

Summary
Leishmania mexicana has a large family of cyclin‐dependent kinases (CDKs) that reflect the complex interplay between cell cycle and life cycle progression. Evidence from previous studies indicated that Cdc2‐related kinase 3 (CRK3) in complex with the cyclin CYC6 is a functional homologue of the major cell cycle regulator CDK1, yet definitive genetic evidence for an essential role in parasite proliferation is lacking. To address this, we have implemented an inducible gene deletion system based on a dimerised Cre recombinase (diCre) to target CRK3 and elucidate its role in the cell cycle of L. mexicana. Induction of diCre activity in promastigotes with rapamycin resulted in efficient deletion of floxed CRK3, resulting in G2/M growth arrest. Co‐expression of a CRK3 transgene during rapamycin‐induced deletion of CRK3 resulted in complementation of growth, whereas expression of an active site CRK3 T178E mutant did not, showing that protein kinase activity is crucial for CRK3 function. Inducible deletion of CRK3 in stationary phase promastigotes resulted in attenuated growth in mice, thereby confirming CRK3 as a useful therapeutic target and diCre as a valuable new tool for analyzing essential genes in Leishmania.
Introduction
The leishmaniases, diseases caused by protozoan parasites of the genus Leishmania, have diverse clinical manifestations dependent on the species and host immune response. Leishmaniasis is a substantial public health issue, causing an estimated 40,000 deaths annually and approximately 0.2–0.4 and 0.7–1.2 million visceral and cutaneous manifestations of the disease respectively (Alvar et al., 2012). Existing drug therapies are problematic because of high treatment costs, toxicity and undesirable administration routes, making the development of novel and effective drug therapies to expand the current repertoire crucial. Phenotypic strategies to identify drug targets in the mammalian infective amastigote life cycle stage are of particular importance for drug discovery programs.
As unicellular organisms, Leishmania depend on stringent control of cellular division to propagate and maintain infection. Protein kinases elicit pronounced effects on the Leishmania cell cycle by regulation of cell signalling pathways, and a number of protein kinases have been identified that are essential for promastigote viability (Dacher et al., 2014; Wang et al., 2005). The cyclin‐dependent kinases (CDK) are of particularly interest because of their pivotal roles as cell cycle regulators. The use of CDK inhibitors in cancer therapy (Cicenas and Valius, 2011; Knapp and Sundström, 2014) and the relative expansion of this protein family in Leishmania relative to other unicellular organisms distinguishes them as suitable drug targets. In particular, the CDK‐related kinase CRK3 has been demonstrated as being important for regulation of the L. mexicana promastigote cell cycle by existing genetic manipulation techniques and cell cycle arrest following treatment with CDK inhibitors (Hassan et al., 2001; Grant et al., 1998; 2004). Recombinant protein kinase activity assays (Gomes et al., 2010) and yeast recovery mutants (Wang et al., 1998) have provided further validation of CRK3 as a drug target, leading to the identification and synthesis of a number of CRK3 inhibitors (Cleghorn et al., 2011; Goyal et al., 2014; Grant et al., 2004; Řezníčková et al., 2015; Walker et al., 2011). Regulation of CRK3 expression in L. mexicana is desirable to further assess its function in both procyclic promastigote and amastigote life cycle stages, however, no system exists for conditional deletion of essential genes. Recent application of plasmid shuffle methodology has addressed this issue by enabling the generation of partial null mutants to further study essentiality and important residues within coding sequences (Dacher et al., 2014; Morales et al., 2010), however, the gene is not deleted and this prevents phenotyping of a null mutant.
To address this limitation, we have implemented a rapamycin‐inducible gene deletion system using a dimerised Cre recombinase (diCre) (Andenmatten et al., 2013; Collins et al., 2013; Jullien et al., 2003) to target CRK3 and elucidate its role in the cell cycle of L. mexicana. L. mexicana is generally diploid (Rogers et al., 2011) and both CRK3 alleles were replaced with a ‘floxed’ CRK3 open reading frame and the diCre coding sequence through promastigote transfection and homologous recombination. This system was used to conditionally delete CRK3 during promastigote growth and so prove that CRK3 mediates the transition through G2/M. Induced loss of CRK3 was complemented by expression of a CRK3 transgene but not by expression of an inactive site (T178E) CRK3 mutant, showing that protein kinase activity is crucial for CRK3 function. Significantly, conditional deletion of CRK3 in stationary phase promastigotes and subsequent attenuation during murine infection demonstrates that CRK3 activity is essential for establishing infection. This system represents a new method to directly assess whether a gene is essential to parasite viability and provides novel insight into the function of essential genes in Leishmania.
Results
DiCre activity is tightly regulated in L. mexicana promastigotes and amastigotes
To test the activity of diCre in L. mexicana promastigotes, a reporter cell line was generated by integration of a loxP‐flanked GFP into the ribosomal locus: [SSU GFP Flox]. This cell line was transfected with a diCre construct containing the two dimerizable Cre recombinase subunits with the homologous flanks of crk3 to generate the heterozygous line (Δcrk3::DICRE/CRK3 [SSU GFP Flox]). Integration of the diCre construct at the CRK3 locus was confirmed by PCR analysis (Fig. S1A). No effect on the growth of SSU GFP Flox or Δcrk3::DICRE/CRK3 [SSU GFP Flox] was observed in the presence of the dimerization ligand, rapamycin, up to the highest dose of 250 nM (Fig. S1B). GFP excision following incubation with increasing concentrations of rapamycin was investigated by PCR using specific primers flanking GFP. A single 1.45 kb PCR product, the floxed GFP fragment, was detected in the absence of rapamycin, whilst a 0.69 kb PCR product, representing the excised locus, was detected following rapamycin treatment only (Fig. 1A), indicating tight regulation of diCre activity. Δcrk3::DICRE/CRK3 [SSU GFP Flox] and [SSU GFP Flox] promastigotes grown for 5 days in the presence or absence of increasing concentrations of rapamycin were analyzed by flow cytometry to measure levels of GFP expression (Fig. 1B). Treatment of Δcrk3::DICRE/CRK3 [SSU GFP Flox] promastigotes with greater than 5 nM rapamycin resulted in substantial loss of GFP expression compared with the untreated controls, whilst GFP expression in [SSU GFP Flox] was the same following growth in all concentrations of rapamycin. GFP loss in Δcrk3::DICRE/CRK3 [SSU GFP Flox] promastigotes grown in the presence or absence of 100 nM rapamycin for 5 days was further assessed by Western blotting of total protein extracts using anti‐GFP antibody (Fig. 1C). Rapamycin‐treated promastigotes had considerably reduced GFP compared with the untreated controls, thereby demonstrating that gene loss results in reduced protein expression. These data also demonstrate that expression of diCre from the CRK3 locus is sufficient to efficiently excise the GFP transgene at rapamycin concentrations above 5 nM, and that no background diCre activity can be detected in the absence of ligand. About 100 nM of rapamycin was chosen as the optimum concentration to induce diCre activity in promastigotes whilst having no effect on in vitro cell growth.
Figure 1.
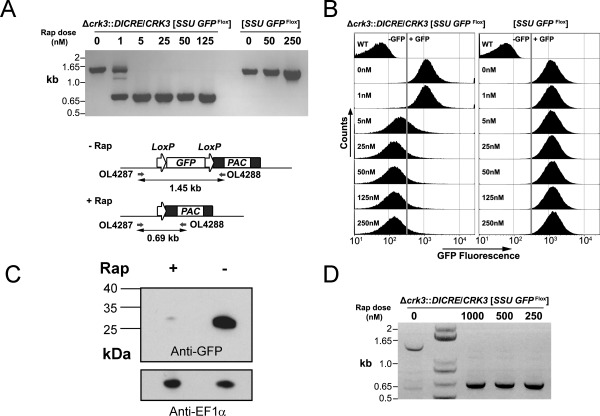
Validation of inducible diCre in L. mexicana: conditional deletion of GFP in promastigotes and amastigotes.
