density estimates (number per mm2) ± SE for the corpus callosum region and grouping.
Volumetric Estimates
The volume of the corpus callosum of one brain hemisphere (bound by bregma coordinates −0.36 to −0.72 mm lateral) was determined by quantitative light microscopy using the Cavalieri method. In brief, sagittal sections containing the corpus callosum regions were stained for LFB (taking every tenth serial section). An average of 5 sections were collected per animal. Mounted sections were viewed at low magnification and montage images of the entire corpus callosum region were electronically captured using a DP72 digital camera attached to a motorized Olympus BX63 digital photomicroscope. Digital images were digitally outlined on each section using the Cell SENS Olympus software package. All analyses were carried out blind to study group assignment. For each animal, the volume of the corpus callosum region (bound by bregma coordinates −0.36 to −0.72 mm lateral) was subsequently derived by multiplying the calculated total surface area by the mean distance between the series of sections. Data are presented as mean volume (in mm3) ± SE per group.
Lipidomic Analyses
Lipids were extracted using the Folch method from the cortex as previously described (44), with addition of di-14:0 fatty acid containing internal standards for phosphatidylethanolamine (PE), phosphoinositols (PI), phosphoglycerols (PG), phosphatidylcholine (PC), phosphates, and 14:0 LPG, LPC and LPE, di-17:0 PS, 14:LPE and LPC and 24:0 LPC. Lipidomic runs were performed in triplicate using SCID LC/MS as described (44, 45). Identification and calculation of concentrations of specific lipid molecular species within each lipid class (LPC, LPE, PC, PI, and sphingomyelin [SM] levels) were calculated from the averaged MS spectrum across the chromatographic peak for each lipid class using LipidomeDB software. Summed mass spectra for each lipid class in an LC/MS run were analyzed using LipidomeDB online (http://lipidome.bcf.ku.edu:9000/Lipidomics/) to identify and quantify (with reference to added internal standards) each lipid molecular species. Each sample was analyzed in triplicate. The biological significance of changes in each lipidomic profile was evaluated using Lipid map pathway tools (www.lipidmaps.org/pathways/) Last accessed: 05-09-2016. Moreover, phospholipid classes were grouped according to the degree of unsaturation for the molecular species. For PC, ratios were calculated using docosahexaenoic acid (DHA)-containing species, PC (38:6, 16:0/22:6) and PC (40:7, 18:1/22:6), to arachidonic acid (AA)-containing species, PC (36:4, 16:0/20: 4), PC (38:4, 18:0/20:4), and PC (38:5, 18:1/20:4). For PE, a ratio of DHA-containing species, PE (40:6, 18:0/22:6) and ether PE (ePE; 40:6, o-18:0/22:6), to AA-containing species, PE (38:4, 18:0/20:4) and ePE (38:4, o-18:0/20:4), was calculated. For PI, a ratio of DHA-containing PI (40:6, 18:0/22:6) to AA-containing PI (36:4, 16:0/20:4) and PI (38:4, 18:0/20:4) species was examined. Group differences were determined using either ANOVA or the χ2 test based on the type of variable. When parametric assumptions were not met, values were log transformed for parametric analyses. Nonparametric testing was only used when transformation was found to be unsatisfactory. Principal Component Analysis (PCA) was used to minimize multicollinearity and achieve dimension reduction, as used routinely for the evaluation of lipidomic data (44, 45). The Kaiser-Meyer-Olkin (KMO) and Bartlett’s tests for sphericity were used to ensure sampling adequacy for PCA (KMO value of >0.6 and Bartlett p < 0.05). Variables with eigenvalues of ≥1 were retained and PCA component was extracted using varimax with Kaiser normalization for rotation to simplify and clarify the data structure. In order to perform mixed linear modeling (MLM) regression analysis on each component (the outcome measure), the Anderson-Rubin method was used for exporting uncorrelated scores while adjusting for random (human) factor and assess independent fixed (diagnostic and replication). Following MLM, multiple-test corrections were performed with the Benjamin-Hochberg procedure to control for the false discovery rate. Individual lipids were analyzed by MLM to identify lipids specifically altered by the study treatment.
Statistical Analysis
The relationships between mTBI and sham animals for Western blotting, IHC, ELISA, neurobehavioral, and CBF data were assessed using either a parametric T test or nonparametric Mann-Whitney U test with a predefined criterion of p < 0.05 to assess group differences. Data were assessed by the Kolmogorov-Smirnov test to evaluate whether data sets fitted the normal Gaussian distribution. All analyses were performed with SPSS 21.0 (IBM Corp., Armonk, New York) and Graph Pad prism (La Jolla, California) statistical software.
RESULTS
Neurobehavioral Observations
We examined behavioral outcomes (6 months post-first injury) in our mouse model of chronic repeated TBI using the 3-chamber test (social interaction and social novelty recognition or memory), open field (sensorimotor and anxiety), and elevated plus maze (anxiety). No impairments in social interaction were observed in sham and injured animals, that is, both groups of animals twice as frequently entered Stranger I chamber compared with empty chamber (Supplementary Fig. 1A). There was a trend (p = 0.058) for diminished social memory in the injured group, with animals 3 times more frequently entering Stranger I chamber compared with new novel Stranger II chamber (Supplementary Fig. 1B). Sham animals behaved normally in the social novelty recognition test, frequenting Stranger II chamber >46% more than Stranger I chamber (Supplementary Fig. 1B). In both tests for anxiety, injured animals demonstrated more disinhibitory behavior than shams, with a trend toward increased exploration (ie, time and frequency of entries) in the center zone of the open-field and open arms of the elevated plus maze tests (Supplementary Fig. 1E, F). However, statistical analyses of these outcome measures did not reach significance. Minimal changes were observed in the sensorimotor activity during the open field test; this was typified by a small trend toward decrease in motor activity in injured compared with sham animals (Supplementary Fig. 1D).
Notably, 5 animals in the injured group showed dermatitis around the skin of the face (around the eye region) and in some cases around the body, approximately 2 to 3 months into the study, following the first injury. This was probably due to lack of grooming and restrained use of front limbs following injury in these mice. Two of the animals that had severe dermatitis had to be euthanized; the other 3, which only had a milder form of dermatitis, were also subsequently removed from the in vivo imaging and behavioral tests; this reduced the sample sizes.
Cerebral Blood Flow
Using a laser Doppler imaging system to assess changes in the CBF of injured animals compared with their sham counterparts (Fig. 1A), a reduction in CBF in the occipital cortex was identified in injured compared with sham animals (p < 0.01) (Fig. 1B). No change was observed in the frontal cortex of either sham or injured animals; however, a trend toward decrease was observed in the entire cortex readings (Fig. 1B).
FIGURE 1.
