

Abstract
Lung cancer is associated with very poor prognosis and considered one of the leading causes of death worldwide. Here, we present highly potent and selective bio-hybrid RNAi-peptide nanoparticles that can induce specific and long-lasting gene therapy in inflammatory tumour associated macrophages (TAMs), via an immune modulation of the tumour milieu combined with tumour suppressor effects. Our data prove that passive gene silencing can be achieved in cancer cells using regular RNAi NPs. When combined with M2 peptide-based targeted immunotherapy that immuno-modulates TAMs cell-population, a synergistic effect and long-lived tumour eradication can be observed along with increased mice survival. Treatment with low doses of siRNA (ED50 0.0025-0.01 mg/kg) in a multi and long-term dosing system substantially reduced the recruitment of inflammatory TAMs in lung tumour tissue, reduced tumour size (∼95%) and increased animal survival (∼75%) in mice. Our results suggest that it is likely that the combination of silencing important genes in tumour cells and in their supporting immune cells in the tumour microenvironment, such as TAMs, will greatly improve cancer clinical outcomes.
Keywords: tumour-associated macrophages, RNAi, tumour targeting, nanoparticles, lung cancer
Graphical abstract
1. Introduction
It is now widely accepted that cancer progression and metastasis arise from a complex and multi-step process consisting of transformation, growth and invasion of cancer cells within the tumour microenvironment.[1,2] Our understanding of the wide variety of cell populations that constitute the tumour microenvironment has considerably expanded over the last decade. Malignant solid tumours are known to contain an abundant population of macrophages within the infiltrating leukocytes. These leukocytes constitute an M2 marker for anti-inflammatory/regulatory macrophages, referred to as TAMs, markers of poor prognosis.[3] TAMs represent the “cellular arm” of cancer immune system, playing a crucial role in many aspects of tumour growth and development.[4] In fact, TAMs have been recently identified as hallmarks in cancer progression, metastasis and resistance to therapy.[5,6]
TAMs represent, therefore, an appealing target for cancer therapy. The most common anti-tumour strategies aiming to target TAMs are inhibition of macrophage recruitment, tumour-promoting activity and survival, or enhancement of tumuoricidal activity of macrophages.[7] This approach has not yet been translated into efficient clinical therapies, mainly because of the nonselective nature of the method, which can compromise the immune system in general. A possible solution is the development of a unique system with high selectivity towards disease cells while sparing healthy cells.
Previous studies, describing the use of immune-modifying microparticles targeting a scavenger receptor (MARCO) to modulate monocyte trafficking to sites of inflammation, have shown promising results in models such as myocardial infarction, autoimmune encephalitis, colitis and peritonitis.[8] Several other ligands, targeting macrophage membrane receptors, such as mannose or folate have been reported in TAMs.[9-11] However, receptors for mannose and folate are also expressed in normal epithelial cells and in all dendritic cells. Therefore, we used herein a peptide sequence identified by phage display, designated as M2pep, to selectively and preferentially bind to murine TAMs, avoiding binding to other leukocytes.[12]
Gene therapy has been receiving increasing attention in tumour eradication, especially when encompassing targeting capabilities. In particular, short interfering RNA (siRNA) has shown promising potential as a molecular approach to down-regulate specific gene expression in cancer cells.[13,14] Although siRNA delivery has been gaining momentum in the past decade, the development of efficient delivery vehicles for in vivo applications, especially for systemic delivery to immune cells, has remained a major obstacle in translating siRNA into therapeutics.[15,16] Recent advances in the development of nanomaterials for in vivo siRNA delivery in cancer therapies [17] allowed the production of highly potent, specific and biocompatible nanoparticle delivery vehicles.[18-23] Nevertheless, the development of clinical nanoformulations capable of selectively delivering siRNA to both cancer cells and immune cells remains challenging.[24]
Here, we hypothesized that peptide functionalized gold nanoparticles (AuNPs) for siRNA delivery could silence VEGF mRNA in the inflammatory tumour M2 macrophages and in lung cancer cells to enhance tumour inhibition in a lung cancer orthotopic murine model (Figure 1). In fact, throughout tumour progression, TAMs express high angiogenic factors, such as VEGF, which promote cancer progression and metastasis.[3,25,26] Consequently, combined therapy against lung cancer cells and TAMs can simultaneously repress cancer development. The main goal was to develop a combination approach in which targeted RNAi nanoparticles can be used to transform tumour-associated immune cells from an immunosuppressive to an immunostimulatory cell type in vivo, along with cancer cells inhibition. Although passive silencing can be achieved in cancer cells using regular RNAi NPs,[18,23] the targeting peptide immunotherapy have the capacity to inhibit both TAMs and cancer cells by targeting VEGF pathway in both cell population, stimulating a host immune response that results in long-lived tumour inhibition. Our hybrid approach combines targeted immunotherapy and cancer cells inhibition to induce long-lasting responses. The production of anti-tumour immune memory may elicit dramatic tumour regression and disease control for extended periods. By targeting these specific immune cells (TAMs), we are also inhibiting migration and consequently adverse function of these cells and their progeny. Rather than silencing just a gene in cancer cells, we aim to eradicate completely this immune cell population from the inflammatory site.