A. Gene excision analyzed by PCR amplification. Schematic (lower) shows the SSU GFP Flox locus and the recombination event expected after treatment with rapamycin (Rap). (upper) PCR amplification with oligonucleotides 4287 and 4288 from experimental (Δcrk3::DICRE/CRK3 [SSU GFP Flox]) and control [SSU GFP Flox] promastigotes at 5 days post‐treatment with different concentrations of rapamycin.
B. Flow cytometry assessment of GFP intensity of experimental and control promastigotes incubated in the presence or absence of rapamycin for 5 days.
C. Western blotting analysis with anti‐GFP and anti‐EF1α loading control antibodies of protein extracted from experimental promastigotes grown for 5 days in the presence or absence of 100 nM rapamycin.
D. PCR analysis of GFP Flox loss (as described in A) in amastigotes after 24 h rapamycin treatment (0–1000 nM), followed by 120 h infection in bone‐marrow derived macrophages. Lane 2 contains a 1 kb+ DNA ladder.
To test diCre functionality in amastigotes, infectious promastigotes of the experimental line Δcrk3::DICRE/CRK3 [SSU GFP Flox] were inoculated into BALB/c footpads and amastigotes purified from the resulting lesion. Ex vivo amastigotes retained high levels of green fluorescence and were incubated with rapamycin for 24 h in Schneider's medium prior to infection of bone‐marrow derived macrophages. Efficient excision of GFP Flox was detected by PCR amplification of a 0.69 kb fragment representative of GFP loss in all rapamycin treated samples (Fig. 1D) and GFP‐ (non‐fluorescent) amastigotes were observed by comparing images obtained through fluorescence live cell imaging (Fig. S1C). Residual GFP+ amastigotes were still visible by microscopy (Fig. S1C) and could be detected by flow cytometry (Fig. S1D); this was possibly because of the slow replication rate of amastigotes leading to a low rate of GFP turnover. These data demonstrate inducible diCre activity in amastigotes.
Inducible deletion of CRK3 in L. mexicana promastigotes
The functional and efficient levels of diCre‐mediated excision of GFP underpinned the development of a system for conditional deletion of essential genes. Gateway recombineering was used to flank appropriate diCre and loxP expression constructs with gene‐specific, homologous flanks (Fig. S2). Plasmids were generated by this method to replace the two alleles of CRK3, an essential gene in L. mexicana (Hassan et al., 2001) (Fig S3A). The first allele of CRK3 was replaced with DICRE (Δcrk3::DICRE/CRK3) and the second allele of CRK3 was subsequently replaced with a floxed C‐terminal GFP‐tagged CRK3 version (Δcrk3::DICRE/Δcrk3::CRK3 Flox; Figs. 2A and S3B). In addition, an mCherry red fluorescent protein coding sequence was incorporated downstream from the floxed CRK3‐GFP to facilitate flow cytometry and microscopy analysis. Transfection resulted in multiple clones with the expected genetic modifications, as confirmed by PCR analysis (Fig. S3B).
Figure 2.
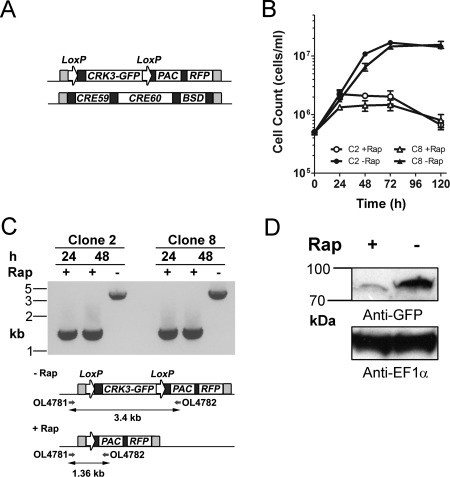
Generation of a CRK3 conditional deletion cell line.
A. Schematic showing the replacement of endogenous CRK3 to generate Δcrk3::DICRE/Δcrk3::CRK3 Flox. One allele contains a loxP flanked CRK3‐GFP coding sequence with mCherry red‐fluorescent protein cassette (RFP) and puromycin drug selectable marker (PAC). The other allele contains genes encoding both diCre subunits (CRE59, CRE60) each linked with rapamycin binding domains (not shown: FKBP12 and FRB respectively) and a blasticidin resistance cassette (BSD). Each construct was flanked with 500 bp arms of homology (light grey) by Gateway recombination to facilitate integration at the CRK3 locus. All coding sequences are flanked by regulatory elements (dark grey). L. mexicana parasites were transfected sequentially with the diCre construct and floxed CRK3 to confer resistance to blasticidin and puromycin antibiotics respectively.
B. Clones 2 and 8 promastigotes were seeded at a density of 5 × 105 cells mL−1 and grown in the presence or absence (+/−) of 100 nM rapamycin for 5 days. Cell density was determined by counting at 24 h intervals and mean ± SD of triplicate values was plotted.
C. (lower) A schematic representation of the floxed CRK3 locus after excision. PCR amplification shows the primers binding upstream of the 5′ CRK3 homologous flank and within the PAC cassette. (upper) PCR amplification of clones 2 and 8 at 24 h and 48 h +/−100 nM rapamycin treatment was conducted and the resulting amplicons resolved on an agarose gel.
D. Western blotting analysis with anti‐GFP and anti‐EF1α loading control antibodies of protein extracted from experimental clone 2 promastigotes grown for 4 days in the presence or absence of 100 nM rapamycin.
The growth of promastigotes from two Δcrk3::DICRE/Δcrk3::CRK3 Flox clones were assessed following diCre‐mediated excision induced with 100 nM rapamycin (Fig. 2B). Cells were counted over the course of 5 days, revealing a pronounced growth defect and reduction in cell number in rapamycin‐treated cells compared with uninduced controls. PCR analysis of promastigotes grown in the presence or absence of 100 nM rapamycin for 24 and 48 h confirmed efficient loss of the CRK3 gene (Fig. 2C) by the amplification of a single 1.36 kb DNA fragment for both rapamycin‐treated clones. The retention of the 3.4 kb amplicon containing the CRK3 gene in both untreated clones is evidence that no background diCre activity can be detected in the absence of rapamycin. To test for loss of the CRK3‐GFP protein, total protein extracts of clone 2 promastigotes grown for 96 h in the presence or absence of 100 nM rapamycin were analysed by Western blot analysis with anti‐GFP antibody (Fig. 2D) Very low levels of protein were detected in the treated promastigotes compared to the untreated cells, confirming that the conditional gene loss leads to reduced protein levels. Treatment with 100 nM rapamycin did not result in any noticeable effect on L. mexicana promastigote growth (Fig. S1B), however the pronounced growth arrest arising from loss of the essential gene could possibly result in cellular stress that synergizes with rapamycin. These data show that this is a viable genetic manipulation strategy and that loss of CRK3 resulted in growth arrest and reduced cell numbers, both phenotypes consistent with loss of an essential gene.
