

Abstract
Immunohistochemistry is a commonly used clinical and research lab detection technique for investigating protein expression and localization within tissues. Many semi-quantitative systems have been developed for scoring expression using immunohistochemistry, but inherent subjectivity limits reproducibility and accuracy of results. Furthermore, the investigation of spatially overlapping biomarkers such as nuclear transcription factors is difficult with current immunohistochemistry techniques. We have developed and optimized a system for simultaneous investigation of multiple proteins using high throughput methods of multiplexed immunohistochemistry and multispectral imaging. Multiplexed immunohistochemistry is performed by sequential application of primary antibodies with secondary antibodies conjugated to horseradish peroxidase or alkaline phosphatase. Different chromogens are used to detect each protein of interest. Stained slides are loaded into an automated slide scanner and a protocol is created for automated image acquisition. A spectral library is created by staining a set of slides with a single chromogen on each. A subset of representative stained images are imported into multispectral imaging software and an algorithm for distinguishing tissue type is created by defining tissue compartments on images. Subcellular compartments are segmented by using hematoxylin counterstain and adjusting the intrinsic algorithm. Thresholding is applied to determine positivity and protein co-localization. The final algorithm is then applied to the entire set of tissues. Resulting data allows the user to evaluate protein expression based on tissue type (ex. epithelia vs. stroma) and subcellular compartment (nucleus vs. cytoplasm vs. plasma membrane). Co-localization analysis allows for investigation of double-positive, double-negative, and single-positive cell types. Combining multispectral imaging with multiplexed immunohistochemistry and automated image acquisition is an objective, high-throughput method for investigation of biomarkers within tissues.
Keywords: Basic Protocol, Issue 110, multispectral imaging, immunohistochemistry, quantitative pathology, co-localization, chromogen, automated pathology
Introduction
Immunohistochemistry (IHC) is a standard lab technique for detection of protein within tissue, and IHC is still widely used in both research and diagnostic pathology. The evaluation of IHC staining is often semi-quantitative, introducing potential bias into interpretation of results. Many semi-quantitative approaches have been developed which incorporate both staining intensity and staining extent into final diagnosis 1-4. Other systems include scoring intensity and subcellular location in order to better localize expression 5. Incorporation of average scores from multiple viewers is often utilized in order to minimize the effects of single viewer bias 6. Despite these efforts, subjectivity in analysis still remains, particularly when evaluating the extent of staining 7. Protocol standardization and minimizing subjectivity from human input is paramount to creating accurate, reproducible IHC results.
There are other options besides IHC for determining protein expression within tissues. Within the research setting, immunohistochemistry has traditionally been viewed as a means to examine protein localization 8, while other techniques such as immunoblotting are viewed as gold standard for investigating protein expression. Determining tissue or cell compartment-specific expression is difficult without incorporating advanced techniques such as cell fractionation or laser capture microdissection 9,10. The use of fluorescent antibodies on tissue slides offers a reasonable compromise, but background autofluorescence due to NADPH, lipofuscins, reticular fibers, collagen, and elastin can make accurate quantitation difficult 11.
Automated computational pathology platforms are a promising direction for more objective quantitation of pathology staining 12-15. Combining multispectral imaging with tissue microarrays facilitates high-throughput analysis of protein expression in large sample sizes. With these techniques, analysis of protein co-localization, staining heterogeneity, and tissue and subcellular localization is possible while substantially reducing both inherent biases and time necessary for analysis, while returning data in a continuous rather than categorical format 16. Therefore, the purpose of this study was to demonstrate the utility of and methodology for performing multiplexed immunohistochemistry with analysis, using multispectral imaging software.
This protocol is written for manual, multiplex immunohistochemical staining of a single tissue section slide with four optimized monoclonal antibodies. As a representative experiment, nuclear anti-rabbit estrogen receptor alpha (ERα) and androgen receptor (AR) are multiplexed with membrane-bound anti-mouse CD147 and membrane-bound anti-mouse E-cadherin. Any antibody of choice may be substituted for the antibodies listed herein, but each combination of antibodies requires separate optimization. Pre-treatment for all the antibodies must be identical. The AR and CD147 antibodies should be optimized individually and then as a cocktail. Each antibody is detected using a biotin-free polymer system and one of 4 unique chromogens.
Protocol
NOTE: The protocol herein describes staining and analysis of a tissue microarray (TMA), described previously 12,17,18. The 4 µm thick TMA section was obtained from a paraffin block using a standard microtome.
NOTE: A spectral library for the 4 chromogens and counterstain should be created for image quantitation. In order to do this, the optimized protocol for each individual antibody should be run with one antibody per slide, minus the final counterstain. A fifth slide should be stained with hematoxylin to generate the 5 images needed to create the spectral library.
1. Multiplex Immunohistochemistry
Bake slides in a 60 °C oven for 20 min. Using a chemical fume hood, immerse slide in 100% xylene. Repeat for 2 changes of xylene. Immerse slide in 100% ethanol for 5 min. Repeat with fresh 100% ethanol.
