

Summary
Background
For second-line antiretroviral therapy, WHO recommends a boosted protease inhibitor plus nucleoside or nucleotide reverse transcriptase inhibitors (NRTIs). However, concerns about toxicity and cross-resistance motivated a search for regimens that do not contain NRTIs. We aimed to assess whether boosted lopinavir plus raltegravir would be non-inferior to boosted lopinavir plus NRTIs for virological suppression in resource-limited settings.
Methods
A5273 was a randomised, open-label, phase 3, non-inferiority study at 15 AIDS Clinical Trials Group (ACTG) research sites in nine resource-limited countries (three sites each in India and South Africa, two each in Malawi and Peru, and one each in Brazil, Kenya, Tanzania, Thailand, and Zimbabwe). Adults with plasma HIV-1 RNA concentrations of at least 1000 copies per mL after at least 24 weeks on a regimen based on a non-NRTI inhibitor were randomly assigned (1:1) to receive oral ritonavir-boosted lopinavir (100 mg ritonavir, 400 mg lopinavir) plus 400 mg raltegravir twice a day (raltegravir group) or to ritonavir-boosted lopinavir plus two or three NRTIs selected from an algorithm (eg, zidovudine after failure with tenofovir and vice versa; NRTI group). Randomised group assignment was done with a computer algorithm concealed to site personnel, and stratified by HIV-1 RNA viral load, CD4 cell count, and intention to use zidovudine, with the groups balanced by each site. The primary endpoint was time to confirmed virological failure (two measurements of HIV-1 RNA viral load >400 copies per mL) at or after week 24 in the intention-to-treat population. Non-inferiority (10% margin) was assessed by comparing the cumulative probability of virological failure by 48 weeks. This trial was registered with ClinicalTrials.gov, NCT01352715.
Findings
Between March 13, 2012, and Oct 2, 2013, we randomly assigned 515 participants: 260 to the raltegravir group and 255 to the NRTI group; two participants in the raltegravir group and one in the NRTI group were excluded from analyses because of ineligibility. By the end of follow-up (October, 2014), 96 participants had virological failure (46 in the raltegravir group and 50 in the NRTI group). By 48 weeks, the cumulative probability of virological failure was 10·3% (95% CI 6·5–14·0) in the raltegravir group and 12·4% (8·3–16·5) in the NRTI group, with a weighted difference of −3·4% (−8·4 to 1·5), indicating that raltegravir was non-inferior, but not superior, to NRTIs. 62 (24%) participants in the raltegravir group and 81 (32%) in the NRTI group had grade 3 or higher adverse events; 19 (7%) and 29 (11%), respectively, had serious adverse events. Three participants in each group died, all from HIV-related causes.
Interpretation
In settings with extensive NRTI resistance but no available resistance testing, our data support WHO’s recommendation for ritonavir-boosted lopinavir plus NRTI for second-line antiretroviral therapy. Ritonavir-boosted lopinavir plus raltegravir is an appropriate alternative, especially if NRTI use is limited by toxicity.
Funding
National Institutes of Health.
Introduction
Suppression of HIV replication with combination antiretroviral therapy decreases morbidity and mortality in people with HIV.1 As of June, 2015, about 37 million people were living with HIV, most of them in resource-limited settings, and an estimated 15 million people were on antiretroviral therapy.2 With increasing evidence about the benefits of antiretroviral therapy for all, WHO is recommending that all people with HIV should receive antiretroviral therapy.3,4 Treatment failure has been reported to occur in about 20% of individuals receiving first-line antiretroviral therapy in a range of resource-limited settings, suggesting that the need for second-line antiretroviral therapy will continue to grow.5,6 In 2015, more than 500 000 people in resource-limited settings received second-line antiretroviral therapy; a number that WHO is projecting will steadily increase over the next 10 years.7
WHO recommends a regimen of a ritonavir-boosted protease inhibitor plus two nucleoside or nucleotide reverse transcriptase inhibitors (NRTIs) for second-line treatment. However, concerns that retaining NRTI in second-line regimens could lead to cumulative toxicity or lower efficacy due to cross-resistance have motivated three large international randomised studies of NRTI-sparing antiretroviral therapy consisting of ritonavir-boosted lopinavir plus raltegravir, an inhibitor of HIV integrase. Results of two of these studies have been published.8–10 EARNEST was an open-label trial in sub-Saharan Africa that aimed to recapitulate a public health approach by using clinician-selected NRTIs without resistance testing and a primary composite endpoint of good HIV disease control.8 SECOND-LINE was done in mainly middle-income and high-income countries, 73% of the participants had study entry resistance testing, and the study had a primary endpoint of HIV suppression lower than 200 copies per mL.9,10 Both trials found the efficacy and safety of ritonavir-boosted lopinavir plus raltegravir to be largely similar to ritonavir-boosted lopinavir plus NRTIs. These findings validated both WHO’s and NRTI-sparing approaches, but the authors emphasised different interpretations, perhaps reflecting the availability of resources in their settings. The investigators for EARNEST concluded that there was no advantage to replacing NRTIs with raltegravir, whereas those for SECOND-LINE concluded that the ritonavir-boosted lopinavir plus raltegravir provided a simple, effective, safe, and tolerable second-line antiretroviral therapy.
Methods
Study design and participants
A5273 was a randomised, phase 3, open-label, non-inferiority study of second-line antiretroviral therapy at 15 ACTG research sites in South Africa and India (three centres each), Malawi and Peru (two each), and Zimbabwe, Kenya, Tanzania, Thailand, and Brazil. 11 sites provided access to research and primary, secondary, or tertiary medical care and four sites were dedicated research sites. We recruited clinically stable, HIV-1-infected adults (≥18 years) who had confirmed plasma HIV-1 RNA concentrations of 1000 copies per mL or higher (two consecutive measurements at least 1 week apart) after at least 24 weeks on an initial regimen containing a non-nucleoside reverse transcriptase inhibitor. The first RNA concentration could be obtained from medical records up to 90 days before study entry. The second measurement had to be obtained within 45 days before study entry and done in a Division of AIDS (DAIDS)-approved laboratory.
Entry criteria are listed in the protocol. Exclusion criteria included previous exposure to protease inhibitors and known broad NRTI resistance (defined as participants with resistance patterns that, in the investigators opinion, would not allow selection of an effective NRTI combination). Prospective resistance testing was not done as part of the study and was not routinely done at study sites to manage first-line antiretroviral therapy failure. Other exclusion criteria included pregnancy, active hepatitis B infection, and tuberculosis needing rifampicin treatment (because of adverse drug interactions with ritonavir). HLA-B*5701 testing followed local standard of care and was not done as part of the study. The study was approved by ethics committees at each site and written informed consent was obtained from each participant.
Procedures
Participants were assigned to receive either oral ritonavir-boosted lopinavir (100 mg ritonavir, 400 mg lopinavir; Abbott Laboratories; Abbott Park, IL, USA) plus 400 mg raltegravir (Merck Sharp & Dohme; North Wales, PA, USA) twice a day (raltegravir group), or ritonavir-boosted lopinavir (same dose as above) plus the best available two or three NRTIs selected by the site investigator before randomisation from an algorithm approved by the study team (NRTI group). For the NRTI group, if tenofovir disoproxil fumarate was not included in the participant’s first-line regimen then emtricitabine plus tenofovir disoproxil fumarate or emtricitabine, tenofovir disoproxil fumarate, plus zidovudine were preferred. If tenofovir disoproxil fumarate was included in the first-line regimen then lamivudine plus zidovudine or emtricitabine, tenofovir disoproxil fumarate, plus zidovudine were preferred. Acceptable alternative NRTI combinations were predefined and included two-drug or three-drug combinations of zidovudine, lamivudine or emtricitabine, abacavir, and tenofovir disoproxil fumarate. NRTIs provided by the study were fixed-dose combination 200 mg emtricitabine and 300 mg tenofovir disoproxil fumarate (Gilead Sciences; Foster City, CA, USA), and the following fixed-dose combinations and drugs from GlaxoSmithKline (Uxbridge, Middlesex, UK): 300 mg abacavir, 150 mg lamivudine, and 300 mg zidovudine; 300 mg abacavir and 150 mg lamivudine; 600 mg abacavir and 300 mg lamivudine; 150 mg lamivudine and 300 mg zidovudine; 300 mg abacavir; 600 mg abacavir; 150 mg lamivudine; 300 mg lamivudine; and 300 mg zidovudine. Other locally provided, US Food and Drug Administration (FDA)-prequalified, NRTIs could be used with the approval of the study team. NRTI substitutions due to intolerance were allowed. Substitution of ritonavir-boosted lopinavir or raltegravir was not allowed.