Cell cycle analysis of CRK3‐deficient promastigotes
Previous attempts to impair CRK3 function in Leishmania by treatment with protein kinase inhibitors may have resulted in off‐target effects (Cleghorn et al., 2011; Efstathiou et al., 2014; Grant et al., 2004; Jorda et al., 2011; Reichwald et al., 2008; Řezníčková et al., 2015). Here the utilization of diCre mediated gene deletion enabled the effect of CRK3 depletion on the cell cycle to be investigated. Firstly, microscopic analysis of the cells at 96 h post‐induction showed an accumulation of large, aberrant cells with altered organelle homeostasis as evidenced by the presence of cells with multiple flagella (Fig. 3A). DAPI labelling of such multi‐flagellated cells to visualize cellular DNA revealed the presence of enlarged nuclei indicative of an arrest in mitosis. Interestingly, cells were also observed that lacked a nucleus but retained the kinetoplast (‘zoids’), a cell cycle defect observed previously by the treatment of promastigotes with CDK inhibitors (Grant et al., 2004). Secondly, flow cytometry was performed to determine the overall DNA content of Δcrk3::DICRE/Δcrk3::CRK3 Flox promastigotes grown in the presence or absence of 100 nM rapamycin for 72 and 96 h (Fig. 3B). This analysis showed that conditional deletion of CRK3 resulted in the accumulation of cells with 4C DNA content, associated with cell cycle arrest at G2/M, whilst an increasing population of cells with DNA content <1C indicates the accumulation of zoids. Finally, to assess the rate of cell death occurring in CRK3‐deficient cells a viability assay was performed on promastigotes after growth in the presence or absence of 100 nM rapamycin for 72 h (Figs. 3C and S4). After 72 h the proportion of propidium iodide positive cells (PI+) was around 40% indicating a high level of cell death, which likely resulted from the accumulation of anucleated zoids at this time point. Flow cytometry analysis of cell size (using forward scatter) was in agreement with the microscopy analysis and showed that CRK3 deficient cells were substantially larger than cells retaining the gene (Fig. S4). Taken together, these data provide evidence that CRK3 plays an essential role in regulating mitosis in replicating promastigotes.
Figure 3.
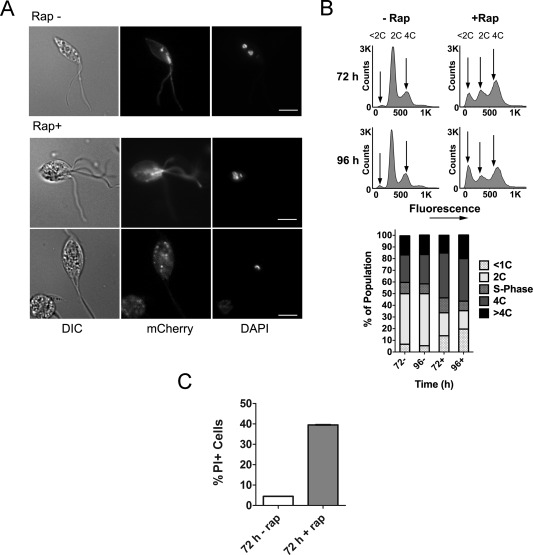
Analysis of CRK3 deficient promastigotes.
A. Representative images of cells grown in the absence (top) or presence (bottom two rows) of 100 nM rapamycin for 96 h. Promastigotes (clone 2) were stained with DAPI to observe nuclear and kinetoplast content alongside mCherry expression by fluorescence microscopy. Scale bar represents 5 μm.
B. (upper) DNA content analysis of clone 2 promastigotes at 72 and 96 h post‐treatment. Cells were fixed with methanol and stained with propidium iodide for flow cytometry analysis of 100,000 cells to examine nuclear content. Arrows indicate the positions of cells in G1 phase (2C), in G2/M (4C) and low DNA content associated with increased incidence of <1C zoids. (lower) Graphical representation of the DNA content of each population based on the flow cytometry plots.
C. The viability of cells grown in the absence (−) or presence (+) of 100 nM rapamycin for 72 h. Promastigotes (clone 2) were incubated with 5 µg mL−1 propidium iodide (PI) for 15 min and analyzed by flow cytometry. A heat lysed (HL) control in which half the sample was lysed by incubation at 70°C for 3 min was included to enable an appropriate live/dead gate to be drawn. Numbers represent the percentage of cells assessed as PI positive (PI+) based on the HL control. Data shown are the means of three technical replicates, data are representative of two independent experiments.
Active CRK3 is required for cell cycle progression in promastigotes
We demonstrated that diCre could be used to efficiently delete a floxed copy of CRK3, so we exploited the efficiency of this system to further study gene function through complementation. Such a system was established by expressing a histidine‐tagged CRK3 (CRK3his) (Hassan et al., 2001) transgene in Δcrk3:DICRE/Δcrk3::CRK3 Flox promastigotes. No significant difference in growth was noted in the presence or absence of rapamycin over a 5 day period (Fig. 4A). Efficient excision of floxed CRK3 in the induced culture was confirmed by PCR amplification of the diagnostic 1.36 kb fragment by 24 h post‐treatment with 100 nM rapamycin (Fig. 4B). The proliferation of promastigotes, despite loss of floxed CRK3, indicates CRK3 transgene complementation in the induced Δcrk3 cell line. Previous studies have shown that recombinant L. mexicana CRK3T178E protein lacks H1 kinase activity (Gomes et al., 2010) and an L. major CRK3 T178E mutant fails to complement a cdc2‐33(ts) yeast mutant (Wang et al., 1998). To test whether active CRK3 is required for cell growth, we exploited this complementation approach by generation of the cell line Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] expressing a T‐loop residue mutated version of CRK3 from the ribosomal locus. Growth curves indicate that expression of the CRK3 T178E transgene failed to complement the loss of CRK3 Flox following induction with rapamycin (Fig. 4A and B) thereby demonstrating that CRK3T178E cannot rescue loss of active CRK3. The overall growth rate of both complementation mutants was reduced relative to the parental line (Table 1) and may explain the growth arrest at 72 h following excision of CRK3 in Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] compared with a more rapid onset of growth arrest in the parental line (Fig. 2B). These data show that active CRK3 is required for parasite growth. The CRK3 deficient cells were analyzed by flow cytometry and fluorescence microscopy showing that Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] cells were blocked in G2/M (Fig. 4C) and were multi‐nucleate and aberrant (Fig. 4D). These data are in agreement with the phenotype observed following excision of CRK3 in wild‐type cells (Fig. 3A and B), thereby indicating the importance of the T‐loop in regulating CRK3 activity. Based on these results, we conclude that transgene complementation can be used to confirm the specificity of conditional deletion of essential genes and also to probe the function of genes following mutagenesis.
Figure 4.
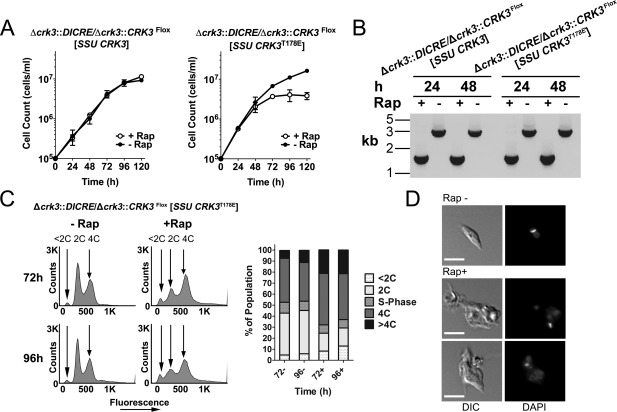
CRK3 wild type and active site mutant complementation assays.
A. Wild type complemented (Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3], left graph) and mutant complemented (Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E], right graph) cell lines were seeded as promastigotes at 1 × 105 cells mL−1 and grown +/− 100 nM rapamycin for 5 days. Cell density was determined by counting at 24 h intervals and the mean ± SD of triplicate values was plotted.
B. The resulting amplicons generated by PCR amplification of each cell line at 24 and 48 h after growth +/− 100 nM rapamycin.
C. (left) DNA content analysis of Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] promastigotes after methanol fixation and staining with propidium iodide for flow cytometry analysis (100,000 cells) to examine nuclear content. Arrows indicate the positions of cells in G1 phase (2C), in G2 (4C) and low DNA content associated with increased incidence of <2C zoids. (right) Graphical representation of the DNA content of each population based on the flow cytometry analysis.
D. Representative images of Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] promastigotes grown in the absence (top) or presence (bottom two rows) of 100 nM rapamycin for 96 h. Parasites were stained with DAPI to detect nuclear and kinetoplast DNA by fluorescence microscopy. Scale bar represents 5 μm.
Table 1.
