

Abstract
Retinoic acid receptors inhibit chondrogenesis, but their ability to block the cartilaginous scaffold of heterotopic endochondral ossification has not been explored. A study in mice shows that agonists of retinoic acid receptor-γ potently inhibit heterotopic endochondral ossification, suggesting therapeutic potential in people with this condition (pages 454–460).
Heterotopic endochondral ossification (HEO), the formation of bone in soft tissues through cartilage anlagen, can lead to catastrophic disability and enormous human misery. Conditions that predispose to HEO range from the extremely rare genetic disorder fibrodysplasia ossificans progressiva (FOP) to relatively common causes such as athletic injuries, total joint arthroplasties, traumatic brain injuries, strokes, paralysis, high-velocity war wounds and endstage valvular heart disease1–5. In all of these conditions, metamorphosis of soft connective tissue into heterotopic bone occurs by a process of endochondral ossification.
The process of HEO resembles the process by which the normotopic skeleton forms during embryogenesis but differs in its induction by an inflammatory trigger. Inflammation leads to tissue destruction and activation of mesenchymal stem cells (MSCs) that differentiate to build a second skeleton of heterotopic bone under the influence of increased bone morphogenetic protein (BMP) signaling1–6.
Attempts to effectively prevent and treat HEO have been frustrating, if not elusive. Steroidal and nonsteroidal anti-inflammatory medications have produced equivocal results, most likely because inflammatory events that initiate HEO may not be clinically apparent until after the induction process is complete. Radiation and high-dose bisphosphonates have limited application and potential long-term side effects. Further, the potential of dorsomorphin-like small-molecule signal transduction inhibitors of BMP receptors is presently limited by the nonspecific nature of available compounds, their inability to completely suppress HEO, the rebound phenomenon that occurs after cessation of use in animal models and a myriad of off-target effects7. In patients with sporadic HEO, bone can be removed surgically, but the recurrence rate is high; in FOP, surgery is anathema, as recurrence is ubiquitous. There is thus a vast, unmet clinical need in the treatment of HEO.
In a landmark study in this issue of Nature Medicine, Shimono et al.8 report a new approach to block HEO: not before induction, but once the inflammation events leading to HEO have already begun and possibly even ceased. The authors build on well-established findings that retinoic acid is a potent skeletal teratogen that inhibits chondrogenesis, a crucial function they exploit to sabotage heterotopic chondrogenesis before the end stage of disabling HEO is reached9,10. In vitro studies and mouse models show that both the prechondrogenic and chondrogenic stages of HEO are extremely sensitive to the inhibitory effects of retinoic acid receptor-γ (RAR-γ) agonists, which block BMP signaling and the skeletogenic potential of progenitor cells. These findings provide new opportunities to derail HEO in sporadic conditions as well as in FOP.
In their mouse experiments8, the authors employed a comprehensive approach to stimulating HEO3,4,6,7 using genetically engineered MSC implantation, BMP induction of HEO and a conditional transgenic mouse that forms FOP-like HEO to show that RAR-γ agonists potently inhibit HEO. Remarkably, when RAR-γ agonists are discontinued, no substantial rebound effect occurs, indicating that the RAR-γ effect may be irreversible. Additionally, RAR-γ agonists were effective in inhibiting HEO during a wide treatment window that includes the prechondrogenic fibroproliferative phase up to, but not including, the ossification phase8.
Whether in an adult with traumatic brain injury or in a child with a flare-up of FOP, new episodes of HEO are often not clinically apparent until the prechondrogenic fibroproliferative lesion has formed—a stage that is beyond the scope of any currently available treatment and that occurs perhaps as long as ten days after the inflammatory induction phase1,3,4,7. The tantalizing findings of Shimono et al.8 suggest that successful, long-term inhibition of HEO may be possible even a week or more after the inflammatory induction events have occurred, an achievement that has not yet been realized by any other class of medications.
Notably, the authors also show that RAR-γ agonists redirect cell fate decisions in prechondrogenic MSCs to a non-osseous lineage8, an observation with wide-reaching implications for skeletal oncology, vascular biology and tissue engineering6. Might it be possible, for example, to alter the course of chondrogenic tumors, inhibit HEO that occurs in end-stage valvular heart disease and atherosclerosis5 and more precisely model genetically engineered chondro-osseous replacement parts6?
Taken together, the work of Shimono et al.8 provides a tour de force in identifying a potent, orally available class of compounds that can block HEO by inhibiting the cartilaginous scaffold and by diverting mesenchymal stem cells to a more benign soft-tissue fate, while avoiding the rebound effect seen in other classes of experimental medications.
The remarkable findings of the study shed light on issues regarding the biology of HEO and how RAR-γ agonists derail the progression of this disabling metamorphosis. Most importantly, the formation of heterotopic bone requires participation of the BMP signaling pathway2–4,6.
How might RAR-γ agonists impair HEO from a constitutively active BMP type I receptor, as in FOP or in the FOP-like transgenic mouse model in which the constitutively active ACVR1 (also known as ALK2) receptor is conditionally activated by inflammation2–4,6,7? The answer lies, at least in part, with an unusual mechanism of action. The authors show that RAR-γ agonists regulate BMP signaling post-translationally by promoting the proteosome-regulated degradation of BMP pathway-specific phosphorylated Smads (signaling molecules downstream of the BMP receptors)8, a finding supported by another recent study11.
The authors also speculate that RAR-γ signaling stimulates Wnt–β-catenin signaling and remind us that Wnt–β-catenin signaling potently inhibits chondrogenesis12,13. RAR-γ agonists may therefore sabotage the cartilaginous scaffold of HEO by both inhibiting BMP signaling and stimulating Wnt–β-catenin signaling in prechondrogenic and chondrogenic cells (Fig. 1).