Immerse slide in 95% ethanol for 3 min. Repeat with fresh 95% ethanol. Immerse slide in 70% ethanol for 3 min. Immerse slide in distilled water for 3 min. Repeat with fresh distilled water. Tap slide vertically to drain water from the slide. Apply 200 µl of ready-to-use endogenous peroxidase blocker for 5 min.
Drain peroxidase blocker from slide and immerse in 45 ml ready-to-use retrieval buffer (pH less than 7.0). Place slide container in a pressure cooker and start program set to 124 °C for 4 min. Allow pressure cooker to cool for 20 min. before opening.
Remove container of retrieval buffer and allow to cool additional 10 min. Rinse slide in distilled water for 10 min. Drain water from the slide and carefully delineate the tissue on the slide with a hydrophobic barrier pen.
Add 200 µl of phosphate buffered saline (PBS).Transfer slide to a flat receptacle with lid (incubation chamber) containing moist gauze.
Prepare 1 liter of 1x Tris-buffered saline with Tween-20 (TBST) by diluting 50 ml of 20x Tris buffered saline containing 1% Tween-20 and 950 ml distilled water.
Drain PBS from slide and apply 200 µl of 1x TBST. Incubate 2 min. Drain TBST from slide and return slide to incubation chamber. Apply 200 µl of ready-to-use universal protein block, close chamber and incubate for 7 min.
Drain protein block and return slide to incubation chamber. Dilute anti-ERα antibody 1:400 by adding 0.6 µl of antibody to 239.4 µl of antibody diluent. Apply 200 µl of diluted antibody to slide and close chamber.
Incubate slide at room temperature for 2 hours. Drain antibody from slide and rinse with TBST followed by immersion for 5 min in TBST.
Drain TBST from slide and return slide to incubation chamber. Apply 200 µl of ready-to-use goat anti-rabbit horseradish peroxidase polymer and close chamber. Incubate for 30 min. at room temperature. Rinse slide with TBST and place slide in container of TBST for 5 min.
Prepare brown horseradish peroxidase-3,3'-diaminobenzidine (HRP-DAB) chromogen by adding 8 µl of DAB chromogen to 250 µl DAB substrate buffer and mixing well.
Drain TBST from slide, return slide to incubation chamber and apply 200 µl of brown HRP-DAB chromogen for 6 min. Thoroughly rinse the slide with distilled water and place slide in a container of distilled water.
Mix the denaturing solution by combining 50 µl of the first elution reagent with 150 µl of the second buffer reagent. Drain water off the slide and apply diluted denaturing solution to the slide for 3 min. at room temperature. Decant and rinse denaturing reagent with TBST. Immerse slide in TBST for 5 min.
Prepare AR/CD147 antibody cocktail by diluting 4.2 µl of anti-AR antibody and 2.8 µl of anti-CD147 antibody with 203 µl of antibody diluent. Drain TBST from the slide, return slide to incubation chamber and apply 200 µl of AR/CD147 cocktail to the slide. Close chamber and incubate for 1 hr at room temperature. Rinse slide with TBST and immerse in TBST for 5 min.
Drain TBST, return slide to incubation chamber and apply 200 µl of ready-to-use goat anti-rabbit alkaline phosphatase polymer. Close chamber and incubate slide for 30 min. at room temperature. Rinse slide with TBST and immerse in TBST for 5 min.
Prepare red alkaline phosphatase chromogen by adding 3.2 µl of red chromogen to 250 µl red chromogen buffer and mix well.
Drain TBST from slide and return slide to incubation chamber. Apply 200 µl of diluted red alkaline phosphatase chromogen to the slide, close chamber and incubate for 7 min.
Drain chromogen and thoroughly rinse slide with distilled water. Place slide in a container of distilled water for 5 min. Drain water from the slide, rinse with TBST and return to incubation chamber. Apply 200 µl goat anti-mouse-HRP polymer to the slide and close chamber. Incubate for 30 min. at room temperature.
Drain polymer from the slide and rinse with TBST. Place slide in TBST for 5 min. Prepare 200 µl of purple HRP-linked chromogen by adding 3.2 µl of the stabilizing reagent to 250 µl of the buffer and mix well. Add 3.2 µl of purple chromogen and mix well, then 3.2 µl of the hydrogen peroxide reagent and mix well.
Drain TBST off slide and apply 200 µl of prepared purple chromogen to slide and incubate for 7 min. at room temperature. Rinse slide with distilled water and place slide in a container of distilled water for 5 min.
Mix the denaturing solution by mixing 50 µl of the first elution reagent with 150 µl of the second buffer reagent. Drain water off the slide and apply diluted denaturing solution to the slide for 3 min. at room temperature.
Rinse denaturing reagent off with TBST. Place slide in container of TBST for 5 min. Dilute anti-E-cadherin antibody to 1:200 by adding 1.1 µl antibody to 218.9 µl antibody diluent. Drain the TBST from the slide, return slide to incubation chamber and apply 200 µl of E-cadherin to the slide. Close chamber and incubate 30 min. at room temperature.
Rinse slide with TBST and place slide in container of TBST for 5 min. Drain the TBST from slide, return slide to incubation chamber and apply 200 µl goat anti-mouse HRP polymer. Incubate at room temperature for 30 min. Rinse slide with TBST and place slide in container of TBST for 5 min.