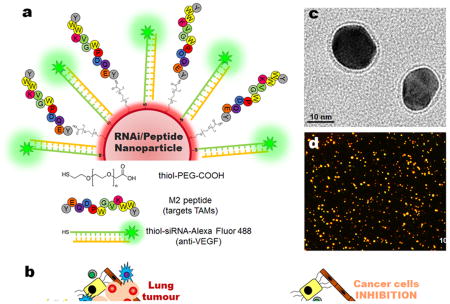
Figure 1. Nanoparticle-based strategy to deliver RNAi for VEGF silencing in both tumour-associated macrophages (TAMs) and lung cancer cells.

(a) Gold nanoparticles (AuNPs, ∼15 nm) functionalized with thiolated-PEG-COOH conjugated to TAMs-targeting peptide (M2pep) and thiolated anti-VEGF siRNA labelled with Alexa Fluor® 488 (b) Schematic of the outcome of the proposed combined silencing therapy (immunotherapy targeting TAMs and cancer cells) in vivo via highly specific and potent nanoparticles administered directly to bronchial airways. Transmission electron microscopy (TEM) with negative staining (c) and dark-field light scattering microscopy (d) images of RNAi-M2pep-AuNPs.
So far, and to the best of our knowledge, this is the first proof-of concept showing the synergistic effect of the dual targeted immunotherapy in cancer via RNAi/peptide nanoparticles. The developed nanodelivery system show the highest efficacy (ED50 0.0025-0.01 mg/kg) yet reported in the literature through targeting both immune cells and cancer cells in an in vivo cancer model. Our data show excellent tolerability and no innate immune response to the foreign siRNA nor the nanoparticles.
2. Results and Discussion
2.1. Nanoparticle design and functionalization
The NPs used in this study consist of a ∼15 nm gold core decorated with thiolated-PEG-COOH polymer and thiolated anti-VEGF siRNA labelled with Alexa Fluor® 488 (Figure 1a).
The specific anti-VEGF siRNA was identified in an in vitro screening from twenty candidate sequences (Supplementary Table S1 and Supplementary Fig. S1). The most efficient siRNA duplex with the sequence 5′: CCCACAUACACACAUAUAUUU (sense) and 3′: UUGGGUGUAUGUGUGUAUAUA (antisense) was selected for subsequent nanoparticle functionalization and in vitro and in vivo testing. See Supplementary Figure S2 for PEG optimization and quantification and Supplementary Figure S3 for M2Pep quantification.
The NPs were further functionalized with M2pep (peptide that selectively targets TAMs and not other leukocytes or alveolar macrophages[12]) via the PEG linker (RNAi-M2pep-AuNPs) and images from dark-field microscopy showed an excellent nanoparticle's dispersion and the images from transmission electron microscopy (TEM) showed an average diameter of the gold core of 14.9±2.5 nm (Figures 1c and 1d). The final siRNA:NP ratio was around 50:1 and the M2pep:NP was 30:1 (see Supplementary Information Table S2). The mean particle diameter of RNAi-M2pep-AuNPs was 23.2 (±2.8) nm with an SPR peak at 526 nm, as measured by DLS and UV-Vis extinction profiles and negative charge (zeta-potential -30.0±1.7 mV) (Supplementary Information Figure S4 and Supplementary Information Table S2).
The mean particle diameter of RNAi-M2pep-AuNPs was 23.2 (±2.8) nm with an SPR peak at 526 nm, as measured by DLS and UV-Vis extinction profiles and negative charge (zeta-potential -30.0±1.7 mV)] to explain particles optical characterization and the hydrodynamic radius determination.