Participants entered the study and were randomly assigned treatment within 45 days of screening. Subsequent on-study investigations occurred at weeks 4, 12, and 24 after entry, and every 12 weeks thereafter. Each participant was followed up until week 96 or 52 weeks after the last participant was enrolled, whichever was earliest. Visits included clinical and laboratory assessments (plasma HIV-1 RNA viral load, CD4 cell counts, complete blood counts, and comprehensive chemistry and lipid panels) and assessment for disease complications and adverse drug effects. Adherence counselling was done by site staff members at study entry according to each site’s standard of care, at any visit deemed necessary by the site, and during the follow-up visit after virological failure. At study entry and at weeks 4, 24, 48, 72, and 96, an adherence questionnaire11 was completed by the participant or with the assistance of the study coordinator. Laboratory testing was completed in DAIDS-approved laboratories. Site staff and investigators graded signs, symptoms, and laboratory events according to the DAIDS Toxicity Scale (version 1.0);12 they received training on recognition and management of abacavir hypersensitivity. AIDS-defining illnesses were those accepted by the Centers for Disease Control and WHO.13,14 Serious non-AIDS diagnoses (based on ACTG appendix 60 diagnostic codes15) were adjudicated by the study Co-chairs, one Vice-chair, and a Co-investigator masked to study treatment; a minimum of two people in the adjudication group had to agree with the designation, and disagreements were resolved by discussion with other people in the adjudication group. Serious non-AIDS events were those that required hospitalisation or were medically severe.
We measured plasma HIV-1 RNA after the first screening test using the Abbott HIV-1 RealTime assay (Abbott Laboratories) at laboratories certified by the DAIDS Virology quality assurance programme. An independent reviewer masked to study treatment adjudicated decisions on virological failure. Full-length reverse transcriptase (codons 1–560) and protease (codons 1–99) genotyping with a quality assurance programme-certified assay was done at the University of Pittsburgh (PA, USA) and the Lancet Laboratory (Johannesburg, South Africa) on stored samples from study entry for all participants and on confirmation samples for participants with study-defined virological failure. Integrase (codons 11–288) genotyping was done on paired samples from study entry and virological failure. Major mutations were defined according to the International Antiviral Society (IAS) USA July, 2014, list and susceptibility scores according to the Stanford algorithm version 7.0.16,17
Randomisation and masking
We used a computer algorithm at the ACTG’s Statistical and Data Management Center to randomly assign participants (1:1) to receive ritonavir-boosted lopinavir plus raltegravir or ritonavir-boosted lopinavir plus NRTI. Randomisation was stratified by screening plasma HIV-1 RNA viral load (<100 000 copies per mL or ≥100 000 copies per mL), CD4 count (<100 cells per μL or ≥100 cells per μL), and intention to use zidovudine (if subsequently assigned to receive NRTI) with use of permuted blocks with a block size of four. The algorithm tracked previous allocations by site; if a preset threshold for imbalance was reached, then the computer allocated the next participant at that site to the group with fewer allocations. Site personnel completed online checklists to confirm eligibility before group allocation and had access to the participant’s allocation details only once these checklists were complete. The study team was masked to the randomisation details and laboratory personnel who ran the viral load assays were masked to treatment allocation. The independent investigator who confirmed the primary outcome was masked to treatment assignment. Participants and other site personnel were not masked to group allocation.
Outcomes
The prespecified primary endpoint was time to virological failure defined as two consecutive plasma HIV-1 RNA viral load measurements higher than 400 copies per mL at or after week 24, irrespective of the time between the two measurements. For participants who discontinued the study for any reason, a final viral load was recorded as virological failure if more than 400 copies per mL and less than 0·5 log10 lower than baseline at week 4; more than 400 copies per mL and less than 1 log10 lower than baseline at week 12; and un confirmed concentration of more than 400 copies per mL as of that study week at or after week 24. Sites were instructed to schedule a confirmatory sample drawn at least 1 week after the initial test showing a viral load higher than 400 copies per mL and, if possible, no later than the next scheduled study visit and before the end of study participation. Prespecified secondary endpoints were change in CD4 cell count; new drug-resistance mutations; time to first grade 3 or higher adverse event; time to discontinuation of randomised treatment for toxicity; time to new AIDS-defining event, death, and targeted serious non-AIDS event; incidence and duration of hospitalisations; and change in fasting lipid concentrations. Additional supportive exploratory endpoints were time to virological failure or earliest treatment discontinuation; HIV-1 RNA concentrations of 400 copies per mL or lower; and liver function tests. We also assessed self-reported adherence.
Statistical analysis
Sample size was based on assessing non-inferiority of the raltegravir arm compared with the NRTI arm at a prespecified margin of 10% with a one-sided 2·5% type I error rate. The analysis plan included assessment for raltegravir arm superiority (upper bound of the CI <0%) if non-inferiority was found. When the study was designed, no published results were available from randomised trials of ritonavir-boosted lopinavir and raltegravir. We estimated the sample size based on estimates from other antiretroviral studies and the opinion of the study team. The target sample size of 600 participants (300 per group) was based on assumed virological failure of 25% in the raltegravir group and 28% in the NRTI group, and would provide at least 90% power for the comparison of the primary endpoint. Sample size was re-evaluated on Aug 7, 2013, due to availability of data from other international studies of second-line therapy,8,9,18 and the ACTG made a decision about revising the protocol enrolment and follow-up that was communicated to the core team on Aug 22, 2013, and the study sites on Aug 28, 2013, and followed by a protocol amendment. These data updated the assumptions of virological failure to 15% for the raltegravir group and 18% for the NRTI group, which provided the study with at least 90% power with a sample size of 480 participants (240 per group).
Baseline values were the closest measurement at or before study entry but after screening. During follow-up, non-overlapping visit windows and measurements closest to each window’s centre were used. Participants were considered to have completed the protocol if they had a week 96 visit or a visit within the study closeout period. Discontinuation of randomised treatment was defined as permanent discontinuation of raltegravir, ritonavir-boosted lopinavir, or NRTI, or intentional addition of two or more NRTIs without study NRTI discontinuation. Efficacy analyses used an intention-to-treat approach, censoring participants at their last visit. In the primary analysis (comparison between groups of the cumulative probability of virological failure by week 48) we used the Kaplan-Meier method with Greenwood’s variance weighted by randomisation stratification factors.19 More specifically, inference regarding non-inferiority was based on the differences in the 48 week cumulative incidence of virological failure with 95% CIs stratified by randomisation stratification factors. The inverse of the stratum-specific variance was used for the stratum weights.20 Subgroup analyses for the primary endpoint were prespecified for the randomisation stratification factors, sex, race (black vs non-black), country, HIV-1 subtype, and drug resistance at study entry; two-way interactions were evaluated based on the Wald test. Comparison of secondary endpoints between the study groups used stratified t test for continuous outcomes, stratified log-rank test for time-to-event out comes, and stratified Cochran-Mantel-Haenszel (CMH) test for binary outcomes. Safety analyses used an as-treated approach, censoring participants at the earlier of dis continuation of randomised treatment or their last contact.