Comparisons of the growth rates of conditional CRK3 deletion lines measured during logarithmic growth.
| Experimental Line | Doubling Time (hrs) |
|---|---|
| Δcrk3::DICRE/Δcrk3::CRK3 Flox | 10.6 |
| Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3] | 15.5 |
| Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T78E] | 13.6 |
CRK3 is essential for in vivo infection of murine hosts
The lack of a conditional system to regulate expression of essential genes is a major obstacle for in vivo studies of essentiality, with such studies having crucial applications for drug target validation. To address this we tested if CRK3 activity is essential for survival of the parasite over the course of in vivo infection. Monitoring infection by detection of the light signal emitted from bioluminescent Leishmania using an in vivo imaging system (IVIS) is an established, longitudinal and noninvasive method to correlate signal with pathogen load (Lang et al., 2005; Lecoeur et al., 2007; Talmi‐Frank et al., 2012; Vasquez et al., 2015). To assess the outcome of CRK3 loss on the proliferation of L. mexicana in vivo, bioluminescent lines were generated by transfection of L. mexicana wild‐type and Δcrk3::DICRE/Δcrk3::CRK3 Flox promastigotes with a ribosomal integration construct encoding red‐shifted firefly luciferase, Ppy RE9H (Branchini et al., 2010; McLatchie et al., 2013). Both lines were bioluminescent as determined by luciferase expression assays on logarithmic stage promastigotes. The resulting Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] cell line produced five fold higher bioluminescence compared with the wild‐type [SSU RE9H] control (Fig. S4). Footpad bioluminescence detected with an in vivo imaging system (IVIS) correlated well with parasite burden in mice infected with L. mexicana expressing Ppy RE9H (Fig. 5A; y = 4.8 + 0.43x, R 2 = 0.743 and p < 0.0001). The slope of the linear regression line (0.43) revealed smaller increases in bioluminescence with increasing parasite burden. This may be related to tissue absorbance of light in vivo or limited substrate availability with increasing numbers of amastigotes within the lesion. Nevertheless, these data show that parasite burdens can be predicted from bioluminescence and that IVIS could be used for the non‐invasive monitoring of parasite growth in mice over 10 weeks of infection. Following treatment of Δcrk3::DICRE/Δcrk3::CRK3 Flox stationary phase promastigotes with rapamycin for 24 h the amplification of a 1.36 kb fragment (Fig. 5B) indicated that the majority of parasites had successfully excised floxed CRK3. The presence of small amounts of a 3.4 kb amplicon corresponding to the intact floxed CRK3 gene, however, also suggested that some parasites had retained the gene. These stationary phase Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] promastigotes either rapamycin treated (+Rap) or not treated (−Rap) were then inoculated into the footpads of BALB/c mice. The in vivo bioluminescence in footpads of mice infected with the rapamycin‐treated Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] was significantly reduced compared to the uninduced control by 5 weeks post‐infection (p < 0.001) and this continued up to 9 weeks post‐infection (p < 0.005) (Fig. 5C and D). From 5 to 9 weeks the bioluminescence from footpads infected with rapamycin‐treated parasites increased 100 fold and was likely because of the proliferation of parasites that had not responded to rapamycin treatment and persisted in the lesion. To investigate this possibility, viable amastigotes were purified from the lesions of four mice at 10 weeks post‐infection and analyzed for the presence of CRK3 Flox by PCR after a single round of in vitro culture (Fig. 5E). A 3.4 kb PCR product containing CRK3 was amplified from all samples, indicating the persistence of parasites that had escaped diCre mediated excision of CRK3.
Figure 5.
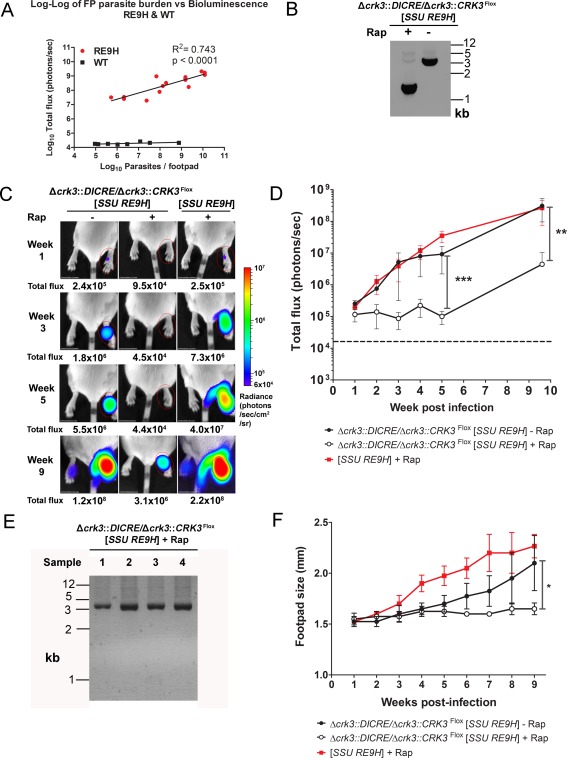
CRK3 conditional deletion in stationary phase promastigotes and in vivo infection.
A. Correlation between in vivo bioluminescence (total flux in photons per second) and parasite burdens from the same infected footpads. BALB/c mice were infected with L. mexicana WT or Ppy RE9H‐expressing stationary phase promastigotes and imaged weekly using an in vivo imaging system (IVIS). At 2, 4, 6 and 8 weeks post‐infection mice were sacrificed after imaging and parasite burdens in infected footpads determined using limiting dilution assays. Each point shows the total flux and parasite burden from the footpad in one mouse (n = 3–4 mice per time point). Linear regression line and R2 was calculated from the log transformed data.
B. PCR amplification of the floxed CRK3 locus of Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] stationary phase promastigotes after incubation in the presence (+) or absence (−) of 1 μM rapamycin for 24 h.
C. Control (−) or 24 h rapamycin‐treated (+) stationary phase promastigotes were inoculated into the footpads of BALB/c mice. The total flux (photons/s) emitted from the infected footpad region of interest (ROI) was quantified weekly.
D. The total flux measured from infected footpads was plotted over 9 weeks of infection. Data shown represent the mean flux and SD from groups of four mice. The dotted line indicates the average background flux emitted from uninfected footpads measured 1 week post infection (n = 12). A significant difference in the mean total flux emitted between the footpads of mice infected with untreated and rapamycin‐induced parasites was observed at 5 and 9 weeks post‐infection (2‐way ANOVA, ***p ≤ 0.001; **p ≤ 0.005).
E. PCR amplification of the floxed CRK3 locus of Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] + Rap after purification of amastigotes from the footpads of 10‐week infected mice. Cells were propagated in vitro to obtain sufficient genomic DNA for PCR analysis.
F. Footpad sizes were recorded by weekly caliper measurement. Data shown represent the mean footpad size and SD from groups of four mice (Unpaired t‐test *p ≤ 0.05).
The ability of CRK3 deficient promastigotes to establish infection was further assessed by measuring footpad sizes at weekly intervals (Fig. 5F). The footpad sizes of mice infected with either untreated or rapamycin‐treated Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] parasites were similarly low until about 4 weeks post‐infection. Subsequently, footpads containing untreated parasites increased steadily over the course of infection, whilst those infected with rapamycin‐treated Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] remained low until 9 weeks post‐infection. Comparison of the bioluminescence and lesion sizes suggest that there is a delay in lesion development despite parasite proliferation and that the lesions only increase significantly when parasite load reaches a certain level (equating to bioluminescence ≈107 photons/s); in the case of the untreated parasites this occurred from about 5 weeks while for rapamycin‐treated parasites this level of parasite burden had still not been reached by 9 weeks. Altogether these data show that loss of active CRK3 impairs the establishment of infection in vivo, and that a later resurgence of parasites likely results from a small population of cells which previously escaped CRK3 conditional deletion.