Prepare black HRP-linked chromogen by adding 8 µl of chromogen to 250 µl buffer. Apply 200 µl chromogen to slide and develop for 2 min. Thoroughly rinse slide with distilled water and immerse in container of distilled water.
Dilute hematoxylin by adding 40 µl hematoxylin to 200 µl distilled water. Drain water from slide and apply 200 µl of diluted hematoxylin to slide for 30 seconds. Rinse slide thoroughly running tap water for 2 min., then rinse in distilled water for 30 seconds. Place slide in a 60 °C oven for 15-30 min.
Immerse slide in freshly prepared 100% xylene for 30 seconds, wipe excess xylene off and add one drop of permanent mounting media. Coverslip with a No. 1.5 coverslip.
2. Automated Image Acquisition and Analysis
Load stained slides onto an automated slide scanner. Based on the diameter, separation, and number of TMA cores, create an automated scanning protocol according the manufacturer's instruction manual.
Open multispectral imaging software to build a spectral library from control slides stained with individual chromogens and the hematoxylin stained slide. Open an image cube acquired from a control slide and select four to five positively stained areas to optically define that chromogen. Repeat with image cubes from other control slides until a complete spectral library representing all chromogens is created, and then save the spectral library.
Begin a new project within multispectral imaging software. Select "Multispectral (.im3)" for the image format option and "Brightfield" for the sample format. First, configure the project by choosing desired options: "Segment Tissue", "Find Features", "Phenotyping", "Score" and "Export". At this point, the image resolution can be changed to expedite analysis time if desired.
3. Tissue Segmentation
Import the previously created spectral library and select all chromogens to be included in analysis.
Open image cubes to be included in the training data set by selecting the "Open Image Cube" option. To ensure training accuracy, select at least 18% of the total number of images to be analyzed. Choose images that represent all disease states to increase segmentation accuracy. This portion of images is called the training set of images.
White balance images in the training set by selecting the eye dropper tool and choosing an area in one image that is white.
Select "Advance" button to move to tissue segmentation. Use the "Tissue Categories" panel to choose tissue types to be analyzed (i.e. "tissue" and "non-tissue"). For more accurate protein tissue localization, multiple tissue categories can be used (i.e. "epithelia," "stroma," and "non-tissue").
Begin creating the algorithm and defining tissue categories by drawing around groups of cells within training images. When finished with one tissue category, repeat for other tissue categories. Be sure to choose groups of cells within images that are characteristic of that tissue category type.
Repeat this process for all images within the training set of images.
Select components (chromogens) to be included in training for the "Tissue Segmenter". When performing analysis of brightfield immunohistochemistry images, all components are normally included in training. Include abundant negative staining images in the training set to avoid bias during this step.
Choose an appropriate "Pattern Scale" for training the tissue segmenter. Under 20X magnification, a large pattern scale is typically appropriate. When working with tissues with a fine architecture under higher magnification, a smaller pattern scale is more appropriate.
Select the "Train Tissue Segmenter" button to start training the tissue segmenter. Observe a pop-up box displaying accuracy reflecting the proportion of pixels within training regions that are properly classified. NOTE: The software continually attempts to improve the accuracy of the algorithm until manually stopped. We have previously demonstrated that training on 18% of images results in 97% training accuracy 12.
After the tissue segmenter is trained, choose an appropriate segment resolution. Segment resolution corresponds to time required to segment images, with coarse resolution requiring less time and fine resolution requiring more time.
Segment the entire training set of images by clicking "Segment Images". Allow the software adequate time to apply the algorithm to all training images. When finished, review the training set to find any misclassified tissue with the current training algorithm.
To fine tune the tissue segmentation process, add, edit, or remove tissue training regions. Use the "trim edges" option if pixels extend on the edges of one tissue category into another. If small groups of pixels are misclassified, adjust the threshold on the minimum segment size.
When edits are completed, select "Segment Images", this will re-train the tissue segmenter to create a new algorithm for tissue segmentation. The previous algorithm is automatically saved and can be returned to if needed.
When confident with tissue segmentation algorithm results, advance to cell segmentation by selecting the "Advance" button.
4. Cell Segmentation
Choose the subcellular compartments to be included in cell segmentation. Ensure that "Nuclei" is already selected to choose "Cytoplasm" or "Membrane". NOTE: "Membrane" segmentation should only be chosen when a membrane-specific protein marker was included in IHC, such as E-cadherin.
Within the "Nuclei" tab, there are several options. Begin by choosing appropriate settings for nuclear segmentation (see step 4.3). Choose whether individual or all tissue categories will be included in segmentation.
- Select an approach for nuclear segmentation
- Select the counterstain "Pixel-Based (Threshold)" approach for a simplified method for obtaining good results without knowing much about image processing settings. NOTE: This often makes a good first choice. When selecting an approach, "Pixel-Based (Threshold)" is more appropriate when there is a reliable nuclear counter-stain, while "Object-Based" should be used if there is a lack of consistent nuclear counterstain.