To evaluate the stability of the formulated NPs in serum, we investigated in vitro the release of siRNA from both RNAi- (passive targeting) and RNAi-M2pep-AuNPs (active targeting), by incubating the NPs in cell culture medium with 10% serum (pH 7.4, 37 °C) for 96 hours and measuring the fluorescence intensity of Alexa Fluor® 488 labelled siRNA (Figure 2a). Over the first 48 hours, the NPs released siRNA at an average rate of approximately 0.5% per hour. This rate increased to approximately 1% per hour over the following 48 hours. The cumulative release of siRNA for the 96 hours was ∼1.3% per hour. This experiment allowed us to estimate the release of siRNA before reaching the tumour site, and it was found that both RNAi- and RNAi-M2pep-AuNPs are stable at a physiological pH in serum over an extended period of time, with only approximately 25% siRNA release over the first 48 h. The stability of the peptide in cell culture medium with 10% serum (pH 7.4, 37 °C) for 96 hours was also assessed. No significant peptide release was observed during 96 hours (Supplementary Information Figure S5).
Figure 2. In vitro siRNA release from RNAi- and RNAi-M2pep-AuNPs and silencing efficiency in stably transfected A549-luciferase-C8 human lung adenocarcinoma cells. (a) siRNA release from the NPs in cell medium with serum during 96 hours.

(b) The silencing effect was expressed as a concentration-dependent decrease in VEGF relative expression. RNAi- and RNAi-M2pep-AuNPs showed potent silencing effects in A549-luciferase-C8 cells with an ED50 of 1 nM, which corresponds to 0.01 mg/kg for in vivo administration. Data points represent group mean ± SD (n = 3, ** P < 0.01, *** P <0.005). (c-d) Ultrastructural TEM images of A-549-luciferase C8 cells incubated with 0.05 mg/kg of RNAi-M2pep-AuNPs. Scale bars: 1 μm (c) and 0.5 μm (d).
2.2. Efficient and selective in vitro and in vivo delivery of siRNA
We next assessed in vitro the cellular uptake and gene silencing efficiency in A549-luciferase-C8 human lung adenocarcinoma cells (with stable transfection of the North American Firefly Luciferase gene expressed from the CMV promoter) of both RNAi- and RNAi-M2pep-AuNPs. Confocal images of cellular uptake of NPs formulated with Alexa Fluor® 488 labelled siRNA (Supplementary Information Figure S6a) showed that both RNAi- and RNAi-M2pep-AuNPs can enter lung cancer cells, with a notable increase in uptake for RNAi-M2pep-AuNPs and an apparent predominance in perinuclear regions, as also reported elsewhere.[27] The ability of both RNAi- and RNAi-M2pep-AuNPs to silence VEGF was evaluated in lung cancer cells, and both systems (passive silencing via RNAi AuNPs and active silencing via RNAi-M2pep-AuNPs) provided a robust knockdown in a dose dependent manner (Figure 2b), with a median effective dose (ED50) between 0.25 and 1 nM (equivalent to 0.0025 and 0.01 mg/kg). Specifically, more than 85% silencing was observed at the mRNA level by qPCR at a dose of 5 nM (0.05 mg/kg) up to 72 hours of incubation (Supplementary Information Figure S6b). All the subsequent in vitro and in vivo studies were performed with 0.05 mg/kg of siRNA conjugated on the surface of the NPs.
