

Abstract
Advances in gene sequencing techniques have led to a dramatic increase in the number of signaling cascade and cytoskeletal assembly mutations associated with malformations of cortical development and epilepsy. At the forefront of this research are novel mutations found in regulators of the PI3K/AKT/mTOR cascade and tubulin-associated malformations of cortical development. However, there is limited understanding of the consequences of these newly discovered germline and somatic mutations on cellular function or how these changes in cell biology may lead to areas—large or small—of malformed cortex and recurrent spontaneous seizures. We summarize and discuss what is currently known in this field in an effort to shine light on vast gaps in our knowledge of relatively common causes of cortical malformations.
Progress in understanding how gene mutations produce malformations of cortical development (MCD) and epilepsy is occurring at a rapid pace. The evolving aims of MCD gene discovery include identifying causative mutations and correlating the mutational effect to an epilepsy phenotype. However, greater understanding of intracellular signaling cascades has permitted an approach to go beyond genotype–phenotype correlation and determine the consequences of mutations on cell and network function as a strategy to define precision treatment modalities. Of particular relevance, mutations in upstream and downstream regulators within the phosphotidylinositol-3 kinase/protein kinase B/mechanistic or mammalian target of rapamycin (PI3K/Akt/mTOR) pathway (1) and components of the cytoskeletal system (2) have emerged as playing key roles in producing MCDs most closely associated with intractable epilepsy, as well as neurobehavioral disabilities.
While neuropathological and radiographic criteria exist to classify MCD subtypes, we will focus on the effects of mutations on cell signaling and the functional attributes of mutations causing MCD. We will summarize some of the more recently discovered gene mutations and how they may interfere with PI3K/Akt/mTOR signaling and cytoskeleton assembly to produce aberrations in morphology, polarity, growth, migration, and excitability that ultimately lead to MCD and epilepsy. For purposes of defining mechanistic effects, malformations will be divided into two broad categories: mTORopathies (1) and tubulinopathies (2).
mTORopathies
MCD caused by mutations in genes that encode protein regulators of mTOR activity—for example, PI3K, Akt3, and mTOR—have been termed “mTORopathies” (1) (Figure 1). Animal models suggest that mutations in these genes disrupt normal proliferation, growth, migration, and laminar destiny of neuroglial progenitor cells during cortical development. The paradigm disorder to understand mTORopathies is tuberous sclerosis complex (TSC) in which inherited or sporadic loss-of-function mutations in the mTOR regulators TSC1 or TSC2 are associated with focal MCD (tubers) that are highly associated with both infantile spasms and intractable epilepsy (3, 4). The phenotypic spectrum of MCDs linked to mTOR cascade mutations is broad and ranges from small focal cortical dysplasias (FCD type II) to widespread hemispheric and whole brain abnormalities, for example, hemimegalencephaly (HME) and megalencephaly (ME), respectively. Some mTOR-related MCD may be very small and difficult to visualize with neuroimaging techniques—for example, “bottom-of-the-sulcus” FCD—and are often revealed only by postoperative neuropathological examination of resected tissue. Despite these phenotypic variations, the central tenet for conceptualizing mTORopathies is that multiple mutations in distinct genes within the PI3K/Akt/mTOR cascade culminate in several common phenotypic features, including histological evidence for cascade activation, abnormal neuronal morphology, aberrant cortical laminar structure, neuronal hyperexcitability, and seizures. Indeed, the net cellular effects of PI3K/Akt/mTOR pathway mutations seems to funnel through mTOR as a common signaling node as evidenced by in vitro and in vivo data, demonstrating reversibility of both structural and functional effects of these mutations (e.g., hyperexcitability) by pharmacological mTOR inhibition with rapamycin or related compounds.
FIGURE 1.