- Select the "Pixel-Based (Threshold)" approach when there is a reliable nuclear counterstain. NOTE: This approach is purely pixel-based. This approach can also be used for other image analysis needs that can be satisfied with a simple threshold, such as detecting all pixels within a tissue category that stain positive for an IHC stain.
- Select the Object-Based approach if the nuclear counterstain does not provide consistent and specific staining of nuclear objects, and more advanced morphometry-based approaches are needed to detect nuclei.
- Select "Components for Nuclear Segmentation", "Nuclear Size", and "Clean-up" if user wishes to further define nuclei segmentation settings.
- Select cytoplasm shape parameters
- Select the "Advance" button to move to cytoplasm segmentation. NOTE: There are several options within cytoplasm segmentation to choose from. The option termed "inner distance to nucleus" is pixel based. It reflects the distance between the outside of the nucleus and the inside of the cytoplasm. Increasing this value decreases the probability that nuclear signal crosses over into the cytoplasm compartment.
- Next select, "outer distance to nucleus". NOTE: This is also pixel based. The outer distance to nucleus reflects the distance between the outside of the nucleus and the outside of the cell. This value can be set large or small enough to include or exclude membrane signals if desired.
- Next select, "minimum size". NOTE: The minimum size, pixel based, reflects the minimum cytoplasm sample size to be included in analysis. If the cytoplasm size is smaller in a particular cell such as when cells are tightly packed together, this cytoplasm segmentation is excluded from analysis.
- Select the next option, "Component" with "Primary", "Secondary", and select "Tertiary" as secondary options. NOTE: Here one may choose a specific cytoplasm marker. Similar to nuclear segmentation, if a specific cytoplasmic IHC marker has been used, this component can be used to define the cytoplasm by finding a minimum threshold value. This assists in defining the cytoplasm but is not necessary for acceptable accuracy.
- Moving to the last of the three tabbed options is "Membrane". Here, the user defines the membrane compartment with a protein marker. Choose "Primary", "Secondary", and/or "Tertiary" for each membrane specific marker used. Adjust "Full Scale OD" to find a minimum threshold or a positive for each marker on cell membranes.
- Continuing under the "Membrane" tab and select the option "Maximum Cell Size". NOTE: This is the distance to membrane value, in pixels, which specifies the maximum size of the cells. A value of 12 is normally appropriate for images taken at 20X magnification.
- After selecting all the options, select "Segment Image" or "Segment All". Apply the settings to images and observe them "individually" or in "Gallery" mode. NOTE: Adjust settings and thresholds if necessary, and re-segment until satisfied with cell segmentation results in the training set of images.
5. Phenotyping of Cells
NOTE: Accurate cell segmentation is required in order to obtain accurate cell phenotyping, and the phenotyping feature is trainable.
Use the "Add" button to add phenotypes to the "Phenotypes" list. Users can select a color for the phenotype.
Click on "Edit Phenotypes" to assign a phenotype to a cell. Click on a particular cell to bring up a drop-down menu, allowing user to choose the phenotype and then move on. Identify at least five (5) cells in each phenotype in order to proceed. Choosing 25 or more cells of each phenotype is needed to achieve optimal results.
6. Scoring IHC and Co-Localization
Choose "Advance" to move to the "Score IHC or IF" step. Choose a "Tissue Category" to score. NOTE: Only one tissue category can be scored during a particular analysis, but if scoring results in another tissue category are desired, the analysis can be repeated later with different settings.
Choose a desired "Scoring" type. NOTE: Positivity creates two bins (positive and negative) for a component of interest, while double positivity is used for co-localization analysis. Other options include 4-bin, 10-bin, and 50-bin analyses for a marker of choice.
Choose the cell "Compartment" to be used in scoring analysis.
Similar to finding a minimum threshold for the primary nuclear component, select "View Component Data" and move cursor over the training images to find appropriate optical density minimum thresholds of staining for positive cells for the component(s) of interest.
If intensely stained cells are to be excluded, lower the threshold max to an appropriate level. Otherwise, use the auto button to choose a threshold maximum for analysis.
Choose training images to score in order to validate thresholding values. NOTE: The percentage of cells within a particular bin will be returned and can be compared by visual inspection or manual counting. Once complete, select "Advance" to proceed to "Export" step.
7. Applying the Algorithm and Batch Analysis
Test the algorithm created through tissue segmentation, cell segmentation, and scoring values by exporting the data for the training set. Follow prompts to create a new folder for the export directory. Select images and tables to be created and included in analysis. Perform the analysis by selecting "Export for All".
Tables are exported in tab-separated text files and can be opened in most data analysis programs.
When analysis is complete, view cell segmentation and scoring data for images with both high and low staining to evaluate accuracy of settings.
When satisfied with settings, apply the algorithm created from the training set of images to the entire set of images through batch analysis. Click on the "Batch Analysis" tab and the opened algorithm will be copied from the active project.
Choose a new export directory and select which images and tables are to be included in the analysis. Under the input files option, select "Add Images" and choose all images to be included in the batch analysis.
Perform the analysis by selecting the "Run" option. As with the analysis of the training set, this step will require a variable amount of time, depending on particular settings and number of images included in the analysis.