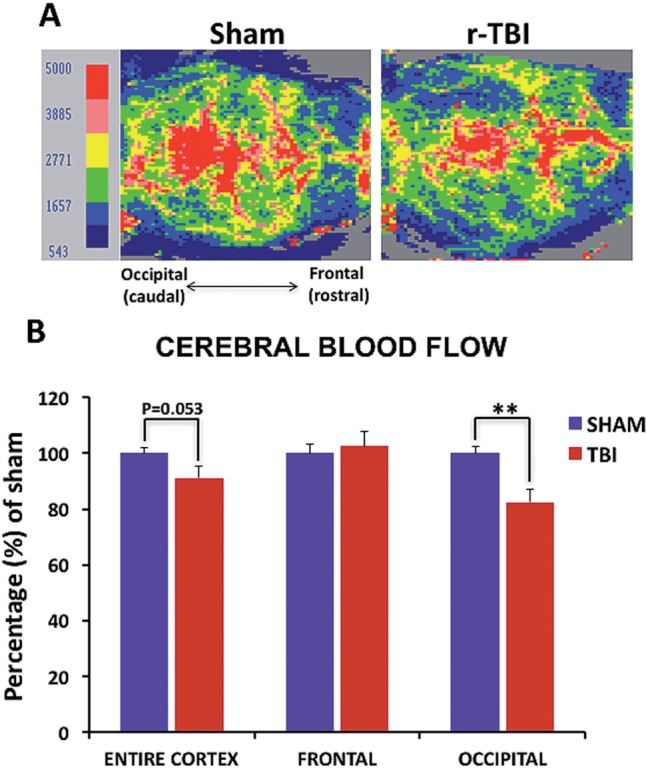
Cerebral blood flow in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A) Two-dimensional color-coded cerebral microvascular blood flow maps recorded using the laser Doppler imager show flow data representing variation of regional blood flow in the cortex of sham and injured animals. (B) Quantitative analysis of mean cerebral blood flow, obtained from 6 measurements per animal, demonstrates significant reduction in the occipital cortex of injured compared with sham animals and a trend towards decrease in the entire cortex, with no change observed in the frontal cortex. Data are presented as percentage of sham. Blood flow velocity was visualized as a 2-dimensional color-coded map, and expressed in an arbitrary perfusion unit as cerebral blood flow flux. N = 6 (injured) or 8 (shams); **p < 0.01.
Neuropathology of Chronic Repetitive mTBI
Macroscopic Pathology
The injury paradigm was consistent with a mild injury, with no evidence of macroscopic pathology in r-TBI exposed mice. Specifically, there were no skull fractures or subdural hemorrhages and no gross pathological changes observed in any animals studied (Supplementary Fig. 2A, B).
White Matter Pathology
Prior examination of acute 24 hour post-last injury brain tissue sections from this model depicted an exaggerated rise in APP-positive axonal profiles in injured animals (212 positive profiles per mm2) accompanied by an intense GFAP and Iba1 immunostaining in the corpus callosum (data not shown).
At the chronic time point, there was a persistent increase in APP-positive axonal profiles (Fig. 2A–F) and increased extents of GFAP (Fig. 3) and Iba1 (Fig. 4) immunoreactivities were observed in the body and splenium of the corpus callosum in injured versus sham animals. These changes were, however, less prominent when compared with the 24-hour time point (data not shown).
FIGURE 2.

Axonal injury in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A–L′) Immunohistochemical staining for amyloid precursor protein ([APP] axonal injury) demonstrated an increase in the corpus callosum (white arrows) versus sham; this was localized within the body (C, D) and the splenium (E, F) of the corpus callosum. High-power micrographs show APP accumulation along the length of damaged axons (I, I′), and in axonal bulbs or swellings (J, J′, K, K′) throughout the entire length of the corpus callosum of injured animals. Increased APP staining was observed in the cell body of pyramidal neurons in layer III of the parietal cortex of injured versus sham animals (G, H). High-power micrograph (L, L′) shows increase APP staining in the cell body and dendrites of injured neurons. CC, corpus callosum; HP, hippocampus; SPL, splenium.
FIGURE 3.

Astrogliosis in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A–L). Immunohistochemical staining for GFAP (astrogliosis) demonstrates a dramatic increase along the length of the corpus callosum of injured animals compared with shams (A, B). Astroglial activation was not present in the cortex of sham or injured animals (A, B). White matter regions surrounding the ventricles also showed increased GFAP staining in the injured (C′) compared with sham animals (C). There are hypertrophic astrocytes in the body (D, E) and splenium (G, H) of the corpus callosum of injured compared with sham animals. A parallel increase in astroglial marker S100ß was also observed in the cell body of astroglial cells in the corpus callosum of injured compared with sham animals (insets of D and E). Hypertrophic astrocytes demonstrated large cell body and prominent thick processes in these regions (F vs F′, I vs I′). Prominent and hypertrophic perivascular astrocyte endfeet (strongly stained with GFAP) are seen along the length of small blood vessels in injured animals (J). High-power micrograph of periventricular region is shown in (K). GFAP staining was increased in the cerebellar molecular cell layer in injured animals (L). CC, corpus callosum; VTR, ventricles; CEREB, cerebellum; Hippo, hippocampus.
FIGURE 4.

Microgliosis in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A–L′) Immunohistochemical staining for Iba1 (microgliosis) demonstrated no differences in the cortex of sham and injured animals (A, B). Microglial cells in the cortex had a quiescent morphology in both sham and injured animals (C, C′). The body (D) and the splenium (E–K) of the corpus callosum showed marked immunostaining of Iba1 in injured animals (see body [C, D] and splenium [E, F and J, K] of the corpus callosum) compared with their sham counterparts. Higher-power micrographs of activated microglial cells demonstrate a prominent cell body and short thick processes in injured animals (F vs F′, I vs I′, L vs L′). CC, corpus callosum.
Axonal swellings immunopositive for APP (Fig. 2I, I′, J, J′, K, K′) were observed in the corpus callosum of injured animals; there were also hypertrophic astroglia (Fig. 3F, F′, I, I′) and activated microglia (Fig. 4F, F′, I, I′, L, L′) in the rTBI animals. These qualitative changes were confirmed by quantitative analyses of segmented profiles showing significant increases in APP-positive profile density (5-fold), Iba1 (2-fold), and GFAP (1.85-fold) immunoreactivity levels in the corpus callosum of injured compared with sham animals (Fig. 6A–C).
FIGURE 6.

Quantitative assessment of GFAP, Iba1, and APP immunostaining and corpus callosum volume in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). GFAP and Iba1 immunostaining are significantly upregulated in the corpus callosum of injured (r-TBI) vs. sham animals (A, B). No changes in GFAP or Iba1 were observed in other regions (gray matter) analyzed (A, B). (C) APP-positive spherical profiles in the corpus callosum were increased by ∼6-fold in the corpus callosum of injured versus sham animals. (D) The volume of the subregion of the corpus callosum bound by bregma (−0.36 to − 0.72 mm lateral) was significantly reduced by ∼30% in injured versus sham animals. N = 6 (sham/injured) for (A)–(C) and n = 4 (sham/injured) for (D); *p < 0.05; **p < 0.01; ***p < 0.001. CC, corpus callosum; P-Cortex, parietal cortex; DG, dentate gyrus.