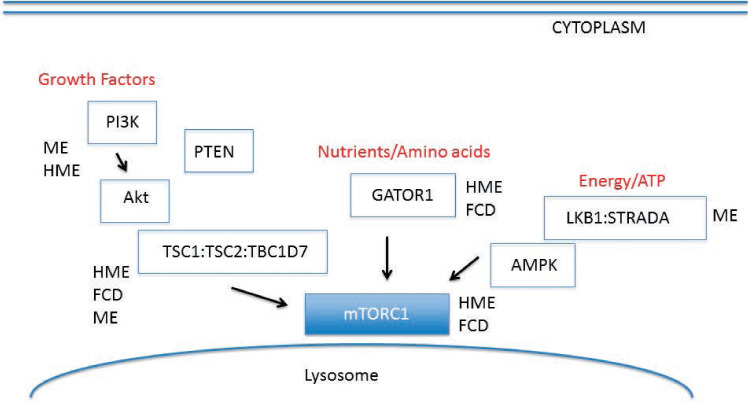
Schematic of PI3K/Akt/mTOR signaling cascade. Multiple protein pathways converge to modulate mTOR signaling in response to growth factors, nutrient cues such as amino acids, and cellular energy levels (ATP). The GATOR1 complex includes DEPDC5 and NPRL3. Much of mTOR signaling regulation occurs in the cytoplasm or in proximity to the lysosomal membrane. MCD subtypes have been linked to gene mutations within components of the cascade, that is, focal cortical dysplasia (FCD), megalencephaly (ME), and hemimegalencephaly (HME). Modified from Crino (1).
HME and ME
The initial intracellular signaling component of the PI3K/Akt/mTOR pathway is PI3K, which transduces extracellular signals from a number of growth factors, for example, insulin-like growth factor-1. Several recent studies have identified gain-of-function somatic mutations in phosphatidylinositol 3-kinase catalytic active isoform (PI3KCA) in HME and gain-of-function somatic or germline mutations in PI3KCA or phosphatidylinositol 3-kinase regulatory subunit beta (PI3KR2) in ME-associated syndromes (5, 6). The PIK3CA gene encodes the p110alpha protein (the catalytic subunit of PI3K) while PI3KR2 is the p85 regulatory component of PI3K. HME and ME are associated with altered cortical cytoarchitecture and may be linked with intractable epilepsy, and intellectual and psychomotor disability. Interestingly, disruption of gyral structure and laminar organization tend to be more severe in HME than ME and, as such, epilepsy is more common and typically intractable, whereas individuals with ME often have more significant cognitive and behavioral deficits than those with HME. However, in some cases termed “dysplastic megelancephaly,” the gyral architecture of both hemispheres is severely disorganized. Most individuals with HME will require epilepsy surgery to treat intractable seizures. In HME, histological examination reveals that the affected hemisphere has large areas of dyslamination with dysmorphic neurons, balloon cells, and white matter heterotopia that may be similar to those found in FCD. Because fewer individuals with ME are eligible for epilepsy surgery than those with HME, the spectrum of neuropathological findings in ME is less well defined than in HME. Patients with ME may exhibit additional phenotypic abnormalities depending on the associated syndrome (e.g., polymicrogyria, cutaneous vascular malformations, polydactyly) (7). Mouse models expressing activating PIK3CA mutations (e.g., H1047R and E545K) recapitulate human pathological features, including brain enlargement and cortical malformations, and treatment with PI3K inhibitors ameliorates seizures in these animals (8).
Exactly how gain-of-function mutations in PI3K isoform genes produce HME and ME—as well as the functional consequences of these mutations on the affected neuroglial progenitor cells—has not been fully determined. HME brain tissue containing a known PI3K mutation exhibits increased phospho-activation of Akt3, an AKT variant primarily expressed in brain, and increased phosphorylation of ribosomal S6 protein (phospho-S6), a common readout for increased downstream mTOR-complex 1 (mTORC1) signaling activity, on histological examination (6). Thus, PI3K mutations lead to enhanced cascade activation signaling downstream to mTOR, and a growing body of evidence suggests that mTOR activation is therefore a likely cause of the abnormal cellular growth, migration, polarization, and metabolism that produces a disorganized cortex in HME and ME patients.
