

Abstract
Loss of plasma membrane asymmetry is a hallmark of apoptosis, but lipid bilayer asymmetry and loss of asymmetry can contribute to numerous cellular functions and responses that are independent of programmed cell death. Exofacial exposure of phosphatidylserine occurs in lymphocytes and mast cells after antigenic stimulation and in the absence of apoptosis, suggesting that there is a functional requirement for phosphatidylserine exposure in immunocytes. In this review we examine current ideas as to the nature of this functional role in mast cell activation. Mechanistically, there is controversy as to the candidate proteins responsible for phosphatidylserine translocation from the internal to external leaflet, and here we review the candidacies of mast cell PLSCR1 and TMEM16F. Finally we examine the potential relationship between functionally important mast cell membrane perturbations and phosphatidylserine exposure during activation.
Keywords: mast cells, membrane lipids, phosphatidylserine, TMEM16F
Abbreviations
- ABCA
ABC binding cassette family A
- CRAC
calcium release activated channel
- GPMV
giant plasma membrane vesicle
- ITIM
immunoreceptor tyrosine based inhibitory motif
- PLA2
phospholipase A2
- PLSCR
phospholipid scramblase
- PMA
phorbol 12,13-myristate acetate
- RBL
rat basophilic leukemia
- RFU
relative fluorescence units
- ROI
region of interest
- TMEM
transmembrane protein
- WGA
wheat germ agglutinin
Non-apoptotic Phosphatidylserine Exposure in Immune System Cells
In eukaryotic cells, the internal and external leaflets of the lipid bilayer display an asymmetric lipid distribution and a feature of this asymmetry is the presence of acidic (anionic) phospholipids such as phosphatidylserine (PS) on the inner leaflet of the membrane.1-4 Phospholipid asymmetry is maintained by: (1) flippases that move lipids between membrane leaflets in an energy-dependent or-independent manner, primarily from external to internal leaflet, (2) floppases that transport lipids from the cytoplasmic to the external lamellum in an energy-dependent manner, and (3) scramblases usually move lipids between both internal and external leaflets, are driven by a pre-existing transbilayer lipid gradient, are energy dependent and can be activated by divalent cations.1,3-9
The best understood example of phospholipid asymmetry loss occurs during apoptosis.2-4,6,9-13 Here, PS is translocated to the outer leaflet of the plasma membrane making the cell a target for scavenger receptors on phagocytes. This critical event in the apoptotic cascade has been the main focus for the study of PS externalization, and there is a significant literature on this highly effective apoptosis marker. Aided by the identification of Annexin-V (AnnV) binding as a high affinity marker of the presence of PS, the role of PS as a marker of apoptosis seems well-established.2-4,6,9-13 There is, however, a growing literature on the externalization of PS in the absence of apoptosis that is refocusing attention on PS and the enzymes that control its plasma membrane distribution.14-21
PS exposure as a hallmark of apoptosis has greatly overshadowed the study of non-apoptotic PS externalization, but these processes have some distinct differences. While apoptotic PS exposure occurs over a timecourse of hours, in non-apoptotic, activating cells, transient PS exposure occurs over a time course of seconds to a few minutes. Transient PS movement to the extracellular leaflet has been described during sperm capitation, myotube development, macrophage-mediated phagocytosis and during activation of T lymphocytes, B lymphocytes, mast cells and neutrophils, all in the absence of apoptosis.14-21 These studies suggest that PS exposure may be a normal component of immunocyte activation in a role entirely distinct from the presentation of a marker of cell death to the scavenging machinery.
In T cells, Elliott et al. describe that non-apoptotic PS exposure occurs during early TCR signaling and that it is constitutively opposed by the transmembrane tyrosine phosphatase CD45.17 CD45 (also known as LCA, the lymphocyte common antigen) is best known for its dephosphorylation of src kinases such as csk, which in turn maintains the ‘off’ status in a lymphocyte until superceded by antigen-TCR engagement. Further insights from this study show that non-apoptotic PS exposure plays a role in the lymphocyte moving to sites of inflammation.17 Data from this study reveal that both PS translocation and shedding of the homing receptor CD62L occurred very quickly after P2×7 stimulation in T-cells. Exposure to the anion exchange inhibitor 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) completely inhibited this shedding and further supports the involvement of PS redistribution in T-cell homing.17
In mast cells, non-apoptotic PS exposure was documented by Ulrich Blank's laboratory in 2000. FcεRI ligation led to reversible, transient and rapid, appearance of Annexin-V positivity in the plasma membrane of non-apoptotic mast cells, and that this phenomenon is associated with mast cell degranulation.18 FcεR1 stimulations lasting for 30 minutes caused a pronounced increase in the surface expression of AnnV binding sites. Specificity for PS was confirmed by the decrease in fluorescence with the addition of excess unlabelled AnnV. Twenty-four hours after PS exposure was observed in FcεR1-stimulated cells, there was no detectable difference in the number of apoptotic nuclei between stimulated and unstimulated cells, indicating that PS exposure in mast cells is also reversible, and not an inevitable marker of apoptosis.18,19,22,23 Data from our laboratory echos and extends those of the Blank studies and show the temporal relationship between mobilization of [Ca2+]i and PS exposure (Fig. 1A), noting that there is a marked lag between calcium elevation and AnnV-positivity, a lag especially pronounced in antigen-stimulated cells. Further experiments suggest that this lag is defined by more than a simple quantitative relationship with the attained concentration of cytosolic free calcium (the initial rate and time-to-onset of PS flipping induced by ionomycin even at low doses is always faster than that of antigen, and even if the Fluo-4 signals are equivalent). We also examined the features of PS exposure, asking first whether AnnV-positivity is initially occurring simultaneously at all points on the membrane or whether it nucleates in discrete locations. Figure 1B and C show that PS exposure is initially limited to membrane patches, and that the interspersed regions of membrane gain PS-positivity at a slower but consistent rate until a uniformly intense PS signal covers the entire membrane circumference. Taken together these data support a model where PS exposure is initially accomplished by an (probably calcium-regulated) enzymatic activity localized at discrete positions, and that subsequently this exposure is transmitted either laterally or initiated by a secondary signal to the remaining un-flipped areas of the membrane.
Figure 1.

(See previous page). Characteristics of phosphatidylserine exposure in activating model mast cells. (A) Kinetics of PS exposure and intracellular free calcium mobilization in RBL2H3 mast cells. Live cell images of Fluo-4 loaded (4 μM for 30 min) RBL2H3 cells (stimulated with PMA/ionomycin (500 nM/500 nM) or via FcεRI (IgE anti-DNP/250 ng/ml KLH-DNP in the presence of 0.1 μg/ml Alexa 568-Annexin V and 1 mM external CaCl2) were captured using laser scanning confocal microscopy (Nikon Ti INSPIRE, 150 nm optical sections gathered every 30s). Whole cell and membrane regions of interest (ROI) were analyzed for average fluorescence intensity in the FITC (Fluo-4) and Texas Red (Alexa 568-Annexin) channels. Scale bar in B is 7 microns. (B) Patchy presentation of AnnV positive regions in activated RBL2H3. Left Panel. Maximum intensity projection (NIS Elements, Nikon, San Diego, CA) of 10 150 nm optical sections for cell stimulated and stained as in A, image captured at 450s. Right Panel. Intensity profile of Alexa 568-Annexin V fluorescence in a selected 3 micron length of membrane at the indicated time points after stimulation as in A. (C) Process of PS exofacial exposure in activating RBL2H3 cell. Intensity surface plots were used to visualize Alexa 568-Annexin V fluorescence (NIS Elements) of a single 150 nm z disc from RBL2H3 cell stimulated via FcεRI (IgE anti-DNP/250 ng/ml KLH-DNP in the presence of 0.1 μg/ml Alexa 568-Annexin V and 1 mM external CaCl2). Time after initial exposure to stimulus is indicated in seconds.
In a number of cell systems, an explicit link between levels of intracellular free calcium (Ca2+i) and the exofacial exposure of PS have been shown.2,9,17,18,22,24-26 In addition to the calcium dependence of several candidate enzymes in the PS exposure pathway, the treatment of cells (T cells, mast cells, thymocytes and platelets) with calcium ionophore is necessary and sufficient to cause PS exposure.2,9,17,18,22,24-26 Given that receptor-mediated calcium signals in these non-excitable cells are biphasic, comprising a calcium release from intracellular stores followed by an influx across the plasma membrane that are experimentally separable, we asked whether non-apoptotic PS exofacial exposure in model mast cells could be accomplished in the absence of extracellular calcium. PS exposure is undetectable in a mast cell model unless extracellular calcium is present, suggesting the involvement of transmembrane calcium selective cation channels in its control (LS, EU, unpublished and 17,18,22-24,27). A further complication arises, especially relevant to the exploration of candidate flippases, when we consider data from Scott syndrome patients that suggests there are distinct mechanistic aspects to non-apoptotic calcium-induced and apoptotic PS exposure.28-31
Functional Consequences of Non-apoptotic PS Exposure
Given the complex mechanisms that have evolved to locate and translocate PS asymmetrically in the PM, it seems key to discuss the properties that PS confers upon the membranes in which it is found. These are summarized here and shown schematically in Figure 2.