S100ß immunoreactivity (an astroglial marker) was also notably increased in the cell body of astrocytes in the corpus callosum of injured compared with sham animals (Fig. 3D, E, insets). Activated IBA1+ microglia were devoid of CD45 and MHC II immunostaining in injured animals within the corpus callosum (data not shown). An increase in cellularity was observed in the corpus callosum of injured animals compared with shams using Cresyl violet staining (Fig. 5E, F).
FIGURE 5.

Luxol fast blue (LFB) and Cresyl violet staining in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A–D) LFB stain shows thinning of the corpus callosum in the injured (r-TBI) compared with sham animal; staining intensity appears similar in both sham and injured animals. (E, F) Cresyl violet staining shows a marked increase in cellularity within this region in injured compared with sham animals. CC, corpus callosum; Hippo, hippocampus. Panels (C)–(F) are higher-magnification images of the areas in the boxes in panels (A) and (B).
An increase in GFAP immunoreactivity was also observed in the white matter regions of the walls of the lateral ventricles (Fig. 3C, C′, K). Notably, perivascular astrocytes in the white matter of injured animals were prominently stained for GFAP (Fig. 3J).
No changes were observed in the intensity of LFB staining (Fig. 5A, B) or myelin basic protein immunostains (data not shown) in the corpus callosum of injured compared with sham animals but there was significant thinning of the corpus callosum following stereological analyses of the volume difference between sham and injured animals. A 30% reduction was observed in the corpus callosum volume of injured compared with sham animals (Figs. 5C, D and 6D).
Glial Response (Gray Matter)
There was a paucity or GFAP and Iba1 immunostaining in the cortex of injured compared with sham animals (Figs. 3A, B and 4A–C, C′, respectively). This was confirmed by quantitative analyses (Fig. 6A, B). There was a notable increase in Iba1 and GFAP (Fig. 3L) immunoreactivity in the molecular cell layer (and white matter region) of the cerebellum of injured animals. Intracellular APP immunostaining was increased in the cell body of pyramidal neurons in layers II-IV of the parietal cortex (Fig. 2G, H).
Quantification of and Immunolocalization of Tau
Gray Matter (Cerebral Cortex)
In a substudy, analysis of brain tissue (gray matter) from mice subjected to this same injury paradigm but killed 24 hours after the last injury showed a 1.6-fold increase in total tau expression by immunoblotting in injured versus sham mice. However, IHC did not show any changes for all other tau species analyzed (data not shown).
For the chronic time point postinjury (ie, the focus of this manuscript), immunoblotting for total tau (DA9) also showed an increase in injured compared with sham animals in the gray matter (Fig. 7A); however, the chronic evaluation also revealed increases in pTau Thr231 (RZ3) and tau oligomers (TOC-1) (Fig. 7B, E, F). This was confirmed by IHC analysis for TOC-1 and RZ3 antibodies on serial sections of sham and injured animals; superficial layers of the cortex showed a mild increase in the immunoreactivity of TOC-1 and RZ3 (Fig. 8A–D).
FIGURE 7.

Quantitative assessment of total tau, pTau, and conformational tau species in gray matter by Western blotting in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A) Representative immunoblots for different tau antibodies. (B–H) Total tau (DA9), pTau Thr231 (RZ3), and tau oligomer (TOC-1) levels are significantly greater in injured compared with sham animals (B, E, F). The conformational tau antibody MC1 raised against neurofibrillary tangles was marginally increased in injured animals compared with sham animals (G). No changes were observed in pTau S396/404 (PHF1), pTau S202 (CP13), and tau-positive N-terminal phosphatase-activated domain (TNT) levels between sham and injured animals (C, D, H). Data are expressed as ratio of GAPDH or ß-actin levels. N = 6–8 (sham/injured); *p < 0.05; **p < 0.01; ***p < 0.001.
FIGURE 8.

Immunohistochemical assessment of conformational tau antibody TOC1 and phosphorylated tau antibody RZ3 (pThr231) in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A, B) There is an increase in anti-TOC-1 immunostaining in the cortex of injured (r-TBI) compared with sham animals. (C, D) There was also an increase in pTau Thr231 (RZ3) immunostaining in pyramidal neurons in layers II-IV of the cortex of injured versus sham animals. High-power micrographs insets B and D were taken from injured animals (white boxes).
Intriguingly, no change was observed by immunoblotting for pTau 396/404 (PHF1) or S202 (CP13) in sham and injured animals (Fig. 7C, D). This was also confirmed by IHC (Supplementary Fig. 3A–F, I–K).
Changes in conformational tau species positive for paired helical filaments and for the N terminal region of the phosphatase activating domain (PAD) were detected using the MC1 and TNT antibodies, respectively. Immunoblotting showed no changes between sham and injured animals in the gray matter for either MC1 or TNT antibodies (Fig. 7A, G, H). Likewise, IHC confirmed no augmentation in the immunoreactivity of MC1 and TNT in injured compared with sham animals (Supplementary Fig. 4A–G).
Argyrophilic-positive neurofibrillary and glial tangles were examined using the Gallyas silver stain. The cortices of both sham and injured animals were devoid of Gallyas staining (Supplementary Fig. 3G, H, L). Caspase-cleaved truncated tau fragment (Tau C3—Asp 421) showed no changes between sham and injured animals by IHC (data not shown).
White Matter
Changes in total tau (DA9), pTau (PHF1, CP13), PAD-positive tau (TNT), and conformational tau species (TOC-1, MC1) were examined by immunoblotting or immunohistochemistry in the white matter of sham versus injured animals, but no changes were observed (Fig. 9A–F). Corresponding IHC showed no changes in the white matter region of the corpus callosum.
FIGURE 9.

Quantitative assessment of total tau, pTau, conformational tau species, α-synuclein and neurofilament 200kda (NFL) in the white matter by Western blotting in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A–H) Representative immunoblots for different protein markers are shown in (A). No changes were observed in total tau (DA9), pTau S202 (CP13), and tau-positive N-terminal phosphatase-activated domain (TNT) levels between sham and injured animals (B–D, F). A marginally significant increase was observed in tau oligomer (TOC-1) levels in injured compared with sham animals (E). No changes were observed in α-synuclein or neurofilament 200kda (NFL) levels between sham and injured animals (G, H). Data are expressed as ratio of ß-actin or total protein levels. N = 6–8 (injured/sham).