Somatic mutations in AKT3 are also associated with HME (9). As with studies examining PI3K, brain tissue resections from patients with AKT mutations show enhanced phospho-S6 levels, suggesting these mutations play a role in impaired cell size and morphology. The net effect of enhanced mTOR signaling, whether due to PI3K or Akt activation, is increased mRNA translation and protein synthesis leading to hypertrophy of the cell body (cytomegaly). Exactly which mRNAs and proteins culminate in increased cell size and which specific cellular components comprise the enlarged soma remain to be defined. Emerging data also demonstrate a role for AKT3 mutations in impaired neuronal migration through the AKT3-FOXG1-reelin pathway (10). Reelin is a secreted protein integral to neuronal migration during corticogenesis and mutations in the RELN gene have been closely linked to lissencephaly (11). Gain-of-function AKT3 mutations engineered in mice to create a focal MCD model led to aberrant reelin expression and impaired migration of the affected neuron. Surprisingly, surrounding neurons unaffected by the AKT3 mutation also showed impaired migration, presumably via non-cell–autonomous effects (10), that is, effects on migration and lamination not directly caused by the mutation. A similar study examining Tsc2 loss or overexpression of Rheb (the regulatory protein that lies between TSC and mTOR) on Reelin-Dab1 signaling also found impaired neuronal migration and, as in the previous study, demonstrated the ability to recover the migration defect using rapamycin (12). Though not directly examined in either of these studies, it is likely that mutations in PI3K confer the same types of molecular changes due to the proximity of PI3K to AKT3 within the molecular cascade. Of course, a pivotal unanswered question is how mutations affecting the same cellular pathway yield very different effects on brain structure, for example, HME versus TSC versus FCD, yet nonetheless confer an epilepsy phenotype either directly through changes in excitability or indirectly through abnormal network formation.
STE20-related kinase adaptor α (STRADα) deletions were found in patients with the autosomal-recessive developmental disorder polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE), also referred to as “Pretzel Syndrome.” STRADα negatively regulates mTORC1 through the energy sensor 5' AMP-activated protein kinase (AMPK), and deletion of STRADα promotes constitutive mTORC1 activation (13). As found in brain tissue specimens obtained from PI3K- or AKT3-associated HME or FCD, PMSE brain tissue shows increased phospho-S6 expression compared to controls, and there is widespread cytomegaly. STRADα shRNA-transfected neural progenitor cells also exhibit cytomegaly (14). Depletion of STRADα in vitro and in vivo dramatically impaired neuronal migration and establishment of cell polarity. These cellular effects were prevented or reversed with rapamycin treatment, and rapamycin has been successfully used to treat seizures in PMSE patients (15).
Germline mutations in phosphatase and tensin homolog PTEN) (have also been shown to cause ME in the context of larger multisystem PTEN Hamartoma Tumor Syndromes, for example, Cowden Syndrome (16). PTEN is a tumor suppressor that inhibits growth and proliferation signals by inhibiting PI3K. Brain specimens obtained from patients with known PTEN mutations show increased AKT activity, and in vitro examination of PTEN mutations revealed increased phospho-S6 staining due to downstream activation of mTOR, as well as abnormal cytoarchitecture (17). Animal models of Cowden Syndrome show increased brain size and weight (18), along with abnormal electrographic activity and seizures (16). mTOR inhibitor administration is effective in correcting the cellular abnormalities observed in vitro and in vivo in mice and, interestingly, is also effective in reducing seizure frequency (19, 20). Recently, mutations in TBC1D7 (21), a binding partner of the TSC1-TSC2 complex, have been associated with ME, although less is known about the role of this gene in brain function.
FCD
Increasing evidence has demonstrated a pathogenic role for germline and somatic mTOR component mutation in FCD. For example, loss-of-function mutations in the nitrogen permease regulator 2-like protein (NPRL2), nitrogen permease regulator 3-like protein (NPRL3), and DEP domain containing DC 5 (DEPDC5) genes that code for proteins comprising the GAP activity towards rags 1 (GATOR1) heteromeric protein complex (22, 23) have been identified in patients with epilepsy and, in some cases, FCD. Mutations in NPRL2 and NPRL3 have been found in patients with familial focal cortical epilepsy and patients with sporadic focal cortical dysplasia (22). DEPDC5 mutations have been found in a number of different epilepsies, including nocturnal frontal lobe epilepsy, autosomal dominant nocturnal frontal lobe epilepsy (24), familial focal epilepsy with variable foci (25), sporadic focal cortical dysplasia with “bottom-of-the-sulcus” dysplasia, FCD type IIa (26), and nonlesional neocortical epilepsy. More than 20 missense and nonsense loss-of-function mutation variants have been identified. GATOR1 complex proteins may be a frequent cause of focal epilepsies and, as more data emerges, may suggest a unique subset of mTORopathies classified functionally as “GATORopathies.”
Highlight Points.
MCD-related mutations are being discovered at a rapid pace but understating how they impact cellular biology lags behind.
Somatic mutations in the central PI3K/AKT/mTOR pathway—PI3K and AKT specifically—have been linked to diffuse MCDs.