To estimate activity of the selected NRTIs for each participant randomised to the NRTI group, we calculated an NRTI genotypic sensitivity score (GSS) by assigning each NRTI in the second-line regimen a score of 0, 0·25, 0·5, 0·75, or 1 for high-level, intermediate, low-level, potential low-level resistance, or susceptibility, respectively, based on Stanford algorithm version 7.0, and summing these scores to give a maximum of 2 or 3, depending on the number of NRTIs in the regimen. The study was monitored by a data and safety monitoring board convened by the National Institute of Allergy and Infectious Diseases; as prespecified, they met twice during the study. We used SAS version 9.2 for all statistical analyses. The trial was registered with ClinicalTrials.gov, number NCT01352715.
Role of the funding source
The funders of the study had an oversight role in the development and monitoring of the study but had no role in the conduct, analyses, and conclusions of the study. PK, an author of this study and a medical monitor from NIH/NIAID, had a role in the design, data interpretation, manuscript revision, and intellectual contribution; however, his views were his own and did not represent the funder’s views. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between March 13, 2012, and Oct 2, 2013, we enrolled and randomly assigned 515 participants (figure 1). Of the 82 participants who prematurely discontinued the study, 31 were at sites unable to continue study participation due to loss of their ACTG network affiliation. 12 (2%) participants discontinued the study before week 48. 23 (4%) discontinued study therapy prematurely. Median follow-up was 87 weeks (IQR 71–96) and this did not differ by groups (85 weeks [70–96] in raltegravir group vs 88 [71–96] in NRTI group; Wilcoxon rank-sum p=0·84). The study closeout period was from Sept 17 to Oct 29, 2014.
Figure 1. Trial profile.
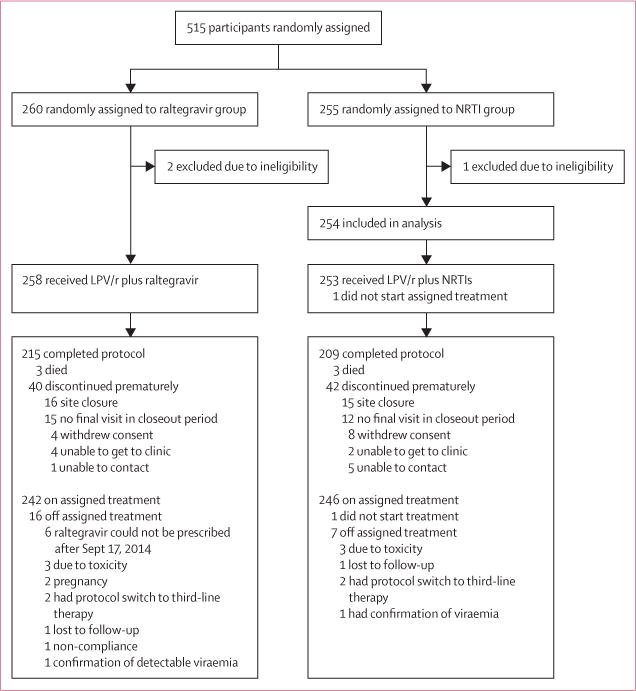
LPV/r=ritonavir-boosted lopinavir. NRTI=nucleoside or nucleotide reverse transcriptase inhibitor.
Characteristics at study entry were balanced between groups (table 1). Antiretroviral therapy exposure was similar between study groups with a median duration of first-line NNRTI-containing triple antiretroviral therapy regimen of 4·0 years (IQR 2·2–6·1); 4·1 years (2·2–6·3) in the raltegravir group and 4·0 years (2·2–6·0) in the NRTI group. The last NNRTI-containing regimen before enrolment was lamivudine, zidovudine, and nevirapine for 124 (24%) participants; lamivudine, tenofovir disoproxil fumarate, and efavirenz for 122 (24%); stavudine, lamivudine, and nevirapine for 109 (21%); lamivudine, tenofovir disoproxil fumarate, and nevirapine for 61 (12%); lamivudine, zidovudine, and efavirenz for 56 (11%); and other combinations for the remaining 40 (8%). Only 19 (4%) participants had locally provided resistance test results available at time of study entry. Emtricitabine and tenofovir disoproxil fumarate was chosen for most (n=173; 68%) of participants in the NRTI group.
Table 1.
Baseline characteristics
| Raltegravir group (n=258) | NRTI group (n=254) | |
|---|---|---|
| Age (years) | 39 (34–44) | 38 (33–43) |
|
| ||
| Men | 124 (48%) | 128 (50%) |
|
| ||
| Race | ||
| White, non-Hispanic | 0 | 1 (<1%) |
| Black, non-Hispanic | 164 (64%) | 162 (64%) |
| Hispanic | 11 (4%) | 9 (4%) |
| Asian or Pacific Islander | 83 (32%) | 82 (32%) |
|
| ||
| Body-mass index (kg/m2) | 23 (20–27) | 22 (19–25) |
|
| ||
| Plasma HIV-1 RNA | ||
| Log10 copies per mL | 4·6 (0·8) | 4·5 (0·9) |
| ≥100 000 copies per mL at screening | 84 (33%) | 82 (32%) |
|
| ||
| CD4 counts (cells per μL) | 178 (170) | 182 (160) |
|
| ||
| Nadir CD4 counts (cell per μL) | 122 (136) | 113 (96) |
|
| ||
| Time on first-line regimen (years) | 4·1 (2·2–6·3) | 4·0 (2·2–6·0) |
|
| ||
| Past AIDS-defining event | 70 (27%) | 80 (31%) |
|
| ||
| HIV-1 subtype* | ||
| A1 | 21/244 (9%) | 26/246 (11%) |
| B | 10/244 (4%) | 8/246 (3%) |
| C | 201/244 (82%) | 195/246 (79%) |
| Other | 12/244 (5%) | 17/246 (7%) |
|
| ||
| NRTI selected for use at study entry† | ||
| Emtricitabine and tenofovir‡ | – | 173 (68%) |
| Lamivudine and zidovudine | – | 49 (19%) |
| Lamivudine and abacavir | – | 21 (8%) |
| Emtricitabine, tenofovir‡, and zidovudine | – | 8 (3%) |
| Lamivudine, zidovudine, and abacavir | – | 2 (1%) |
|
| ||
| Local resistance test available before study entry | 10 (4%) | 9 (4%) |
|
| ||
| Genotypic mutations at baseline§ | 244 (95%) | 246 (97%)† |
| At least one NRTI mutation | 231/244 (95%) | 238/246 (97%) |
| Met184Val/Ile | 217/244 (89%) | 221/246 (90%) |
| Lys65Arg | 56/244 (23%) | 51/246 (21%) |
| ≥1 TAM | 113/244 (46%) | 124/246 (50%) |
| ≥3 TAMs | 59/244 (24%) | 60/246 (24%) |
| Gln151Met | 3/244 (1%) | 7/246 (3%) |
| 69 insertion/deletion | 0 | 4/246 (2%) |
| Complex resistance (Lys65Arg, ≥3 TAMs, Gln151Met, 69 insertion/deletion) | 115/244 (47%) | 119/246 (48%) |
| NRTI genotypic sensitivity score† | ||
| <1 | – | 117/245 (48%) |
| ≥1 | – | 128/245 (52%) |
|
| ||
| ≥1 NNRTI mutation | 238/244 (98%) | 238/246 (97%) |
Data are median (IQR), n (%), mean (SD), or n/N (%). NRTI=nucleoside or nucleotide reverse transcriptase inhibitor. NNRTI=non-nucleoside/nucleotide reverse transcriptase inhibitor. TAM=thymidine analogue mutation.
HIV subtype for 490 participants with HIV resistance tests at study entry.
One participant did not start treatment in NRTI arm.
Tenofovir disoproxil fumarate.
Codon mixtures are counted as mutant.