Figure 2.
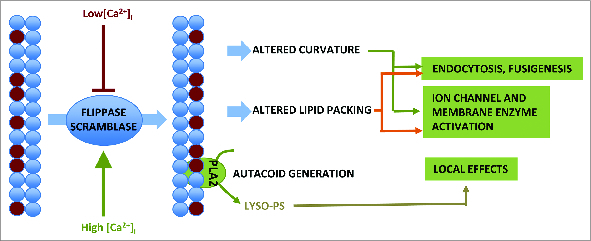
Schematic summary of the functional consequences of exofacial PS exposure.
Endocytosis and fusigenesis
PS forms a poly-anionic surface to which poly-cationic molecules, including cytoskeletal proteins such as spectrin, proteins involved in endo and exocytosis (e.g., K-Ras, Rab, Rac1) can bind.32,33 Docking of endosomes and lysosomes is thought to involve PS on their cytosolic leaflet. Flippase-mediated movement of PS to the inner leaflet of the plasma membrane is thought to be permissive for endocytosis.15,21,34 A parallel role for PS externalization during exocytosis has been postulated.15
Acquisition of membrane curvature
Asymmetric lipid distribution has been postulated as both a cause and effect of the increased membrane curvature necessary for vesicle formation.21,35-37 Causally, the presence of ‘conical’ lipids in a leaflet such as PS (which has a smaller head group than tail cross-sectional area) will tend to promote negative curvature, while depleting PS and raising local relative concentrations of PE and PC would promote membrane curling through the lateral pressures exerted by an excess of unsaturated fatty acid chains.35 Conversely, as a membrane curves, extra space in the stretched outer leaflet creates a void into which inner leaflet lipids flip down an energy/concentration gradient.21,38
Transmembrane protein regulation
Lipid packing, specifically the local ratios of PE, PS and PC, generates interfacial forces that are sufficient to deform transmembrane protein structures. Membrane tension is the sum of the lateral and transverse forces affecting the membrane and can change rapidly in response to environmental forces such as hydrostatic pressure that bends or stretches the lipid bilayers.39,40 Alterations in membrane tension also result from the insertion, removal, and modification of membrane-associated molecules as well as from intracellular and extracellular mechanical stimuli such as osmotic pressure and forces initiated by the cytoskeleton. The ability of cells to increase and decrease membrane tension results in changes in membrane curvature and is one of the mechanisms used to modulate the coordinated membrane changes associated with exocytosis, endocytosis and vesicle formation.8,21,26,34,41
Some models of ion channel activation postulate that membrane dilation (during swelling or exocytosis) would tend to decrease the lateral pressure placed on the protein structure by the membrane, causing dilation and opening of the channel.42-45 Similar scenarios would be postulated to arise from changes in lateral membrane pressure resulting from altered distribution of membrane phospholipids, and may explain the apparent connection between PS concentration and calcium channel activation. Other enzymes, such as PLA2, can be controlled by lateral pressure of the lipid bilayer, relating their activation status to membrane stretch and membrane flipping.46-49 New data also suggest that PS redistribution (in this case mediated by PLSCR phospholipid scramblases) is important in CD4 co-receptor signaling, and permits the secretory leukocyte protease to regulate this transmembrane protein.50
PS and lyso-PS signaling
Exogenous PS has been described as a suppressor of immunocyte activation through activity at the CD300a-type ITIM-containing inhibitory receptors.51-55 Conversely, exogenous PS 56-58 applied at fairly high concentrations, was recognized as a stimulator of mast cell activation in papers dating back to the 1970s. Lyso-PS, which is released from exposed PS at least in apoptotic cells, may activate Gαq heterotrimeric protein coupled pathways via the GPCR GPR34c.59,60 Lyso-PS stimulates histamine release from mast cells, and is formed via the action of PS-specific PLA2 on PS.61,62
Altered internal leaflet phospholipid composition
A possible additional consequence of PS exposure to extracellular leaflets of the membrane concerns the altered composition of the internal leaflet it leaves behind. The inner leaflet depleted of PS might have a higher concentration per linear unit distance of other signaling lipids (e.g., PC, the major membrane substrate for the generation of PI (4,5) P2 by phospholipase C). This would change the substrate:enzyme ratio for activities such as PLC. If intentional inward flipping of PC accompanies outward flipping of PS/PE, then the substrate pool for PLC would be even more increased.34 Complex experiments would be required to elucidate whether this has any effect on the threshold and activity of PC-dependent downstream signaling pathways and the cellular responses they control.
Clearly, each of these potential roles of PS has direct relevance to the process of mast cell activation, which involves membrane reorganization, exocytosis and endocytosis, and the activation of calcium entry channels by diverse upstream signals
Enzyme Candidates for Catalysis of Phospholipid Exposure in Mast Cells
Flippases, floppases and scramblases in mast cells
Flippases (ATP-dependent aminophospholipid translocases such as the type IV P-type ATPases, transport aminophospholipids from the extracellular to cytoplasmic leaflets (e.g., ATP8). Floppases, primarily of the ATP-binding cassette (ABC) family of ATPases (especially ABCA1) have been proposed as ATP-dependent proteins that move lipids from the cytosolic to the extracellular leaflets.63-65 The latter could potentially have a role in the PS exposure in mast cells that is the focus of this article, but (see below) molecular genetic data seems to refute this role. The former do not have an activity consistent with mediating PS exposure and are not treated in detail here.
Scramblase candidates
(a) The phospholipid scramblase (PLSCR1) was first described by Basse, in a study where this 37 kDa protein could reconstitute PS exposure activity in liposomes.66-68 Several studies demonstrated that siRNA knockdown was associated with a loss of PS exposure capacity and it was convincingly shown to be calcium regulated in mast cells. PLSCR1 has been described as a target for antigen-receptor mediated tyrosine phosphorylation in mast cells, and there may be some tyrosine phosphorylation-dependence to PLSCR1 activity, with receptor-activated kinases responding to calcium entry (e.g., via P2 × 7) and CD45 constitutively opposing this activating phosphorylation.23
Later studies by Marc Benhamou's laboratory showing that mast cells deficient in PLSCR1 were defective in secretory granule exocytosis supported a role for both PS exposure in secretion (see above) and for PLSCR1 in PS exposure.19,22,23,69 Over-expression of the PLSCR scramblase and the resulting confusion in the basal membrane asymmetry of mast cells was shown by Kato et al. to interfere with subsequent degranulation responses to pharmacological stimulation.70 Exocytosis was inhibited both when the asymmetry of the phospholipids was altered before cell stimulation and after exposure of calcium ionophore and PMA in scramblase overexpressing cells.70
The position of PLSCR1 as a seemingly excellent candidate for scramblase activity took a series of serious blows in subsequent studies. Calcium-dependent PS exposure was normal in PLSCR1−/− cells.68,71 PLSCR1 has been shown to be localized in the nucleus and to act as a transcription factor; observations at odds with the idea that it is a plasma membrane flippase.72-78 These studies clearly require some reconciliation: We note that PLSCR1 is a member of a 5 protein family, and further studies would be needed to discern whether compensatory upregulation of other family members plays a role in the phenotypes observed.79 Similarly, the dominant negative and siRNA based studies in mast cells did not control for bystander effects on other family members and also cannot fully exclude off-target effects. The transcriptional activity of PLSCR1 perhaps provides a relatively straightforward reconciliation: PLSCR1 is not a scramblase but it regulates the transcriptional activity of genes that are.71,72 In 2005, Zhou et al. demonstrated that PLSCR1 is able to transcriptionally activate the inositol 1,4,5-triphosphate receptor type 1 gene (IP3R1).80 In a model mast cell line we can reproduce the expression and calcium dependent tyrosine phosphorylation of PLSCR122,23 (Fig. 3A and B), and we observe its primary location in structures that resemble nuclear bodies81,82 (Fig. 3C).
Figure 3.