AKT mutations may impact both the MTOR and REELIN pathway contributing to both impaired cell size and migration and to non-cell autonomous effects.
Somatic mutations in PI3K/AKT/mTOR pathway regulators lead to both diffuse (ME) and focal (FCD) malformations.
Emerging data demonstrate the involvement of myriad cytoskeletal genes in MCD formation and epilepsy.
The exact mechanism by which GATOR 1 proteins lead to MCDs and epilepsy remains to be defined, but these proteins are pivotal regulators of PI3K/AKT/mTOR pathway activity in response to nutrient status of the cell, in particular, amino acid levels. GATOR1 regulates mTOR in response to changes in amino acid levels by modulating the localization of mTORC1 to the lysosomal surface by activating the RAG GTPases on the lysosomal membrane; leucine and arginine are of particular importance in this mechanism (27). Loss-of-function mutations in GATOR1 promote constitutive mTOR activation as evidenced by enhanced phosphorylation of another mTOR substrate, 4E-BP1, in Drosophila oocytes (28). mTOR hyper-activation due to functional loss of an inhibitory regulator (e.g., TSC1, TSC2, STRADα) can lead to cellular hypertrophy, abnormal cellular morphology, impaired cellular migration, and a lack of defined cellular polarity (4, 14, 29). Brain tissues from patients with known DEPDC5 or NPRL3 mutations exhibit cytomegalic phospho-S6 positive neurons (26, 30) and may be classified as FCD IIa, although the effects of gene knockdown or knockout in vitro or in vivo have not been assessed, and the molecular effects of GATOR1 mutations on brain development remain largely undefined. Furthermore, an upstream, inhibitory, heteromeric complex that modulates GATOR1 (known as GATOR 2) has been recently described (31). We speculate that future studies might identify mutations in GATOR2 components associated with MCD.
Securing the mechanistic framework for mTORopathies, gain-of-function somatic mutations in the MTOR gene itself have been recently identified in patients with FCD type IIa and IIb (32). These specimens exhibit phospho-S6 labeled balloon cells and dysmorphic neurons. MTOR mutations found in patients with FCD type IIa and IIb that were examined were associated with increased activity of 4E-BP1 in vitro (33). In a separate study, in vivo examination of FCD-associated somatic mTOR mutations by in utero electroporation of the mutant construct into fetal mice revealed abnormal cortical lamination and cytomegalic neurons, resulting in spontaneous seizures. These cellular deficits were subsequently corrected with rapamycin treatment (34), suggesting a possible role for mTOR inhibition in the treatment of seizures associated with MTOR mutations.
Tubulinopathies and Cytoskeleton-Related MCDs
Though not a signaling cascade per se, the cytoskeletal system is a complex and dynamic orchestra of protein subunits that assemble and disassemble in response to signaling cues to permit cell motility, polarity, and cytoplasmic transport. Not surprisingly, mutations in cytoskeletal genes can have devastating effects on neuronal function leading to severe MCD and epilepsy (2). The first CNS cytoskeleton-related MCD gene, LIS1, was discovered in 1993 in association with Miller–Dieker lissencephaly syndrome, and its clinical relevance and biological implications have been well characterized (35). Over the past decade, additional evidence has emerged for a larger spectrum of cytoskeleton-related MCDs in the form of so-called “tubulinopathies” reflecting de novo mutations in the tubulin (TUB) alpha (A), beta (B) and gamma (G) genes TUBA1A, TUBA8, TUBB2A, TUBB2B, TUBB3, TUBB5, and TUBG1 (36). These primarily missense mutations can lead to a wide range of associated MCDs including polymicrogyria (TUBA1A [37]), microcephaly (TUBB2B, TUBB5, TUBG1 [38]), and microlissencephaly (TUBB3 [39]). In addition, mutations in TUBB4A have been observed in a few patients with cerebellar, basal ganglia, and putamen malformations and disorganization (40). Most patients, regardless of mutation specificity, have a decreased brain size (microcephaly), agenesis of the corpus callosum, and a highly disorganized cortical structure (41). These mutations do not necessarily fit into orderly genotype–phenotype categories, and there is often significant overlap between mutation, MCD type, and clinical phenotype.