490 (96%) participants had full protease and reverse transcriptase sequences available from study entry (table 1). Overall, 96% had at least one NRTI mutation, 97% had at least one NNRTI mutation, and 95% had both. Most participants had extensive resistance (table 1). Except for the four participants with 69 insertion detected only in the NRTI group, resistance at study entry seemed similar between groups. The median NRTI GSS in the NRTI group was 1·00 (IQR 0·25–1·00; range 0–2). Major protease inhibitor mutations were rare; none were expected to affect susceptibility to ritonavir-boosted lopinavir.
By week 48, the cumulative probability of virological failure was 10·3% (95% CI 6·5–14·0) in the raltegravir group and 12·4% (8·3–16·5) in the NRTI group (figure 2). The primary analysis weighted by randomisation stratification factors showed a small difference of −3·4% (95% CI −8·4 to 1·5) favouring raltegravir and indicating non-inferiority, but not superiority, to NRTIs.
Figure 2. Cumulative probability of virological failure.
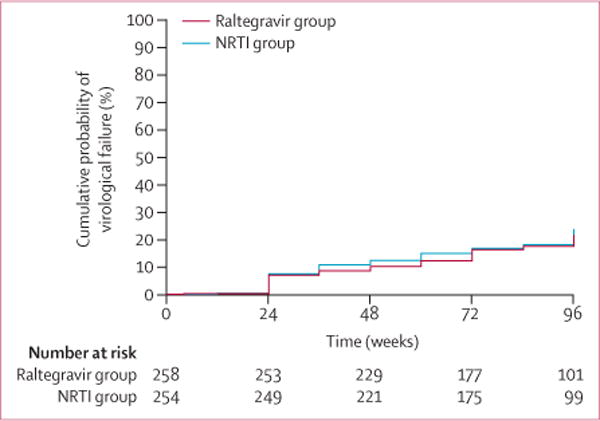
Time is weeks since study entry. LPV/r=ritonavir-boosted lopinavir.
NRTI=nucleoside or nucleotide reverse transcriptase inhibitor.
At week 48, 92% (95% CI 87–95) of participants in the raltegravir group and 91% (87–95) in the NRTI group had plasma HIV-1 RNA concentrations lower than 400 copies per mL (missing data ignored; stratified CMH p=0·91). The increase in CD4 count was 199 (95% CI 181–218) in the raltegravir group and 190 (171–209) in the NRTI group. The mean increase in CD4 count from study entry to week 48 was 195 cells per μL (182–208), with no difference between groups (weighted t test p=0·33).
Cumulative probability of a composite of virological failure or discontinuation of randomised study treatment showed a similar result as our findings for the primary endpoint (difference of −3·4%, 95% CI −8·6 to 1·7). Additionally, analyses treating missing data as failure (like an FDA snapshot analysis) and only including those on randomised study treatment gave similar results (data not shown). Post-hoc analyses at weeks 24, 48, and 96 using thresholds of 200 copies per mL and 40 copies per mL were consistent with the primary analysis (data not shown).
We observed no difference between the probability of virological failure between groups by any of the stratification factors (figure 3). A strong inverse association was detected between extent of NRTI resistance at study entry, as measured by multiple prespecified criteria, and probability of virological failure by 48 weeks. In the NRTI group, participants with a GSS of less than 1 (more resistance) had a lower probability of virological failure than did those with GSS of 1 or more (difference –8·4%, 95% CI –16·6 to –0·3; p=0·042). Similarly, having three or more IAS NRTI mutations was associated with a lower probability of virological failure in both groups (hazard ratio [HR] 0·45, 95% CI 0·30–0·70; p=0·00031). Also, having complex resistance at study entry (Lys65Arg, ≥3 thymidine analogue mutations [TAM], Gln151Met, or 69 insertion/deletion) was associated with lower probability of virological failure in both groups (HR 0·49, 95% CI 0·31–0·76; p=0·0013). Finally, presence of Met184Val/Ile at study entry was associated with a lower risk of virological failure compared to absence of Met184Val/Ile (0·41, 0·25–0·67; p=0·0004) and presence of both Lys65Arg and Met184Val/Ile at entry was associated with even lower risk of virological failure (0·19, 0·08–0·44; p=0·0001). These associations remained after adjustment for multiple confounding factors (baseline HIV-1 RNA concentration, week 4 self-reported adherence, first-line NRTI exposure, and country).
Figure 3. Difference in the cumulative probability of virological failure at week 48 between groups for prespecified subgroups.
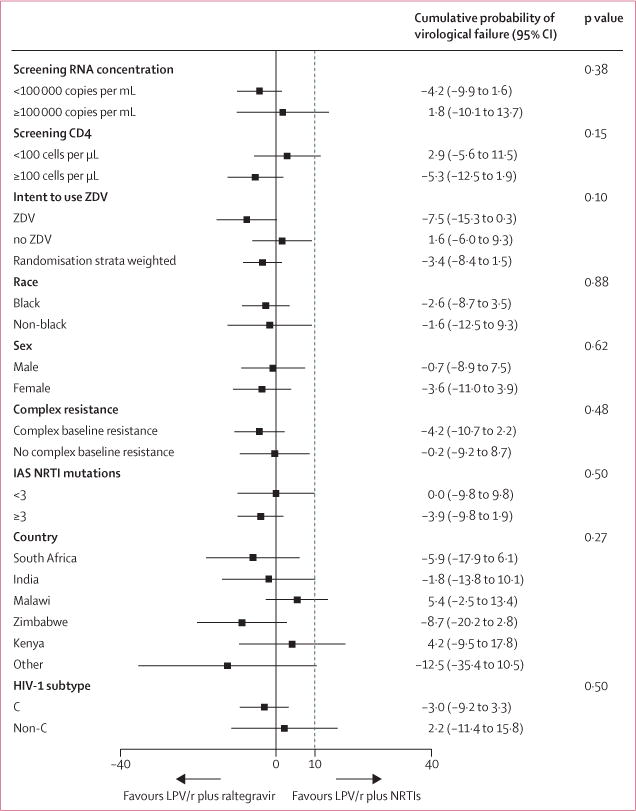
LPV/r=ritonavir-boosted lopinavir. NRTI=nucleoside or nucleotide reverse transcriptase inhibitors. ZDV=zidovudine.
IAS=International AIDS Society. The dotted line represents the prespecified non-inferiority margin.
Of the 96 participants (46 in the raltegravir group, 50 in the NRTI group) who had virological failure by the end of follow-up (Oct 27, 2014), paired study entry and failure samples were available for genotype analysis for 88% (table 2). 13 (29%) of 45 participants with available samples in the NRTI group developed new resistance mutations. Ten (26%) of 39 in raltegravir group developed new resistance mutations, including integrase mutations in all ten, with Asn155His being most frequent.
Table 2.
HIV resistance testing at virological failure
| Raltegravir group (n=46) | NRTI group* (n=50) | |
|---|---|---|
| Any mutation† | 39 (85%) | 45 (90%) |
| New NRTI mutations | ||
| Met184Val/Ile | 1/39 (3%) | 1/45 (2%) |
| Lys65Arg | 0 | 0 |
| TAMS | 0 | 4/45 (9%) |
| Gln151Met | 0 | 0 |
| Thr69Asn | 0 | 1/45 (2%) |
| Major protease inhibitor mutations | ||
| Met46Ile | 0 | 3/45 (7%) |
| Leu76Val | 0 | 2/45(4%) |
| Val82Ala | 0 | 2/45(4%) |
| Integrase mutations | ||
| Thr66Ala | 1/39 (3%) | 0 |
| Thr97Ala | 3/39 (8%) | 0 |
| Tyr143Cys/Arg | 2/39 (5%) | 0 |
| Asn155His | 6/39 (15%) | 0 |
Codon mixtures are counted as mutant. NRTI=nucleoside reverse transcriptase inhibitor. TAM=thymidine analogue mutation.
