

Abstract
Background
Apart from inducing apoptosis in tumor cells, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) influences inflammatory reactions. Murine colon ascendens stent peritonitis (CASP) represents a model of diffuse peritonitis. Recently, it has been demonstrated that administration of exogenous TRAIL not only induces apoptosis in neutrophils but also enhances survival in this model. The aim of this study was to examine the impact of genetic TRAIL deficiency on the course of CASP.
Methods
Peritonitis was induced in 6- to 8-week-old female TRAIL−/− mice as well as in wild-type mice. The sepsis severity score and survival of mice were monitored. Bacterial loads in blood as well as in the lymphoid organs were examined. Additionally, the number of apoptotic cells within the lymphoid organs was determined.
Results
As early as 8 hours postinduction of CASP, TRAIL−/− mice were significantly more affected by sepsis than wild-type mice, as measured by the sepsis severity score. However, during the further course of sepsis, TRAIL deficiency led to significantly decreased sepsis severity scores, resulting in an enhanced overall survival in TRAIL−/− mice. The better survival of TRAIL−/− mice was accompanied by a decreased bacterial load within the blood. In marked contrast, the number of apoptotic cells within the lymphoid organs was highly increased in TRAIL−/− mice 20 hours after induction of CASP.
Conclusion
Hence, exogenous and endogenous TRAIL is protective during the early phase of sepsis, while endogenous TRAIL appears to be detrimental in the later course of this disease.
Keywords: CASP, mice, sepsis, TRAIL, inflammation
Introduction
Despite recent advances in the diagnosis and therapy of sepsis, the mortality due to sepsis and septic shock still remains high.1,2 Sepsis has been defined as an overwhelming inflammatory response to bacterial invasion. Consequently, proinflammatory cytokines such as interleukin (IL)-6,3,4 platelet-activating factor,5 IL-1, and tumor necrosis factor (TNF) predominate in this phase.6 It has been postulated that in response to this hyperinflammatory reaction, a compensatory anti-inflammatory response syndrome is mounted, which is characterized by anti-inflammatory cytokines such as IL-10,6 increased lymphocyte apoptosis, and a shift from T-helper 1 to T-helper 2 subpopulations.7–10 However, recent studies call into question this traditional understanding of pro- versus anti-inflammatory immune response in sepsis.11,12 Moreover, early postoperative sepsis seems to be characterized by the coexistence of pro- and anti-inflammatory cytokine production, resulting in a primary mixed antagonistic response syndrome.12 However, sepsis severity seems to be correlated with a disproportional elevation of anti-inflammation relative to proinflammation.12 Accordingly, Novotny et al12 have shown that murine colon ascendens stent peritonitis (CASP) caused by 16G stent represents a model of high-grade sepsis that causes a systemic cytokine rise until 12 hours after sepsis onset. Due to the disproportionate elevation of the anti-inflammatory response, the primary mixed antagonistic response syndrome shifts toward a predominance of anti-inflammatory cytokines during the course of sepsis in this model.12
TNF-related apoptosis-inducing ligand (TRAIL) is known to induce apoptosis mainly in transformed cells, but it also has an impact on inflammatory reactions. Thus, TRAIL influences the activation of T-cells13 and is important for natural killer (NK) cell-mediated killing.14,15 Using the 16G CASP model, we have previously shown that exogenous TRAIL is highly protective in murine sepsis when administered between 1 hour and 48 hours following induction of 16G CASP.16 The improved survival of TRAIL-treated septic mice was accompanied by decreased bacterial loads within the septic organs, whereas the cytokine levels remained unchanged.16 Furthermore, we have detected increased fractions of apoptotic neutrophils, resulting in a decreased number of neutrophils within the septic organs and less tissue damage in TRAIL-treated animals during sepsis.17 These results are supported by findings in other models of sepsis.18 However, there are also reports indicating that TRAIL is involved in immune unresponsiveness following sepsis.19 Consequently, the authors conclude that TRAIL neutralization might be beneficial for the recovery of cellular immunity in septic patients.19 If, however, on the one hand, administration of a supraphysiological dose of recombinant TRAIL improved bacterial elimination, thereby diminishing septic hyperinflammation, but on the other hand, increased susceptibility to secondary infections, this would restrict the possible clinical application of TRAIL in septic patients.
The aim of this study was to investigate the impact of endogenous TRAIL on the course of murine sepsis using the model of CASP. For these purposes, TRAIL-knockout (TRAIL−/−) mice were used and compared to respective wild-type mice.
Materials and methods
Mice
Eight- to 12-week-old female C57BL/6 mice with body weights of 20–25 g were obtained from Charles River Laboratories (Sulzfeld, Germany) and were allowed to adapt to the new environment for about 14 days. TRAIL−/− C57BL/6 mice were obtained from Amgen (Seattle, WA, USA).20 TRAIL knockout was confirmed by polymerase chain reaction. Mice received food and water ad libitum. Animal rooms had a 12:12-hour circadian cycle and were maintained at constant temperature and humidity. All animal studies had been approved by the ethics committee for animal care of Mecklenburg–Vorpommern, Germany, prior to initiation of the experiments (LALLF M- V/TSD/7221.3-1.1-049/11 and LALLF M-V/TSD/7221.3-1.1-030/08). The experiments were done according to the German Animal Welfare Act (TierSchG).
In vivo TRAIL treatment
Recombinant soluble TRAIL (Biomol GmbH, Hamburg, Germany) was given intravenously at a concentration of 1 µg/g (wt/wt) 1 hour, 24 hours, and 48 hours after induction of CASP. TRAIL was dissolved in sterile saline solution at a final concentration of 0.1 µg/µL.