The functional implications of TUB mutations on the cell can be divided into three main categories: decrease in heterodimer production, decreased interactions with motor proteins, and loss of microtubule assembly/stability. Studies examining mutations in TUBA1A, TUBB2B, and TUBB3 have demonstrated a decrease in production of alpha and beta tubulin heterodimers due to an inability of the subunits to properly interact with protein-folding chaperones. Contradictory studies examining the overexpression of these same misfolded proteins suggest they will incorporate into microtubules to form normal structures (37). Less is known about the effects of TUB mutations in the human cortex since fewer patients with TUB mutations are treated with epilepsy surgery than are, for example, mTORopathies.
Microtubules provide scaffolding upon which motor proteins can transport molecular cargo. Single nucleotide mutations in TUBB2B and TUBB3 have been shown to interfere with kinesin binding in vivo and in vitro (38). Whether this interaction occurs directly between the tubulin protein and kinesin or is indirect is unknown, as is how impaired transport may affect excitability. Microtubules are highly dynamic structures that are in near constant flux, a property that is particularly important for neuronal migration. Frequent findings when examining various TUB mutations are changes that affect the ability of microtubules to elongate. These mutations affect either the nucleotide binding site or the binding surfaces between alpha/beta subunits. Therefore, these mutations prevent efficient microtubule elongation and hamper cellular migration (39). Lastly, mutations are also found in regions coding for the lateral aspects of alpha/beta heterodimers. Lateral components are involved in interacting with microtubule-associated proteins that stabilize microtubules and prevent degradation or breaking (42). This, again, has dramatic implications for neuronal migration as well as molecular transport and axon guidance, as has been demonstrated in vitro (34).
Additional mutations in genes encoding cytoskeletal proteins have been linked to MCDs and epilepsy; these include KIF1A (43), ARFGEF2 (44), and FLNA (45). Filamin A (FLNA) is an actin-binding protein that crosslinks actin filaments and anchors the actin cytoskeleton. FLNA mutations are associated with periventricular nodular heterotopia and often refractory seizures. FLNA mutations (45) may exert their pathophysiological effect by preventing proper organization of radial glia in the ventricular zone, resulting in migration defects in vivo (46). Doublecortin (DCX) is a microtubule-associated protein expressed by neuronal precursor cells, and DCX mutations are associated with subcortical band heterotopia in females and X-linked lissencephaly in males, both of which are frequently associated with severe and intractable epilepsy. DCX enhances polymerization and bundling of microtubules and also interacts with Lis1, also known as platelet-activating factor acetylhydrolase IB subunit alpha, binds to DCX and TUBA1A, and plays an important role in regulating the motor protein dynein. Mutations in LIS1 are associated with lissencephaly and the microdeletion Miller–Dieker syndrome. While the precise mechanism of epileptogenesis induced by mutations in cytoskeletal genes is not fully understood, seizure activity in Lis1 mouse models is associated with increased glutamate-mediated excitation and an increased pool of vesicles at the presynaptic site (47). Similarly, in a Lis1-conditional knock-out mouse, induced LIS1 deficiency enhanced excitatory input to granule cells, even in the absence of neuronal disorganization, suggesting that LIS1 mutations may have direct effects on excitatory synaptic transmission (48). Conversely, mouse models of tubulin gene mutations do not exhibit spontaneous seizures. Thus, future studies will be needed to define the links between mutations in, for example, TUB genes and epileptogenesis so that more targeted treatment strategies can be developed.
Conclusions and Future Directions
We posit that the next important phase of understanding the molecular mechanisms causing MCD is to examine individual gene mutations in the context of cellular signaling cascades or relevant cellular functions as a strategy to implement targeted or “precision” therapeutic approaches. Indeed, understanding the intricacies of the PI3K/AKT/mTOR pathway has led to a greater understanding of how rapamycin and related compounds may be used to treat intractable seizures. As deep gene sequencing techniques improve and the ability to identify somatic mutations in small populations of cells increases, the spectrum of known cell signaling cascade mutations leading to MCDs is likely to increase. Future endeavors should focus on rapidly assessing the functional significance of these mutations in animal models and searching for mechanisms to obviate the effects of these mutations based on their role in normal cellular functioning.
Footnotes
Editor's Note: Authors have a Conflict of Interest disclosure which is posted under the Supplemental Materials (207.1KB, docx) link.