Not available for one participant who did not start treatment in NRTI arm.
Number of participants with paired study entry and failure genotypes.
Self-reported adherence was similar by randomised group at each visit overall and among those with virological failure. Self-reported adherence decreased over time, with 399 (78%) of 509, 352 (72%) of 490, and 147 (62%) of 237 of participants reporting never skipping medications at weeks 4, 48, and 96, respectively. Adherence was the lowest in participants with virological failure, with only 53% reporting never skipping antiretroviral therapy.
Three (1%) participants in each group prematurely discontinued randomised study treatment due to toxic effects and 21 (8%) in the NRTI group substituted one or more NRTIs. Grade 3 or higher signs and symptoms and grade 3 or higher neurological events of many types occurred (table 3), with grade 3 or higher laboratory abnormalities found in more than a quarter of participants. More chemistry and haematological abnormalities occurred in the NRTI group than the raltegravir group; but more metabolic abnormalities occurred in the raltegravir group, mostly in LDL and total cholesterol. At week 48, the mean difference between groups for total cholesterol was 15·4 mg/dL (95% CI 8·3–22·6; p<0·0001), for LDL cholesterol was 7·9 mg/dL (2·0–13·8; p=0·0088), and for triglycerides was 43·6 mg/dL (18·2–69·1; p=0·0078). Time to first grade 3 or higher adverse event at least one grade higher than at study entry was shorter in the NRTI group than in the raltegravir group (stratified log-rank p=0·040; figure 4).
Table 3.
Grade 3 or 4 adverse events, deaths, AIDS-defining events, non-AIDS-defining events, and hospitalisations
| Raltegravir group (n=258) | NRTI group (n=254) | |
|---|---|---|
| Clinical adverse events | 27 | 46 |
| Participants with at least one event | 18 (7%) | 27 (11%) |
| General body | 10 (4%) | 15 (6%) |
| Pain | 5 (2%) | 5 (2%) |
| Cachexia | 4 (2%) | 5 (2%) |
| Asthenia, fatigue, or malaise | 0 (0%) | 2 (1%) |
| Fever | 1 (<0·5%) | 4 (2%) |
| Circulatory or cardiac | 1 (<0·5%) | 0 |
| Haematology | 2 (1%) | 0 |
| Gastrointestinal | 6 (2%) | 7 (3%) |
| Diarrhoeal | 6 (2%) | 7 (3%) |
| Skin | 0 | 1 (<0·5%) |
| Neurological | 1 (<0·5%) | 7 (3%) |
| Special senses | 0 | 1 (<0·5%) |
| Other | 0 | 2 (1%) |
| Multiple attribution | 1 (<0·5%) | 2 (1%) |
| Laboratory adverse events | 185 | 197 |
| Participants with at least one event(%) | 62 (24%) | 71 (28%) |
| Chemistry | 18 (7%) | 35 (14%) |
| Low albumin | 1 (<0·5%) | 0 |
| High alkaline phosphatase | 1 (<0·5%) | 1 (<0·5%) |
| Low bicarbonate | 2 (1%) | 0 |
| High or low calcium | 2 (1%) | 0 |
| High carbon dioxide | 0 | 1 (<0·5%) |
| Low phosphorus | 11 (4%) | 23 (9%) |
| High or low potassium | 3 (1%) | 6 (2%) |
| High or low sodium | 4 (2%) | 7 (3%) |
| Haematology | 9 (3%) | 14 (6%) |
| Low absolute neutrophil count | 3 (1%) | 7 (3%) |
| Low haemoglobin | 1 (<0·5%) | 6 (2%) |
| Low platelets | 5 (2%) | 3 (1%) |
| Low white blood cells | 0 | 2 (1%) |
| Hepatic | 8 (3%) | 6 (2%) |
| Metabolic | 38 (15%) | 23 (9%) |
| High fasting LDL cholesterol | 17 (7%) | 6 (2%) |
| High fasting total cholesterol | 18 (7%) | 10 (4%) |
| High fasting triglycerides | 12 (5%) | 13 (5%) |
| High non-fasting glucose | 3 (1%) | 1 (<0·5%) |
| Endocrine | 3 (1%) | 5 (2%) |
| Pancreatic | 0 | 1 (<0·5%) |
| Renal | 3 (1%) | 3 (1%) |
| Urinalysis | 0 | 2 (1%) |
| Any* | 62 (24%) | 81 (32%) |
| Events | ||
| Deaths† | 3 | 3 |
| AIDS-defining illnesses | 13 (n=12) | 14 (n=14) |
| Bacterial pneumonia‡ | 1 | 1 |
| Cervical cancer | 0 | 1 |
| Cryptococcal meningitis | 0 | 1 |
| Herpes simplex virus disease§ | 1 | 1 |
| HIV-associated dementia | 1 | 0 |
| Tuberculosis | 10 | 10 |
| Number of serious non-AIDS-defining events¶ | 6 (n=5) | 5 (n=5) |
| Acute gastrointestinal or diarrhoeal syndrome | 0 | 1 |
| Bacterial sepsis | 3 | 0 |
| Cerebrovascular accident (stroke) | 0 | 1 |
| Acute myocardial infarction | 1 | 0 |
| Deep-vein thrombosis | 1 | 0 |
| Depression | 0 | 1 |
| Genitourinary and renal system disease or disorder | 1 | 2 |
| Number of hospitalisations | 16 (6%) | 23 (9%) |
| Incidence (per 100 person-years) | 5·2 | 8·3 |
Adverse events do not add to the total of the category since participants can have multiple events across categories. NRTI=nucleoside or nucleotide reverse transcriptase inhibitors.
These events also had to be at least one grade higher than baseline and occur while on randomised therapy.
All deaths were considered HIV-related; primary causes of death were tuberculosis, sepsis, Kaposi’s sarcoma, acute respiratory failure, acute gastroenteritis, and stroke.
Two separate episodes of bacterial pneumonia within 12 months.
Herpes simplex virus lesions persisting longer than 30 days.
To be considered serious, these events had to either result in hospitalisation or be of moderate or higher severity.
Figure 4. Cumulative probability of an adverse event.
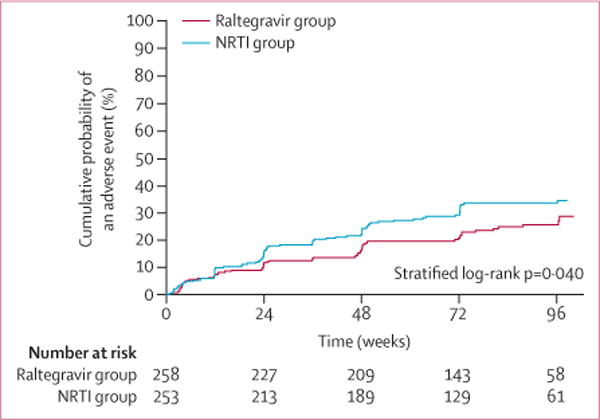
Includes all participants who started randomised study drug; time is weeks since first dose of study treatment. LPV/r=ritonavir-boosted lopinavir.
NRTI=nucleoside or nucleotide reverse transcriptase inhibitors.
Three participants in each group died; all deaths occurred in participants starting the study with CD4 cell counts lower than 50 cells per μL (table 3). 27 new AIDS-defining diagnoses occurred in 26 (5%) participants with no difference by group (stratified log-rank p=0·66). Tuberculosis was the most commonly reported AIDS-defining diagnosis (n=20). 11 new serious non-AIDS diagnoses occurred in ten (2%) participants with no difference by group (stratified log-rank p=0·95); the most common was bacterial sepsis (table 3). Incidence of hospitalisation and total hospital days did not significantly differ by group (9% in the NRTI group and 6% in the raltegravir group). 48 (9%) participants had at least one serious adverse event: 29 (11%) in the NRTI group and and 19 (7%) in the raltegravir group. Five women in each group became pregnant.