CASP surgery
CASP surgery was performed as previously described.21 In brief, mice were anesthetized with intraperitoneal administration of ketamine (100 µg/g [wt/wt]) and xylazine (8 µg/g [wt/wt]). The ascending colon was exposed, and a catheter (16-gauge (16G), Venflon; BOC Ohmeda AB, Helsinborg, Sweden) was inserted through the antimesenteric wall into the lumen of the ascending colon and was fixed with two sutures (7/0 Ethilon thread; Ethicon, Norderstedt, Germany). The inner needle of the stent was removed and the stent was cut 2 mm above the puncture site. To ensure proper intraluminal positioning of the stent, stool was milked from the cecum into the stent until a small amount appeared above the stent. Fluid replacement was performed by injecting 0.5 mL of sterile saline solution into the peritoneal cavity before closing the abdominal wall (4/0 Polyester). Postoperatively, mice received buprenorphine (0.1 mg/kg [wt/wt], intramuscularly) to reduce suffering.
Survival analyses and determination of the sepsis severity score
Peritonitis was induced in TRAIL−/− mice and respective wild-type mice. Disease severity (n=20 per group) and survival (n=23 for wild-type mice, n=30 for TRAIL−/− mice) were monitored every 4 hours for 10 days. According to the sepsis severity score reported by Zantl et al,22 disease severity was scored on the basis of general appearance, breathing frequency, and spontaneous as well as provoked behavior. Scoring points reaching from 0 (healthy) to 3 (severe alterations) were assigned to every mouse on each occasion. Mice reaching a severity score that indicated a disease point “of no return” were euthanized by cervical dislocation under deep anesthesia.
Cytokine analyses
TRAIL−/− mice (n=5) and respective wild-type controls (n=5) were sacrificed under general anesthesia by cervical dislocation. Spleens were taken and splenocytes were isolated by passing the organs through a 100 µm nylon mesh (BD Falcon™ cell strainer; BD Biosciences, San Jose, CA, USA). Cells were stored on ice and washed three times with 2% fetal calf serum (FCS) (Biochrom AG, Berlin, Germany) in phosphate-buffered saline (PBS). Cells were maintained in RPMI-1640 medium supplemented with 10% FCS, 100 U/mL of penicillin, and 100 µg/mL of streptomycin. Tissue culture reagents were obtained from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). Lipopolysaccharide (LPS) from Escherichia coli (Sigma-Aldrich, St Louis, MO, USA) was used for stimulation at a final concentration of 1 µg/mL. Cell cultures were maintained in a humidified incubator at 37°C with 5% CO2 for 48 hours.
Supernatants were collected and stored at −80°C until analysis. For cytokine detection, a commercially available cytometric bead array kit (BD Biosciences) was used according to the manufacturer’s instructions. Fold changes were calculated as the ratio of cytokine concentrations of LPS-stimulated cultures to nonstimulated controls.
Proliferation assay of CD4+CD25− cells
CD4+CD25− conventional T-cells were purified from the spleens of wild-type mice using the MACS® cell-sorting system (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s instructions. CD4+CD25−FoxP3− expression was confirmed to be >95% by fluorescence-activated cell sorting (FACS) analysis. Respective antibodies were purchased from BD Biosciences.
T-cells were stimulated with 1 µg/mL plate-bound anti-CD3 antibody and 1 µg/mL of anti-CD28 antibody in RPMI-1640 medium supplemented with 2 mM L-glutamine, 100 units/mL penicillin, 100 µg/mL streptomycin, and 10% heat-inactivated FCS for 72 hours. Recombinant murine TRAIL was added to the cultures at a concentration of 100 ng/mL. Controls received equal volumes of medium. Proliferation was detected using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based proliferation kit (Sigma-Aldrich) according to the manufacturer’s instructions. The optical densities (ODs) of samples were separately measured using a microplate reader at a wave length of 570 nm. The results of the proliferation assay were expressed as a stimulation index, which was calculated by dividing the mean OD of stimulated cultures by the OD of the nonstimulated (control) cultures.
Bacteriology
For quantification of the bacterial load, CASP was performed in TRAIL−/− mice (n=8) and respective wild-type controls (n=16) as described herein. For control purposes, a subgroup of wild-type mice received TRAIL treatment following CASP. The livers, lungs, kidneys, and spleens were removed under general anesthesia 20 hours after CASP surgery. Removed organs were placed in 5 mL of ice-cold sterile saline solution and were homogenized for 30 seconds at a rate of 2,500 rpm by using a tissue homogenizer (T25 Ultra-Turrax Basic; IKA, Staufen, Germany). Homogenates were plated at different dilutions on blood-containing agar (Columbus 5% SB; BD Biosciences) and incubated for 18 hours at 37°C. The number of colony-forming units was calculated for each organ. Whole blood was collected in ethylenediaminetetraacetic acid vials (Becton Dickinson, Franklin Lakes, NJ, USA), 10 µL was plated onto agar plates for analysis of bacterial loads, and colonies were counted after an incubation period of 18 hours. Results were normalized to 1 mL of whole blood.
Detection of neutrophils within the organs of septic mice
For detection of neutrophils, thymus and spleens of TRAIL−/− mice and respective wild-type mice (n=5 per group) were taken 20 hours following CASP. Organs were snap-frozen in liquid nitrogen. Frozen sections (5 µm) were cut for further studies. To visualize neutrophils, sections were incubated with rat anti-Ly6G (clone RB6-8C5, rat IgG2a kappa; BD Pharmingen) and an appropriate fluorescein isothiocyanate (FITC)-labeled secondary antibody (FITC mouse anti-rat IgG2b; BD Pharmingen). Slides were analyzed by confocal laser scanning microscopy using an Olympus IX70 microscope (Olympus America Inc., Center Valley, PA, USA). Staining was analyzed using the imaging program Metamorph (Visitron, Puchheim, Germany).