Neurofilament, Synaptophysin, Transactive Response DNA-Binding Protein (TDP-43), α-Synuclein, and Amyloid
Damage to large caliber myelinated axons in the gray matter was examined using neurofilament (200kda) protein marker (46); no changes were observed in sham compared with injured animals (Supplementary Fig. 5A, B). Synaptic integrity was determined using the presynaptic vesicle marker synaptophsyin; we did not observe any injury-dependent changes in its expression (Supplementary Fig. 5A, C).
No changes were observed in TDP-43 or α-synuclein levels in the gray matter of sham and injured animals (Supplementary Fig. 5A, D, E). Brain tissue from both sham and injured animals was devoid of Congo-red staining and 4G8 immunoreactivity (data not shown).
Vascular Response
Levels of Claudin-5 and ZO1 were below limits of detection by Western blotting using commercially available antibodies. No changes were observed in laminin or occludin levels in the gray matter between injured compared with sham animals (Supplementary Fig. 5F, G). Immunohistochemistry for laminin and collagen IV-positive vessels showed no overt damage to the vasculature and an apparently similar vascular density between both shams and injured animals in different brain regions (Supplementary Fig. 6A, A′–D, D′).
No evidence of extravasation of endogenous mouse IgG staining was observed in the cortical regions beneath the impact site in injured mice (Supplementary Fig. 6E, E′) and there was no evidence of microhemorrhages in the brain tissue of injured (or sham) animals examined by Prussian blue staining (Supplementary Fig. 6F, F′).
Brain Phospholipid Species Profile
No changes were observed in PC and phosphatidylinositol (PI) lipid species in injured versus sham animals (Supplementary Fig. 7A, B). However, increases in total PE and SM lipid species were observed in injured compared with sham animals (Supplementary Fig. 7C, D). There was an increase in saturated fatty acid classes of PE and SM lipid species in injured animals compared with shams (Supplementary Fig. 7C, D). The mono-unsaturated fatty acid class of SM lipid species was also observed to significantly increase in injured compared with sham animals (Supplementary Fig. 7D). Only PE lipid species showed an increase in their classes of polyunsaturated fatty acids in injured compared with sham animals (Supplementary Fig. 7C).
We also measured AA- and DHA-containing lipid species (Supplementary Fig. 8A–C). A significant increase in AA containing lipid species for PC and PE was observed in injured compared with sham animals (Supplementary Fig. 8A, C). There was no change in DHA-containing lipid species for PI (Supplementary Fig. 8B). We compared the AA:DHA ratio for PC, PE, PI lipid species. A significant increase in the AA:DHA ratio was observed for PE lipid species in injured versus sham animals (Supplementary Fig. 8D).
Neuroendocrine and Inflammatory Cytokine Profiles in the Plasma
There was significant decrease in IL-1β and IL-5 inflammatory cytokines in the plasma of injured compared with sham animals (Fig. 10A, B). No significant changes were observed with the other inflammatory cytokines measured (GM-CSF, IFN-γ, TNF, IL-10); however, a trend toward decrease in the injured group was noted in all cases (Fig. 10C–F). The changes in inflammatory cytokines correlated with an increase in the neuroendocrine marker corticosterone in the plasma of injured compared with sham animals (Fig. 10G).
FIGURE 10.

Inflammatory cytokine profile and corticosterone levels in the plasma in an hTau mouse model of chronic repeated traumatic brain injury (r-TBI). (A, B) There was a significant decrease in IL-1ß and IL-5 observed in the plasma of injured versus sham animals. (C–F) There was a trend toward decrease in GM-CSF, IFN-γ, TNF, and IL-10 in injured versus sham animals but these did not reach statistical significance. N = 10–12 (sham/injured). (G) There was a significant increase in corticosterone in the plasma of injured versus sham animals. N = 6–8 (sham/injured); *p < 0.05; **p < 0.01.
DISCUSSION
We have developed a new model with the intent to explore the chronic effects of repetitive, concussion-like mild TBI over a prolonged period in hTau mice, which express all 6 human tau isoforms. In this first characterization, we report persistent increases in total tau, tau oligomers, and pTau (Thr231) species a 2–3 months after the cessation of injury; this may represent developing tau pathology in our model. Further studies examining extended chronic time points will be valuable to confirm progressive TBI-dependent tauopathy in this model because it may have relevance to studies in CTE in humans exposed to a history of repetitive concussion/mTBI. In this first characterization we also report behavioral (ie, anxiety, cognition, sensorimotor) and neuropathological abnormalities induced by exposure to chronic repetitive mTBI, and biochemistry of central and peripheral biomarkers.
Neuropathological Changes in Gray and White Matter After Chronic r-mTBI
The most conspicuous histopathological features were localized to the corpus callosum, consistent with observations with this same mTBI injury in our previous repetitive injury paradigm in which 5 mTBI injuries were delivered with a 48-hour interval between each injury (29, 33).
These features included pronounced and persistent ongoing axonal degeneration typified by increase APP-positive profiles, axonal swellings, bulbs, and varicosities that were associated with corpus callosum thinning at chronic time points after the last injury. Previous studies that have examined the effects of APP immunoreactivity in mouse models of repetitive mTBI typically show an initial rapid increase in APP staining (47–48), which we observed at the acute 24 hour post-last injury time point in this study (a 30-fold increase vs controls). However, in other animal models these effects diminish at subacute time points postinjury, with only nonexistent or minor differences seen between injury and age-matched controls (47–48). In contrast, with the present model, we observed a persistent ongoing increase in APP-positive profiles following injury compared with shams, although these effects were significantly less prominent when compared with the acute effects observed 3 to 4 months earlier. Such interlaboratory differences could be attributed to variations in experimental models and injury paradigms. For example, our experimental device uses a flat tip impactor whereas other groups mentioned typically use a rubber tip impactor that may absolve the force of injury on the site of impact, making the injury more diffuse and less severe. We have also focused on chronicity of hits in this study at a level that has not been previously examined by most groups. Because mild head injury increases the vulnerability of the brain to a second concussive impact (47), it is plausible that in our current model, by design, there is a limited temporal window of recovery, resulting in the pronounced ongoing axonal degeneration evident months after injury. Other features observed in the corpus callosum included marked astroglial and microglial activation. These degenerative white matter features observed in our model echo appearances documented in descriptions of human autopsy (49) and in vivo imaging (50) studies in white matter regions such as the internal capsule in TBI survivors, and at necropsy examination of brain tissue from a variety of repetitive mTBI animal models (28, 31, 33, 36, 39, 51).