Discussion
Our findings from nine resource-limited countries show that both NRTIs and raltegravir, when added to ritonavir-boosted lopinavir, provide high virological efficacy with more than 90% of participants having HIV-1 RNA concentrations lower than 400 copies per mL at week 48. Our primary analysis showed raltegravir to be non-inferior, but not superior to, NRTI therapy. Immunological response was robust in both groups with a mean increase of 195 CD4 cells per mL gained from study entry to week 48.
The virological suppression rate in the NRTI group is particularly impressive given the high prevalence of resistance at study entry to NRTIs (table 1) and the selection of NRTIs without aid of resistance testing in 96% of participants. Although the study’s NRTI selection algorithm recommended zidovudine for participants who had virological failure with tenofovir disoproxil fumarate and vice versa (to take advantage of the antagonistic pathways for thymidine analogues and tenofovir resistance),21 most participants had only one (range zero to two) active NRTI in their regimen and only a few (21%) were prescribed zidovudine. Overall, efficacy of the NRTI group was not compromised by extensive resistance at study entry and did not increase with more active NRTIs in the regimen. In fact, we observed a strong inverse association between the extent of NRTI resistance at study entry and probability of virological failure; a finding that was consistent in multiple analyses with different measures to quantify NRTI resistance including NRTI GSS (<1 vs >1), number of IAS NRTI mutations (>3 vs <3), or complex NRTI resistance (presence vs absence of Lys65Arg, ≥3 TAMs, Gln151Met, or 69 insertion/deletion). An inverse association between NRTI resistance and probability of virological failure was also reported by the SECOND-LINE22 and EARNEST23 trials.
Our results confirm that ritonavir-boosted lopinavir provides substantial activity in second-line antiretroviral therapy.24 Additional virological activity might be attributable to NRTIs, either as partly active drugs despite genotypical evidence of resistance or possibly by maintaining reduced viral fitness. Lys65Arg and Met184Val/Ile reduce replication capacity and their effects might be additive.25 Consistent with this, participants with Lys65Arg and Met184Val/Ile at study entry had a lower hazard of virological failure compared with participants with no Met184Val/Ile or with Met184Val/Ile alone. We hypothesise that resistance that arose during first-line therapy could have rendered the virus hypersusceptible to protease inhibitors and might partly explain the favourable response, despite extensive NRTI resistance, as well as to the raltegravir group that included ritonavir-boosted lopinavir in the regimen. This possibility is being investigated. Phenotypic hyper susceptibility to lopinavir or ritonavir and other protease inhibitors has been described in association with reduced replication capacity in viral isolates with wild-type protease.26
The inverse association between resistance at study entry and virological failure on second-line antiretroviral therapy could also be explained if limited resistance is a marker of non-adherence during first-line antiretroviral therapy that continued during second-line antiretroviral therapy. Participants with virological failure in this study had low self-reported adherence, suggesting that suboptimal adherence was likely a major contributor to virological failure. Similar to other clinical trials of boosted protease inhibitors, major lopinavir resistance mutations were rarely detected during virological failure. However, mutations in gag or gp41 of envelope have been reported to affect susceptibility to protease inhibitor in the absence of protease inhibitor mutations.27 To investigate this possibility, genotypic analyses of gag and env comparing entry and virological failure samples are in progress.
Similar proportions of participants with virological failure in each group had newly detected mutations, but the raltegravir group more frequently had integrase mutations whereas the NRTI group had either protease inhibitor or NRTI mutations (table 2). Which resistance patterns will be less challenging to treat depends on availability of specific antiretroviral agents and strategies for regimen sequencing.
Two other large international randomised clinical trials have previously reported efficacy findings similar to ours.8–10 SECOND-LINE, in middle-income to high-income countries, found virological efficacy of ritonavir-boosted lopinavir plus raltegravir to be non-inferior to that of ritonavir-boosted lopinavir plus NRTIs, with about 82% and 75% of participants maintaining HIV-1 RNA concentrations lower than 200 copies per mL at 48 weeks and 96 weeks, respectively. EARNEST was done in five low-income sub-Saharan African countries and although it used a composite clinical and laboratory primary endpoint, 86% of participants in both groups had HIV-1 RNA viral loads less than 400 copies per mL at 96 weeks. In both studies, significant NRTI resistance at entry was found; however, the raltegravir group was non-inferior to the NRTI group. Although the efficacy results were similar in these studies of second-line antiretroviral therapy, ours include data from nine low-income and middle-income countries on three continents.
Significant, albeit modest, increases in concentrations of total cholesterol, LDL cholesterol, and triglycerides were noted in the raltegravir group (table 3). We observed no differences in HDL cholesterol or total:HDL cholesterol ratio (data not shown). These observations partly reflect the previously reported lipid-lowering effect of tenofovir disoproxil fumarate.28,29 SECOND-LINE showed similar differences for total and LDL cholesterol.9 EARNEST did not report lipid outcomes.8 It will be important to determine whether the lipid advantages observed in the NRTI group translate into clinical benefit in persons with hyperlipidaemia or other major risk factors for coronary artery disease. We observed a shorter time to any grade 3 or 4 toxicity (at least one grade higher than baseline) in the NRTI group (figure 4). SECOND-LINE reported more grade 3 and 4 adverse events in their NRTI group than in their raltegravir group, although overall adverse event rates were not different between groups. EARNEST observed no differences in grade 3 or 4 adverse events overall or those attributed to a study drug. In our study, no single type of toxicity predominated. This contrasts with the data from SECOND-LINE where gastrointestinal events were the predominant toxicity and were more common with NRTI use. Of note, zidovudine, a common cause of gastrointestinal adverse events, was used by 45% of the NRTI group in SECOND-LINE compared with 21% in our study (table 1). Types of adverse events in EARNEST were distributed among various categories. Cultural differences in self-reported signs and symptoms and varying assessment methods could explain the poor concordance of clinical adverse event rates among the three studies. Absence of a predominant NRTI-associated toxicity profile across studies makes it difficult to suggest a specific population for whom ritonavir-boosted lopinavir plus raltegravir could offer short-term or long-term safety benefits. Similarly, we cannot address whether the difference in toxic effects would continue to favour the raltegravir group with longer-term follow-up, although long-term toxicities of NRTIs are of potential concern with therapies that might be taken for decades.
A strength of our study is that it was done at diverse sites in low-to-middle-income countries in Africa, Asia, and Latin America with little access to resistance testing, making it generalisable to settings where most people with HIV reside. One weakness of this study, SECOND-LINE, and EARNEST is that the open-label design might have led to bias in assessment of symptoms and signs. However, our primary endpoint and several of our secondary endpoints were objective laboratory measurements on masked specimens. Adherence self-reporting might overestimate adherence and wording of adherence questions might influence responses (although we used a validated questionnaire that has been used previously in international studies). We could not assess adherence at baseline since participants could enter the study even if they had stopped antiretroviral medication after detection of first-line virological failure. Rates of AIDS-defining illnesses and serious toxic effects were low, making it more challenging to show differences between groups. Lipoatrophy, a side-effect of some NRTIs, was not assessed by quantitative methods, potentially attenuating our capacity to detect a difference between groups in terms of this side-effect.
In summary, A5273 adds clear and generalisable findings, which, in combination with EARNEST and SECOND-LINE, provide solid evidence to recommend ritonavir-boosted lopinavir plus NRTI or raltegravir for second-line antiretroviral therapy. Availability, cost, potential toxic effects, and plans for third-line antiretroviral therapy will be important determinants of the preferred strategy at the country level. Our data support the current WHO recommendation for a boosted protease inhibitor plus NRTIs as second-line HIV therapy after failing non-NRTI-based regimens in resource-limited settings, even when there is no access to resistance testing, irrespective of NRTI resistance. The high virological efficacy achieved in this setting suggests that health-care systems with limited resources might be best served by focusing on optimisation of drug availability and adherence, rather than resistance testing after first-line failure. The explanation for the observation that more extensive NRTI resistance at study entry was associated with better second line therapy outcome is under investigation.