Detection of apoptotic cells within lymphoid organs
Peritonitis was induced in TRAIL−/− and wild-type animals. Mice were sacrificied 6 hours and 20 hours after CASP. Cells of the thymus (n≥5 per group) and the spleen (n≥5 per group) were isolated by passing the organs through a 100 µm nylon mesh (BD Falcon cell strainer; BD Biosciences). Cells were stored on ice and washed three times with 2% FCS in PBS. Then, an erythrocyte lysis buffer containing 155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM ethylenediaminetetraacetic acid was added. A total of 105 cells was fixed in 70% ethanol and incubated with 0.025 M sodium citrate and 0.067 M disodium phosphate at pH 7.8 at room temperature. The pellets were washed with PBS plus 5% FCS, resuspended in 30 µL RNase (1 mg/mL; Qiagen, Venlo, the Netherlands), and stained with 25 µg/mL propidium iodide (Sigma Aldrich). Apoptosis was measured by flow cytometry (FACSCalibur™; BD Biosciences). The hypodiploid DNA peaks in single variable DNA histograms were identified.
To confirm these data via immunohistochemistry, organs of respective septic mice were harvested 20 hours after induction of CASP. Frozen sections of 5 µm thickness were cut. Apoptosis was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining using an Apopt-Tag Fluorescein kit (Chemicon International, Temecula, CA, USA) according to the manufacturer’s instructions. The number of TUNEL-positive cells was counted using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Statistical methods
Survival analyses were performed using the Kaplan–Meier method. Differences in survival were analyzed by a log-rank test. Continuous variables were tested for Gaussian distribution using an omnibus K2 test. Data were compared using a Student’s t-test or a Mann–Whitney test if data were not normally distributed. For all calculations, Graph Pad Prism (version 6.00 for Macintosh; GraphPad Software, La Jolla, CA, USA) was used. A P-value <0.05 was considered statistically significant. Data are expressed as mean ± standard deviation.
Results
TRAIL deficiency significantly increased sepsis severity at 8 hours following induction of CASP
To examine the impact of TRAIL deficiency on sepsis severity, the sepsis severity score was determined every 4 hours for 100 hours. As early as 8 hours following induction of 16G CASP, the sepsis severity score reached only 2.9±0.95 in wild-type mice (n=20). However, the score increased to 7.2±0.4 in TRAIL−/− mice (n=20; P=0.0006) at the same time point, indicating that these mice were significantly more affected by sepsis at this early time point when compared to wild-type mice (Figure 1A).
Figure 1.

TRAIL deficiency improved survival in CASP.
Notes: (A) Peritonitis was induced in wild-type and TRAIL−/− mice (n=20 per group). Sepsis severity scores were determined every 4 hours for 100 hours according to Zantl et al.22 Mean values and standard errors of the mean at respective time points are shown for wild-type (black) and TRAIL−/− mice (gray). Eight hours after induction of CASP, TRAIL−/− mice displayed highly significantly increased sepsis severity scores when compared to wild-type mice. In marked contrast, when compared to wild-type mice, sepsis severity was decreased in TRAIL−/− mice from 16 hours until 72 hours following CASP. A representative result of two experiments is shown. *P<0.05; **P<0.01; ***P < 0.001. (B) Peritonitis was induced in wild-type and TRAIL−/− mice (n≥23 per group). An additional group of wild-type mice received TRAIL treatment after induction of CASP (wild type + TRAIL). Survival was monitored every 4 hours. Kaplan–Meier curves are shown for wild-type mice (black), TRAIL-treated wild-type mice (dashed line), and TRAIL−/− mice (gray). TRAIL deficiency significantly improved survival. As previously shown,16 TRAIL treatment highly significantly improved survival after CASP. *P<0.05; **P<0.01 when compared to septic wild-type controls.
Abbreviations: CASP, colon ascendens stent peritonitis; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
TRAIL deficiency significantly decreased sepsis severity from 20 hours until 72 hours following induction of CASP
Twelve hours after induction of 16G CASP, the situation was inversed: the sepsis severity score was now slightly higher in wild types than in TRAIL−/− mice (6.7±0.6 versus 5.6±0.6; P=0.2). This trend increased, reaching significance at 20 hours following CASP (7.7±0.4 in wild-type mice versus 6.2±0.4 in TRAIL−/− mice; P=0.0093) (Figure 1A). The difference between wild-type and TRAIL−/− mice further increased up to 52 hours following CASP, reaching a Severity Score of 5.2±1.3 in wild-type mice compared to 1.4±0.2 in TRAIL−/− mice (P=0.0198) and remained significant until 72 hours following CASP.
Thus, the initially increased sepsis severity in TRAIL−/− mice is followed by a significantly decreased sepsis severity during the further course of sepsis.
TRAIL deficiency significantly improved survival of mice after sepsis
Peritonitis was induced in TRAIL−/− mice and respective wild-type controls (n=43 for wild-type mice, n=30 for TRAIL−/− mice). A subgroup of wild-type mice (n=20) was treated with TRAIL. While 22% of wild-type mice survived 16G CASP, the survival was 48% in TRAIL−/− mice. Thus, TRAIL deficiency significantly improved survival in CASP (chi-square test: 6.5; P=0.01) (Figure 1B). As previously shown, the administration of supraphysiological amounts of recombinant TRAIL improved survival of CASP.16 In detail, survival was 75% in TRAIL-treated wild-type mice (Figure 1B).
TRAIL deficiency significantly reduced bacterial dissemination within the blood during sepsis
Twenty hours following induction of 16G CASP, TRAIL−/− mice displayed significantly lower amounts of bacteria within the blood when compared to wild-type mice (n=8 per group). Thus, there were 213.6±21.3 colony-forming units in wild-type mice, whereas there were only 26.5±21.2 in TRAIL−/− mice (P=0.01, Figure 2A). As previously shown,16 TRAIL treatment of septic wild-type mice reduced bacterial loads within the blood (P=0.001, Figure 2A) and within the spleen (P=0.02, Figure 2B). In TRAIL−/− mice, there were also trends toward lower bacterial loads in septic spleens, lungs, and livers, although not reaching significance. In detail, there were 212.1±21.0 colony-forming units in septic spleens of wild-type mice compared to 29.0±21.7 in spleens of TRAIL−/− mice (P=0.1). The lungs of septic TRAIL−/− mice showed 29.9±21.6 colony-forming units, whereas there were 212.1±21.0 within septic lungs of wild-type mice (P=0.4). Within the livers of septic wild-type mice we found 211.7±21.1 colony-forming units, whereas there were 28.9±21.6 colony-forming units in livers of TRAIL−/− mice (P=0.1).