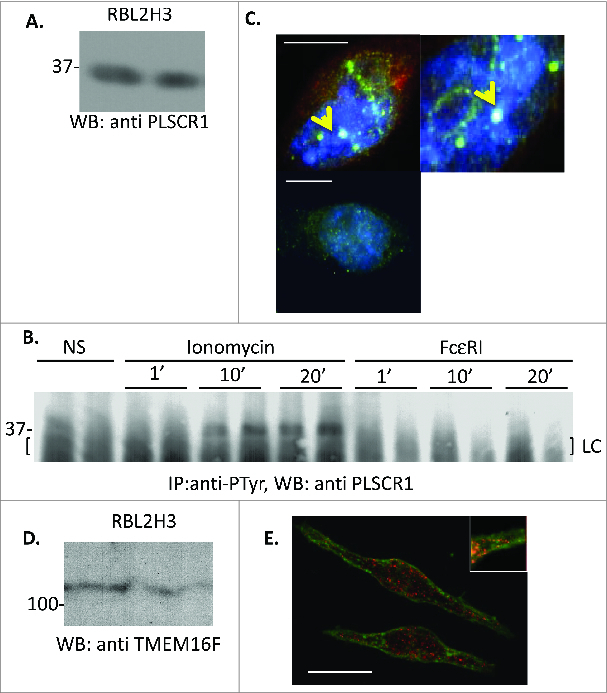
PLSCR1 expression and localization in RBL2H3 (A and B). PLSCR1 expression and calcium-induced tyrosine phosphorylation in RBL2H3. (A) Total cellular lysates were extracted from RBL2H3 (ATCC CRL-2256) in a buffer containing 50 mM Hepes pH 7.4, 250 mM NaCl, 20 mM NaF, 10 mM iodoacetamide, 0.5%(w/v) Triton X100, 1 mM PMSF (phenylmethylsulfonylfluoride), 500 mg/ml aprotinin, 1.0 mg/ml leupeptin and 2.0 mg/ml chymostatin. Following acetone precipitation, 10 μg of protein were loaded in duplicated lanes of a 10% SDS-PAGE gel run under reducing conditions. Following electrotransfer to PVDF membrane, a Western blot using 0.1 μg/ml anti rat PLSCR1 (mouse monoclonal 13A6, 1 h, RT in 0.05% Tween 20 and 0.1% BSA) was performed. Developing antibody was goat anti-rat IgG conjugated to HRP (Amersham, Piscataway, NJ), and signal was visualized using Enhanced Chemiluminescence (Amersham) and Kodak Biomax film. Duplicate lanes shown. (B) Cells were stimulated for the indicated times (in min) with 500 nM ionomycin, or 200 ng/ml KLH-DNP. For the latter, cells were pre-incubated for 16 h with 0.1 μg/IgE anti-DNP (clone SPE-7, Sigma, St. Louis, MO). Total lysates from 10 million RBL2H3 per lane were produced using a high salt (see above) lysis buffer. NaCl was normalized to 75 mM before pre-clearing (murine IgG2b) and immunoprecipitation with 1 μg per lane of anti-phosphotyrosine (clone 4G10, Cell Signaling Technologies, Danvers, MA). Western blotting for the presence of PLSCR1 was performed as described above. (C) Immunofluorescent localization of PLSCR1 in RBL2H3. Cells were grown on glass coverslips, fixed in 0.4% (w/v) paraformaldehyde (30 min, RT) and permeabilized (0.4% Triton X100 for 4 min). After blocking with 0.75% (w/v) Fish skin gelatin, immunocytochemistry was performed with anti-PLSCR1 (0.1 μg/ml for 45 min) followed by washing and secondary antibody staining with Alexa-conjugated IgG. Lower panel shows matched exposure of staining were primary antibody was omitted but all other conditions were equivalent. Counterstains were ER Tracker and DAPI (4 μM/30 min and 10 nM for 4 min, Molecular Probes Eugene, OR). Three separate cell nuclei are shown at left (scale bar 4 microns) and digitally zoomed 2X is shown at right. Arrows indicate putative nuclear bodies. Images were acquired through a Plan Apo VC 100X 1.40 oil objective (Nikon). Imaging was performed on a Nikon Ti Eclipse C1 epi-fluorescence and confocal microscopy system. Pinhole size was 60 microns. (D) TMEM16F expression in RBL2H3. Western blotting protocol as in Figure 1, with probing antibody anti-TMEM16F (0.1 μg/ml G-14, Santa Cruz Biotechnology, Dallas, TX). Duplicate lanes shown. (E) Immunofluorescent localization of TMEM16F in RBL2H3. Staining and imaging protocols as in Figure 1 with Alexa-488 conjugated wheat germ agglutinin and Alexa 568 anti-rabbit IgG (Molecular Probes). Scale bar is 8 microns.
(b) ABCA1. ABCA1 was initially identified as a receptor on macrophages important for the engulfment of apoptotic cells. However, a role for ABCA1 as a lipid translocator was suggested by Chimini and Williamson who found that efficient clearance of apoptotic cells required transbilayer distribution of PS on the membrane of both the phagocyte and the dying cells.83 Chimini et al. proposed ABCA1 as a scramblase, based on Abca1 deficient mice and ABCA1 over-expressing cells. These data suggested that Abca1 promoted calcium- induced exposure of PS at the plasma membrane. Conversely, the evidence supporting dismissal of the ABCA1 as a candidate for scramblase and/or floppase activity was presented by Williamson et al.84 Phospholipid movements were studied in Abca1 −/- mice and in patients with Tangier disease, as well as in normal and ABCA1-expressing HeLa cells.84 In cells with Ca2+-induced scramblase activity, no differences in the rate of PS externalization were detectable. Additionally, the data indicated that PS-specific externalization in these cells was ABCA1-independent and that ABCA1 was not the aminophospholipid translocase whose activity is responsible for exposing PS.67,84,85 These data suggest that ABCA1 is not the sole arbiter of PS translocation.
(c) TMEM 16F. Mutations in the TMEM16F (Ano6) gene have been identified in patients with Scott syndrome, a rare hereditary disorder that can cause severe bleeding due to defective phospholipid scrambling.29 Scott Syndrome has also been attributed to missense mutations in ABCA1,86 so it is possible that there are heritable mutations in more than one gene that impair PS translocation. ABCA1 and TMEM16F may both contribute to PS exposure with neither demonstrating sufficiency.
Phospholipid scrambling in human platelets 87,88 has been shown to occur via both TMEM16F-independent and TMEM16F-dependent pathways.28,29,89 The question remains as to whether whether TMEM16F is a regulatory protein or if it is a scramblase in its own right.90 In their review, Kunzelmann et al. conclude that current studies are unable to present solid evidence that TMEM16F (also known as Ano6) is a phospholipid scramblase or if it serves a regulatory function.29 For example, Tmem16F deficient platelets exhibit reduced but not absent PS exposure in response to ionophore.31
TMEM is part of a multi-gene family, of which 5 can scramble lipids and 2 have been established as chloride channels. TMEM16F is one of the latter, and has been proposed as having a yet undemonstrated calcium channel activity in addition to its ability to act as a flippase.9,30,91 Electrophysiologically, a chloride exporting ion channel has a profound influence upon the plasma membrane potential (ΔΨ) and hence the driving force for calcium entry across the plasma membrane. An additional explanation could be that TMEM16F influences the driving force for calcium entry by either receptor regulated or basally active calcium channels present in the plasma membrane and it is these that provide the calcium that drives PS exposure. In thymocytes, a role for the calcium channel P2X7 has been proposed, which would again be influenced by chloride-channel mediated alterations in ΔΨ.17 Whole cell patch clamping is needed to reconcile this question, and a recent paper showing that genetic elimination of TMEM16F caused a concomitant loss of an outwardly rectifying, voltage dependent cation channel that was calcium activated from megakaryocytes.31 This non-selective cation channel (NSCC) has an extremely small unit conductance (∼0.5pS) and a pharmacological profile with some overlap with the TRP channels. Moreover, in a striking result, Yang et al. identified a single amino acid variance (D409G) between the TMEM16A chloride channel and TMEM16F that could confer cationic selectivity.31 Moreover, a recent study by Malvezzi showed that a purified fungal TMEM16F can reconstitute a cation channel with PK/PCl of ∼1.5.90 Thus TMEM16F is a calcium permeant channel, is calcium activated and is necessary for PS exposure in some cell systems. Yang et al. demonstrate calcium permeability, but it is the calcium sensitivity of TMEM16F, not the permeability that is affected by the D409G mutation.31 It is still not clear if the calcium dependency of PS exposure involves calcium permeation through TMEM16F itself.