RNAi- and RNAi-M2pep-AuNPs were next tested for the ability to deliver anti-VEGF siRNA to a lung cancer orthotopic murine model (BALB/c nude), expressing A549-luciferase-C8 human lung adenocarcinoma cells. To improve NP distribution in the lungs, and avoid liver/spleen non-specific accumulation, RNAi- and RNAi-M2pep-AuNPs were administered via intratracheal instillation directly to lung tumours (bronchial airways). Intratracheal instillation is a non-invasive and highly efficacious route of administration with high clinical relevance, especially in lung cancer treatment. Inductively coupled plasma mass spectrometry (ICP-MS) showed that after 7 days of treatment, both RNAi- and RNAi-M2pep-AuNPs accumulate specifically, and almost exclusively, in lung tissue (with a 5-10 fold increase for RNAi-AuNPs and a 15-20 fold increase for RNAi-M2pep-AuNPs when compared to other organs, such as heart, liver, spleen, kidney, intestine) (Supplementary Information Figure S7). Other organs showed residual accumulation, consistent with previous work reported for AuNPs.[28] These results showed that RNAi-M2pep-AuNPs accumulated in the tumour (lung) after administration and that minimal diffusion from the tumour to other organs in the body had occurred. Ultrastructural TEM images of lung tumour tissue from treated mice further revealed that the RNAi-M2pep-AuNPs were taken up by the A549 human lung adenocarcinoma cells, and were mainly localized in intracellular organelles, such as endosomes and/or lysosomes (Figures 2c,d). Nevertheless, less than 25% of RNAi-M2pep-AuNPs were colocalized in the lysosomes within macrophages from lung cancer mice, at days 1, 7 and 14 of exposure (Supplementary Information Figure S8), proving that around 75% of the RNAi-M2pep-AuNPs could escape lysosomal compartments and deliver siRNAs to cytoplasm.
Having established that RNAi-M2pep-AuNPs accumulated primarily in lung tumours, the evaluation of macrophage specific targeting via the M2pep-functionalized AuNPs was assessed by bronchoalveolar lavage (BAL) of lungs from mice treated with both RNAi- and RNAi-M2pep-AuNPs. BAL fluid cell patterns reflect inflammatory cell profiles in affected lung tissues and may be used to investigate inflammatory parameters in infection and neoplasms progression.[29] BAL studies indicate that RNAi-M2pep-AuNPs mediate intracellular delivery of cargo specifically in macrophages via active targeting of TAMs. Representative microscopy images of BAL cells from lung lavage show that RNAi-M2pep-AuNPs exclusively accumulate in macrophages at days 1, 7 and 14 after NPs instillation, whereas no accumulation in macrophages was observed for RNAi-AuNPs (Figure 3a). Representative light microscopy images of BAL cells from mice treated with RNAi- and RNAi-M2pep-AuNPs compared to RNAi-Controlpep-AuNPs (5 nM of siRNA) for day 14 validates the specificity of the M2 peptide towards TAMs, when compared to a control peptide (Supplementary Information Figure S9). In addition, an approximately 98% (P <0.005) change in uptake was observed for RNAi-M2ep-AuNPs after 14 days of exposure, when compared to the uptake of RNAi-AuNPs only (Supplementary Information Figure S10). To evaluate siRNA delivery into macrophages and VEGF expression at the same time, an immunohistochemical assay was performed over a period of 14 days (Figure 3b). Fluorescently labelled siRNA on the surface of RNAi-M2pep-AuNPs was observed in the cytoplasm of the macrophages with an increased fluorescence of 90% from day 1 to day 14. Concomitantly, a ∼80% decrease in VEGF expression can be seen from day 1 to day 14 (Figure 3c).
Figure 3. Efficient and selective in vitro and in vivo delivery of siRNA from functionalized AuNPs.

(a) Representative light microscopy images of BAL cells, specifically murine macrophages recovered from bronchoalveolar lavage illustrating cells from mice treated with RNAi- and RNAi-M2pep-AuNPs (0.05 mg/kg of siRNA) for days 1, 7 and 14. Black spots on macrophages treated with only RNAi-M2pep-AuNPs represent internalized NPs. Scale bars: 15 μm. (b) Immunofluorescence microscopy images of recovered macrophages from BAL fluid show RNAi-M2pep-AuNPs (green) internalization and VEGF (red) expression during 14 days of exposure. Nuclei are stained with DAPI (blue). Scale bars: 10 μm. These pictures are a composite of stylized images captured with a confocal microscope. (c) Bar graph depicting normalized fluorescence intensity (NFI) of RNAi-M2pep-AuNPs internalization and VEGF expression for days 1, 3, 5, 7, 11 and 14 of exposure (d) Silencing activity of RNAi- and RNAi-M2pep-AuNPs (0.05 mg/kg of siRNA) of recovered macrophages from treated mice, compared to RNAicontrol-AuNPs and RNAicontrol-M2pep-AuNPs. Data points represent group mean ± SD (n = 6, ** P < 0.01, *** P <0.005).
qPCR amplification of mRNA from FACS-sorted lung macrophages (CD11b+F4/80hi TAMs), isolated after intratracheal instillation, confirmed the efficient RNA interference (RNAi) mechanism, as indicated by decreased VEGF expression upon systemic treatment with RNAi-M2pep-AuNPs (∼75% silencing in macrophages at day 14, Figure 3d). No VEGF knockdown in macrophages was detected upon treatment with RNAi-AuNPs, from day 1 to day 14 (Figure 3d). qPCR blot analysis of VEGF from sorted lung macrophages after treatment with RNAi-M2pep-AuNPs confirms a decrease in expression, from as low as 0.25 nM to a ∼92% reduction with 5 nM of siRNA conjugated on NPs, when compared to RNAi-AuNPs (Supplementary Information Figure S11).