Figure 2.

TRAIL deficiency decreased bacterial loads within the blood during CASP.
Notes: TRAIL deficiency did not alter the number of infiltrating neutrophils during sepsis. Peritonitis was induced in wild-type mice (n=16 per group) and TRAIL−/− mice (n=8 per group). A subgroup of wild-type mice (n=8) received TRAIL treatment following CASP induction. (A and B) 20 hours following CASP, bacterial loads within the spleen and the blood were determined. Mean values and standard errors of the means of the binary logarithms of the CFUs are shown. As previously shown,16 TRAIL treatment significantly decreased bacterial loads within the blood and the spleen following CASP. In septic TRAIL−/− mice, bacterial loads within the blood (A) were significantly decreased when compared to septic wild-type mice. Bacterial load within the spleen (B) was also reduced in septic TRAIL−/− mice but failed to attain statistical significance. (C–F) Neutrophils were detected by Ly6G-staining via immunohistochemistry (n=5 per group). Mean values and standard errors of the means of the number of neutrophils per HPF within the (C) spleens and (D) thymi of septic mice are depicted. (E and F) Scale bars =50 µm. Representative Ly6G-stained sections of septic wild-type (E) and TRAIL−/− (F) mice are shown. *P<0.05 when compared to septic wild-type control; **P<0.01 when compared to septic wild-type control.
Abbreviations: CASP, colon ascendens stent peritonitis; CFUs, colony-forming units; HPF, high-power field; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
TRAIL deficiency did not alter the number of infiltrating neutrophils but significantly decreased the number of apoptotic neutrophils during sepsis
Spleens and thymi of septic TRAIL−/− and septic wild-type mice were taken 20 hours after CASP and immunohistochemistry was performed. Within the spleen, the number of neutrophils per high-power field (HPF) did not differ between septic TRAIL−/− and septic wild-type mice (61±9.5 per HPF in wild-type mice versus 67.8±10.7 per HPF in TRAIL−/− mice, n=5 per group; P=0.65; Figure 2C). Regarding the number of neutrophils per HPF within the thymus, there were no differences between septic wild-type and septic TRAIL−/− mice (21.3±3.2 versus 25.6±6.7; P=0.6; Figure 2D–F). However, the fraction of apoptotic neutrophils was significantly decreased in TRAIL−/− mice 20 hours following CASP (3.1%±0.7% in wild-type mice versus 1.5%±0.2% in TRAIL−/− mice; P=0.048).
TRAIL deficiency significantly increased sepsis-induced apoptosis within the lymphoid organs 20 hours after induction of CASP
For detection of apoptotic cells, propidium iodide cell cycle analyses were performed in spleens and thymi 6 hours and 20 hours following induction of CASP. In untreated mice, the fraction of apoptotic cells did not significantly differ between wild-type and TRAIL−/− mice. However, there was a trend toward a decreased fraction of apoptotic cells in TRAIL−/− mice when compared to wild-type mice (P=0.096 for spleens and P=0.061 for thymi). As early as 6 hours following CASP, sepsis led to induction of apoptosis within the spleen and within the thymus in wild-type as well as in TRAIL−/− mice. In untreated wild-type mice, the fractions of apoptotic cells were 2.5%±0.8% within the thymus and 3.7%±0.9% within the spleen. Six hours following CASP, there were 11.0%±1.1% apoptotic cells within the thymus (P=0.0001) and 5.3%±0.4% (P=0.1) apoptotic cells within the spleen in wild-type mice. In TRAIL−/− mice, the fraction of apoptotic cells increased from 0.25%±0.03% to 11.68%±1.83% within the thymus (P=0.0001) and from 2.1%±0.33% to 5.8%±0.65% within the spleen 6 hours following CASP. As early as 6 hours after induction of CASP, there were no significant differences between wild-type and TRAIL−/− mice with respect to the fraction of apoptotic cells. In marked contrast, 20 hours after induction of CASP, the fraction of apoptotic cells was significantly increased in spleens and thymi of TRAIL−/− mice when compared to respective septic wild-type controls (P=0.0395 in spleens [Figure 3A] and P=0.0242 in thymi [Figure 3B]).
Figure 3.

TRAIL deficiency increased sepsis-induced apoptosis.
Notes: Apoptosis was detected in untreated wild-type mice and TRAIL−/− mice. Additionally, 16G CASP was performed in wild-type and TRAIL−/− mice and apoptosis was detected 6 hours and 20 hours following CASP. (A and B) Propidium iodide cell cycle analyses were performed in (A) spleens and (B) thymi of untreated wild-type mice (n=8) and untreated TRAIL−/− mice (n=9), as well as in wild-type and TRAIL−/− mice 6 hours and 20 hours following CASP (n≥5 per group). The fraction of apoptotic cells within the organs was determined by FACS analysis. (A) Mean values and standard errors of the means of the fraction of apoptotic cells within the spleen are shown. (B) Mean values and standard errors of the means of the fraction of apoptotic cells within the thymus are shown. (C–H) TUNEL staining of apoptotic cells was performed. Sections were examined by fluorescent microscopy, and the number of apoptotic cells per HPF was counted using ImageJ. (C–F) Representative TUNEL-stained sections of spleens are shown. Original magnifications: ×20; scale bars =50 µm. (C) Spleen of an untreated wild-type mouse; (D) spleen of an untreated TRAIL−/− mouse; (E) spleen of a wild-type mouse 20 hours following CASP induction; (F) spleen of a TRAIL−/− mouse 20 hours after induction of CASP. (G) Quantification of TUNEL stain within the spleen was performed for untreated wild-type (n=5) and untreated TRAIL−/− mice (n=5), as well as for wild-type mice, 20 hours following CASP (n=5) and for TRAIL−/− mice 20 hours following CASP (n=5). Mean values and standard errors of the mean of the number of apoptotic cells per HPF are shown. (H) TUNEL stainings within the thymi of wild-type and TRAIL−/− mice without treatment as well as 20 hours following CASP were performed. The mean values of the apoptotic cells per HPF and the standard errors of the means are shown. *P<0.05; **P<0.01.