Research in context.
Evidence before this study
After failure of first-line regimens consisting of a non-nucleoside reverse transcriptase inhibitor plus two nucleoside or nucleotide reverse transcriptase inhibitors (NRTIs) in resource-limited settings, second-line regimens are generally selected without resistance testing and few different drugs are generally available. Thus, NRTIs might have cross-resistance to those used in initial therapy, which could potentially result in less effective second-line therapy. When this study was being planned, no adequately powered, randomised studies had been completed in resource-limited settings to compare an NRTI-sparing regimen with the WHO-recommended second-line regimen of a boosted protease inhibitor plus NRTIs. We were aware of two planned and subsequently completed studies. EARNEST was a three-group study that compared ritonavir-boosted lopinavir plus NRTIs, ritonavir-boosted lopinavir plus raltegravir, and ritonavir-boosted lopinavir monotherapy in five sub-Saharan African countries with a composite endpoint of good disease control and without resistance testing to guide NRTI selection. SECOND-LINE assessed the same two combination regimens in middle-income and high-income countries with a pure virological endpoint and three-quarters of the NRTIs selected with resistance-testing results. In EARNEST, ritonavir-boosted lopinavir monotherapy was inferior. However, both EARNEST and SECOND-LINE showed ritonavir-boosted lopinavir plus raltegravir to be non-inferior and not superior to ritonavir-boosted lopinavir plus NRTIs, and pronounced these two regimens as viable alternatives for second-line therapy in resource-limited settings. We searched PubMed for papers published in English and Spanish with no date restrictions in September and October, 2015, using terms that included “secondline and HIV” or “secondline and antiretroviral” and “randomized clinical trial”. We also read the references of US and WHO antiretroviral therapy guidelines and the abstracts of major HIV research meetings, including the Conference on Retroviruses and Opportunistic Infections, International AIDS Conferences, and International AIDS Society Meetings for the previous 4 years, and asked all of the masthead authors for relevant research for this manuscript.
Added value of this study
Results from this, the AIDS Clinical Trials Group (ACTG) A5273 SELECT study, confirms findings from EARNEST and SECOND-LINE and expands the breadth of evidence by providing data from a robust clinical trial in nine low-income and middle-income countries in Africa, Asia, and South America where resistance testing is not generally used for selection of second-line antiretroviral therapy (96% of our participants enrolled did not have access to resistance tests). Our findings support current WHO guidance for second-line therapy in resource-limited settings.
Implications of all the available evidence
The evidence indicates that ritonavir-boosted lopinavir plus NRTIs and ritonavir-boosted lopinavir plus raltegravir are both acceptable options for second-line antiretroviral treatment in resource-limited settings. Although concern about cross-resistance has been a major driver of interest in NRTI-sparing options for settings without resistance-testing capacity, evidence has accumulated showing that efficacy of ritonavir-boosted lopinavir plus NRTI for second-line ART in resource-limited settings is not significantly compromised by previous NRTI resistance. Accordingly, drug availability, cost, and potential toxic effects should drive decisions about which strategy to prioritise over others at the country level.
Here we report results of AIDS Clinical Trials Group (ACTG) A5273, the Second-Line Effective Combination Therapy (SELECT) study; which was done exclusively in low-income and middle-income countries in Africa, Asia, and Latin America without use of genotyping to select NRTIs. We hypothesised that the combination of ritonavir-boosted lopinavir plus raltegravir is non-inferior to one of ritonavir-boosted lopinavir plus the best-available NRTIs.
Acknowledgments
This study was supported National Institute of Allergy and Infectious Diseases (award number U01AI068636), National Institute of Mental Health (NIMH), and the National Institute of Dental and Craniofacial Research (NIDCR). The study was also funded via the following grants: AI069432, AI069438, AI069481, 2UM1AI069423 (including a subcontract to the Pittsburgh Virology Specialty Laboratory), UM1 AI069471, and UM1 AI068634 (ACTG Statistical and Data Management Center) and the Investigator-Initiated Studies Program of Merck Sharp & Dohme. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, or Merck Sharp & Dohme. Abbott Laboratories, Gilead Sciences, GlaxoSmithKline, and Merck Sharp & Dohme provided the study drugs. We thank the study participants, the other members of the A5273 team, especially Lara Hosey and Jennifer Rothenberg, and the staff members at the virology and genotyping laboratories for their work in testing for the primary endpoint and the genotyping. In addition, we thank Raphael J Landovitz for his support in reviewing all virological endpoints and the contributions of the staff at the sites and grants supporting their work, including grants UM1AI069423, UM1AI069423, UM1 AI069436, UM1 AI108568, UM1AI068636, AI069463, AI068638, AI069497, UM1AI069453, U01AI069484, UM1AI069518, UM1AI069417, UM1AI069476, 5UM1AI069399, and UM1 AI068636.
Footnotes
Contributors
AMLR contributed to the literature search, study design, data collection and interpretation, and manuscript drafting and revision. LJH contributed to the figures, data analysis and interpretation, and manuscript drafting and revision. BT contributed to the literature search, study design, data interpretation, and manuscript revision. CLW contributed to the literature search, study design, virological resistance studies, data interpretation, and manuscript drafting and revision. LZ contributed to the data analysis and interpretation and manuscript revision. PK contributed to the study design, data interpretation, and manuscript revision. NK contributed to participant recruitment, data collection and interpretation, and manuscript revision. MCH contributed to participant recruitment, data collection and interpretation, and manuscript revision. BJ contributed to data management and manuscript revision. JWM contributed to the study design, virological resistance studies, data interpretation, and manuscript revision. ACC contributed to the literature search, study design, data interpretation, and manuscript drafting and revision. The members of the study group were responsible for study oversight and played other important roles for the study at their sites, reviewed the study results, manuscript, and provided input and other intellectual contributions.
The ACTG A5273 Study Group
S Poongulali, Chennai Antiviral Research and Treatment Clinical Research Center (CRS), Chennai, India; Metch Matoga, Malawi CRS, Lilongwe, Malawi; Anthony Chisada, Parirenyatwa CRS, Harare, Zimbabwe; Fatma Faraj Some, Moi University CRS, Eldoret, Kenya; Umesh G Lalloo, Durban International CRS, Durban, South Africa; Mohammed Siddique Rassool, University of Witwatersrand Helen Joseph CRS, Johannesburg, South Africa; Dileep Babasaheb Kadam, Byramjee Jeejeebhoy Government Medical College CRS, Puni, India; Lerato Mohapi, Soweto ACTG CRS, Soweto, South Africa; Venance Maro, Kilimanjaro Christian Medical Center CRS, Moshi, Tanzania; Mulinda Nyirenda, Blantyre CRS and Johns Hopkins Research Project Blantyre, Malawi; Raman Raghunathrao Gangakhedkar, Pune CRS, Pune, India; Sandra Wagner Cardoso, Instituto de Pesquisa Clinica Evandro Chagas-FIOCRUZ, Rio de Janeiro, Brazil; Khuanchai Supparatpinyo, Chiang Mai University, Chiang Mai, Thailand; Mey Leon, Barranco CRS, Lima, Peru; John MacRae, San Miguel CRS, Lima, Peru.
Declaration of interests
AMLR has received salary support from Johnson & Johnson (Peru) and Merck Sharp & Dohme (Peru). BT has received research grants and honoraria from ViiV, and honoraria from Gilead Sciences, GlaxoSmithKline, and Janssen. CLW has received honoraria from Abbvie/Abbott, Celera, and Merck Sharp & Dohme. JWM has received funds from Gilead Sciences, support from Cocrystal Pharma, and has a patent for 3’Azido Purine Nucleotide Prodrugs for treatment of viral infections (number 8, 815, 829). ACC has received research support from Bristol-Myers Squibb, Merck Sharp & Dohme, and Roche Molecular Systems; and honoraria from Merck Sharp & Dohme for data and safety monitoring board membership. All other authors declare no competing interests.