These studies suggest that TMEM16F should be investigated in the mast cell system to ascertain whether it is positioned to mediate PS exposure under non-apoptotic conditions. Interestingly, we previously observed that TMEM16 family members emerged from a bioinformatic screen that our laboratory was performing to attempt to identify novel calcium channels in mast cells (Turner and Stokes, unpublished). Armed with these data suggesting that this family is present in mast cells, we revisited TMEM16F to ascertain if its position and expression in mast cell is compatible with a possible role in PS exposure in this cell type. We asked if TMEM16F was positioned in mast cells to mediate membrane PS exposure. Our data show that TMEM16F protein is present in mast cells, and that it is partially plasma membrane localized in clusters but is also present in a number of intracellular locations (Fig. 3D and E). Thus TMEM16F is perhaps better positioned to directly mediate PS exposure in the mast cell than PLSCR1.
(d) Perforin. Perforin is a pore-forming enzyme that is critical in cell-mediated cytotoxicity, forming pores through which Granzyme B exerts killing activity. The size of perforin-induced pores varies with the prevalent membrane lipid composition. In CD8+ T cells perforin pore structures are associated with membrane lipid ‘flip-flop’ and PS exposure, with the latter occurring independently of any calcium influx through the perforin pores.92 These data suggest that, intriguingly, perforin may control its own lipid microenvironment, initiating flip-flop that results in an optimal environment for its activity that also corresponds to enrichment in exofacial PS.92 Mast cells and basophils produce Granzyme B, but not perforin.93 Thus endogenous perforin production is unlikely to be a source of PS flipping in mast cells. However, mastocytoma cells are sensitive to T cell-derived perforin-mediated lysis, suggesting that perforin-mediated PS exposure could occur during the particular circumstance of cytotoxic killing of mast cells or basophils.94
Membrane Rearrangements and PS Exposure
Mast cells represent a system in which to study the impact of phospholipid asymmetry and PS externalization may have on processes such as exosomal and endosomal exchange as well as the dynamic membrane changes that accompany antigen receptor activation.18,22,69 The signaling events and functional responses that follow antigen receptor engagement in mast cells are critical to inflammation. Massive membrane rearrangements occur after mast cell activation by a range of stimuli. These comprise (1) exocytosis of secretory vesicles,95-97 (2) exosome formation,98-100 and (3) the formation of large degranulation channels101-103 which may correspond to the poorly characterized Giant Plasma Membrane Vesicles (GPMV).104,105 Here we will examine the relationship and possible enabling roles for PS exposure in each of these membrane dependent events, noting (as mentioned above) that PS exposure fundamentally changes membrane curvature, fusigenicity and interactions with cytoskeletal elements. All of these are potentially important in the genesis of membrane rearrangements following mast cell activation.
PS exposure and secretory granule exocytosis
Blank hypothesized that the ‘patches’ of Annexin V decorated PS observed in RBL2H3 are associated with secretory vesicle exocytosis.18 Based on the results of several kinetic experiments, Martin et al. present compelling data for a strong association between surface expression of PS and the release of β-hexosaminidase in RBL2H3 cells.18 Both β-hexosaminidase secretion and Ann-V binding site expression were markedly inhibited when cells were stimulated in presence of EGTA. Similarly, when cells were incubated with bisindolylmaleimide and wortmannin, β-hexosaminidase secretion and Ann-V binding site expression exhibited dose-dependent inhibition. Additionally, when stimulated with ionomycin, the cells revealed a strong relationship between PS externalization and β-hexosaminidase release. Similarly, a study by Kato suggested an important link between exocytosis and lipid bilayer asymmetry.70
PS exposure and exosome formation
Mast cells release large numbers of exosomes that contain inflammatory mediators, cytokines and mRNA. While not explicitly studied in the mast cell system, there are links between PS exposure and exosome formation and release. For example, in a study in the COS-1 fibroblast line, PS was found to be important for the retrograde trafficking of recycling endosomes via interaction with evectin-2.80 Depletion of evectin-2 or blocking of intracellular PS suppressed the return of recycling endosomes to the Golgi. Elliott et al., and Chang et al., have both shown that shed microparticles from platelets retain high levels of exposed PS.24,106
PS exposure and GPMV formation
GPMVs (Giant Plasma Membrane Vesicles) are large plasma membrane vesicles that have been documented to form in diverse cell types in response to cell stress. In mast cells these may have a particular physiological manifestation as the large ‘degranulation channels’ postulated by the Dvorak laboratory,102,103 and others, that form in the absence of apoptosis and provide an extracellular environment for the dense cores of released secretory granules. GPMV are induced by diverse stimuli.107,108 but strikingly their formation seems to be always dependent upon large intracellular free calcium fluxes.105,107,109,110 We have reproduced these observations in a model mast cell line. Interestingly, we note that the GPMV are annexinV–positive, indicating that they bear exofacially-flipped PS (unpublished data). Isolated GPMV formed by formaldehyde/DTT treatment of RBL2H3 were also positive for externalized PS.110 In the apoptotic situation, PS exposure is associated with developing membrane blebs, the cell-death correlate of live cell GPMVs. Elliott et al. suggest that PS exposure depends on cell shrinkage, membrane buckling and decreased lipid packing, all conditions conducive to bleb/GPMV formation. While they observed in apoptotic B cells that PS exposure occurred preferentially at the sites of bleb formation,34 we do not see obvious localized PS exposure in GPMV formation in non-apoptotic mast cells. It is an interesting connection that a component of cell shrinking and bleb formation is chloride efflux,34 one of the molecular activities of TMEM16F.29,87,111
Summary and Open Questions
How is non-apoptotic PS exposure important in mast cells?
Non-apoptotic PS exposure is a feature of activating mast cells. Pharmacological and immunological stimuli induce this biological process, and its explicit contributions to mast cell activation are likely to be as a permissive step in the dramatic membrane reorganizations that are necessary for exocytosis and degranulation channel formation. The possibility of regulating this pathway to intervene in the progress of pro-inflammatory responses is presently remote, but would absolutely depend on unequivocal identification of the direct mediators of PS exposure. In this regard novel methods for PS sensing112 may become important, as will loss-of-function experiments targeting the correct enzyme activity to dyregulate PS exposure.
Which protein or proteins accomplishes PS scrambling?
PLSCRs may play an indirect regulatory role, suggested by prior studies and the network analysis shown here, but TMEM16F appears to be a candidate for direct involvement in PS exposure. Our new data show that this protein is present in mast cells, and future experiments will determine if it is causally involved in non-apoptotic PS exposure and hence functional responses in this cell type. A larger open question concerns the multiplicity of proteins that have some link to the scrambling mechanism, but where no one protein seems to be completely quantitatively responsible for the degree of PS exposure.
How is PS exposure laterally transmitted from (presumably) a discretely localized enzymatic flipping activity?
There is an information gap on the presumed lateral transmission mechanism of PS exposure. Assuming that each scramblase protein is responsible for a relatively small area (our data and published studies suggest that neither TMEM16F or ABCA1 are uniform or ubiquitous in the plasma membrane, but are located in clusters) then there must be some lateral transmission of PS flipping from the enzymatic locus. Indeed Blank's early data suggested that there was some initial localization to PS exposure which then became uniform throughout the PM.18 Whether this reflects diffusion of PS laterally (over extremely fast timescales) from a nucleating site of enzyme activity or a diffusible second messenger component to the signal that activates a protein-independent flipping is an intriguing question.
How does non-apoptotic flipping avoid engagement of phagocytosis and clearance mechanisms?
When cell injury occurs, phospholipid asymmetry is lost and exposed PS becomes a signal of cell damage. Recruitment of macrophages to clear dead or dying cells is an essential component of the processes of infection clearance and wound resolution. The presence of PS in bacterial and fungal membranes underscores the evolutionary origins of exposed PS as a ‘danger’ signal to the innate immune system. PS activates innate pathways both in situ in the plasma membrane, and is metabolized to lyso-PS which acts over distance as an autacoid. If, indeed, there is truly a non-apoptotic physiological need for PS exposure, there this infers an avoidance mechanism that prevents T cells, B cells and mast cells that exhibit these responses from being phagocytosed and cleared as if they were indeed apoptotic. Further investigations as to whether this reflects quantitative, qualitative of kinetic differences between apoptotic and non-apoptotic PS exposure are required.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Carl Sung for microscopy support.
Funding
This work was funded by Chaminade University BRIC Program (NIH P20 MD006084), Hawaii State INBRE Program (NIH 2P20GM103466) and the IMUA III EPSCOR Program (NSF 0903833) and the AFRL Research Collaboration Program FA8650-13-C-5800.