Abbreviations: CASP, colon ascendens stent peritonitis; FACS, fluorescence-activated cell sorting; HPF, high-power field; TNF. tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Additionally, apoptotic cells were visualized in spleens and thymi of untreated TRAIL−/− and wild-type mice by immunohistochemistry using TUNEL staining 20 hours following CASP. The number of TUNEL-positive cells per HPF was determined. In untreated mice, the fraction of apoptotic cells did not differ between TRAIL−/− mice and their respective wild-type controls (P=0.28 for spleens, P=0.88 for thymi; Figure 3C–H). When comparing septic TRAIL−/− to septic wild-type mice 20 hours following CASP, the fraction of apoptotic cells was significantly higher in spleens and thymi of septic TRAIL−/− mice (P=0.01 for spleens, Figure 3A; P=0.03 for thymi, Figure 3C–H). Thus, 20 hours following CASP, TRAIL deficiency significantly increased sepsis-induced apoptosis within the lymphoid organs, whereas there were no significant differences as early as 6 hours following CASP. In untreated animals, TRAIL−/− mice displayed a trend toward decreased fractions of apoptotic cells within the lymphoid organs.
Cytokine levels following stimulation by LPS did not significantly differ between TRAIL−/− and wild-type splenocytes
Splenocytes of untreated TRAIL−/− mice and respective wild-type controls were harvested. Cultures of splenocytes were stimulated through addition of LPS, and the levels of cytokines (IL-6, TNFα, IL-12, IL-10, and interferon [IFN]γ) and monocyte chemotactic protein (MCP-1) within the supernatants were measured. Fold changes were calculated as the ratio of cytokine concentrations of LPS-stimulated cultures to nonstimulated controls. There were no significant differences between levels of cytokines/chemokines in nonstimulated splenocytes of wild-type mice compared to splenocytes of TRAIL−/− mice (Table 1). In splenocytes of wild-type mice, LPS stimulation significantly induced the release of IL-6, TNFα, MCP-1, and IFNγ. However, there were no significant differences between LPS-stimulated TRAIL−/− splenocytes and LPS-stimulated splenocytes of respective wild-type mice. Nevertheless, in LPS-stimulated TRAIL−/− splenocytes, there was a trend toward increased concentrations of TNFα and IFNγ when compared to LPS-stimulated wild-type splenocytes (Table 1). For TNFα, IFNγ, and IL-6, fold changes were increased in TRAIL−/− mice when compared to wild-type mice. However, fold changes of IL-10 levels were higher in wild-type mice when compared to TRAIL−/− mice. Remarkably, LPS stimulation did not induce the release of IL-12, neither in wild-type mice nor in TRAIL−/− mice.
Table 1.
In vitro cytokine response of harvested splenocytes of wild-type mice and TRAIL−/− mice to LPS stimulation
| Cytokine/chemokine | Wild-type splenocytes
|
TRAIL-/- splenocytes
|
P-value | Wild-type splenocytes + LPS
|
TRAIL−/− splenocytes + LPS
|
P-value | Fold change
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentration (pg/mL) | n | Concentration (pg/mL) | n | Concentration (pg/mL) | n | Concentration (pg/mL) | n | Wild-type + LPS/wild-type mice | TRAIL−/− LPS/TRAIL−/− | |||
| TNFα | 17.83±1.17 | 11 | 21.36±2.60 | 12 | 0.4398 | 355.3±63.03 | 12 | 532.6±76.14 | 12 | 0.0858 | 19.93 | 24.93 |
| IFNγ | 3.14±0.15 | 12 | 3.45±0.14 | 12 | 0.1168 | 26.22±10.26 | 12 | 44.64±13.56 | 12 | 0.0805 | 8.35 | 13.05 |
| IL-6 | 17.18±4.71 | 10 | 11.56±0.47 | 10 | 0.7102 | 136.3±34.60 | 10 | 171.8±10.14 | 10 | 0.1121 | 7.93 | 14.86 |
| MCP-1 | 71.81±16.49 | 5 | 65.59±4.56 | 5 | 0.6667 | 143.7±57.18 | 5 | 135.0±4.83 | 5 | 0.8831 | 2.00 | 2.06 |
| IL-12 | 22.77±2.27 | 5 | 22.02±1.91 | 5 | 0.5000 | 22.04±1.54 | 5 | 24.68±1.49 | 5 | 0.2222 | 0.97 | 1.12 |
| IL-10 | 26.31±1.44 | 5 | 31.90±3.72 | 5 | 0.8016 | 133.8±40.35 | 5 | 104.1±6.14 | 5 | 0.9444 | 5.09 | 3.26 |
Notes: Splenocytes of wild-type mice and TRAIL−/− mice were cultured for 48 hours with or without LPS. Levels of TNFα, IFNγ, IL-6, MCP-1, IL-12, and IL-10 in supernatants were determined. Shown are the mean concentrations ± SEMs of respective cytokines/chemokines in the supernatants of LPS-stimulated and unstimulated cultures of splenocytes harvested from TRAIL−/− mice and wild-type controls. P-values were obtained by either comparing unstimulated wild-type splenocytes versus unstimulated TRAIL−/− splenocytes or by comparing LPS-stimulated wild-type splenocytes to LPS-stimulated TRAIL−/− splenocytes.