References
- 1.Granich R, Gupta S, Hersh B, et al. Trends in AIDS deaths, new infections and ART coverage in the top 30 countries with the highest AIDS mortality burden; 1990–2013. PLoS One. 2015;10:e0131353. doi: 10.1371/journal.pone.0131353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UNAIDS. “15 by 15” A Global Target Achieved. http://www.unaids.org/sites/default/files/media_asset/UNAIDS_15by15_en.pdf (accessed Sept 8, 2015).
- 3.INSIGHT START Study Group. Lundgren JD, Babiker AG, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015;373:795–807. doi: 10.1056/NEJMoa1506816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Guideline on when to start antiretroviral therapy and on pre-exposure prophylaxis for HIV. 2015 Sep; http://www.who.int/hiv/pub/guidelines/earlyrelease-arv/en/ (accessed Oct 13, 2015). [PubMed]
- 5.Campbell TB, Smeaton LM, Kumarasamy N, et al. Efficacy and safety of three antiretroviral regimens for initial treatment of HIV-1: A randomized clinical trial in diverse multinational settings. PLoS Med. 2012;9:e1001290. doi: 10.1371/journal.pmed.1001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tilghman M, Tsai D, Buene TP, et al. Pooled nucleic acid testing to detect antiretroviral treatment failure in HIV-infected patients in Mozambique. J Acquir Immune Defic Syndr. 2015;70:256–61. doi: 10.1097/QAI.0000000000000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization. Forecasting demand for antiretrovirals: Preliminary analyses from the GPRM data adult formulations. 2015 Mar 23; http://www.who.int/hiv/amds/ARV-forecasting-meeting-2015/en/ (accessed Sept 11, 2015).
- 8.Paton NI, Kityo C, Hoppe A, et al. Assessment of second-line antiretroviral regimens for HIV therapy in Africa. N Engl J Med. 2014;371:234–47. doi: 10.1056/NEJMoa1311274. [DOI] [PubMed] [Google Scholar]
- 9.SECOND-LINE Study Group. Ritonavir-boosted lopinavir plus nucleoside or nucleotide reverse transcriptase inhibitors versus ritonavir-boosted lopinavir plus raltegravir for treatment of HIV-1 infection in adults with virological failure of a standard first-line ART regimen (SECOND-LINE): a randomized, open-label, non-inferiority study. Lancet. 2013;381:2091–99. doi: 10.1016/S0140-6736(13)61164-2. [DOI] [PubMed] [Google Scholar]
- 10.Amin J, Boyd MA, Kumarasamy N, et al. Raltegravir non-inferior to nucleoside based regimens in second-line therapy with lopinavir/ritonavir over 96 weeks: a randomised open label study for the treatment of HIV-1 infection. PLoS One. 2015;10:e0118228. doi: 10.1371/journal.pone.0118228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chesney MA, Ickovic JR, Chambers DB, et al. Self-reported adherence to antiretroviral medications among participants in HIV clinical trials: the AACTG adherence instruments. Patient Care Committee & Adherence Working Group of the Outcomes Committee of the Adult AIDS Clinical Trials Group (AACTG) AIDS Care. 2000;12:255–66. doi: 10.1080/09540120050042891. [DOI] [PubMed] [Google Scholar]
- 12.Division of AIDS. Toxicity scale. 2004 Dec; Version 1.0. Clarification August 2009. http://rsc.tech-res.com/safetyandpharmacovigilance/gradingtables.aspx (accessed April 14, 2015)
- 13.Centers for Disease Control. Revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Morb Mortal Wkly Rep. 1993;1992;41:17. [PubMed] [Google Scholar]
- 14.WHO. Interim WHO clinical staging of HIV/AIDS and HIV/AIDS case definitions for surveillance. Geneva: World Health Organization; 2005. (WHO/HIV/2005.02). [Google Scholar]
- 15.Appendix 60. http://www.hptn.org/web20documents/HPTN052/Appendix60V1.123Feb2007.pdf (accessed Sept 11, 2015).
- 16.Wensing AM, Calvez V, Gü nthard HF, et al. 2014 Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22:642–50. [PMC free article] [PubMed] [Google Scholar]
- 17.Stanford Genotypic Resistance Interpretation Algorithm. Version 7. 0 (last updated 02/27/14). http://sierra2.stanford.edu/sierra/servlet/JSierra (accessed Sept 11, 2015).
- 18.Gross R, Zheng L, La Rosa A, et al. Partner-focused adherence intervention for second-line antiretroviral therapy: a multinational randomized trial (ACTG A5234) Lancet HIV. 2015;2:e12–19. doi: 10.1016/S2352-3018(14)00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenwood M. Reports on Public Health and Medical Subjects. Vol. 33. London: HM Stationery Office; 1926. The natural duration of cancer; pp. 1–26. [Google Scholar]
- 20.Prentice RL, Kalbfleisch JD. Hazard rate models with covariates. Biometrics. 1979;35:25–39. [PubMed] [Google Scholar]
- 21.Parikh UM, Zelina S, Sluis-Cremer N, Mellors JW. Molecular mechanisms of bidirectional antagonism between K65R and thymidine analog mutations in HIV-1 reverse transcriptase. AIDS. 2007;21:1405–14. doi: 10.1097/QAD.0b013e3281ac229b. [DOI] [PubMed] [Google Scholar]
- 22.Boyd MA, Moore CL, Molina J-M, et al. Baseline HIV-1 resistance, virological outcomes, and emergent resistance in the SECOND-LINE trial: an exploratory analysis. Lancet HIV. 2015;2:e42–51. doi: 10.1016/S2352-3018(14)00061-7. [DOI] [PubMed] [Google Scholar]
- 23.Paton N, Kityo C, Thompson J, et al. Impact of NRTI cross-resistance on second-line PI + NRTI therapy outcomes in Africa; Conference on Retrovirus and Opportunistic Infections; Seattle, WA, USA. Feb 23–26, 2015; 2015. 119 (abstr) [Google Scholar]
- 24.Kumarasamy N, Aga E, Ribaudo HJ, et al. Lopinavir/ritonavir monotherapy as second-line antiretroviral treatment in resource-limited settings: week 104 analysis of AIDS Clinical Trials Group (ACTG) A5230. Clin Infect Dis. 2015;60:1552–58. doi: 10.1093/cid/civ109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frankel FA, Invernizzi CF, Oliveira M, Wainberg MA. Diminished efficiency of HIV-1 reverse transcriptase containing the K65R and M184V drug resistance mutations. AIDS. 2007;21(6):665–75. doi: 10.1097/QAD.0b013e3280187505. [DOI] [PubMed] [Google Scholar]
- 26.Martinez-Picado J, Wrin T, Frost SD, et al. Phenotypic hypersusceptibility to multiple protease inhibitors and low replicative capacity in patients who are chronically infected with human immunodeficiency virus type 1. J Virol. 2005;79:55907–13. doi: 10.1128/JVI.79.10.5907-5913.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rabi SA, Laird GM, Durand CM, et al. Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J Clin Invest. 2013;123:3848–60. doi: 10.1172/JCI67399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moyle GJ, Orkin C, Fisher M, et al. A randomized comparative trial of continued abacavir/lamivudine plus efavirenz or replacement with efavirenz/emtricitabine/tenofovir DF in hypercholesterolemic HIV-1 infected individuals. PLoS One. 2015;10:e0116297. doi: 10.1371/journal.pone.0116297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Souza SJ, Luzia LA, Santos SS, Rondó PH. Lipid profile of HIV-infected patients in relation to antiretroviral therapy: a review. Rev Assoc Med Bras. 2013;59:186–98. doi: 10.1016/j.ramb.2012.11.003. [DOI] [PubMed] [Google Scholar]