References
- 1. Bevers EM, Comfurius P, Dekkers DW, Zwaal RF. Lipid translocation across the plasma membrane of mammalian cells. Biochim Biophys Acta 1999; 1439:317-30; PMID:10446420; http://dx.doi.org/ 10.1016/S1388-1981(99)00110-9 [DOI] [PubMed] [Google Scholar]
- 2. Bevers EM, Comfurius P, Zwaal RF. Regulatory mechanisms in maintenance and modulation of transmembrane lipid asymmetry: pathophysiological implications. Lupus 1996; 5:480-7; PMID:8902787 [DOI] [PubMed] [Google Scholar]
- 3. Sims PJ, Wiedmer T. Unraveling the mysteries of phospholipid scrambling. Thromb Haemost 2001; 86:266-75; PMID:11487015 [PubMed] [Google Scholar]
- 4. Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci 2005; 62:971-88; PMID:15761668; http://dx.doi.org/ 10.1007/s00018-005-4527-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bevers EM, Comfurius P, Dekkers DW, Harmsma M, Zwaal RF. Regulatory mechanisms of transmembrane phospholipid distributions and pathophysiological implications of transbilayer lipid scrambling. Lupus 1998; 7 Suppl 2:S126-31; PMID:9814689; http://dx.doi.org/ 10.1177/096120339800700228 [DOI] [PubMed] [Google Scholar]
- 6. Bevers EM, Comfurius P, Dekkers DW, Harmsma M, Zwaal RF. Transmembrane phospholipid distribution in blood cells: control mechanisms and pathophysiological significance. Biol Chem 1998; 379:973-86; PMID:9792430 [PubMed] [Google Scholar]
- 7. Bevers EM, Williamson PL. Phospholipid scramblase: an update. FEBS Lett 2010; 584:2724-30; PMID:20302864; http://dx.doi.org/ 10.1016/j.febslet.2010.03.020 [DOI] [PubMed] [Google Scholar]
- 8. Devaux PF. Phospholipid flippases. FEBS Lett 1988; 234:8-12; PMID:3292284; http://dx.doi.org/ 10.1016/0014-5793(88)81291-2 [DOI] [PubMed] [Google Scholar]
- 9. Suzuki J, Nagata S. Phospholipid scrambling on the plasma membrane. Methods Enzymol 2014; 544:381-93; PMID:24974298; http://dx.doi.org/ 10.1016/B978-0-12-417158-9.00015-7 [DOI] [PubMed] [Google Scholar]
- 10. Fadok VA, Chimini G. The phagocytosis of apoptotic cells. Semin Immunol 2001; 13:365-72; PMID:11708892; http://dx.doi.org/ 10.1006/smim.2001.0333 [DOI] [PubMed] [Google Scholar]
- 11. Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem 2001; 276:1071-7; PMID:10986279; http://dx.doi.org/ 10.1074/jbc.M003649200 [DOI] [PubMed] [Google Scholar]
- 12. Fadok VA, Henson PM. Apoptosis: giving phosphatidylserine recognition an assist-with a twist. Curr Biol 2003; 13:R655-7; PMID:12932346; http://dx.doi.org/ 10.1016/S0960-9822(03)00575-X [DOI] [PubMed] [Google Scholar]
- 13. Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol 2001; 155:649-59; PMID:11706053; http://dx.doi.org/ 10.1083/jcb.200108080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Das A, Slaughter BD, Unruh JR, Bradford WD, Alexander R, Rubinstein B, Li R. Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat Cell Biol 2012; 14:304-10; PMID:22344035; http://dx.doi.org/ 10.1038/ncb2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Devaux PF. Is lipid translocation involved during endo- and exocytosis? Biochimie 2000; 82:497-509; PMID:10865135; http://dx.doi.org/ 10.1016/S0300-9084(00)00209-1 [DOI] [PubMed] [Google Scholar]
- 16. Dong B, Zhou Q, Zhao J, Zhou A, Harty RN, Bose S, Banerjee A, Slee R, Guenther J, Williams BR, et al. Phospholipid scramblase 1 potentiates the antiviral activity of interferon. J Virol 2004; 78:8983-93; PMID:15308695; http://dx.doi.org/ 10.1128/JVI.78.17.8983-8993.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elliott JI, Surprenant A, Marelli-Berg FM, Cooper JC, Cassady-Cain RL, Wooding C, Linton K, Alexander DR, Higgins CF. Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat Cell Biol 2005; 7:808-16; PMID:16025105; http://dx.doi.org/ 10.1038/ncb1279 [DOI] [PubMed] [Google Scholar]
- 18. Martin S, Pombo I, Poncet P, David B, Arock M, Blank U. Immunologic stimulation of mast cells leads to the reversible exposure of phosphatidylserine in the absence of apoptosis. Int Arch Allergy Immunol 2000; 123:249-58; PMID:11112862; http://dx.doi.org/ 10.1159/000024451 [DOI] [PubMed] [Google Scholar]
- 19. Pastorelli C, Veiga J, Charles N, Voignier E, Moussu H, Monteiro R, Benhamou M. Phospholipid scramblase, a new effector of FcepsilonRI signaling in mast cells. Mol Immunol 2002; 38:1235-8; PMID:12217389; http://dx.doi.org/ 10.1016/S0161-5890(02)00069-X [DOI] [PubMed] [Google Scholar]
- 20. Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A 2011; 108:19246-51; PMID:22084121; http://dx.doi.org/ 10.1073/pnas.1114799108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu P, Baldridge RD, Chi RJ, Burd CG, Graham TR. Phosphatidylserine flipping enhances membrane curvature and negative charge required for vesicular transport. J Cell Biol 2013; 202:875-86; PMID:24019533; http://dx.doi.org/ 10.1083/jcb.201305094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Amir-Moazami O, Alexia C, Charles N, Launay P, Monteiro RC, Benhamou M. Phospholipid scramblase 1 modulates a selected set of IgE receptor-mediated mast cell responses through LAT-dependent pathway. J Biol Chem 2008; 283:25514-23; PMID:18579528; http://dx.doi.org/ 10.1074/jbc.M705320200 [DOI] [PubMed] [Google Scholar]
- 23. Pastorelli C, Veiga J, Charles N, Voignier E, Moussu H, Monteiro RC, Benhamou M. IgE receptor type I-dependent tyrosine phosphorylation of phospholipid scramblase. J Biol Chem 2001; 276:20407-12; PMID:11259432; http://dx.doi.org/ 10.1074/jbc.M100790200 [DOI] [PubMed] [Google Scholar]
- 24. Elliott JI, Mumford AD, Albrecht C, Collins PW, Giddings JC, Higgins CF, Tuddenham EG, McVey JH. Characterisation of lymphocyte responses to Ca2+ in Scott syndrome. Thromb Haemost 2004; 91:412-5; PMID:14961172 [PubMed] [Google Scholar]
- 25. Taylor SR, Gonzalez-Begne M, Dewhurst S, Chimini G, Higgins CF, Melvin JE, Elliott JI. Sequential shrinkage and swelling underlie P2´7-stimulated lymphocyte phosphatidylserine exposure and death. J Immunol 2008; 180:300-8; PMID:18097031; http://dx.doi.org/ 10.4049/jimmunol.180.1.300 [DOI] [PubMed] [Google Scholar]
- 26. Wolfs JL, Comfurius P, Bevers EM, Zwaal RF. Influence of erythrocyte shape on the rate of Ca2+-induced scrambling of phosphatidylserine. Mol Membr Biol 2003; 20:83-91; PMID:12745928 [PubMed] [Google Scholar]
- 27. Dekkers DW, Comfurius P, Bevers EM, Zwaal RF. Comparison between Ca2+-induced scrambling of various fluorescently labelled lipid analogues in red blood cells. Biochem J 2002; 362:741-7; PMID:11879203; http://dx.doi.org/ 10.1042/0264-6021:3620741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kmit A, van Kruchten R, Ousingsawat J, Mattheij NJ, Senden-Gijsbers B, Heemskerk JW, Schreiber R, Bevers EM, Kunzelmann K. Calcium-activated and apoptotic phospholipid scrambling induced by Ano6 can occur independently of Ano6 ion currents. Cell Death Dis 2013; 4:e611; PMID:23618909; http://dx.doi.org/ 10.1038/cddis.2013.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kunzelmann K, Nilius B, Owsianik G, Schreiber R, Ousingsawat J, Sirianant L, Wanitchakool P, Bevers EM, Heemskerk JW. Molecular functions of anoctamin 6 (TMEM16F): a chloride channel, cation channel, or phospholipid scramblase? Pflugers Arch 2014; 466:407-14; PMID:23748496; http://dx.doi.org/ 10.1007/s00424-013-1305-1 [DOI] [PubMed] [Google Scholar]