Abbreviations: IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; MCP, monocyte chemotactic protein; SEM, standard error of the mean; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
TRAIL impaired the proliferative capacity of murine CD4 cells
To assess the impact of TRAIL on CD4 cell proliferation, effector T-cells were stimulated with anti-CD3/anti-CD28 and cultured with and without TRAIL (n=5 per group).
In nonstimulated splenocytes, the stimulation index was 1.0±0.14 without TRAIL and 0.9±0.16 in TRAIL-treated cell cultures, indicating that TRAIL stimulation alone had no significant effect on T-cell proliferation. As expected, CD3/CD28 stimulation led to strong proliferation of effector T-cells. However, TRAIL significantly decreased the proliferation induced by CD3/CD28 stimulation (stimulation index: 3.4±0.2 without TRAIL treatment versus 2.2±0.1 in TRAIL-treated cell cultures; P=0.0025) (Figure 4). This indicates that TRAIL can inhibit effector T-cell proliferation induced by T-cell receptor stimulation.
Figure 4.
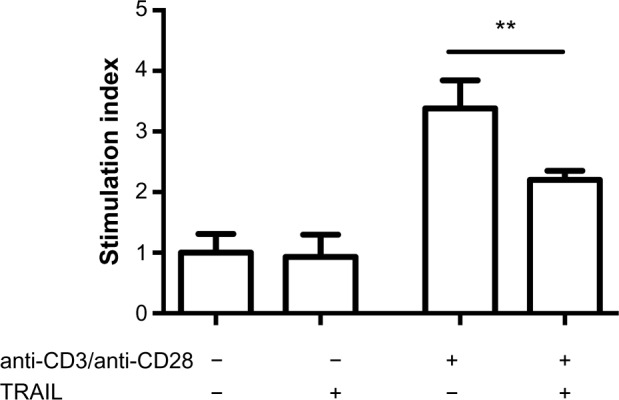
TRAIL impaired the proliferative capacity of murine CD4 cells.
Notes: To assess the impact of TRAIL on CD4 cell proliferation, effector T-cells were subjected to anti-CD3/anti-CD28 stimulation with and without TRAIL. T-cell proliferation was determined by the MTT assay. The results are expressed as SI, which is calculated by dividing the mean optical density of stimulated cultures by the optical density of the nonstimulated (control) cultures. Bars represent mean values and standard errors of the mean. TRAIL significantly decreased proliferation induced by anti-CD3/anti-CD28 stimulation (P=0.0025). **P<0.01.
Abbreviations: MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; SI, stimulation index; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
Discussion
Despite recent advances in the understanding of TRAIL function, the potential clinical benefits of TRAIL in the treatment of sepsis remain the topic of ongoing discussions.16,18,19 As previously shown, the administration of supraphysiological amounts of recombinant TRAIL improved survival of mice after CASP.16 This protective effect of exogenously administered TRAIL was accompanied by an increased fraction of apoptotic neutrophils, and depletion of neutrophils abrogated the TRAIL-induced survival benefit.17 In marked contrast to the induction of apoptosis in septic neutrophils, TRAIL treatment led to an overall decrease in sepsis-induced apoptosis within the lymphoid organs.16 In accordance with our previously reported results, McGrath et al18 found that in zymosan-induced peritonitis, a model of neutrophilic inflammation, TRAIL−/− mice displayed enhanced inflammatory responses compared to wild-type mice. This was accompanied by a decreased fraction of apoptotic neutrophils within the peritoneal lavage. Corroborating these findings, exogenous TRAIL accelerated the apoptotic death of neutrophils in TRAIL−/− as well as in wild-type mice.18 Altogether, these results strongly indicate a dampening effect of exogenous TRAIL on inflammation, which was beneficial in several inflammation models, in particular in those dominated by neutrophils.
Other groups, in contrast, have reported detrimental effects of TRAIL in several inflammatory settings.19,23 Particularly immune suppression following sepsis appears to be mediated by TRAIL. Gurung et al19 have shown that TRAIL impaired the outcome of secondary bacterial infections following sepsis. The authors reported that this effect was dependent upon CD8+ T-cells. It was concluded that TRAIL-expressing regulatory CD8+ T-cells were also causally related to immune unresponsiveness after sepsis.19 Additionally, TRAIL–receptor costimulation inhibited T-cell activation and proliferation.24 This implies that TRAIL neutralization, rather than TRAIL administration, may be therapeutic at later stages of the disease course of sepsis, acting by restoring the cellular immune response.
In addition to CD4 cells that express the death receptor (DR)5, there are myeloid-derived suppressor cells (MDSCs) that express high amounts of TRAIL receptors and are highly sensitive to TRAIL-induced apoptosis.25 Although most data regarding these cells have come from cancer studies, the number of MDSCs has been shown to be increased during sepsis also.26 However, the role of MDSCs in sepsis has been controversial.
To resolve the conflicting interpretations of the role of TRAIL in sepsis, analyses were performed in this study in TRAIL−/− mice to investigate the impact of endogenous TRAIL on murine peritonitis. In our recent studies, exogenous TRAIL had been administered as early as 1 hour after induction of CASP, resulting in good protection during the early course of sepsis. The observed effect was mediated via induction of neutrophil apoptosis and was targeted at the very early hyperinflammatory phase of sepsis.17 Nevertheless, a secondary detrimental effect of TRAIL in murine peritonitis could not be excluded based on these data due to the short half-life of TRAIL.
In this study, peritonitis was induced in TRAIL−/− mice. In line with previously reported results, TRAIL−/− mice displayed lower levels of apoptotic neutrophils 20 hours following CASP induction. However, in this study, TRAIL deficiency did not alter the total number of neutrophils within the lymphoid organs during sepsis. According to the results in TRAIL-treated septic mice, the decrease in the fraction of apoptotic neutrophils is accompanied by significantly increased numbers of apoptotic cells within the septic organs. As previously shown, sepsis-induced apoptosis mainly affects T-cells.27,28 Accordingly, in this study, the highest amounts of apoptotic cells were detected within the thymus during sepsis, indicating that the increase of apoptotic cells in TRAIL−/− mice was due to higher amounts of apoptotic T-cells.