2.3. VEGF silencing inhibits lung inflammatory TAMs and cancer cells in mice
It is well known that VEGF stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via monocyte/macrophage recruitment and invasion.[30,31] Recent work described the administration of lipid-based nanoparticles silencing a chemokine receptor CCR2, and showed rapid blood clearance, accumulation in the spleen and bone marrow, and localization to monocytes in vivo, reducing inflammatory effects.[32] To our knowledge, a system describing the active targeting of TAMs in vivo with the ability to impair tumour progression and increase mice survival, via the simultaneous reduction of inflammatory immune TAMs and cancer cells in mice, has not been described so far.
To evaluate the silencing activity of RNAi- and RNAi-M2pep-AuNPs in different organs, the VEGF expression level in different tissues was evaluated after intratracheal administration to lung tumour bearing mice. RNAi-M2pep-AuNPs showed significant silencing in the lung (tumour) compared to that seen in the liver, heart and kidney (Figure 4a), after 21 days of exposure. These data prove that we can also achieve VEGF silencing in lung cancer cells. Moreover, since an increased invasion of TAMs critically promotes angiogenesis and tumour progression6, being intimately correlated with patient survival.[33,34] we next investigated the effect of anti-VEGF RNAi treatment with RNAi- and RNAi-M2pep-AuNPs in populations of immune cells from blood, lung tissue and lung lavage from lung cancer mice (Figure 4b). Treatment with RNAi-M2pep-AuNPs reduces the number of macrophages in both lung tissue and lung lavage, when compared to RNAi-AuNPs only, after 21 days of exposure. No difference was observed in the number of neutrophils and lymphocytes in blood, lung tissue and lung lavage from both RNAi- and RNAi-M2pep-AuNPs. Once we targeted a recruitment mechanism,[35] the remaining pulmonary and blood immune cells persisted mostly unaffected. More importantly, the RNAi-M2pep-AuNPs were orders of magnitude more specific for gene silencing in tumour-associated macrophages (Figure 4a), reducing TAMs subset in lung tissue and in lung lavage fluid (Figure 4b), demonstrating high tissue and cell selectivity. Flow cytometry analysis in lung tissue tumours that at 10 days of treatment, 81.8% of TAMs exposed to M2-targeted NPs internalized them, whereas only 3.7% of TAMS exposed to non-targeted NPs internalized them (Figure 4c). The FACS analysis was performed with previously sorted TAMs with antibodies against CD11b and F4/80hi and probed with PE/Cy5 Streptavidin and Streptavidin-Allophycocyanin-Cy7.
Figure 4. VEGF silencing reduces inflammatory TAMs in mice (21 days of exposure).

(a) VEGF silencing in lung, liver, heart, kidney after treatment with RNAi- and RNAi-M2pep-AuNPs, in comparison to the sham group (no NPs treatment) and RNAicontrol-AuNPs and RNAi control-M2Pep-AuNPs. (b) Number of macrophages, neutrophils and lymphocytes in blood, lung tissue and lung lavage from untreated mice (sham) and mice treated with RNAi- or RNAi-M2pep-AuNPs. (c) CD11b F4/80hi expression on TAMs (CD11b+F4/80hi) for mice treated with RNAi- and RNAi-M2pep-AuNPs at days 11 and 21. Plots represent the uptake of RNAi- and RNAi-M2pep-AuNPs in TAMs, depicting the fluorescence intensity of Alexa Fluor® 488-labelled siRNA on NPs (measured in FITC channel) and of CD11b and F4/80hi expression on TAMs (measured in Cy7 channel). The TAMs expressing both CD11b and F4/80hi were previously sorted with antibodies against CD11b and F4/80hi and probed with PE/Cy5 Streptavidin and Streptavidin-Allophycocyanin-Cy7.