- 30. Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem 2013; 288:13305-16; PMID:23532839; http://dx.doi.org/ 10.1074/jbc.M113.457937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012; 151:111-22; PMID:23021219; http://dx.doi.org/ 10.1016/j.cell.2012.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hankins HM, Baldridge RD, Xu P, Graham TR. Role of flippases, scramblases, and transfer proteins in phosphatidylserine subcellular distribution. Traffic 2015; 16(1):35-47; PMID:25284293; http://dx.doi.org/ 10.1111/tra.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Diakowski W, Sikorski A. Brain spectrin exerts much stronger effect on anionic phospholipid monolayers than erythroid spectrin. Biochim Biophys Acta 2002; 1564:403-11; PMID:12175923; http://dx.doi.org/ 10.1016/S0005-2736(02)00476-5 [DOI] [PubMed] [Google Scholar]
- 34. Elliott JI, Sardini A, Cooper JC, Alexander DR, Davanture S, Chimini G, Higgins CF. Phosphatidylserine exposure in B lymphocytes: a role for lipid packing. Blood 2006; 108:1611-7; PMID:16684961; http://dx.doi.org/ 10.1182/blood-2005-11-012328 [DOI] [PubMed] [Google Scholar]
- 35. Paredes-Quijada G, Aranda-Espinoza H, Maldonado A. Shapes of mixed phospholipid vesicles. J Biol Phys 2006; 32:177-81; PMID:19669461; http://dx.doi.org/ 10.1007/s10867-006-9007-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Slater SJ, Kelly MB, Yeager MD, Larkin J, Ho C, Stubbs CD. Polyunsaturation in cell membranes and lipid bilayers and its effects on membrane proteins. Lipids 1996; 31 Suppl:S189-92; PMID:8729117; http://dx.doi.org/ 10.1007/BF02637074 [DOI] [PubMed] [Google Scholar]
- 37. Stebelska K, Dubielecka PM, Sikorski AF. The effect of PS content on the ability of natural membranes to fuse with positively charged liposomes and lipoplexes. J Membr Biol 2005; 206:203-14; PMID:16456715; http://dx.doi.org/ 10.1007/s00232-005-0793-0 [DOI] [PubMed] [Google Scholar]
- 38. Janmey PA, Kinnunen PK. Biophysical properties of lipids and dynamic membranes. Trends Cell Biol 2006; 16:538-46; PMID:16962778; http://dx.doi.org/ 10.1016/j.tcb.2006.08.009 [DOI] [PubMed] [Google Scholar]
- 39. Frolov VA, Shnyrova AV, Zimmerberg J. Lipid polymorphisms and membrane shape. Cold Spring Harb Perspect Biol 2011; 3:a004747; PMID:21646378; http://dx.doi.org/ 10.1101/cshperspect.a004747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frolov VA, Zimmerberg J. Membranes: Shaping biological matter. Nat Mater 2009; 8:173-4; PMID:19229264; http://dx.doi.org/ 10.1038/nmat2390 [DOI] [PubMed] [Google Scholar]
- 41. Williamson P, Bevers EM, Smeets EF, Comfurius P, Schlegel RA, Zwaal RF. Continuous analysis of the mechanism of activated transbilayer lipid movement in platelets. Biochemistry 1995; 34:10448-55; PMID:7654698; http://dx.doi.org/ 10.1021/bi00033a017 [DOI] [PubMed] [Google Scholar]
- 42. Aguilella VM, Verdia-Baguena C, Alcaraz A. Lipid charge regulation of non-specific biological ion channels. Phys Chem Chem Phys 2014; 16:3881-93; PMID:24452437; http://dx.doi.org/ 10.1039/c3cp54690j [DOI] [PubMed] [Google Scholar]
- 43. Ashrafuzzaman M, Tseng CY, Tuszynski JA. Regulation of channel function due to physical energetic coupling with a lipid bilayer. Biochem Biophys Res Commun 2014; 445:463-8; PMID:24530910; http://dx.doi.org/ 10.1016/j.bbrc.2014.02.012 [DOI] [PubMed] [Google Scholar]
- 44. Rosenhouse-Dantsker A, Mehta D, Levitan I. Regulation of ion channels by membrane lipids. Compr Physiol 2012; 2:31-68; PMID:23728970 [DOI] [PubMed] [Google Scholar]
- 45. Senning EN, Collins MD, Stratiievska A, Ufret-Vincenty CA, Gordon SE. Regulation of TRPV1 ion channel by phosphoinositide (4,5)-bisphosphate: the role of membrane asymmetry. J Biol Chem 2014; 289:10999-1006; PMID:24599956; http://dx.doi.org/ 10.1074/jbc.M114.553180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Honger T, Jorgensen K, Stokes D, Biltonen RL, Mouritsen OG. Phospholipase A2 activity and physical properties of lipid-bilayer substrates. Methods Enzymol 1997; 286:168-90; PMID:9309651; http://dx.doi.org/ 10.1016/S0076-6879(97)86011-9 [DOI] [PubMed] [Google Scholar]
- 47. Jorgensen K, Davidsen J, Mouritsen OG. Biophysical mechanisms of phospholipase A2 activation and their use in liposome-based drug delivery. FEBS Lett 2002; 531:23-7; PMID:12401197; http://dx.doi.org/ 10.1016/S0014-5793(02)03408-7 [DOI] [PubMed] [Google Scholar]
- 48. Leidy C, Ocampo J, Duelund L, Mouritsen OG, Jorgensen K, Peters GH. Membrane restructuring by phospholipase A2 is regulated by the presence of lipid domains. Biophys J 2011; 101:90-9; PMID:21723818; http://dx.doi.org/ 10.1016/j.bpj.2011.02.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mouritsen OG, Andresen TL, Halperin A, Hansen PL, Jakobsen AF, Jensen UB, Jensen MO, Jorgensen K, Kaasgaard T, Leidy C, et al. Activation of interfacial enzymes at membrane surfaces. J Phys Condens Matter 2006; 18:S1293-304; PMID:21690842; http://dx.doi.org/ 10.1088/0953-8984/18/28/S12 [DOI] [PubMed] [Google Scholar]
- 50. Py B, Basmaciogullari S, Bouchet J, Zarka M, Moura IC, Benhamou M, Monteiro RC, Hocini H, Madrid R, Benichou S. The phospholipid scramblases 1 and 4 are cellular receptors for the secretory leukocyte protease inhibitor and interact with CD4 at the plasma membrane. PLoS One 2009; 4:e5006; PMID:19333378; http://dx.doi.org/ 10.1371/journal.pone.0005006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Borrego F. The CD300 molecules: an emerging family of regulators of the immune system. Blood 2013; 121:1951-60; PMID:23293083; http://dx.doi.org/ 10.1182/blood-2012-09-435057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lankry D, Rovis TL, Jonjic S, Mandelboim O. The interaction between CD300a and phosphatidylserine inhibits tumor cell killing by NK cells. Eur J Immunol 2013; 43:2151-61; PMID:23640773; http://dx.doi.org/ 10.1002/eji.201343433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Murakami Y, Tian L, Voss OH, Margulies DH, Krzewski K, Coligan JE. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ 2014; 21:1746-57; PMID:25034781; http://dx.doi.org/ 10.1038/cdd.2014.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakahashi-Oda C, Tahara-Hanaoka S, Honda S, Shibuya K, Shibuya A. Identification of phosphatidylserine as a ligand for the CD300a immunoreceptor. Biochem Biophys Res Commun 2012; 417:646-50; PMID:22185693; http://dx.doi.org/ 10.1016/j.bbrc.2011.12.025 [DOI] [PubMed] [Google Scholar]
- 55. Simhadri VR, Andersen JF, Calvo E, Choi SC, Coligan JE, Borrego F. Human CD300a binds to phosphatidylethanolamine and phosphatidylserine, and modulates the phagocytosis of dead cells. Blood 2012; 119:2799-809; PMID:22302738; http://dx.doi.org/ 10.1182/blood-2011-08-372425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tamori-Natori Y, Nojima S. Activation of histamine secretion from rat mast cells by phosphatidylserine analogues resistant to phospholipase A action. J Biochem 1982; 91:1825-8; PMID:6178731 [DOI] [PubMed] [Google Scholar]
- 57. Read GW, Knoohuizen M, Goth A. Relationship between phosphatidylserine and cromolyn in histamine release. Eur J Pharmacol 1977; 42:171-7; PMID:66150; http://dx.doi.org/ 10.1016/0014-2999(77)90357-0 [DOI] [PubMed] [Google Scholar]
- 58. Goth A, Adams HR, Knoohuizen M. Phosphatidylserine: selective enhancer of histamine release. Science 1971; 173:1034-5; PMID:4106549; http://dx.doi.org/ 10.1126/science.173.4001.1034 [DOI] [PubMed] [Google Scholar]