According to the decreased fraction of apoptotic neutrophils and the increase of sepsis-induced overall apoptosis, TRAIL−/− mice displayed increased sepsis severity scores very early in sepsis. However, this was rapidly reversed, and from 20 hours after sepsis induction, TRAIL−/− mice appeared to be less severely affected than wild-type mice. Importantly, survival was significantly better in TRAIL−/− mice, indicating an overall detrimental effect of endogenous TRAIL. In line with the improved survival, bacterial loads in the blood were significantly decreased in septic TRAIL−/− mice when compared to septic wild-type mice. These findings strongly indicate that TRAIL−/− mice were more effective in combating the underlying infection, suggesting that endogenous TRAIL may interfere with bacterial clearance. At first sight, this appears to be in contrast to our previously reported data, which showed that administration of high-dose TRAIL within 1 hour after sepsis induction dramatically reduced bacterial dissemination and apoptosis, as well as increasing survival, making a case for a beneficial role of TRAIL in sepsis.16 The two conflicting observations may be reconciled when taking the time course of sepsis into account: Novotny et al12 have shown that early postoperative sepsis leads to a primary mixed antagonistic response syndrome. However, sepsis severity was positively correlated with an increased elevation of anti-inflammatory response compared to the proinflammatory response.12 Induction of 16G CASP led to a systemic rise of IL-6 and IL-10 until 12 hours after induction of CASP, whereas in 20G CASP, the highest levels of systemic cytokines were observed 3 hours after induction of CASP.12 Due to the disproportionate elevation of the anti-inflammatory response relative to the proinflammatory response, the anti-inflammatory response becomes predominant during the later course of sepsis in 16G CASP.12
Therefore, early intravenous administration of recombinant TRAIL kills neutrophils, resulting in resolution of hyperinflammation.17 Accordingly, the fraction of apoptotic neutrophils was significantly decreased in septic TRAIL−/− mice when compared to respective septic wild-type controls. In agreement with this, lack of endogenous TRAIL, as was studied here, increased sepsis severity very early after disease onset. However, in this study, this effect was not decisive for the outcome. Obviously, apart from endogenous TRAIL, there are other mechanisms that can efficiently control hyperinflammation in the early stage of sepsis.
Later, during the course of severe sepsis – as represented by 16G CASP, however, the anti-inflammatory response becomes dominant.12 Our data and those of others13,19,29,30 indicate that endogenous TRAIL contributes to this state of profound immune suppression. In this study, this was supported by the finding that TRAIL inhibits proliferation of T-effector cells to anti-CD3/anti-CD28. These results are in line with data published by Lehnert et al24 showing that human T-cell activation and proliferation can be suppressed through TRAIL application. Considering cytokine production by splenocytes following LPS stimulation, there were no significant differences between wild-type and TRAIL−/− splenocytes. However, fold changes of proinflammatory cytokines induced by LPS stimulation tended to be higher in TRAIL−/− mice when compared to wild-type mice. Additionally, fold changes of IL-10 were higher in wild-type mice, indicating the anti-inflammatory effects of TRAIL.
Apparently, immune suppression was the dominating effect in this study, explaining the overall beneficial effect of TRAIL deficiency on sepsis survival. In contrast, when applied very early after the onset of sepsis, exogenous TRAIL would not affect the immune-suppressive phase due to its short half-life of 3–5 minutes, explaining the net positive effect of this intervention.
With regard to apoptosis induction, our findings are in agreement with the general antiapoptotic effect of exogenous TRAIL, previously observed by us, 20 hours following CASP.16 However, in the very early course of sepsis, the fraction of apoptotic cells does not significantly differ between TRAIL−/− mice and respective wild-type controls.
Probably the lack of endogenous TRAIL during the early phase of sepsis increased the hyperinflammation and, as a consequence, also increased immune cell apoptosis and sepsis severity in the TRAIL−/− animals. In this study, however, this did not outweigh the beneficial impact of a lower degree of TRAIL-mediated immune suppression in the TRAIL−/− mice, which – in the end – survived better than the wild-type controls.
We therefore propose that TRAIL has an anti-inflammatory – and thereby antiapoptotic – effect in sepsis, which is beneficial during the early phase but detrimental when immune suppression predominates. This ambiguous role of TRAIL in sepsis should be taken into account when considering the possible application of TRAIL for the treatment of clinical sepsis.