Discussion
We have developed an inducible system for the genetic manipulation of essential genes in Leishmania. Inducible diCre was used to demonstrate the requirement for CRK3 activity in the regulation of mitosis. A distinct growth defect was observed 48 h after induced deletion of CRK3 (Fig. 2) resulting in cells arrested in G2/M, as well as an accumulation of zoids and eventually a population of enlarged, multi‐flagellated cells (Fig. 3). This phenotype was rescued by expression of a CRK3 transgene from the ribosomal locus, confirming that loss of CRK3 caused mitotic arrest (Fig. 4). Arrest in G2/M and the accumulation of zoids have previously been reported following incubation of L. mexicana promastigotes with the CRK3 inhibitors flavopiridol (Hassan et al., 2001) and indirubin (Grant et al., 2004), showing correlation between genetic and chemical downregulation of CRK3 activity. In Trypanosoma brucei RNAi knockdown of the syntenic orthologue of CRK3 in the procyclic form also results in G2/M arrest and zoid formation (Tu and Wang, 2004), with the accumulation of such aberrant cells explained by the lack of a checkpoint controlling exit from mitosis and entry in cytokinesis (Hammarton et al., 2003; Ploubidou et al., 1999). Inducible deletion of CRK3 indicates that this checkpoint is also absent in L. mexicana promastigotes, resulting in impairment of mitotic progression, followed by re‐initiation of G1 in the absence of cytokinesis. It appears that these abnormal cells can eventually undergo cytokinesis; however the daughter cell lacks a nucleus and is often multi‐flagellated (see bi‐flagellated zoid in Fig. 3A), whilst the high levels of cell death occurring 72 h after gene loss show that such progeny are not viable.
CRK3 is active at different stages in the cell cycle by forming complexes with cyclin partners such as CYC6 and CYCA, therefore CRK3 deletion could impact the cell cycle at multiple stages. RNAi of the CYC6 in T. brucei procyclic forms results in growth arrest within 48 h of induction and the accumulation of zoids and cells in G2/M (Hammarton et al., 2003). A similar phenotype was found in this study with CRK3 inducible deletion, suggesting that the CRK3:CYC6 complex is involved in regulation of mitosis (Walker et al., 2011). Less is known about the activity of CRK3:CYCA. Protein expression assays of L. donovani CYC1 (the functional orthologue of CYCA) demonstrates an increased abundance during S‐phase (Banerjee et al., 2006) coupled with histone phosphorylation by an active CRK3:CYC1 complex (Maity et al., 2011), which is suggestive of S‐phase kinase activity. Active, recombinant L. mexicana CRK3:CYCA has also been engineered, with phosphorylation of the T‐loop residue T178 by the CDK activating kinase (CAK) Civ‐1 increasing activity (Gomes et al., 2010). The T178 residue is essential for CRK3 activity as T178E mutagenesis inhibits functional rescue in S. pombe (Wang, 1998) and ablates kinase activity in recombinant CRK3T178E:CYCA (Gomes et al., 2010). The necessity of T178 was tested directly in this study, with excision of floxed CRK3 in the Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] line leading to cell cycle arrest in G2/M and zoid formation. The growth rate of this line and Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3] were reduced when compared to Δcrk3::DICRE/Δcrk3::CRK3 Flox (Table 1), indicative of generally reduced growth rate when expressing a transgene. Episomal complementation with CRK3 did not result in an observable growth defect (Hassan et al., 2001), but this may result from the modulation of the number of episomal copies, as has been observed previously following complementation of the essential MCA gene (Ambit et al., 2008). Integration into the 18s rRNA locus results in consistently high levels of expression (Misslitz et al., 2000) leading to nonphysiological levels of CRK3 and subsequent CRK3:CYC6 activity at potentially inappropriate stages of the life cycle.
The reduced growth rate of promastigotes overexpressing CRK3T178E is likely because of a partial dominant negative effect, whereby inactive CRK3T178E binds endogenous CYC6 leading to impaired protein kinase activity even in the presence of active CRK3. This reduced growth rate may explain both the cell cycle arrest at 72 h in the [SSU CRK3 T178E] complemented line (Fig. 4A) compared to arrest at 48 h in Δcrk3::DICRE/Δcrk3::CRK3 Flox (Fig. 2B) and additionally the lower proportion of zoids when analyzed by flow cytometry (Fig. 4C). The accumulation in G2/M suggests that mutation ablates CRK3:CYC6 activity, rather than CRK3:CYCA, where an increase of cells in G1/S might be anticipated. Both induced and uninduced Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU CRK3 T178E] have dramatically reduced flagellum length and are immotile (Fig. 4D). The reduced size of the flagellum and a growth defect are similar phenotypes to those observed in cell lines deficient in ATG5, a key component of the autophagic pathway (Williams et al., 2012). This is likely a result of their impaired ability to salvage material through the autophagic pathway, imparting selection on the parasites to reduce energy through flagellum regression. The partial dominant negative effect of CRK3T178E may also result in metabolic stress in these cells leading to the phenotype observed. The importance of T178 as an active site residue for regulating progression through G2/M implicates upstream modifiers of this residue as essential regulators of the L. mexicana cell cycle. In mammalian cells CDK7 acts as a CAK to regulate CDK1 by phosphorylation at this T‐loop residue, yet no CAK homologues have been identified in the Leishmania genome (Gomes et al., 2010). The identification of potential post‐transcriptional modifiers of the CRK3 T‐loop residue that act in an analogous fashion to CDK7 would therefore yield promising targets for drug discovery. The phenotype of the induced cell line shows the importance of the T‐loop residue for CRK3 activity and mitotic function within the cell, endorsing this complementation assay as a rational approach for active site investigation.
The assessment of gene essentiality for amastigote viability is an important approach in the context of drug target validation as this life cycle stage is the pathologically significant form. The recent utilization of plasmid shuffle has facilitated the study of Leishmania genes involved in life cycle differentiation and essentiality both in amastigote and promastigote forms by the generation of partial null mutants (Dacher et al., 2014; Morales et al., 2010). Retention of an episomal gene in a null mutant cell line after murine infection is a useful approach to assess that gene as necessary to amastigote survival in vivo (Wiese, 1998). Despite such elegant utilization of reverse genetic methods to probe gene function, no method exists for the generation of conditional null mutants during in vivo infection. Our study does not address this lack directly because of the sensitivity of amastigotes to rapamycin, however as diCre activity remains high in stationary‐phase promastigotes CRK3 was efficiently excised (Fig. 5B) to probe the subsequent infectivity of CRK3‐deficient promastigotes. By tracking the progression of infection with reporter parasites expressing the highly sensitive red‐shifted luciferase (Branchini et al., 2010; McLatchie et al., 2013) and by footpad size measurement, we demonstrate that the CRK3‐deficient L. mexicana are unable to proliferate in their mammalian host (Figs. 5C, D, and F). Importantly, the wild‐type line expressing luciferase grows normally in mice following rapamycin treatment, which indicates that lack of growth of the CRK3‐deficient mutant is not a result of the drug treatment. The average light intensities emitted from footpads infected with the wild‐type [SSU RE9H) line and those from footpads infected with the Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] line retaining floxed CRK3 are at similar levels throughout infection, yet mean footpad size is larger in wild‐type [SSU RE9H] infected mice after 3 weeks post‐infection; such disagreement may be a result of the five fold lower signal intensity of the wild‐type [SSU RE9H] compared with the experimental line (Fig. S5) and therefore an overall higher burden of the wild‐type line is likely masked by a reduced bioluminescent signal intensity.
Interestingly, parasite burden as measured by total flux remains consistently above the background intensity (dashed line, Fig. 5D) in those footpads infected with the CRK3‐deficient line, suggestive of the survival of a low number of bioluminescent parasites. The outgrowth of these parasites was observed through an increased bioluminescence signal at 9 weeks post‐infection compared with 5 weeks (Fig. 5C and D). Purification and PCR analysis of these parasites show they retained the floxed CRK3 (Fig. 5E) and that the persistence of signal and subsequent increase are a result of incomplete excision of floxed CRK3 during the 24 h incubation with rapamycin. These data further demonstrate the essentiality of CRK3 activity for establishing infection.