When completed, advance to the "Review/Merge" tab. The batch directory defaults to the export directory from the batch analysis and can be changed if desired. Select "Include All" and select "Merge" to create data sheets with summary data for analysis.
8. Analysis of Exported Data
Open exported tables in data analysis software and remove or hide data columns that are irrelevant to analysis, such as minimum and maximum values for each component. The primary data used in analysis is in the mean optical density column for every component.
Review exported segmentation maps and raw images to determine which samples or TMA cores are to be excluded from analysis. Tissue percentage values assist in determining whether a sample should be included or not, with an arbitrary cutoff of <5% used most often for exclusion criteria.
Remove irrelevant data or rows containing samples to be excluded from data analysis.
Use protein quantification data to investigate changes in disease state or the relationship with clinico-pathological characteristics of disease 12,18-21.
Representative Results
In Figure 1, training is performed on prostate tissues to segment images into epithelial and stromal portions, along with the non-tissue compartment. By using the epithelial membrane marker E-cadherin, cell segmentation was performed to separate the nucleus, cytoplasm, and membrane portions, shown in Figure 2.
In one experiment, we used multiplexed IHC to investigate the expression and localization of AR, ERα, E-cadherin, and CD147, as shown in Figure 3. Using these techniques, we are able to identify cells positive for nuclear expression of both ERα and AR (Figure 3B-3C) despite overlapping colorimetric signals, as shown with green arrows in Figure 3A. We found marked differences in the proportion of double positive stromal cells within different states of prostate disease (Figure 3D). We quantified cell membrane-specific expression of CD147 by using E-cadherin as a marker protein (red arrows in Figure 3A), and we were the first group to investigate membrane specific CD147 expression in prostate tissues. We found a significant decrease in CD147 expression in association with prostate cancer progression (Figure 3E) and found an important association with post-surgical prognosis 19.
There are instances where poor experiment design can lead to inaccurate tissue segmentation. In Figure 4, the algorithm applied to a set of tissues did not accurately segment epithelial and stromal compartments (Figure 4C). In this experiment, α-smooth muscle actin (α-SMA) was used to mark the stromal compartment. Because α-SMA and smooth muscle is decreased or lost in some tumors (Figure 4A), the algorithm created through tissue segmentation (Figure 4B) was unable to accurately differentiate between epithelial and stromal compartments from morphometric data alone. Care should be taken when choosing protein markers for tissue or cell compartments.
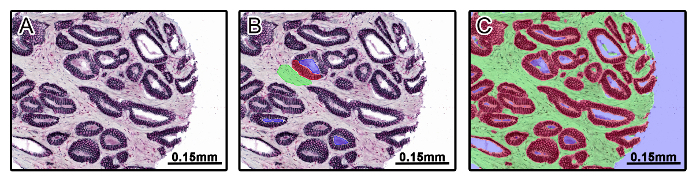 Figure 1: Tissue Segmentation Using Multispectral Imaging Software. A prostate tissue microarray was stained using multiplexed immunohistochemistry for androgen receptor (AR), estrogen receptor-alpha (ERα), E-cadherin, and CD147. A set of training images were imported into multispectral imaging software representing tissue types and disease states of the entire set of images (A). Tissue categories were created including stroma (green), epithelia (red), and non-tissue (blue), and categories were defined by manually drawing on top of training images (B). After drawing on training images, an algorithm for tissue segmentation was created and applied to the training set of images (C). Please click here to view a larger version of this figure.
Figure 1: Tissue Segmentation Using Multispectral Imaging Software. A prostate tissue microarray was stained using multiplexed immunohistochemistry for androgen receptor (AR), estrogen receptor-alpha (ERα), E-cadherin, and CD147. A set of training images were imported into multispectral imaging software representing tissue types and disease states of the entire set of images (A). Tissue categories were created including stroma (green), epithelia (red), and non-tissue (blue), and categories were defined by manually drawing on top of training images (B). After drawing on training images, an algorithm for tissue segmentation was created and applied to the training set of images (C). Please click here to view a larger version of this figure.
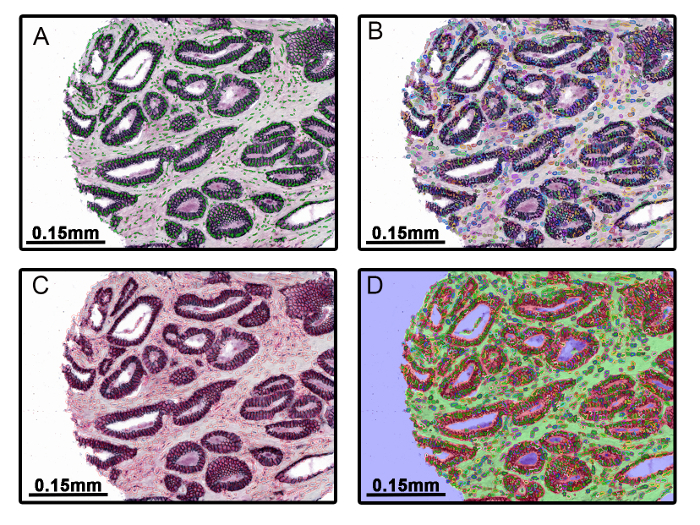 Figure 2: Cell Segmentation into Nuclear, Cytoplasmic, and Membrane Compartments. The nuclear compartment was defined for cell segmentation by setting a minimum threshold for hematoxylin counterstaining (A). The cytoplasm was defined in relation to the nucleus by using pre-set algorithms within the software (B). E-cadherin was used as a membrane marker and a minimum mean OD threshold was applied to define the membrane compartment (C). This technique allows simultaneous quantitation of protein expression in all subcellular compartments (D). Please click here to view a larger version of this figure.