Acknowledgments
The authors would like to express their utmost gratitude to Amgen for the kind donation of the TRAIL−/− mice. Our special thanks go to Kristi Phalmer as well as Ramiro Kasimiro. We also wish to thank Antje Janetzko and Doreen Biedenweg for excellent technical assistance.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Engel C, Brunkhorst FM, Bone HG, et al. Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive Care Med. 2007;33(4):606–618. doi: 10.1007/s00134-006-0517-7. [DOI] [PubMed] [Google Scholar]
- 2.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41(5):1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 3.Remick DG, Bolgos G, Copeland S, Siddiqui J. Role of interleukin-6 in mortality from and physiologic response to sepsis. Infect Immun. 2005;73(5):2751–2757. doi: 10.1128/IAI.73.5.2751-2757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pallua N, von Heimburg D. Pathogenic role of interleukin-6 in the development of sepsis. Part I: study in a standardized contact burn murine model. Crit Care Med. 2003;31(5):1490–1494. doi: 10.1097/01.CCM.0000065724.51708.F5. [DOI] [PubMed] [Google Scholar]
- 5.Opal SM, Dellinger RP, Vincent JL, Masur H, Angus DC. The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C?*. Crit Care Med. 2014;42(7):1714–1721. doi: 10.1097/CCM.0000000000000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4(10):854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 7.Hotchkiss RS, Coopersmith CM, Karl IE. Prevention of lymphocyte apoptosis – a potential treatment of sepsis? Clin Infect Dis. 2005;41(suppl 7):S465–S469. doi: 10.1086/431998. [DOI] [PubMed] [Google Scholar]
- 8.Muenzer JT, Davis CG, Dunne BS, Unsinger J, Dunne WM, Hotchkiss RS. Pneumonia after cecal ligation and puncture: a clinically relevant “two-hit” model of sepsis. Shock. 2006;26(6):565–570. doi: 10.1097/01.shk.0000235130.82363.ed. [DOI] [PubMed] [Google Scholar]
- 9.Muenzer JT, Davis CG, Chang K, et al. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun. 2010;78(4):1582–1592. doi: 10.1128/IAI.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez HG, Gonzalez SM, Londono JM, et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study*. Crit Care Med. 2014;42(4):771–780. doi: 10.1097/CCM.0000000000000100. [DOI] [PubMed] [Google Scholar]
- 11.Osuchowski MF, Craciun F, Weixelbaumer KM, Duffy ER, Remick DG. Sepsis chronically in MARS: systemic cytokine responses are always mixed regardless of the outcome, magnitude, or phase of sepsis. J Immunol. 2012;189(9):4648–4656. doi: 10.4049/jimmunol.1201806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novotny AR, Reim D, Assfalg V, et al. Mixed antagonist response and sepsis severity-dependent dysbalance of pro- and anti-inflammatory responses at the onset of postoperative sepsis. Immunobiology. 2012;217(6):616–621. doi: 10.1016/j.imbio.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 13.Xiao H, Wang S, Miao R, Kan W. TRAIL is associated with impaired regulation of CD4+CD25- T cells by regulatory T cells in patients with rheumatoid arthritis. J Clin Immunol. 2011;31(6):1112–1119. doi: 10.1007/s10875-011-9559-x. [DOI] [PubMed] [Google Scholar]
- 14.Hayakawa Y, Screpanti V, Yagita H, et al. NK cell TRAIL eliminates immature dendritic cells in vivo and limits dendritic cell vaccination efficacy. J Immunol. 2004;172(1):123–129. doi: 10.4049/jimmunol.172.1.123. [DOI] [PubMed] [Google Scholar]
- 15.Collison A, Foster PS, Mattes J. Emerging role of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) as a key regulator of inflammatory responses. Clin Exp Pharmacol Physiol. 2009;36(11):1049–1053. doi: 10.1111/j.1440-1681.2009.05258.x. [DOI] [PubMed] [Google Scholar]
- 16.Cziupka K, Busemann A, Partecke LI, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) improves the innate immune response and enhances survival in murine polymicrobial sepsis. Crit Care Med. 2010;38(11):2169–2174. doi: 10.1097/CCM.0b013e3181eedaa8. [DOI] [PubMed] [Google Scholar]
- 17.Beyer K, Poetschke C, Partecke LI, et al. TRAIL induces neutrophil apoptosis and dampens sepsis-induced organ injury in murine colon ascendens stent peritonitis. PLoS One. 2014;9(6):e97451. doi: 10.1371/journal.pone.0097451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGrath EE, Marriott HM, Lawrie A, et al. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol. 2011;90(5):855–865. doi: 10.1189/jlb.0211062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurung P, Rai D, Condotta SA, Babcock JC, Badovinac VP, Griffith TS. Immune unresponsiveness to secondary heterologous bacterial infection after sepsis induction is TRAIL dependent. J Immunol. 2011;187(5):2148–2154. doi: 10.4049/jimmunol.1101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sedger LM, Glaccum MB, Schuh JC, et al. Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur J Immunol. 2002;32(8):2246–2254. doi: 10.1002/1521-4141(200208)32:8<2246::AID-IMMU2246>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 21.Maier S, Traeger T, Entleutner M, et al. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock. 2004;21(6):505–511. doi: 10.1097/01.shk.0000126906.52367.dd. [DOI] [PubMed] [Google Scholar]
- 22.Zantl N, Uebe A, Neumann B, et al. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun. 1998;66(5):2300–2309. doi: 10.1128/iai.66.5.2300-2309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Unsinger J, Kazama H, McDonough JS, Griffith TS, Hotchkiss RS, Ferguson TA. Sepsis-induced apoptosis leads to active suppression of delayed-type hypersensitivity by CD8+ regulatory T cells through a TRAIL-dependent mechanism. J Immunol. 2010;184(12):6766–6772. doi: 10.4049/jimmunol.0904054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehnert C, Weiswange M, Jeremias I, et al. TRAIL-receptor costimulation inhibits proximal TCR signaling and suppresses human T cell activation and proliferation. J Immunol. 2014;93(8):4021–4031. doi: 10.4049/jimmunol.1303242. [DOI] [PubMed] [Google Scholar]
- 25.Condamine T, Kumar V, Ramachandran IR, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124(6):2626–2639. doi: 10.1172/JCI74056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai D, Qin C, Shu Q. Myeloid-derived suppressor cells in sepsis. Biomed Res Int. 2014;2014:598654. doi: 10.1155/2014/598654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood. 1996;87(10):4261–4275. [PubMed] [Google Scholar]
- 28.Busse M, Traeger T, Potschke C, et al. Detrimental role for CD4+ T lymphocytes in murine diffuse peritonitis due to inhibition of local bacterial elimination. Gut. 2008;57(2):188–195. doi: 10.1136/gut.2007.121616. [DOI] [PubMed] [Google Scholar]
- 29.Condotta SA, Cabrera-Perez J, Badovinac VP, Griffith TS. T-cell-mediated immunity and the role of TRAIL in sepsis-induced immunosuppression. Crit Rev Immunol. 2013;33(1):23–40. doi: 10.1615/critrevimmunol.2013006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anel A, Bosque A, Naval J, et al. Apo2L/TRAIL and immune regulation. Front Biosci. 2007;12:2074–2084. doi: 10.2741/2212. [DOI] [PubMed] [Google Scholar]