In contrast to this ongoing white matter damage, there was no evidence of glial activation or ongoing axonal degeneration in gray matter, or of neuronal cell loss or structural damage to neuronal integrity in injured animals. However, immunoblots on material from gray matter did reveal a persistent increase (by 2-fold) in total tau levels and this was accompanied by a significant increase in pTau Thr231 (RZ3-positive), tau oligomers (TOC-1-positive [52]), and a trend toward increase in tau conformer species (MC1-positive). In support of these observations, patchy immunoreactivity to these significantly altered tau species (RZ3, TOC-1) was shown in the superficial cortical layers of r-mTBI exposed animals. PHF1 (Ser394/404) and CP13 (Ser202) levels remained unchanged with injury. There was no evidence of the distinct perivascular accumulation of pTau immunoreactive neurons or glia, typical of human CTE; neither was there evidence of neuropil threads. At this stage, we do not consider this model to represent a preclinical model of CTE but our findings appear to be among the first to show significant changes in tau oligomer levels at a chronic time point after repetitive mTBI in a mouse model. Small, misfolded tau species or tau oligomers are thought to be the toxic pathogenic species of tau that are resistant to ubiquitination (53–57). They have been demonstrated both in vitro and in vivo to spread in a retrograde and anterograde manner from one neuron to another, within connected anatomical pathways (58). Whether these tau oligomeric species will inevitably form into β-pleated sheet conformation or fibrillary material is unknown. The specific changes in phospho-tau epitope pThr231 levels in our model may also implicate the role of cis-trans conformational pTau changes after TBI in vivo. Cis-trans conformational pTau has been demonstrated to play an important role early in mild cognitive impairment; and they further accumulate in neurofibrillary degenerated neurons in Alzheimer disease and TBI (59, 60). We were not able to analyze these different conformational states in our studies due to the lack of commercially available antibodies that can distinguish between cis from trans pThr231 pro-conformation in tau; therefore, a definitive role of cis versus trans tau forms cannot be made in our study. Further exploration in our model using these antibodies as well as others specific to different tau species at longer time points postinjury may be informative in this regard; this would confirm whether the persistent tau changes that we observe are on a continuum toward progressive TBI-dependent neurofibrillary tau pathology.
The TBI-dependent changes in tau reported herein contrast with our findings in our previous injury paradigm (5 mTBI with a 48-hour interval) in either C57BL/6 or hTau young mice (8–12 weeks old). In those studies we did not observe any persistent changes in tau species following analyses of brain tissue by ELISA and immunohistochemistry, from 24 hours to 12 months postinjury in either wild type or hTau mice ([33], Mouzon B, Bachmeier C, Ferro A, Crynen G, Acker CM, Davies P, Ojo JO, Mullan M, Stewart W, Crawford F. 2016, personal communication). Other groups have also published data reporting absence of tau-dependent changes following repeated injuries at acute to chronic time points (30, 31, 38). Our findings suggest that factors such as chronicity of impacts, age at exposure, and frequency of injuries may play a role in precipitating tau pathologies at chronic time points after mTBI. This observation is supported by data from Petraglia et al using a model of repeated mTBI in wild-type mice, said to recapitulate “subconcussive” injuries; the model involved 6 mTBIs per day (2-hour intervals) for 7 days totaling 42 hits (36). Following this paradigm, the authors reported an increase in murine pTau levels by IHC at 7 days, 1 month, and 6 months postinjury. Intriguingly other models of chronic TBI exposure in younger mice involving fewer total injuries (ranging from 2 to 5 mTBI) (28, 32, 34, 37), and also in a single mouse model of blast-related traumatic injury (61, 62) have reported significant changes in pTau levels immediately postinjury. However, although these studies have reported acute alterations in tau, data on tau with chronic survival are not available from these models.
Thus far, the majority of studies exploring effects of TBI on tau pathology has been conducted in young, wild-type mice, which have a strong bias for the 4R murine tau isoform. Some investigators have suggested that this isoform has a low propensity to form toxic tau aggregates in vivo. Our use of hTau transgenic mice may shed some light on the factors that influence propagation of (human) tau pathology after injury. hTau mice typically develop aggregated, hyperphosphorylated tau by 9 months of age, in a spatial temporally relevant distribution localized to regions such as the entorhinal cortex, subiculum and hippocampus, and by 15 months of age develop thioflavin-S-positive neurofibrillary tangles, loss of neurons and apoptotic damage (41). In our previous work on aged hTau mice (>18 months) exposed to 5 repetitive hits, we demonstrated augmentation of pTau pathology in superficial layers of the cortex and also in the hippocampus by immunohistochemistry (39). Of note, these effects appeared to be more pronounced than those in this study using younger mice (<13 weeks at first injury), perhaps suggesting the importance of an underlying tau pathology at the time of injury as a necessary trigger for precipitating persistent TBI-dependent tauopathy.
In addition to tau, we also investigated other signature proteins related to neurodegeneration in our model. We found no evidence of amyloid pathology in repetitive mTBI injured animals, as determined by histopathology. To date, although the role of amyloid in CTE remains unknown, retrospective case studies have shown that a majority of CTE patients have amyloid plaques at autopsy (13, 25). We also did not observe any changes in the levels of TDP43 after injury. TDP43 has been identified as the major disease protein in frontal temporal lobar degeneration with ubiquitinated inclusions, and in amyotrophic lateral sclerosis. TDP43-immunoreactive intraneuronal and intraglial inclusions are commonly described in CTE and are present in a majority of cases studied (13, 63–65). To our knowledge, this is among one of the first studies to asses TDP-43 levels at a chronic time point in a rodent repetitive mTBI model. Although our findings did not show any correlation between repetitive mTBI and TDP43, we are cautious when interpreting our findings regarding the role of TDP43 in r-mTBI, given that there might be differences between murine versus human TDP43. More studies need to be conducted to evaluate the role of this protein following TBI.
Lipid Dysfunction After Chronic r-mTBI
Lipids are known to play an important role in neurological disorders and CNS injury, and have been implicated in TBI (66). We previously demonstrated TBI-dependent brain lipid changes in a controlled cortical injury model using our lipidomic platform (45), and thus sought to evaluate changes in different lipid species in this new model. Our analyses revealed an increase in total PE and SM lipid species in the cortex of our chronic repetitive mTBI exposed animals, accompanied by increases in saturated, mono or poly-unsaturated fatty acid classes of PE and SM lipid species. The reason for these lipid changes and their role in late TBI pathologies is not clear at this time, but we hypothesize that they may reflect (I) dysregulation in brain lipid metabolism, (II) failure of abluminal lipid transporters, or (III) increased luminal transport from the periphery as free fatty acids or in esterified forms bound to lipoproteins. These high lipid levels after TBI could predispose to oxidative damage (ie, lipid peroxidation), especially under inflammatory conditions. We additionally observed an increase in the ratio of AA (20:4) to DHA (22:6) containing polyunsaturated lipid species in injured animals. AA is metabolized to proinflammatory and vasoactive eicosanoids (such as prostaglandins, leukotrienes, and thromboxane) by lipoxygenase and/or cyclooxygenase pathways (66). DHA, the most abundant polyunsaturated fatty acid, is metabolized by phospholipase A2 enzymes to produce metabolites resolvins and protectins, such as neuroprotectin D1 (67). These metabolites serve as endogenous neuroprotectants that inhibit apoptosis, oxidative stress, and proinflammatory processes (67–70). The consequences of this increase in AA to DHA ratio could, therefore, indicate an increase in proinflammatory versus anti-inflammatory environment at this late time point following exposure to chronic repetitive mTBI.