Figure 2: Cell Segmentation into Nuclear, Cytoplasmic, and Membrane Compartments. The nuclear compartment was defined for cell segmentation by setting a minimum threshold for hematoxylin counterstaining (A). The cytoplasm was defined in relation to the nucleus by using pre-set algorithms within the software (B). E-cadherin was used as a membrane marker and a minimum mean OD threshold was applied to define the membrane compartment (C). This technique allows simultaneous quantitation of protein expression in all subcellular compartments (D). Please click here to view a larger version of this figure.
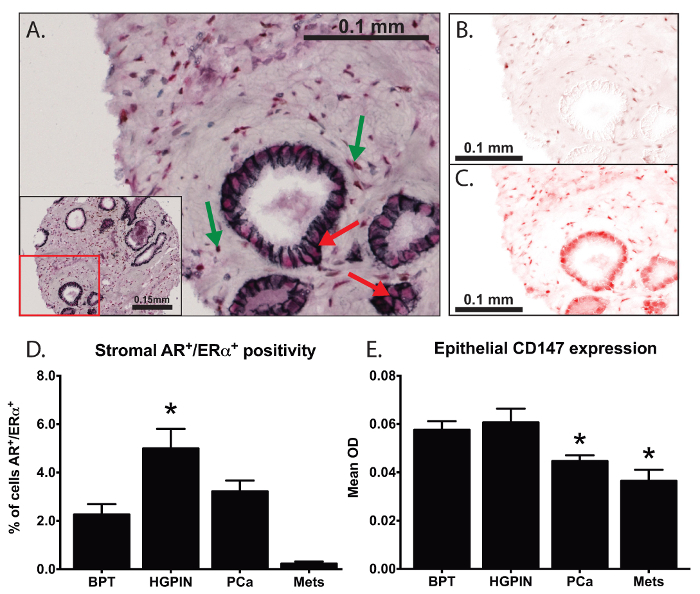 Figure 3: Analysis of Co-localization and Membrane-specific Protein Expression. Multiplexed IHC was used to investigate the expression and localization of androgen receptor (AR), estrogen receptor-alpha (ERα), E-cadherin, and CD147 (A). DAB (brown chromogen) was used to mark AR (B) and a red chromogen used to mark ERα (C). We were able to identify spatially overlapping biomarkers (green arrows in A) and quantify (D) the proportion of cells with nuclear co-localization of ERα and AR within prostate stroma. By using E-cadherin (black chromogen) to define the plasma membrane (red arrows in A), we quantified membrane-specific expression of CD147 within prostate tissues (E) 19. One-way analysis of variance (ANOVA) was used for statistical analysis, error bars reflect standard error of the mean, and asterisks represent p<0.05. Please click here to view a larger version of this figure.
Figure 3: Analysis of Co-localization and Membrane-specific Protein Expression. Multiplexed IHC was used to investigate the expression and localization of androgen receptor (AR), estrogen receptor-alpha (ERα), E-cadherin, and CD147 (A). DAB (brown chromogen) was used to mark AR (B) and a red chromogen used to mark ERα (C). We were able to identify spatially overlapping biomarkers (green arrows in A) and quantify (D) the proportion of cells with nuclear co-localization of ERα and AR within prostate stroma. By using E-cadherin (black chromogen) to define the plasma membrane (red arrows in A), we quantified membrane-specific expression of CD147 within prostate tissues (E) 19. One-way analysis of variance (ANOVA) was used for statistical analysis, error bars reflect standard error of the mean, and asterisks represent p<0.05. Please click here to view a larger version of this figure.
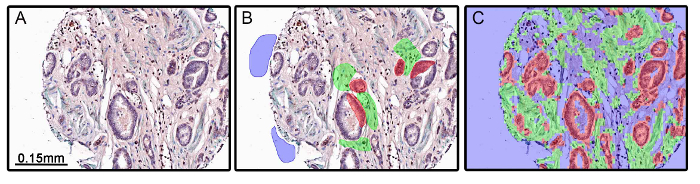 Figure 4: Inadequate Tissue Segmentation Resulting from Experimental Design. Benign and malignant prostate tissues were stained for α-smooth muscle actin (α-SMA; green chromogen), androgen receptor (red chromogen), and androgen receptor variant 7 (DAB chromogen). α-SMA was used as a marker of stroma and is decreased in some tumors, including prostate (A). When training was performed (B) and the tissue segmentation algorithm was applied, the stromal and epithelial compartments were inadequately segmented due to the absence of α-SMA staining (C). Please click here to view a larger version of this figure.