References
- 1.Crino PB. mTOR signaling in epilepsy: insights from malformations of cortical development. Spring Harb Perspect Med. 2015;5:a022442. doi: 10.1101/cshperspect.a022442. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tischfield MA, Cederquist GY, Gupta ML, Jr., Engle EC. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Opin Genet Dev. 2011;21:286–294. doi: 10.1016/j.gde.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han JM, Sahin M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011;585:973–980. doi: 10.1016/j.febslet.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crino PB. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013;125:317–332. doi: 10.1007/s00401-013-1085-x. [DOI] [PubMed] [Google Scholar]
- 5.Riviere JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA, Albrecht B, Armour CM, Armstrong L, Caluseriu O, Cytrynbaum C, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM, Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, Uyanik G, Weksberg R, Zirn B, Beaulieu CL, Finding of Rare Disease Genes (FORGE) Canada Consortium. Majewski J, Bulman DE, O'Driscoll M, Shendure J, Graham JM, Jr, Boycott KM, Dobyns WB. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, Riviere JB, St-Onge J, Ojemann JG, Shendure J, Hevner RF, Dobyns WB. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613–1628. doi: 10.1093/brain/awv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirzaa GM, Riviere JB, Dobyns WB. Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet. 2013;163C:122–130. doi: 10.1002/ajmg.c.31361. [DOI] [PubMed] [Google Scholar]
- 8.Roy A, Skibo J, Kalume F, Ni J, Rankin S, Lu Y, Dobyns WB, Mills GB, Zhao JJ, Baker SJ, Millen KJ. Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy. Elife. 2015;4:e12703. doi: 10.7554/eLife.12703. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baek ST, Copeland B, Yun EJ, Kwon SK, Guemez-Gamboa A, Schaffer AE, Kim S, Kang HC, Song S, Mathern GW, Gleeson JG. An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat Med. 2015;21:1445–1454. doi: 10.1038/nm.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang BS, Duzcan F, Kim S, Cinbis M, Aggarwal A, Apse KA, Ozdel O, Atmaca M, Zencir S, Bagci H, Walsh CA. The role of RELN in lissencephaly and neuropsychiatric disease. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:58–63. doi: 10.1002/ajmg.b.30392. [DOI] [PubMed] [Google Scholar]
- 12.Moon UY, Park JY, Park R, Cho JY, Hughes LJ, McKenna J, III, Goetzl L, Cho SH, Crino PB, Gambello MJ, Kim S. Impaired Reelin-Dab1 signaling contributes to neuronal migration deficits of tuberous sclerosis complex. Cell Rep. 2015;12:965–978. doi: 10.1016/j.celrep.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eggers CM, Kline ER, Zhong D, Zhou W, Marcus AI. STE20-related kinase adaptor protein alpha (STRADalpha) regulates cell polarity and invasion through PAK1 signaling in LKB1-null cells. J Biol Chem. 2012;287:18758–18768. doi: 10.1074/jbc.M111.316422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, Baybis M, Helfferich J, Okochi K, Strauss KA, Crino PB. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med. 2013;5:182ra53. doi: 10.1126/scitranslmed.3005271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilarski R, Stephens JA, Noss R, Fisher JL, Prior TW. Predicting PTEN mutations: an evaluation of Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome clinical features. J Med Genet. 2011;48:505–512. doi: 10.1136/jmg.2011.088807. [DOI] [PubMed] [Google Scholar]
- 17.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D'Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mester JL, Tilot AK, Rybicki LA, Frazier TW, Eng C. Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock-in murine model. Eur J Hum Genet. 2011;19:763–768. doi: 10.1038/ejhg.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S, Parghi D, D'Arcangelo G, Anderson AE. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065–2075. doi: 10.1111/j.1528-1167.2011.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alfaiz AA, Micale L, Mandriani B, Augello B, Pellico MT, Chrast J, Xenarios I, Zelante L, Merla G, Reymond A. TBC1D7 mutations are associated with intellectual disability, macrocrania, patellar dislocation, and celiac disease. Hum Mutat. 2014;35:447–451. doi: 10.1002/humu.22529. [DOI] [PubMed] [Google Scholar]