CBF Outcomes After Chronic r-mTBI
We observed a reduction in CBF in the injured mice that was localized primarily to the regions beneath the impact site around the occipital (/parietal) lobes. Intriguingly, in a study utilizing brain perfusion single photon emission computed tomography (SPECT) imaging in a cohort of >100 active and former American football players, the authors reported a decrease in brain perfusion, with a preponderance for the prefrontal lobe, temporal lobe, occipital lobe, and cingulate gyrus and hippocampus regions in their cohort (71). Although we cannot draw direct comparisons between our mouse CBF and human SPECT data, the similarities in the direction of change in brain perfusion highlight vascular dysfunction as another sequelae of TBI. The reason behind this is unknown, given that we did not observe any focal injury or gross pathological changes in this region. Cerebral glucose metabolism has been linked to reductions in blood flow after TBI and this could possibly be altered in our model (72). Of note, although we did not observe any impairment in blood-brain barrier (BBB) integrity in control or injured animals, we did observe astroglial activation in perivascular regions in injured animals. Though observations in human, autopsy-derived material from patients surviving single moderate to severe TBI demonstrate widespread BBB disruption in a high proportion of both acute and late (>1-year survival) survivors (73), data on vascular pathology in CTE, in particular BBB integrity, are lacking.
Neurobehavioral, Neuroendocrine, and Neuroimmune Outcomes After Chronic r-mTBI
We conducted behavioral analyses of hTau mice at 9 months of age. hTau mice typically do not show any significant impairment in spatial learning and memory until 12 months of age, although 6-month-old mice have been noted by group to show abnormal spatial learning compared with control mice (74, 75). Locomotor function, anxiety levels, and gross motor functions are typically not different from those in age-matched controls (74). Our observations were conducted prior to the appearance of age-related neurobehavioral dysfunction to eliminate underlying confounding phenotypes. Behavioral evaluations report a pattern toward disinhibitory-like behavior and deficits in cognitive performance. The effects were more pronounced compared with our previously reported mTBI paradigms in young mice, which employed exactly the same level and nature of injury but investigated the effects of either a single or a different repetitive injury (5 hits over a 9-day period) (29, 33, 39). Behavioral outcomes in our model could be attributed to underlying neuropathological correlates involving white matter degeneration, and accumulation of abnormal tau in neurons within the gray matter. Both sites of localized pathologies are relevant given their involvement in controlling disinhibitory behavior and cognitive function. Comparable behavioral outcome measures have also been reported in another repetitive mTBI model involving multiple “subconcussive” impacts; the authors observed cognitive deficits, increased risk taking, depression-like behavior, and sleep disturbances at 1 month postexposure (36). However, this model differed from ours in that it was a much more concentrated paradigm with all the impacts occurring within a 1-week period. Human patients exposed to a history of mTBI in contact sports demonstrate a spectrum of neurobehavioral symptoms including postconcussive syndrome and CTE, which involve alterations in mood, neuropsychiatric behavior, and cognition (72, 76–82).
Correlating with the neurobehavioral deficits seen in our model, we also observed an increase in the glucocorticoid steroid hormone corticosterone. Abnormalities in neuroendocrine function and circadian rhythm have been shown after TBI of different severities (83–89). Upregulation in cortisol levels may result in an increased negative feedback to the brain via the HPA axis. This can result in the dysregulation of sympathetic and autonomic nervous system functions implicated in the regulation of the fight-or-flight response (90). Neuroinflammatory cytokine levels in the periphery were also reduced in our model. Glucocorticoids, such as corticosterone, are known to influence a variety of immune-related functions (91), and might dampen down immune-inflammatory cytokine signaling, as seen in our model. What specific effects downregulation in systemic cytokine profiles may have on the injured brain are unknown and warrant further studies in the context of TBI. Our lipidomic studies did implicate a possible increase in baseline proinflammatory response in the cortex based on the modest increase in AA-containing lipid species we found. A closer assessment of microglial cell markers (Iba1, CD45, and MHC II), however, showed a paucity of staining in the cortex in sham and injured animals. These findings are intriguing and may be a unique feature of this animal model. Very few studies exploring systemic inflammation and even brain neuroglial responses after repeated mTBI in humans have been conducted to date; it is therefore very difficult to draw a direct comparison between our model and what happens in humans.
Conclusion
In this study, we explored the effects of chronic repetitive mTBI in a mouse model with a human tau genetic background, with an injury paradigm designed to mimic the effects of repeated mTBI injuries sustained over a prolonged timeframe. Our characterization encompasses the chronic histopathological, biochemical, and neurobehavioral effects of these injuries after many months survival. In this first characterization of our chronic TBI experiments, we confirm a 2-fold increase in total tau levels and mild increases in tau oligomers/conformers and pre-tangle pTau (Thr231) species in the gray matter up to 3 months after cessation of injury, which we anticipate may be on a continuum of progressive TBI-dependent tauopathy. Our data also confirm impairment in CBF (hypoperfusion), and a prominent feature of white matter damage typified by gliosis, axonal injury, and corpus callosum thinning. These changes were also accompanied by increases in lipid species of different fatty acid classes and their secondary messengers. Subtle neurobehavioral deficits typified by disinhibition and deficits in cognitive performance were also observed. Together these data support the relevance of this concussive injury model in studying the consequences of repetitive mTBI in humans, and its usefulness in testing potential neuroprotective therapeutic strategies for chronic repeated mTBI.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr. Gary Laco and Thinh Nguyen for their help with the lipidomic studies, Carlyn Lungmus and Naomi Gail Rafi for their help with the preparation of the manuscript. We would also like to thank Peter Davies and Dr. Lester Binder’s lab for provision of tau antibodies.