Figure 4: Inadequate Tissue Segmentation Resulting from Experimental Design. Benign and malignant prostate tissues were stained for α-smooth muscle actin (α-SMA; green chromogen), androgen receptor (red chromogen), and androgen receptor variant 7 (DAB chromogen). α-SMA was used as a marker of stroma and is decreased in some tumors, including prostate (A). When training was performed (B) and the tissue segmentation algorithm was applied, the stromal and epithelial compartments were inadequately segmented due to the absence of α-SMA staining (C). Please click here to view a larger version of this figure.
Discussion
The use of traditional immunohistochemistry for evaluating protein expression is limited by subjective, semi-quantitative methods of analysis 22,23. Advance platforms have been created for high-throughput analysis of biomarker expression and localization. Detailed segmentation of both tissue and subcellular compartments allows users to study biomarker expression, localization, and co-localization with other markers of interest. In previous studies, we have demonstrated the utility of IHC and multispectral imaging, particularly when used to study proteins localized to the same cellular compartment 18,20,21. When combined with tissue microarrays 24, these techniques allow for faster and more objective quantitation of protein expression than allowed by manual analysis by a pathologist.
One issue with the use of immunohistochemistry for protein quantitation is irreproducibility of results due to the subjective nature of analysis and inherent differences in technique and reagents. A significant advantage of using platforms like multispectral imaging for measuring protein expression is reproducibility of results. Data from computerized pathology platforms correlate highly with manual analysis by a pathologist while returning data in a continuous format and substantially reducing work time, particularly when working with large sample sizes 12,16,25. Studies have shown high overall concordance in results between different multispectral platforms 14. Furthermore, we have previously demonstrated that staining results are highly reproducible even when there is variability in the number of proteins investigated 12. The use of automated pathology platforms for both staining and analysis will not eliminate all discrepancies in results, such as those stemming from antibodies created towards different epitopes, but these platforms do substantially reduce bias and irreproducibility commonly associated with immunohistochemistry.
There are particular steps within the protocol that are essential for returning accurate, reproducible results. Appropriate experimental design through selection of proteins to be included as tissue or subcellular markers is important for accurate segmentation. When performing positivity analysis, the selection of a lower threshold for positive staining has a large effect on the final results. While choosing a threshold for "on/off" proteins such as nuclear transcription factors is rather straightforward, finding a threshold for more heterogeneously expressed proteins is difficult. This should ideally be performed in collaboration with a board-certified genitourinary pathologist or as an averaged score across multiple observers to find the ideal threshold for analysis.
It is important to recognize some limitations of current versions of this technology. When defining the cytoplasm compartment, three approaches can be used: (1) staining with a cytoplasm-specific marker, (2) staining with a membrane-specific marker and using the nucleus-to-membrane marker distance as cytoplasm, and (3) using a drawing approach to manually define the boundaries of the cytoplasm in relation to the nucleus. From our experience, using a membrane-specific marker is the most accurate technique. The manual drawing approach is normally accurate if nuclei are centrally located or if the biomarker of interest is uniformly distributed throughout the cytoplasm. Accurately defining the cytoplasm of stromal cells like fibroblasts and smooth muscle cells remains difficult and should be taken into consideration when designing an experiment.
Another limitation of the technology is the dependence upon nuclei for cell segmentation. If plane of section excludes the nucleus of a particular cell, this cell will not be included in analysis. If there is no visible cytoplasm between adjacent nuclei or clumps of nuclei, these are often recognized as one large nuclear lump, rather than distinct nuclei. Generally speaking, hematoxylin counterstain is normally adequate for acceptable nuclear segmentation, and manipulation of software settings such as maximum threshold for nucleus size can fix most issues with nuclear segmentation.
A final limitation of automated pathology platforms is unreliable segmentation of tissues resulting from poor experimental design. The importance of choosing appropriate tissue and cell biomarkers for answering the question of interest cannot be overstated. As we demonstrated in our representative results, an epithelial or stromal marker that significantly changes expression between disease states can pose problems when creating an algorithm for tissue segmentation. It is worth noting that there are alternative options when difficult analyses like this arise. For example, individual images can be analyzed by manually drawing all tissue compartments, rather than by applying an algorithm from a training set of images. This provides the advantage of segmentation that is perfectly in line with what the user desires, but it also introduces subjectivity and can significantly increase the time required to finish an analysis. Appropriate experimental design is the easiest way to expedite analysis and maximize the accuracy of tissue and cell segmentation.
This technology and protocol have many future applications. Numerous biomarkers for particular disease states have been identified but not validated. High throughput objective analysis with multispectral platforms facilitates validation of these biomarkers. Further, the evaluation of expression and co-localization of multiple proteins can provide insights into poorly-understood signaling pathways. Undoubtedly, reducing the inherent subjectivity associated with analysis of immunohistochemical staining is valuable for understanding the expression and localization of a wide range of protein markers.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors thank the University of Wisconsin Translational Research Initiatives in Pathology laboratory, in part supported by the UW Department of Pathology and Laboratory Medicine and UWCCC grant P30 CA014520, for use of its facilities and services.