- 22.Ricos MG, Hodgson BL, Pippucci T, Saidin A, Sze OY, Heron SE, Licchetta L, Bisulli F, Bayly MA, Hughes J, Baldassari S, Palombo F, Epilepsy Electroclinical Study Group. Santucci M, Meletti S, Berkovic SF, Rubboli G, Thomas PQ, Scheffer IE, Tinuper P, Geoghegan J, Schreiber AW, Dibbens LM. Mutations in the mTOR pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2015;79:120–131. doi: 10.1002/ana.24547. [DOI] [PubMed] [Google Scholar]
- 23.Poduri A. DEPDC5 does it all: shared genetics for diverse epilepsy syndromes. Ann Neurol. 2014;75:631–633. doi: 10.1002/ana.24160. [DOI] [PubMed] [Google Scholar]
- 24.Picard F, Makrythanasis P, Navarro V, Ishida S, de BJ, Ville D, Weckhuysen S, Fosselle E, Suls A, De JP, Vasselon RM, Lesca G, Depienne C, An-Gourfinkel I, Vlaicu M, Baulac M, Mundwiller E, Couarch P, Combi R, Ferini-Strambi L, Gambardella A, Antonarakis SE, Leguern E, Steinlein O, Baulac S. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 2014;82:2101–2106. doi: 10.1212/WNL.0000000000000488. [DOI] [PubMed] [Google Scholar]
- 25.Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, Licchetta L, Provini F, Bisulli F, Vadlamudi L, Gecz J, Connelly A, Tinuper P, Ricos MG, Berkovic SF, Dibbens LM. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–787. doi: 10.1002/ana.24126. [DOI] [PubMed] [Google Scholar]
- 26.Scerri T, Riseley JR, Gillies G, Pope K, Burgess R, Mandelstam SA, Dibbens L, Chow CW, Maixner W, Harvey AS, Jackson GD, Amor DJ, Delatycki MB, Crino PB, Berkovic SF, Scheffer IE, Bahlo M, Lockhart PJ, Leventer RJ. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol. 2015;2:575–580. doi: 10.1002/acn3.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei Y, Lilly MA. The TORC1 inhibitors Nprl2 and Nprl3 mediate an adaptive response to amino-acid starvation in Drosophila. Cell Death Differ. 2014;21:1460–1468. doi: 10.1038/cdd.2014.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YJ, Di NA, Kramvis I, Meikle L, Kwiatkowski DJ, Sahin M, He X. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008;22:2485–2495. doi: 10.1101/gad.1685008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baulac S, Ishida S, Marsan E, Miquel C, Biraben A, Nguyen DK, Nordli D, Cossette P, Nguyen S, Lambrecq V, Vlaicu M, Daniau M, Bielle F, Andermann E, Andermann F, Leguern E, Chassoux F, Picard F. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol. 2015;77:675–683. doi: 10.1002/ana.24368. [DOI] [PubMed] [Google Scholar]
- 31.Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9:1–8. doi: 10.1016/j.celrep.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leventer RJ, Scerri T, Marsh AP, Pope K, Gillies G, Maixner W, Mac-Gregor D, Harvey AS, Delatycki MB, Amor DJ, Crino P, Bahlo M, Lockhart PJ. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurology. 2015;84:2029–2032. doi: 10.1212/WNL.0000000000001594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, Shiina M, Shirozu H, Masuda H, Watanabe K, Ohba C, Tsurusaki Y, Miyake N, Zheng Y, Sato T, Takebayashi H, Ogata K, Kameyama S, Kakita A, Matsumoto N. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol. 2015;78:375–386. doi: 10.1002/ana.24444. [DOI] [PubMed] [Google Scholar]
- 34.Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, Cho YW, Kim S, Kim HM, Kim JA, Kim J, Rhee H, Kang SG, Kim HD, Kim D, Kim DS, Lee JH. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. doi: 10.1038/nm.3824. [DOI] [PubMed] [Google Scholar]
- 35.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller–Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 36.Bahi-Buisson N, Poirier K, Fourniol F, Saillour Y, Valence S, Lebrun N, Hully M, Bianco CF, Boddaert N, Elie C, Lascelles K, Souville I, LIS-Tubulinopathies Consortium. Beldjord C, Chelly J. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain. 2014;137:1676–1700. doi: 10.1093/brain/awu082. [DOI] [PubMed] [Google Scholar]