- 59. Ritscher L, Engemaier E, Staubert C, Liebscher I, Schmidt P, Hermsdorf T, Rompler H, Schulz A, Schoneberg T. The ligand specificity of the G-protein-coupled receptor GPR34. Biochem J 2012; 443:841-50; PMID:22348703; http://dx.doi.org/ 10.1042/BJ20112090 [DOI] [PubMed] [Google Scholar]
- 60. Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR, Kapralov AA, Macphee CH, Kagan VE. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ 2014; 21:825-35; PMID:24464221; http://dx.doi.org/ 10.1038/cdd.2014.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hosono H, Aoki J, Nagai Y, Bandoh K, Ishida M, Taguchi R, Arai H, Inoue K. Phosphatidylserine-specific phospholipase A1 stimulates histamine release from rat peritoneal mast cells through production of 2-acyl-1-lysophosphatidylserine. J Biol Chem 2001; 276:29664-70; PMID:11395520; http://dx.doi.org/ 10.1074/jbc.M104597200 [DOI] [PubMed] [Google Scholar]
- 62. Tamori-Natori Y, Horigome K, Inoue K, Nojima S. Metabolism of lysophosphatidylserine, a potentiator of histamine release in rat mast cells. J Biochem 1986; 100:581-90; PMID:2430954 [DOI] [PubMed] [Google Scholar]
- 63. Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res 2003; 44:233-42; PMID:12576505; http://dx.doi.org/ 10.1194/jlr.R200019-JLR200 [DOI] [PubMed] [Google Scholar]
- 64. Groen A, Romero MR, Kunne C, Hoosdally SJ, Dixon PH, Wooding C, Williamson C, Seppen J, Van den Oever K, Mok KS, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology 2011; 141:1927-37 e1-4; PMID:21820390; http://dx.doi.org/ 10.1053/j.gastro.2011.07.042 [DOI] [PubMed] [Google Scholar]
- 65. Takatsu H, Tanaka G, Segawa K, Suzuki J, Nagata S, Nakayama K, Shin HW. Phospholipid flippase activities and substrate specificities of human type IV P-type ATPases localized to the plasma membrane. J Biol Chem 2014; 289:33543-56; PMID:25315773; http://dx.doi.org/ 10.1074/jbc.M114.593012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Basse F, Stout JG, Sims PJ, Wiedmer T. Isolation of an erythrocyte membrane protein that mediates Ca2+-dependent transbilayer movement of phospholipid. J Biol Chem 1996; 271:17205-10; PMID:8663431; http://dx.doi.org/ 10.1074/jbc.271.29.17205 [DOI] [PubMed] [Google Scholar]
- 67. Stout JG, Basse F, Luhm RA, Weiss HJ, Wiedmer T, Sims PJ. Scott syndrome erythrocytes contain a membrane protein capable of mediating Ca2+-dependent transbilayer migration of membrane phospholipids. J Clin Invest 1997; 99:2232-8; PMID:9151796; http://dx.doi.org/ 10.1172/JCI119397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ. Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem 1997; 272:18240-4; PMID:9218461; http://dx.doi.org/ 10.1074/jbc.272.29.18240 [DOI] [PubMed] [Google Scholar]
- 69. Benhamou M, Blank U. Stimulus-secretion coupling by high-affinity IgE receptor: new developments. FEBS Lett 2010; 584:4941-8; PMID:20851120; http://dx.doi.org/ 10.1016/j.febslet.2010.09.023 [DOI] [PubMed] [Google Scholar]
- 70. Kato N, Nakanishi M, Hirashima N. Transbilayer asymmetry of phospholipids in the plasma membrane regulates exocytotic release in mast cells. Biochemistry 2002; 41:8068-74; PMID:12069598; http://dx.doi.org/ 10.1021/bi016022v [DOI] [PubMed] [Google Scholar]
- 71. Wiedmer T, Zhao J, Nanjundan M, Sims PJ. Palmitoylation of phospholipid scramblase 1 controls its distribution between nucleus and plasma membrane. Biochemistry 2003; 42:1227-33; PMID:12564925; http://dx.doi.org/ 10.1021/bi026679w [DOI] [PubMed] [Google Scholar]
- 72. Ben-Efraim I, Zhou Q, Wiedmer T, Gerace L, Sims PJ. Phospholipid scramblase 1 is imported into the nucleus by a receptor-mediated pathway and interacts with DNA. Biochemistry 2004; 43:3518-26; PMID:15035622; http://dx.doi.org/ 10.1021/bi0356911 [DOI] [PubMed] [Google Scholar]
- 73. Chen CW, Sowden M, Zhao Q, Wiedmer T, Sims PJ. Nuclear phospholipid scramblase 1 prolongs the mitotic expansion of granulocyte precursors during G-CSF-induced granulopoiesis. J Leukoc Biol 2011; 90:221-33; PMID:21447647; http://dx.doi.org/ 10.1189/jlb.0111006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen MH, Ben-Efraim I, Mitrousis G, Walker-Kopp N, Sims PJ, Cingolani G. Phospholipid scramblase 1 contains a nonclassical nuclear localization signal with unique binding site in importin alpha. J Biol Chem 2005; 280:10599-606; PMID:15611084; http://dx.doi.org/ 10.1074/jbc.M413194200 [DOI] [PubMed] [Google Scholar]
- 75. Lott K, Bhardwaj A, Sims PJ, Cingolani G. A minimal nuclear localization signal (NLS) in human phospholipid scramblase 4 that binds only the minor NLS-binding site of importin alpha1. J Biol Chem 2011; 286:28160-9; PMID:21690087; http://dx.doi.org/ 10.1074/jbc.M111.228007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sahu SK, Gummadi SN, Manoj N, Aradhyam GK. Phospholipid scramblases: an overview. Arch Biochem Biophys 2007; 462:103-14; PMID:17481571; http://dx.doi.org/ 10.1016/j.abb.2007.04.002 [DOI] [PubMed] [Google Scholar]
- 77. Wyles JP, Wu Z, Mirski SE, Cole SP. Nuclear interactions of topoisomerase II alpha and beta with phospholipid scramblase 1. Nucleic Acids Res 2007; 35:4076-85; PMID:17567603; http://dx.doi.org/ 10.1093/nar/gkm434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhou Q, Ben-Efraim I, Bigcas JL, Junqueira D, Wiedmer T, Sims PJ. Phospholipid scramblase 1 binds to the promoter region of the inositol 1,4,5-triphosphate receptor type 1 gene to enhance its expression. J Biol Chem 2005; 280:35062-8; PMID:16091359; http://dx.doi.org/ 10.1074/jbc.M504821200 [DOI] [PubMed] [Google Scholar]
- 79. Wiedmer T, Zhou Q, Kwoh DY, Sims PJ. Identification of three new members of the phospholipid scramblase gene family. Biochim Biophys Acta 2000; 1467:244-53; PMID:10930526; http://dx.doi.org/ 10.1016/S0005-2736(00)00236-4 [DOI] [PubMed] [Google Scholar]
- 80. Uchida Y, Hasegawa J, Chinnapen D, Inoue T, Okazaki S, Kato R, Wakatsuki S, Misaki R, Koike M, Uchiyama Y, et al. Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc Natl Acad Sci U S A 2011; 108:15846-51; PMID:21911378; http://dx.doi.org/ 10.1073/pnas.1109101108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Meldi L, Brickner JH. Compartmentalization of the nucleus. Trends Cell Biol 2011; 21:701-8; PMID:21900010; http://dx.doi.org/ 10.1016/j.tcb.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Van Bortle K, Corces VG. Nuclear organization and genome function. Annu Rev Cell Dev Biol 2012; 28:163-87; PMID:22905954; http://dx.doi.org/ 10.1146/annurev-cellbio-101011-155824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Marguet D, Luciani MF, Moynault A, Williamson P, Chimini G. Engulfment of apoptotic cells involves the redistribution of membrane phosphatidylserine on phagocyte and prey. Nat Cell Biol 1999; 1:454-6; PMID:10559991; http://dx.doi.org/ 10.1038/15690 [DOI] [PubMed] [Google Scholar]
- 84. Williamson P, Halleck MS, Malowitz J, Ng S, Fan X, Krahling S, Remaley AT, Schlegel RA. Transbilayer phospholipid movements in ABCA1-deficient cells. PLoS One 2007; 2:e729; PMID:17710129; http://dx.doi.org/ 10.1371/journal.pone.0000729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dekkers DW, Comfurius P, Vuist WM, Billheimer JT, Dicker I, Weiss HJ, Zwaal RF, Bevers EM. Impaired Ca2+-induced tyrosine phosphorylation and defective lipid scrambling in erythrocytes from a patient with Scott syndrome: a study using an inhibitor for scramblase that mimics the defect in Scott syndrome. Blood 1998; 91:2133-8; PMID:9490700 [PubMed] [Google Scholar]