Moreover, tumours that underwent treatment with RNAi-M2pep-AuNPs during 21 days showed a ∼95% reduction (∼30-fold) of TAMs (CD11b+F4/80hi TAMs), while at the same time it demonstrated an increase (∼100-fold) in Alexa Fluor® 488 labelled-siRNA signal (Figure 4c). These results also confirmed the increased accumulation of RNAi-M2pep-AuNPs in lung tissue when compared to the RNAi-AuNPs. This FACS data proves that only RNAi-M2pep-AuNPs can be uptaken by the M2-phenotype macrophages and decrease their population.
Besides NPs delivery into TAMs, confocal images of lung tissues incubated with RNAi- and RNAi-M2pep-AuNPs confirmed that NPs could also be internalized to lung cancer cells within the tumour tissue (Figure 5a). Immunohistochemical staining for VEGF and histological analysis in lung tissue indicated a ∼70% reduction in VEGF expression (Figure 5b) and a reduced (10-fold) presence of TAMs in lungs (Figure 5c), only in mice treated with RNAi-M2pep-AuNPs. To prove that TAMs reduction was not achieved due to toxicity effects, FACS-sorted lung macrophages (CD11b+F4/80hi TAMs), isolated after intratracheal instillation, were subjected to a dose of 5 nM of siRNA on NPs (equivalent to 0.05 mg/kg of siRNA in vivo) for 14 days (Supplementary Information Figure S12). No changes in TAMs viability was observed for either RNAi-or RNAi-M2pep-AuNPs during the 14 days of incubation. Moreover, the sham and RNAi-AuNPs exposed groups exhibited macrophage accumulation at the bronchoalveolar junction and thickened local alveolar walls (Figure 5c). Significant reduction in severe interstitial infiltration of inflammatory cells is noted in the RNAi-M2pep-AuNPs treated group. Sham and RNAi-AuNPs treated groups showed a predominance of mononuclear cells, as well as perivascular and peribronchiolar edema, occurrence of hypercellularity and thickened local alveolar walls and septa and increased number of tumoral clones (arrows in Figure 5d). These histological images confirmed an evident decrease (5-fold) in the incidence and severity of tumour clones in lung from mice treated with RNAi-M2pep-AuNPs, after 21 days of exposure.
Figure 5.

(a) Confocal microscopy images of lung tumour tissue for cellular uptake of RNAi- and RNAi-M2pep-AuNPs formulated with Alexa Fluor® 488-labelled siRNA (green) by lung cancer cells. Nuclei are stained with DAPI (blue). (b) Immunohistochemical staining for VEGF in lung tissue indicating a ∼70% reduction in VEGF expression for RNAi-M2pep-AuNPs. (c) Histological analysis of lung tissue indicating a reduced presence of TAMs in lungs only in mice treated with RNAi-M2pep-AuNPs. Sham and RNAi-AuNPs treated groups present macrophage accumulation at bronchoalveolar junction and thickened local alveolar walls (arrows). (d) H&E staining of lung tumour sections showed no evidence of tumoral clones or any histopathology in RNAi-M2pep-AuNPs-injected tumours. Sham and RNAi-AuNPs treated groups showed peribronchiolar edema, hypercellularity and thickened local alveolar walls and septa and increased number of tumoral clones (arrows). Scale bars: 50 μm. Data points represent group mean ± SD (n = 6, ** P < 0.01, *** P <0.005).
Taken together, treatment with RNAi-M2pep-AuNPs efficiently and selectively reduced the number of TAMs and tumoral clones in the lung via an active targeting to these immune cells population. We showed by immunohistochemical and histological analysis, immune cells counting and flow cytometry, that intratracheal administered RNAi-M2pep-AuNPs deliver siRNA to TAMs and lung cancer cells specifically. Our experiments confirm that the lung TAM reservoir should be targeted when using anti-VEGF RNAi in the treatment of acute inflammation. Our approach has been demonstrated to be very effective and distinct from previous studies using RNAi in cancer. Once we selectively target TAMs and a common excretory factor in this immune cell subset, we interrupt the recruitment and cell migration of TAMs, removing this cell population from the inflammatory tumour sites.