Figure 1.
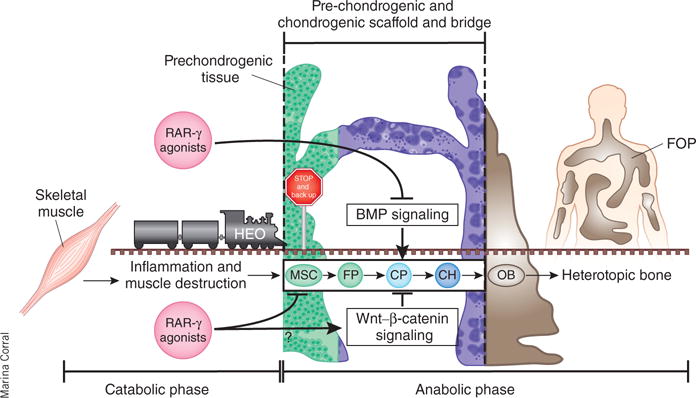
RAR-γ agonists inhibit the cartilaginous scaffold of HEO. The process of HEO involves two major phases: a catabolic phase of inflammation and tissue destruction followed by an anabolic phase of tissue neogenesis involving the formation of a transient cartilaginous scaffold and its replacement with mature heterotopic bone. A key feature of all HEO is the formation of a bridging cartilaginous scaffold that is under control of the BMP and the Wnt–β-catenin signaling pathways. Shimono et al.8 show that RAR-γ agonists inhibit BMP signaling and putatively promote Wnt–β-catenin signaling in cells that build the cartilaginous scaffold, disrupting the bridge and derailing HEO. RAR-γ agonists can reprogram MSCs to a non-HEO soft-tissue fate, effectively backing up the train into the station (skeletal muscle) if it has not yet reached the bridge. The length of the train depicts the well-established finding that contiguous stages of HEO occur simultaneously in different anatomic areas of the lesion. FP, fibroproliferative cells; CP, cartilage progenitor cells; CH, chondrocytes; OB, osteoblasts.
The therapeutic implications of this work in preventing and treating both sporadic and progressive HEO are enormous, but some clinical caveats remain. First, RAR-γ agonists, like the trans-retinoic acid ligands, are teratogenic, and their use in women of childbearing age must be monitored carefully7. Second, the authors predictably show that the RAR-γ agonists delay endochondral bone formation during fracture repair8, suggesting that these agents may have limited applicability in people with intercurrent long-bone fractures in addition to their HEO-prone injuries, as in wounded soldiers and civilians with multiple traumas. Third, as the use of RAR-γ agonists may adversely affect cartilaginous growth plates, additional studies in knock-in mice with the canonical FOP mutation will be necessary before RAR-γ agonists can be considered for long-term use in children. Nevertheless, as Shimono et al.8 indicate, RAR-γ agonists are presently in clinical trials for other disorders, which will probably expedite their application to HEO.
It is difficult to find effective molecular targets for intractable diseases. Successful therapeutic targeting of highly conserved signaling pathways requires exquisite planning and good fortune. The study by Shimono et al.8 combines both. It identifies RAR-γ agonists as a class of compounds that profoundly inhibit the BMP-induced chondrogenesis required for the cartilaginous scaffold of HEO. The beauty of this approach is that it targets not just a seminal signaling pathway but rather a specific pathological process of tissue metamorphosis that requires this specific signaling pathway to cause disabling disease3,4.
The authors have identified a new and powerful class of compounds to derail the cartilaginous scaffold of HEO. Without the cartilaginous scaffold, there is no HEO. With little additional work, these compounds seem ‘RARing’ to go into clinical trials in people, who are desperately waiting for clinical answers.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Contributor Information
Frederick S Kaplan, Email: frederick.kaplan@uphs.upenn.edu, Departments of Orthopedic Surgery and Medicine and the Center for Research in FOR and Related Disorders, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Eileen M Shore, Departments of Orthopedic Surgery and Genetics and the Center for Research in FOP and Related Disorders, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA.
References
- 1.Pignolo RJ, et al. Clin Rev Bone Miner Metab. 2005;3:261–266. [Google Scholar]
- 2.Shore EM, et al. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. erratum 39, 276 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Kaplan FS, et al. Cytokine Growth Factor Rev. 2009;20:399–407. doi: 10.1016/j.cytogfr.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shore EM, Kaplan FS. Nat Rev Rheumatol. 2010;6:518–527. doi: 10.1038/nrrheum.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohler ER, III, et al. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 6.Medici D, et al. Nat Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong CC, et al. Cytokine Growth Factor Rev. 2009;20:409–418. doi: 10.1016/j.cytogfr.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimono K, et al. Nat Med. 2011;17:454–460. doi: 10.1038/nm.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zasloff MA, et al. Clin Orthop Relat Res. 1998;346:121–129. [PubMed] [Google Scholar]
- 10.Hoffman LM, et al. J Cell Biol. 2006;174:101–113. doi: 10.1083/jcb.200604150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng N, et al. Proc Natl Acad Sci USA. 2010;107:18886–18891. doi: 10.1073/pnas.1009244107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy KAM, et al. BMC Biol. 2009;7:67. doi: 10.1186/1741-7007-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasuhara R, et al. J Biol Chem. 2010;285:317–327. doi: 10.1074/jbc.M109.053926. [DOI] [PMC free article] [PubMed] [Google Scholar]