- 86. Albrecht C, McVey JH, Elliott JI, Sardini A, Kasza I, Mumford AD, Naoumova RP, Tuddenham EG, Szabo K, Higgins CF. A novel missense mutation in ABCA1 results in altered protein trafficking and reduced phosphatidylserine translocation in a patient with Scott syndrome. Blood 2005; 106:542-9; PMID:15790791; http://dx.doi.org/ 10.1182/blood-2004-05-2056 [DOI] [PubMed] [Google Scholar]
- 87. Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y. TMEM16F is a component of a Ca2+-activated Cl- channel but not a volume-sensitive outwardly rectifying Cl- channel. Am J Physiol Cell Physiol 2013; 304:C748-59; PMID:23426967; http://dx.doi.org/ 10.1152/ajpcell.00228.2012 [DOI] [PubMed] [Google Scholar]
- 88. Tian Y, Schreiber R, Kunzelmann K. Anoctamins are a family of Ca2+-activated Cl- channels. J Cell Sci 2012; 125:4991-8; PMID:22946059; http://dx.doi.org/ 10.1242/jcs.109553 [DOI] [PubMed] [Google Scholar]
- 89. van Kruchten R, Mattheij NJ, Saunders C, Feijge MA, Swieringa F, Wolfs JL, Collins PW, Heemskerk JW, Bevers EM. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 2013; 121:1850-7; PMID:23303820; http://dx.doi.org/ 10.1182/blood-2012-09-454314 [DOI] [PubMed] [Google Scholar]
- 90. Malvezzi M, Chalat M, Janjusevic R, Picollo A, Terashima H, Menon AK, Accardi A. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat Commun 2013; 4:2367; PMID:23996062; http://dx.doi.org/ 10.1038/ncomms3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010; 468:834-8; PMID:21107324; http://dx.doi.org/ 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- 92. Metkar SS, Wang B, Catalan E, Anderluh G, Gilbert RJ, Pardo J, Froelich CJ. Perforin rapidly induces plasma membrane phospholipid flip-flop. PLoS One 2011; 6:e24286; PMID:21931672; http://dx.doi.org/ 10.1371/journal.pone.0024286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Strik MC, de Koning PJ, Kleijmeer MJ, Bladergroen BA, Wolbink AM, Griffith JM, Wouters D, Fukuoka Y, Schwartz LB, Hack CE, et al. Human mast cells produce and release the cytotoxic lymphocyte associated protease granzyme B upon activation. Mol Immunol 2007; 44:3462-72; PMID:17485116; http://dx.doi.org/ 10.1016/j.molimm.2007.03.024 [DOI] [PubMed] [Google Scholar]
- 94. De Leon M, Jackson KM, Cavanaugh JR, Mbangkollo D, Verret CR. Arrest of the cell cycle reduces susceptibility of target cells to perforin-mediated lysis. J Cell Biochem 1998; 69:425-35; PMID:9620169; http://dx.doi.org/ 10.1002/(SICI)1097-4644(19980615)69:4%3c425::AID-JCB4%3e3.0.CO;2-P [DOI] [PubMed] [Google Scholar]
- 95. Amin K. The role of mast cells in allergic inflammation. Respir Med 2012; 106:9-14; PMID:22112783; http://dx.doi.org/ 10.1016/j.rmed.2011.09.007 [DOI] [PubMed] [Google Scholar]
- 96. Harvima IT, Nilsson G. Mast cells as regulators of skin inflammation and immunity. Acta Derm Venereol 2011; 91:644-50; PMID:21879235 [DOI] [PubMed] [Google Scholar]
- 97. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol 2014; 14:478-94; PMID:24903914; http://dx.doi.org/ 10.1038/nri3690 [DOI] [PubMed] [Google Scholar]
- 98. Carroll-Portillo A, Surviladze Z, Cambi A, Lidke DS, Wilson BS. Mast cell synapses and exosomes: membrane contacts for information exchange. Front Immunol 2012; 3:46; PMID:22566928; http://dx.doi.org/ 10.3389/fimmu.2012.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shefler I, Salamon P, Hershko AY, Mekori YA. Mast cells as sources and targets of membrane vesicles. Curr Pharm Des 2011; 17:3797-804; PMID:22103851; http://dx.doi.org/ 10.2174/138161211798357836 [DOI] [PubMed] [Google Scholar]
- 100. Skokos D, Goubran-Botros H, Roa M, Mecheri S. Immunoregulatory properties of mast cell-derived exosomes. Mol Immunol 2002; 38:1359-62; PMID:12217408; http://dx.doi.org/ 10.1016/S0161-5890(02)00088-3 [DOI] [PubMed] [Google Scholar]
- 101. Behrendt H, Rosenkranz U, Schmutzler W. Ultrastructure of isolated human mast cells during histamine release induced by ionophore A 23187. Int Arch Allergy Appl Immunol 1978; 56:188-92; PMID:74364; http://dx.doi.org/ 10.1159/000232022 [DOI] [PubMed] [Google Scholar]
- 102. Dvorak AM, Colvin RB, Monahan RA. Immunoferritin electron microscopic studies with antibasophil serum of guinea pig basophil degranulation and regranulation in vitro. Clin Immunol Immunopathol 1985; 37:63-76; PMID:4028521; http://dx.doi.org/ 10.1016/0090-1229(85)90136-9 [DOI] [PubMed] [Google Scholar]
- 103. Dvorak AM, Schulman ES, Peters SP, MacGlashan DW, Jr, Newball HH, Schleimer RP, Lichtenstein LM. Immunoglobulin E-mediated degranulation of isolated human lung mast cells. Lab Invest 1985; 53:45-56; PMID:3874321 [PubMed] [Google Scholar]
- 104. Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci U S A 2007; 104:3165-70; PMID:17360623; http://dx.doi.org/ 10.1073/pnas.0611357104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kelly CV, Kober MM, Kinnunen P, Reis DA, Orr BG, Banaszak Holl MM. Pulsed-laser creation and characterization of giant plasma membrane vesicles from cells. J Biol Phys 2009; 35:279-95; PMID:19669579; http://dx.doi.org/ 10.1007/s10867-009-9167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chang CP, Zhao J, Wiedmer T, Sims PJ. Contribution of platelet microparticle formation and granule secretion to the transmembrane migration of phosphatidylserine. J Biol Chem 1993; 268:7171-8; PMID:8463253 [PubMed] [Google Scholar]
- 107. Keller H, Lorizate M, Schwille P. PI(4,5)P2 degradation promotes the formation of cytoskeleton-free model membrane systems. Chemphyschem 2009; 10:2805-12; PMID:19784973; http://dx.doi.org/ 10.1002/cphc.200900598 [DOI] [PubMed] [Google Scholar]
- 108. Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K, Levental I. Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat Protoc 2012; 7:1042-51; PMID:22555243; http://dx.doi.org/ 10.1038/nprot.2012.059 [DOI] [PubMed] [Google Scholar]
- 109. Gaspersic N, Ambrozic A, Bozic B, Majhenc J, Svetina S, Rozman B. Annexin A5 binding to giant phospholipid vesicles is differentially affected by anti-beta2-glycoprotein I and anti-annexin A5 antibodies. Rheumatol (Oxford) 2007; 46:81-6; PMID:16820381; http://dx.doi.org/ 10.1093/rheum-atology/kel200 [DOI] [PubMed] [Google Scholar]
- 110. Johnson SA, Stinson BM, Go MS, Carmona LM, Reminick JI, Fang X, Baumgart T. Temperature-dependent phase behavior and protein partitioning in giant plasma membrane vesicles. Biochim Biophys Acta 2010; 1798:1427-35; PMID:20230780; http://dx.doi.org/ 10.1016/j.bbamem.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rock JR, O'Neal WK, Gabriel SE, Randell SH, Harfe BD, Boucher RC, Grubb BR. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl- secretory channel in mouse airways. J Biol Chem 2009; 284:14875-80; PMID:19363029; http://dx.doi.org/ 10.1074/jbc.C109.000869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kay JG, Grinstein S. Phosphatidylserine-mediated cellular signaling. Adv Exp Med Biol 2013; 991:177-93; PMID:23775696; http://dx.doi.org/ 10.1007/978-94-007-6331-9_10 [DOI] [PubMed] [Google Scholar]