References
- Valdman A, et al. Expression of redox pathway enzymes in human prostatic tissue. Anal Quant Cytol Histol. 2009;31(6):367–374. [PubMed] [Google Scholar]
- Rimm DL, Camp RL, Charette LA, Olsen DA, Provost E. Amplification of tissue by construction of tissue microarrays. Exp Mol Pathol. 2001;70(3):255–264. doi: 10.1006/exmp.2001.2363. [DOI] [PubMed] [Google Scholar]
- Jonmarker S, et al. Expression of PDX-1 in prostate cancer, prostatic intraepithelial neoplasia and benign prostatic tissue. APMIS. 2008;116(6):491–498. doi: 10.1111/j.1600-0463.2008.01020.x. [DOI] [PubMed] [Google Scholar]
- McCarty KS, Miller LS, Cox EB, Konrath J, McCarty KS., Sr Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch Pathol Lab Med. 1985;109(8):716–721. [PubMed] [Google Scholar]
- Volante M, et al. Somatostatin receptor type 2A immunohistochemistry in neuroendocrine tumors: a proposal of scoring system correlated with somatostatin receptor scintigraphy. Mod Pathol. 2007;20(11):1172–1182. doi: 10.1038/modpathol.3800954. [DOI] [PubMed] [Google Scholar]
- Muris JJ, et al. Immunohistochemical profiling of caspase signaling pathways predicts clinical response to chemotherapy in primary nodal diffuse large B-cell lymphomas. Blood. 2005;105(7):2916–2923. doi: 10.1182/blood-2004-07-2716. [DOI] [PubMed] [Google Scholar]
- Jaraj SJ, et al. Intra- and interobserver reproducibility of interpretation of immunohistochemical stains of prostate cancer. Virchows Arch. 2009;455(4):375–381. doi: 10.1007/s00428-009-0833-8. [DOI] [PubMed] [Google Scholar]
- Nakane PK, Pierce GB. Enzyme-labeled antibodies: preparation and application for the localization of antigens. J Histochem Cytochem. 1966;14(12):929–931. doi: 10.1177/14.12.929. [DOI] [PubMed] [Google Scholar]
- Peters TJ. Investigation of tissue organelles by a combination of analytical subcellular fractionation and enzymic microanalysis: a new approach to pathology. J Clin Pathol. 1981;34(1):1–12. doi: 10.1136/jcp.34.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert-Buck MR, et al. Laser Capture Microdissection. Science. 1996;274(5289):998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- Waters JC. Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol. 2009;185(7):1135–1148. doi: 10.1083/jcb.200903097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Hennrick K, Drew S. A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum Pathol. 2013;44(1):29–38. doi: 10.1016/j.humpath.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Rimm DL. C-Path: A Watson-Like Visit to the Pathology Lab. Science Translational Medicine. 2011;3(108) doi: 10.1126/scitranslmed.3003252. [DOI] [PubMed] [Google Scholar]
- Fiore C, et al. Utility of multispectral imaging in automated quantitative scoring of immunohistochemistry. J Clin Pathol. 2012;65(6):496–502. doi: 10.1136/jclinpath-2012-200734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014;70(1):46–58. doi: 10.1016/j.ymeth.2014.08.016. [DOI] [PubMed] [Google Scholar]
- Rizzardi AE, et al. Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn Pathol. 2012;7:42. doi: 10.1186/1746-1596-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman TM, et al. Characterization of fibrillar collagens and extracellular matrix of glandular benign prostatic hyperplasia nodules. PLoS One. 2014;9(10):e109102. doi: 10.1371/journal.pone.0109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman TM, et al. Beta-catenin is elevated in human benign prostatic hyperplasia specimens compared to histologically normal prostate tissue. Am J Clin Exp Urol. 2014;2(4):313–322. [PMC free article] [PubMed] [Google Scholar]
- Bauman TM, Ewald JA, Huang W, Ricke WA. CD147 expression predicts biochemical recurrence after prostatectomy independent of histologic and pathologic features. BMC Cancer. 2015;15(1):549. doi: 10.1186/s12885-015-1559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman TM, et al. Finasteride treatment alters tissue specific androgen receptor expression in prostate tissues. Prostate. 2014;74(9):923–932. doi: 10.1002/pros.22810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson TM, Sehgal PD, Drew SA, Huang W, Ricke WA. Sex steroid receptor expression and localization in benign prostatic hyperplasia varies with tissue compartment. Differentiation. 2013;85(4-5):140–149. doi: 10.1016/j.diff.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CR, Levenson RM. Quantification of immunohistochemistry--issues concerning methods, utility and semiquantitative assessment II. Histopathology. 2006;49(4):411–424. doi: 10.1111/j.1365-2559.2006.02513.x. [DOI] [PubMed] [Google Scholar]
- Matos LL, Trufelli DC, de Matos MG, da Silva Pinhal MA. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomark Insights. 2010;5:9–20. doi: 10.4137/bmi.s2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononen J, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4(7):844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- Ong CW, et al. Computer-assisted pathological immunohistochemistry scoring is more time-effective than conventional scoring, but provides no analytical advantage. Histopathology. 2010;56(4):523–529. doi: 10.1111/j.1365-2559.2010.03496.x. [DOI] [PubMed] [Google Scholar]