This is the first time an essential gene in promastigotes has been studied in vivo by conditional deletion, representing a useful tool to probe gene function. We are validating the feasibility of conditional gene deletion ex vivo and in vivo using rapamycin and nonimmuno‐inhibitory rapamycin analogues (‘rapalogs’), with such work being useful for the future of drug target validation. DiCre activity has been demonstrated in vivo (Jullien et al., 2007), however rapamycin treatment may be a limitation because of influence on the host immune response and on amastigote proliferation. Our attempts to study the effect of CRK3 Flox deletion in lesion‐derived amastigotes grown in axenic culture medium was problematic because of reduced proliferation of both experimental and wild‐type L. mexicana at the relatively low dose of 50 nM rapamycin, therefore the use of rapalogs would be a rational approach for induction of diCre activity if they have reduced binding affinity for Leishmania TORs (Madeira da Silva and Beverley, 2010). A second generation diCre is currently in development and may present an alternative method for inducible gene deletion in vivo. In diCre2, each subunit is fused to mutant FKBP domains that are dimerised by the rapalog AP20187, which is amenable to in vivo use (Collins et al., 2013). Such a system could be applied for use in Leishmania and would complement our existing floxed gene replacement approach.
In conclusion we have developed a highly efficient inducible gene deletion system that when used with transgene complementation allows for the first time the function of essential Leishmania genes to be elucidated. We have applied this approach to show that CRK3 is required for promastigote progression through mitosis, with gene deletion mutants showing a G2/M arrest and an accumulation of zoids, indicative of a lack of a cell cycle checkpoint in cytokinesis. Inducible deletion of CRK3 in stationary phase promastigotes attenuates infection in a murine host, providing further genetic validation of CRK3 as a potential drug target (Gomes et al., 2010; Grant et al., 1998; 2004; Hassan et al., 2001; Walker et al., 2011). Our diCre method provides a powerful tool for analysing genes essential for promastigote proliferation and to the study of the differentiation of promastigotes to amastigotes.
Experimental procedures
Ethics statement
Animal studies were carried out under UK Home Office regulations (Project licence PPL 60/4442).
Parasite culture and transfection
Leishmania mexicana mexicana (MNYC/BZ/62/M379) promastigotes were cultured at 25°C in HOMEM supplemented with 10% heat inactivated foetal calf serum (HI‐FCS) and 1% penicillin/streptomycin (PEN/STREP). Amastigotes were cultured in Schneider's Insect Medium supplemented with 20% HI‐FCS, 1% PEN/STREP and 15 μg mL−1 Hemin at pH5.5. Mid‐log phase L. mexicana promastigotes were transfected with 10 μg of digested DNA by electroporation using the Nucleofector system with the Human T‐Cell kit (Lonza) as described previously (Castanys‐Muñoz et al., 2012). Transgenic cell lines were grown in the presence of appropriate antibiotics at the following concentrations: G418 50 μg mL−1, blasticidin 10 μg mL−1 and puromycin 10 μg mL−1 (InvivoGen).
Construct design and development
A full list and descriptions of all primers (Table S1) and plasmids (Table S2) used in this study are available. To produce a diCre expression vector, the diCre coding sequences Cre59‐FKBP12 and Cre60‐FRB were each flanked by actin and β‐tubulin sequences in array with blasticidin resistance cassette flanked by DHFR‐TS regulatory elements. The sequence was synthesized and sub‐cloned into the pDONR221 vector (GenScript). The backbone of the loxP vector containing the loxP sites flanking a multiple cloning site and other restriction enzyme regions flanked by regulatory elements was synthesized (GenScript). The PAC, mCherry and CRK3‐GFP cassettes were inserted by enzymatic restriction digest mediated ligation, and subsequently sub‐cloned into pDONR221. Addition of CRK3 homology flanking homology was performed by MultiSite Gateway 3‐fragment vector construction (Invitrogen) as per manufacturers’ guidelines. Briefly, flanks were amplified by PCR by Phusion polymerase (New England BioLabs) using oligonucleotides conferring attB recombination sites to the amplicons. Subsequent BP reactions inserted the flanks into appropriate pDONR vectors containing attL sites for site‐specific recombination. An LR reaction resulted in the flanking of diCre and loxP vectors into a pDEST vector for transfection. Finally, complementation plasmids were generated by inserting the CRK3, CRK3 T178E and RE9H genes (Branchini et al., 2010; McLatchie et al., 2013) into a modified version of pGL631 (Misslitz et al., 2000) containing a G418r cassette for SSU integration construct by XhoI & NotI restriction enzyme digestion and ligation.
Induction of diCre mediated gene deletion
All experiments were conducted using cells in the early to mid log stage of exponential growth (between 1 and 5 × 106 cells mL−1) with the exception of the stationary phase inducible gene deletion. Between 1 nM and 1 μM rapamycin (Abcam) was administered by inoculation into the cell culture medium from a 100 μM working stock.
Conditional gene deletion analysis
Taq polymerase (NEB) was used to PCR amplify the regions surrounding GFP Flox and CRK3 Flox using primers shown in Table S1 and a T A calculated using an online T m calculator (New England BioLabs) and 30 cycles for amplification.
Western blot analysis
For western blotting analysis, either 1 × 107 cells were loaded per lane or equal concentrations of protein extract as quantified by Bradford assay of a 10% NuPAGE Bis‐Tris gel (Invitrogen) in MOPS running buffer and transferred onto Hybond‐C nitrocellulose membranes (GE Healthcare). Primary antibodies against GFP were used to detect GFP and CRK3‐GFP expression at 1:1000 whilst anti‐EF1α was used as a loading control at 1:5000. Membranes were washed three times in TBST, incubating for 10 min each time, before incubation with horse radish peroxidase (HRP)‐conjugated secondary rabbit and mouse antibodies at 1:5000 dilution for 1 h at room temperature. After washing three times in TBST, the membrane was treated with an ECL (enhanced chemiluminescence) kit (SuperSignal West Pico Chemoluminescent Substrate, Pierce) according to manufacturer's instructions and then exposed on Kodak photographic film.
Infection of mice
BALB/c mice were purchased from Charles River (MA., USA) and infected in the right footpad with 2 × 106 stationary‐phase L. mexicana promastigotes in 1 × PBS. Lesion size was monitored weekly and Δcrk3::DICRE/CRK3 [SSU GFP Flox] amastigotes were purified before the lesions reached a thickness of 5 mm.
Purification of lesion‐derived amastigotes
Lesion‐derived Δcrk3::DICRE/CRK3 [SSU GFP Flox] amastigotes were purified by homogenizing the extracted lesion in 1xPBS and passing the solution through a 20 μm cell strainer. Amastigotes were pelleted by centrifugation at 2000 g for 10 mins, followed by re‐suspension in culture medium. To prevent cells from clumping together and ensure accurate cell counting, amastigote cultures were first centrifuged at 2000 g for 10 mins and the supernatant removed to leave the pellet in 500 μL volume. The pellet was re‐suspended in this volume by gentle syringing through a blunt 16G needle and the single cell suspension added back to the culture medium. Cell counting was performed by mixing the homogenized culture 1:1 with Trypan blue and cell counting with a Haemocytometer (Neubauer).