- 37.Tian G, Jaglin XH, Keays DA, Francis F, Chelly J, Cowan NJ. Disease-associated mutations in TUBA1A result in a spectrum of defects in the tubulin folding and heterodimer assembly pathway. Hum Mol Genet. 2010;19:3599–3613. doi: 10.1093/hmg/ddq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cederquist GY, Luchniak A, Tischfield MA, Peeva M, Song Y, Menezes MP, Chan WM, Andrews C, Chew S, Jamieson RV, Gomes L, Flaherty M, Grant PE, Gupta ML, Jr, Engle EC. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum Mol Genet. 2012;21:5484–5499. doi: 10.1093/hmg/dds393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tischfield MA1, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Chan WM, Andrews C, Demer JL, Robertson RL, Mackey DA, Ruddle JB, Bird TD, Gottlob I, Pieh C, Traboulsi EI, Pomeroy SL, Hunter DG, Soul JS, Newlin A, Sabol LJ, Doherty EJ, de Uzcátegui CE, de Uzcátegui N, Collins ML, Sener EC, Wabbels B, Hellebrand H, Meitinger T, de Berardinis T, Magli A, Schiavi C, Pastore-Trossello M, Koc F, Wong AM, Levin AV, Geraghty MT, Descartes M, Flaherty M, Jamieson RV, Møller HU, Meuthen I, Callen DF, Kerwin J, Lindsay S, Meindl A, Gupta ML, Jr, Pellman D, Engle EC. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamilton EM, Wolf NI, van der Knaap MS. Reply: TUBB4A novel mutation reinforces the genotype-phenotype correlation of hypomyelination with atrophy of the basal ganglia and cerebellum. Brain. 2015;138:e328. doi: 10.1093/brain/awu243. [DOI] [PubMed] [Google Scholar]
- 41.Romaniello R, Arrigoni F, Bassi MT, Borgatti R. Mutations in alpha- and beta-tubulin encoding genes: implications in brain malformations. Brain Dev. 2015;37:273–280. doi: 10.1016/j.braindev.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Tischfield MA, Engle EC. Distinct alpha- and beta-tubulin isotypes are required for the positioning, differentiation and survival of neurons: new support for the ‘multi-tubulin’ hypothesis. Biosci Rep. 2010;30:319–330. doi: 10.1042/BSR20100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Esmaeeli NS, Madou MR, Sirajuddin M, Fregeau B, McKnight D, Lexa K, Strober J, Spaeth C, Hallinan BE, Smaoui N, Pappas JG, Burrow TA, McDonald MT, Latibashvili M, Leshinsky-Silver E, Lev D, Blumkin L, Vale RD, Barkovich AJ, Sherr EH. De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Ann Clin Transl Neurol. 2015;2:623–635. doi: 10.1002/acn3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bardon-Cancho EJ, Munoz-Jimenez L, Vazquez-Lopez M, Ruiz-Martin Y, Garcia-Morin M, Barredo-Valderrama E. Periventricular nodular heterotopia and dystonia due to an ARFGEF2 mutation. Pediatr Neurol. 2014;51:461–464. doi: 10.1016/j.pediatrneurol.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 45.Lange M, Kasper B, Bohring A, Rutsch F, Kluger G, Ho an S, Spranger S, Behnecke A, Ferbert A, Hahn A, Oehl-Jaschkowitz B, Graul-Neumann L, Diepold K, Schreyer I, Bernhard MK, Mueller F, Siebers-Renelt U, Beleza-Meireles A, Uyanik G, Janssens S, Boltshauser E, Winkler J, Schuierer G, Hehr U. 47 patients with FLNA associated periventricular nodular heterotopia. Orphanet J Rare Dis. 2015;10:134. doi: 10.1186/s13023-015-0331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carabalona A, Beguin S, Pallesi-Pocachard E, Buhler E, Pellegrino C, Arnaud K, Hubert P, Oualha M, Siffroi JP, Khantane S, Coupry I, Goizet C, Gelot AB, Represa A, Cardoso C. A glial origin for periventricular nodular heterotopia caused by impaired expression of Filamin-A. Hum Mol Genet. 2012;21:1004–1017. doi: 10.1093/hmg/ddr531. [DOI] [PubMed] [Google Scholar]
- 47.Greenwood JS, Wang Y, Estrada RC, Ackerman L, Ohara PT, Baraban SC. Seizures, enhanced excitation, and increased vesicle number in Lis1 mutant mice. Ann Neurol. 2009;66:644–653. doi: 10.1002/ana.21775. [DOI] [PubMed] [Google Scholar]
- 48.Hunt RF, Dinday MT, Hindle-Katel W, Baraban SC. LIS1 deficiency promotes dysfunctional synaptic integration of granule cells generated in the developing and adult dentate gyrus. J Neurosci. 2012;32:12862–12875. doi: 10.1523/JNEUROSCI.1286-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]