REFERENCES
- 1.Gedye A, Beattie BL, Tuokko H, et al. Severe head injury hastens age of onset of Alzheimer’s disease. J Am Geriatr Soc 1989;37:970–3 [DOI] [PubMed] [Google Scholar]
- 2.Mortimer JA, Van Duijn CM, Chandra V, et al. Head trauma as a risk factor for Alzheimer’s disease: A collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991;20(suppl. 2):S28–35 [DOI] [PubMed] [Google Scholar]
- 3.Schofield PW, Tang M, Marder K, et al. Alzheimer’s disease after remote head injury: An incidence study. J Neurol Neurosurg Psychiatry 1997;62:119–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo Z, Cupples LA, Kurz A, et al. Head injury and the risk of AD in the MIRAGE study. Neurology 2000;54:1316–23 [DOI] [PubMed] [Google Scholar]
- 5.Plassman BL, Havlik RJ, Steffens DC, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 2000;55:1158–66 [DOI] [PubMed] [Google Scholar]
- 6.Fleminger S, Oliver DL, Lovestone S, et al. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 2003;74:857–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guskiewicz KM, Marshall SW, Bailes J, et al. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery 2005;57:719–26 [DOI] [PubMed] [Google Scholar]
- 8.Lehman EJ, Hein MJ, Baron SL, et al. Neurodegenerative causes of death among retired national football league players. Neurology 2012;79:1970–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat Rev Neurol 2013;9:211–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nordström P, Michaelsson K, Gustafson Y, et al. Traumatic brain injury and young onset dementia: A nationwide cohort study. Ann Neurol 2014;75:374–81 [DOI] [PubMed] [Google Scholar]
- 11.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 2009;68:709–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Omalu B, Bailes J, Hamilton RL, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery 2011;69:173–83 [DOI] [PubMed] [Google Scholar]
- 13.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts GW. Immunocytochemistry of neurofibrillary tangles in dementia pugilistica and Alzheimer’s disease: Evidence for common genesis. Lancet 1988;2:1456–8 [DOI] [PubMed] [Google Scholar]
- 15.Geddes JF, Vowles GH, Robinson SF, et al. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol 1996;22:12–6 [PubMed] [Google Scholar]
- 16.Omalu BI, DeKosky ST, Minster RL, et al. Chronic traumatic encephalopathy in a national football league player. Neurosurgery 2005;57:128–34 [DOI] [PubMed] [Google Scholar]
- 17.Omalu BI, DeKosky ST, Hamilton RL, et al. Chronic traumatic encephalopathy in a national football league player: Part II. Neurosurgery 2006;59:1086–92 [DOI] [PubMed] [Google Scholar]
- 18.Stewart W, McNamara PH, Lawlor B, et al. Chronic traumatic encephalopathy: A potential late and under recognized consequence of rugby union. QJM 2015;109:11–5 [DOI] [PubMed] [Google Scholar]
- 19.Zetterberg H, Hietala MA, Jonsson M, et al. Neurochemical aftermath of amateur boxing. Arch Neurol 2006;63:1277–80 [DOI] [PubMed] [Google Scholar]
- 20.Neselius S, Brisby H, Theodorsson A, et al. CSF-biomarkers in Olympic boxing: Diagnosis and effects of repetitive head trauma. PLoS One 2012;7:e33606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shahim P, Tegner Y, Wilson DH, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol 2014;71:684–92 [DOI] [PubMed] [Google Scholar]
- 22.Olivera A, Lejbman N, Jeromin A, et al. Peripheral total tau in military personnel who sustain traumatic brain injuries during deployment. JAMA Neurol 2015;72:1109–16 [DOI] [PubMed] [Google Scholar]
- 23.Shahim P, Linemann T, Inekci D, et al. Serum tau fragments predict return to play in concussed professional ice hockey players. J Neurotrauma 2016 May 2 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2012;246:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardner RC, Burke JF, Nettiksimmons J, et al. Dementia risk after traumatic brain injury vs nonbrain trauma: The role of age and severity. JAMA Neurol 2014;71:1490–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKee AC, Daneshvar DH, Alvarez VE, et al. The neuropathology of sport. Acta Neuropathol 2014;127:29–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshiyama Y, Uryu K, Higuchi M, et al. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotrauma 2005;22:1134–41 [DOI] [PubMed] [Google Scholar]
- 28.Kane MJ, Angoa-Perez M, Briggs DI, et al. A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods 2012;203:41–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouzon B, Chaytow H, Crynen G, et al. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma 2012;29:2761–73 [DOI] [PubMed] [Google Scholar]
- 30.Mannix R, Meehan WP, Mandeville J, et al. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol 2014;74:65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolton AN, Saatman KE. Regional neurodegeneration and gliosis are amplified by mild traumatic brain injury repeated at 24-hour intervals. J Neuropathol Exp Neurol 2014;73:933–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo J, Nguyen A, Villeda S, et al. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol 2014;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouzon BC, Bachmeier C, Ferro A, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol 2014;75:241–54 [DOI] [PubMed] [Google Scholar]
- 34.Namjoshi DR, Cheng WH, McInnes KA, et al. Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): A novel, surgery-free model of traumatic brain injury. Mol Neurodegener 2014;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ojo J, Bachmeier C, Mouzon B, et al. Chronic ultrastructural changes in the grey and white matter of repeated concussive head injured wild-type and hTau mice. J Neuropathol Exp Neurol 2015;74:1012–35 [DOI] [PubMed] [Google Scholar]
- 36.Petraglia AL, Plog BA, Dayawansa S, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int 2014;5:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Teng Z, Song Y, et al. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab 2015;35:443–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L, Nguyen JV, Lehar M, et al. Repetitive mild traumatic brain injury with impact acceleration in the mouse: Multifocal axonopathy, neuroinflammation, and neurodegeneration in the visual system. Exp Neurol 2016;275:436–49 [DOI] [PubMed] [Google Scholar]
- 39.Ojo JO, Mouzon B, Greenberg MB, et al. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol 2013;72:137–51 [DOI] [PubMed] [Google Scholar]
- 40.Ojo JO, Mouzon BC, Crawford F. Repetitive head trauma, chronic traumatic encephalopathy and tau: Challenges in translating from mice to men. Exp Neurol 2016;275:389–404 [DOI] [PubMed] [Google Scholar]
- 41.Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 2003;86:582–90 [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Belayev L, Zhao W, et al. Neuroprotective effect of treatment with human albumin in permanent focal cerebral ischemia: Histopathology and cortical perfusion studies. Eur J Pharmacol 2001;428:193–201 [DOI] [PubMed] [Google Scholar]
- 43.Paris D, Humphrey J, Quadros A, et al. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: Role of inflammation. Neurol Res 2003;25:642–51 [DOI] [PubMed] [Google Scholar]
- 44.Abdullah L, Evans JE, Bishop A, et al. Lipidomic profiling of phosphocholine containing brain lipids in mice with sensorimotor deficits and anxiety-like features after exposure to Gulf War agents. NeuroMolecular Med 2008;14:349–61 [DOI] [PubMed] [Google Scholar]
- 45.Abdullah L, Evans JE, Ferguson S, et al. Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB J 2014;28:5311–21 [DOI] [PubMed] [Google Scholar]