Macrophage differentiation and amastigote infection
Nondifferentiated monocytes were extracted from the femurs and tibia of BALB/c mice by dissection to remove the bones. RPMI 1640 medium was used to wash the bone marrow out of the intact bones by syringing with a 25G needle. Extracted cells were quantified by dilution in Trypan blue (1:1) and counting with a haemocytometer. Monocytes were seeded at 5 × 105 cells mL−1 in MΦ Medium (DMEM + L‐Glut + 20%FCS + 1% P/S + 30% L‐Cell M) in 8 mL volumes in Petri dishes and incubated at 37°C with 5% CO2 for 3 days to induce differentiation to monocyte‐derived macrophage. After this period the medium was replaced and by day 5 the cells were removed from the dishes using a cell scraper with ice‐ cold RPMI 1640. Bone‐marrow derived macrophages were adhered at a concentration of 5 × 105 cells mL−1 overnight in DMEM medium with 10% HIFCS at 37°C in 5% CO2 onto 8‐chamber tissue culture slides (LAB‐TEK) for microscopic analysis or 12 well plates for DNA extraction and flow cytometry analysis. Macrophages were then infected at a ratio of five parasites per macrophage with lesion‐derived Δcrk3::DICRE/CRK3 [SSU GFP Flox] amastigotes, which had been previously grown in axenic medium in the presence or absence of rapamycin for 24 h. Wells were washed at 24 h post‐infection to remove extracellular parasites and media replenished with DMEM/10% HIFCS. Cells were removed from the plates for DNA extraction and flow cytometry analysis by gentle scraping in ice cold RPMI at the 120 h end time point.
Fluorescence microscopy analysis
For imaging, 2 × 106 parasites were washed in 1 × PBS, re‐suspended in Fluoromount‐G (SouthernBiotech) DAPI infused mounting medium and mounted on glass slides for analysis. Parasite morphology was observed by DIC and mCherry fluorescent imaging, and DNA content observed by DAPI fluorescent imaging using a Delta Vision core (Image Solutions) inverted microscope equipped with mCherry and DAPI filter sets. Images were processed using Photoshop CS (Adobe) image software. GFP expression of intracellular amastigotes was assessed by fluorescent microscopy. Cells were imaged between 24 and 120 h after infection in the DeltaVision Core environmental chamber at 37°C and 5% CO2 upon incubation in 1 × PBS infused with DAPI.
DNA content and GFP expression analysis by flow cytometry
Parasites were prepared for DNA content analysis as described previously (Paul Hassan et al., 2001) with the exceptions that a MacsQuant flow cytometer was used to analyze 100,000 cells per sample. Cell distribution was modelled using FlowJo software (Tree Star). For determining GFP expression of promastigotes and amastigotes by flow cytometry analysis, live cells were washed twice in 1xPBS and passed through a nitex mesh prior to acquisition.
Viability assay
Log‐phase promastigotes were seeded at 5 × 105 cells mL−1 and grown in the presence or absence of 100 nM rapamycin. At 72 h post‐treatment 1 × 107 cells were washed once with 1 × PBS and incubated with 5 ug mL−1 propidium iodide (PI) for 15 minutes at room temperature in the dark. A heat lysed (HL) control in which half the sample was lysed by incubation at 70°C for 3 min was included to enable an appropriate live/dead gate to be drawn. Cells were washed with 1 × PBS and used to acquire 100,000 events per group by flow cytometry using a MacsQuant flow cytometer.
In vivo imaging
For imaging, mice were anaesthetized with 4.0% isofluorane/1.5 L O2 per minute and inoculated by subcutaneous injection with 200 μL of D‐luciferin (15 mg mL−1 in Mg/Ca‐free Dulbecco's modified PBS). Light emission was recorded 10 min after inoculation using an IVIS Spectrum bioluminescence imaging system (PerkinElmer). Imaging was performed with an open emission filter, for 30–60 s exposures, large binning, and 1 f/stop, and captured with a charge‐coupled device (CCD) camera. The absolute unit of photon emission was given as radiance (photons/second/cm2/steradian). Images were analysed using Living Image Software (PerkinElmer) and regions of interest (ROI) of equal size were selected over the infected footpads to quantify the amount of photon emission as total photon flux in photons per second (photons/s).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5. The analysis of significance of the data was performed by 2‐way ANOVA when comparing data from induced (+Rap) and uninduced (−Rap) Δcrk3::DICRE/Δcrk3::CRK3 Flox [SSU RE9H] infections and by paired t‐test when comparing footpad sizes.
Supporting information
Supporting Information
Acknowledgements
We thank Jim Scott and Alana Hamilton for technical support, Ryan Ritchie for imaging assistance, and Bruce Branchini and colleagues (Department of Chemistry, Connecticut College) for the Ppy‐RE9H. SD was supported by a Medical Research Council studentship. This work was supported by the Medical Research Council grant (MR/K019384) and the Wellcome Trust (104976, 104111).
The authors declare no conflict of interest.
References
- Alvar, J. , Vélez, I.D. , Bern, C. , Herrero, M. , Desjeux, P. , Cano, J. , et al (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7: e35671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambit, A. , Fasel, N. , Coombs, G.H. , and Mottram, J.C. (2008) An essential role for the Leishmania major metacaspase in cell cycle progression. Cell Death Differ 15: 113–122. [DOI] [PubMed] [Google Scholar]
- Andenmatten, N. , Egarter, S. , Jackson, A.J. , Jullien, N. , Herman, J.‐P. , and Meissner, M. (2013) Conditional genome engineering in Toxoplasma gondii uncovers alternative invasion mechanisms. Nat Methods 10: 125–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, S. , Sen, A. , Das, P. , and Saha, P. (2006) Leishmania donovani cyclin 1 (LdCyc1) forms a complex with cell cycle kinase subunit CRK3 (LdCRK3) and is possibly involved in S‐phase‐related activities. FEMS Microbiol Lett 256: 75–82. [DOI] [PubMed] [Google Scholar]
- Branchini, B.R. , Ablamsky, D.M. , Davis, A.L. , Southworth, T.L. , Butler, B. , Fan, F. , et al (2010) Red‐emitting luciferases for bioluminescence reporter and imaging applications. Anal Biochem 396: 290–297. [DOI] [PubMed] [Google Scholar]
- Castanys‐Muñoz, E. , Brown, E. , Coombs, G.H. , and Mottram, J.C. (2012) Leishmania mexicana metacaspase is a negative regulator of amastigote proliferation in mammalian cells. Cell Death Dis 3: e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicenas, J. , and Valius, M. (2011) The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol 137: 1409–1418. [DOI] [PubMed] [Google Scholar]
- Cleghorn, L.A.T. , Woodland, A. , Collie, I.T. , Torrie, L.S. , Norcross, N. , Luksch, T. , et al (2011) Identification of inhibitors of the Leishmania cdc2‐related protein kinase CRK3. ChemMedChem 6: 2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, C.R. , Das, S. , Wong, E.H. , Andenmatten, N. , Stallmach, R. , Hackett, F. , et al (2013) Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol Microbiol 88: 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacher, M. , Morales, M.A. , Pescher, P. , Leclercq, O. , Rachidi, N. , Prina, E. , et al (2014) Probing druggability and biological function of essential proteins in Leishmania combining facilitated null mutant and plasmid shuffle analyses. Mol Microbiol 93: 146–166. [DOI] [PubMed] [Google Scholar]
- Efstathiou, A. , Gaboriaud‐Kolar, N. , Smirlis, D. , Myrianthopoulos, V. , Vougogiannopoulou, K. , Alexandratos, A. , et al (2014) An inhibitor‐driven study for enhancing the selectivity of indirubin derivatives towards leishmanial Glycogen Synthase Kinase‐3 over leishmanial cdc2‐related protein kinase 3. Parasit Vectors 7: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes, F.C. , Ali, N.O.M. , Brown, E. , Walker, R.G. , Grant, K.M. , and Mottram, J.C. (2010) Recombinant Leishmania mexicana CRK3:CYCA has protein kinase activity in the absence of phosphorylation on the T‐loop residue Thr178. Mol Biochem Parasitol 171: 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal, S. , Dhanjal, J.K. , Tyagi, C. , Goyal, M. , and Grover, A. (2014) Novel fragment‐based QSAR modeling and combinatorial design of pyrazole‐derived CRK3 inhibitors as potent antileishmanials. Chem Biol Drug Des 84: 54–62. [DOI] [PubMed] [Google Scholar]
- Grant, K.M. , Dunion, M.H. , Yardley, V. , Skaltsounis, A.‐L. , Marko, D. , Eisenbrand, G. , et al (2004) Inhibitors of Leishmania mexicana CRK3 cyclin‐dependent kinase: chemical library screen and antileishmanial activity. Antimicrob Agents Chemother 48: 3033–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, K.M. , Hassan, P. , Anderson, J.S. , and Mottram, J.C. (1998) The crk3 gene of Leishmania mexicana encodes a stage‐regulated cdc2‐related histone H1 kinase that associates with p12cks1. J Biol Chem 273: 10153–10159. [DOI] [PubMed] [Google Scholar]
- Hammarton, T.C. , Clark, J. , Douglas, F. , Boshart, M. , and Mottram, J.C. (2003) Stage‐specific differences in cell cycle control in Trypanosoma brucei revealed by RNA interference of a mitotic cyclin. J Biol Chem 278: 22877–22886. [DOI] [PubMed] [Google Scholar]
- Hassan, P. , Fergusson, D. , Grant, K.M. , and Mottram, J.C. (2001) The CRK3 protein kinase is essential for cell cycle progression of Leishmania mexicana . Mol Biochem Parasitol 113: 189–198. [DOI] [PubMed] [Google Scholar]
- Jorda, R. , Sacerdoti‐Sierra, N. , Voller, J. , Havlíček, L. , Kráčalíková, K. , Nowicki, M.W. , et al (2011) Anti‐leishmanial activity of disubstituted purines and related pyrazolo[4,3‐d]pyrimidines. Bioorganic Med Chem Lett 21: 4233–4237. [DOI] [PubMed] [Google Scholar]
- Jullien, N. , Goddard, I. , Selmi‐Ruby, S. , Fina, J.‐L. , Cremer, H. , and Herman, J.‐P. (2007) Conditional transgenesis using Dimerizable Cre (DiCre). PLoS One 2: e1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien, N. , Sampieri, F. , Enjalbert, A. , and Herman, J. (2003) Regulation of Cre recombinase by ligand‐induced complementation of inactive fragments. Nucleic Acids Res 31: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp, S. , and Sundström, M. (2014) Recently targeted kinases and their inhibitors – the path to clinical trials. Curr Opin Pharmacol 17: 58–63. [DOI] [PubMed] [Google Scholar]
- Lang, T. , Goyard, S. , Lebastard, M. , and Milon, G. (2005) Bioluminescent Leishmania expressing luciferase for rapid and high throughput screening of drugs acting on amastigote‐harbouring macrophages and for quantitative real‐time monitoring of parasitism features in living mice. Cell Microbiol 7: 383–392. [DOI] [PubMed] [Google Scholar]
- Lecoeur, H. , Buffet, P. , Morizot, G. , Goyard, S. , Guigon, G. , Milon, G. , and Lang, T. (2007) Optimization of topical therapy for Leishmania major localized cutaneous leishmaniasis using a reliable C57BL/6 model. PLoS Negl Trop Dis 1: e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira da Silva, L. , and Beverley, S.M. (2010) Expansion of the target of rapamycin (TOR) kinase family and function in Leishmania shows that TOR3 is required for acidocalcisome biogenesis and animal infectivity. Proc Natl Acad Sci U S A 107: 11965–11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity, A.K. , Goswami, A. , and Saha, P. (2011) Identification of substrates of an S‐phase cell cycle kinase from Leishmania donovani . FEBS Lett 585: 2635–2639. [DOI] [PubMed] [Google Scholar]
- McLatchie, A.P. , Burrell‐Saward, H. , Myburgh, E. , Lewis, M.D. , Ward, T.H. , Mottram, J.C. , et al (2013) Highly sensitive In Vivo imaging of Trypanosoma brucei expressing “Red‐Shifted” luciferase. PLoS Negl Trop Dis 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misslitz, A , Mottram, J.C. , Overath, P. , and Aebischer, T. (2000) Targeted integration into a rRNA locus results in uniform and high level expression of transgenes in Leishmania amastigotes. Mol Biochem Parasitol 107: 251–261. [DOI] [PubMed] [Google Scholar]
- Morales, M. A. , Watanabe, R. , Dacher, M. , Chafey, P. , Osorio y Fortéa, J. , Scott, D. a , et al (2010) Phosphoproteome dynamics reveal heat‐shock protein complexes specific to the Leishmania donovani infectious stage. Proc Natl Acad Sci U S A 107: 8381–8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploubidou, A. , Robinson, D.R. , Docherty, R.C. , Ogbadoyi, E.O. , and Gull, K. (1999) Evidence for novel cell cycle checkpoints in trypanosomes: kinetoplast segregation and cytokinesis in the absence of mitosis. J Cell Sci 112 (Pt 2): 4641–4650. [DOI] [PubMed] [Google Scholar]
- Reichwald, C. , Shimony, O. , Dunkel, U. , Sacerdoti‐Sierra, N. , Jaffe, C.L. , and Kunick, C. (2008) 2‐(3‐aryl‐3‐oxopropen‐1‐yl)‐9‐tert‐butyl‐paullones: a new antileishmanial chemotype. J Med Chem 51: 659–665. [DOI] [PubMed] [Google Scholar]
- Řezníčková, E. , Popa, A. , Gucký, T. , Zatloukal, M. , Havlíček, L. , Bazgier, V. , et al (2015) 2,6,9‐Trisubstituted purines as CRK3 kinase inhibitors with antileishmanial activity in vitro. Bioorg Med Chem Lett 25: 2298–2301. [DOI] [PubMed] [Google Scholar]
- Rogers, M.B. , Hilley, J.D. , Dickens, N.J. , Wilkes, J. , Bates, P.A. , Depledge, D.P. , et al (2011) Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania . Genome Res 21: 2129–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmi‐Frank, D. , Jaffe, C.L. , Nasereddin, A. , and Baneth, G. (2012) Leishmania tropica experimental infection in the rat using luciferase‐transfected parasites. Vet Parasitol 187: 57–62. [DOI] [PubMed] [Google Scholar]
- Tu, X. , and Wang, C.C. (2004) The involvement of two cdc2‐related Kinases (CRKs) in Trypanosoma brucei cell cycle regulation and the distinctive stage‐specific phenotypes caused by CRK3 depletion. J Biol Chem 279: 20519–20528. [DOI] [PubMed] [Google Scholar]
- Vasquez, M.A. , Iniguez, E. , Das, U. , Beverley, S.M. , Herrera, L.J. , Dimmock, J.R. , and Maldonado, R.A. (2015) Evaluation of α,β‐unsaturated ketones as antileishmanial agents. Antimicrob Agents Chemother 59: 3598–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, R.G. , Thomson, G. , Malone, K. , Nowicki, M.W. , Brown, E. , Blake, D.G. , et al (2011) High throughput screens yield small molecule inhibitors of Leishmania CRK3:CYC6 cyclin‐dependent kinase. PLoS Negl Trop Dis 5: e1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Melzer, I.M. , Kruse, M. , Sander‐Juelch, C. , and Wiese, M. (2005) LmxMPK4, a mitogen‐activated protein (MAP) kinase homologue essential for promastigotes and amastigotes of Leishmania mexicana . Kinetoplastid Biol Dis 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Dimitrov, K. , Garrity, L.K. , Sazer, S. , and Beverley, S.M. (1998) Stage‐specific activity of the Leishmania major CRK3 kinase and functional rescue of a Schizosaccharomyces pombe cdc2 mutant. Mol Biochem Parasitol 96: 139–150. [DOI] [PubMed] [Google Scholar]
- Wiese, M. (1998) A mitogen‐activated protein (MAP) kinase homologue of Leishmania mexicana is essential for parasite survival in the infected host. EMBO J 17: 2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R.A.M. , Smith, T.K. , Cull, B. , Mottram, J.C. , and Coombs, G.H. (2012) ATG5 is essential for ATG8‐dependent autophagy and mitochondrial homeostasis in Leishmania major . PLoS Pathog 8: e1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information