- 46.Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec 1970;167:379–87 [DOI] [PubMed] [Google Scholar]
- 47.Laurer HL, Bareyre FM, Lee VM, et al. Mild head injury increasing the brain’s vulnerability to a second concussive impact. J Neurosurg 2001;95:859–70 [DOI] [PubMed] [Google Scholar]
- 48.Longhi L, Saatman KE, Fujimoto S, et al. Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery 2005;56:364–74 [DOI] [PubMed] [Google Scholar]
- 49.Johnson VE, Stewart JE, Begbie FD, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013;136:28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol 2011;70:374–83 [DOI] [PubMed] [Google Scholar]
- 51.Shitaka Y, Tran HT, Bennett RE, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol 2011;70:551–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patterson KR, Remmers C, Fu Y, et al. Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem 2011;286:23063–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res 1989;477:90–9 [DOI] [PubMed] [Google Scholar]
- 54.Köpke E, Tung YC, Shaikh S, et al. Microtubule-associated protein tau. Abnormal phosphorylation of a nonpaired helical filament pool in Alzheimer’s disease. J Biol Chem 1993;268:24374–84 [PubMed] [Google Scholar]
- 55.Uchihara T, Nakamura A, Yamazaki M, et al. Evolution from pretangle neurons to neurofibrillary tangles monitored by thiazin red combined with Gallyas method and double immunofluorescence. Acta Neuropathol 2001;101:535–9 [DOI] [PubMed] [Google Scholar]
- 56.Kovacech B, Skrabana R, Novak M. Transition of tau protein from disordered to misordered in Alzheimer’s disease. Neurodegener Dis 2010;7:24–7 [DOI] [PubMed] [Google Scholar]
- 57.Braak H, Tredici KD. Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr Opin Neurol 2012;25:708–14 [DOI] [PubMed] [Google Scholar]
- 58.Wu JW, Herman M, Liu L, et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 2013;288:1856–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura K, Zhen Zhou X, Ping Lu K. Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion 2013;7:117–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kondo A, Shahpasand K, Mannix R, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015;523:431–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goldstein LE, Fisher AM, Tagge CA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 2012;4:134–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huber BR, Meabon JS, Martin TJ, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis 2013;37:309–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2010;69:918–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.King A, Sweeney F, Bodi I, et al. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer’s disease. Neuropathology 2010;30:408–19 [DOI] [PubMed] [Google Scholar]
- 65.McKee A, Montine T, Alvarez V, et al. Distinctive patterns of tau and TDP-43 in a former professional football player and marine as compared to 3 siblings. J Neuropathol Exp Neurol 2011;70:517 [Google Scholar]
- 66.Adibhatla RM, Hatcher JF. Role of lipids in brain injury and diseases. Future Lipidol 2007;2:403–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ariel A, Serhan CN. Resolvins and protectins in the termination program of acute inflammation. Trends Immunol 2007;28:176–83 [DOI] [PubMed] [Google Scholar]
- 68.Marcheselli VL, Hong S, Lukiw WJ, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 2003;278:43807–17 [DOI] [PubMed] [Google Scholar]
- 69.Bazan N. The onset of brain injury and neurodegeneration triggers the synthesis of docosanoid neuroprotective signaling. Cell Mol Neurobiol 2006;26:901–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim H-Y. Novel metabolism of docosahexaenoic acid in neural cells. J Biol Chem 2007;282:18661–5 [DOI] [PubMed] [Google Scholar]
- 71.Amen DG, Newberg A, Thatcher R, et al. Impact of playing American professional football on long-term brain function. J Neuropsychiatry Clin Neurosci 2011;23:98–106 [DOI] [PubMed] [Google Scholar]
- 72.Jordan BD. The clinical spectrum of sport-related traumatic brain injury. Nat Rev Neurol 2013;9:222–30 [DOI] [PubMed] [Google Scholar]
- 73.Hay JR, Johnson VE, Young AM, et al. Blood brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J Neuropathol Exp Neurol 2015;74:1147–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polydoro M, Acker CM, Duff K, et al. Age-dependent impairment of cognitive and synaptic function in the hTau mouse model of tau pathology. J Neurosci 2009;29:10741–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phillips M, Boman E, Osterman H, et al. Olfactory and visuospatial learning and memory performance in two strains of Alzheimer’s disease model mice—a longitudinal study. PLoS One 2011;6:e19567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mez J, Stern RA, McKee AC. Chronic traumatic encephalopathy: Where are we and where are we going? Curr Neurol Neurosci Rep 2013;13:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Millspaugh JA. Dementia pugilistica. US Naval Med Bull 2013;35:297–303 [Google Scholar]
- 78.Critchley M. Medical aspects of boxing, particularly from a neurological standpoint. Br Med J 1957;1:357–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grahmann H, Ule G. Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica and traumatic encephalopathy of boxers) [In German]. Psychiatr Neurol (Basel) 1957;134:261–83 [PubMed] [Google Scholar]
- 80.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med 1973;3:270–303 [DOI] [PubMed] [Google Scholar]
- 81.Hall RC, Chapman MJ. Definition, diagnosis and forensic implications of postconcussional syndrome. Psychosomatics 2005;46:195–202 [DOI] [PubMed] [Google Scholar]
- 82.Shively S, Scher AI, Perl DP, et al. Dementia resulting from traumatic brain injury: What is the pathology? Arch Neurol 2012;69:1245–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuan XQ, Wade CE. Neuroendocrine abnormalities in patients with traumatic brain injury. Front Neuroendocrinol 1991;12:209–30 [PubMed] [Google Scholar]
- 84.Bondanelli M, Ambrosio M, Zatelli M, et al. Hypopituitarism after traumatic brain injury. Eur J Endocrinol 2005;152:679–69 [DOI] [PubMed] [Google Scholar]
- 85.Rothman M, Arciniegas D, Filley C, et al. The neuroendocrine effects of traumatic brain injury. J Neuropsychiatry Clin Neurosci 2007;19:363–73 [DOI] [PubMed] [Google Scholar]
- 86.Behan LA, Phillips J, Thompson CJ, et al. Neuroendocrine disorders after traumatic brain injury. J Neurol Neurosurg Psychiatry 2008;79:753–9 [DOI] [PubMed] [Google Scholar]
- 87.Krahulik D, Zapletalova J, Frysak Z, et al. Dysfunction of hypothalamic-hyperphysical axis after traumatic brain injury in adults. J Neurosurg 2009;113:581–4 [DOI] [PubMed] [Google Scholar]
- 88.Tanriverdi F, Unluhizarci K, Kelestimur F. Pituitary function in subjects with mild traumatic brain injury: A review of literature and proposal of a screening strategy. Pituitary 2010;13:146–53 [DOI] [PubMed] [Google Scholar]
- 89.Baxter D, Sharp DJ, Feeney C, et al. Pituitary dysfunction after blast traumatic brain injury: The UKBIOSAP study. Ann Neurol 2013;74:527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harbuz M. Neuroendocrine function and chronic inflammatory stress. Exp Physiol 2002;87:519–25 [DOI] [PubMed] [Google Scholar]
- 91.Wohleb ES, McKim DB, Sheridan JF, et al. Monocyte trafficking to the brain with stress and inflammation: A novel axis of immune-to brain communication that influences mood and behavior. Front Neurosci 2015;8:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.