2.4. Silencing TAMs hinders lung cancer progression and enhances mice survival
Cancer of the lung and bronchus continue to be the most common cause of cancer death, with more than one-quarter of all deaths due to lung cancer in 2014.[36] Previous studies reported successful tumour growth inhibition by therapeutic siRNA silencing using RGD-nanoparticles in a lung cancer mouse model.[20] Nevertheless, targeting of inflammatory immune cells was not achieved with this system.
We hypothesized that RNAi-M2pep-AuNPs would enhance the likelihood of observing a tumour size reduction, followed by an increase in mice survival by knocking down the VEGF protein in ∼90% of TAMs, thus reducing their recruitment to the inflammatory lung tumour milieu. Accordingly, BALB/c nude mice bearing A549-luciferase-C8 human lung adenocarcinoma tumours were treated with both RNAi- and RNAi-M2pep-AuNPs (dose of 0.05 mg/kg of siRNA in three and six doses treatment). Using live animal bioluminescence imaging, a 10-fold decrease in luciferase activity (Figure 6b) and in tumour size (Figure 6c) was observed in the RNAi-M2pep-AuNPs treated tumours 36 days post-administration, compared to sham group (n = 6, P < 0.005) and a 6-fold decrease when compared to RNAi-AuNPs group. Monitoring the change in tumour size as a function of time after treatment with RNAi-M2pep-AuNPs revealed a significant decrease in tumour growth (P<0.005) 22 day post NP treatment (Figure 6c). Animals treated with 6 doses of RNAi-M2pep-AuNPs up to the 36 days of treatment show complete tumour regression (Figure 6b). At 36 days, animals treated with RNAi-M2pep-AuNPs show a tumour size of 8.1±9.6%, compared to 52.6±14.9% for RNAi-AuNPs and 100±15.5% for sham groups. In addition, control siRNA sequences on NPs with (RNAicontrol-M2pep-AuNPs, 96.1±8.8%) and without M2 peptide (RNAi control-AuNPs, 98.0±7.4%) produced no effect on tumour size. Taken together, our results prove that a combination approach by silencing VEGF in both TAMs and cancer cells using M2 targeted siRNA-NPs (RNAi-M2pep-AuNPs) is far more efficient in tumour size reduction than the just silencing VEGF in cancer cells using RNAi-AuNPs only via passive targeting.
Figure 6. Silencing tumour-associated macrophages and cancer cells hinders lung cancer progression and enhances mice survival.

(a) Live imaging of BALB/c nude mice bearing A549-luciferase-C8 human lung adenocarcinoma tumours non-treated (Sham) and treated with both RNAi- and RNAi-M2pep-AuNPs and with RNAicontrol-M2pep-AuNPs (dose of 0.05 mg/kg of siRNA). Representative imaging of individual mice from each treated group (n = 6 animals) is shown at days 18 and 36, with the same scale of photon flux indicating luciferase activity (tumour) and epi-fluorescence from the NPs bearing siRNAs labelled with Alexa Fluor 488. (b) Bioluminescence and epi-fluorescence analysis of ex vivo lungs from non-treated and treated mice. (c) Relative tumour size in BALB/c nude mice bearing lung tumour xenografts treated with the RNAi- and RNAi-M2pep-AuNPs, compared to untreated mice (sham), RNAicontrol-AuNPs and RNAicontrol-M2Pep-AuNPs. The mice were treated through repeated intratracheal instillation at 0.05 mg/kg of siRNA on days 14, 18 and 21. The tumour volume was monitored by luciferase bioluminescence (n = 6, * P < 0.05, *** P <0.005). (d-e) Kaplan-Meier survival curves of untreated control mice (sham, in black) and for RNAi-AuNPs (gray) and RNAi-M2pep-AuNPs (green), using two dosage cohorts: a first trial with an administration of NPs at days 14, 18 and 21 (d) and a second trial on days 14, 18, 21, 26, 28 and 36 (e).
The safety of all nanoformulations was confirmed by delivering the same NP doses as the ones for in vitro studies. No in vivo toxicity or other physiological complications were observed in all the animal groups for 46 days post NP exposure (dose of 0.05 mg/kg of siRNA in six doses treatment cohort) as indicated by the maintenance of stable body weight (Supplementary Information Figure S13), suggesting that the NP treatments were not toxic at the administered dose. A major concern associated with siRNA delivery nanovectors is the induction of type I interferon and inflammatory cytokines.[37] To assess the tolerability of the NPs, and to prove that immunostimulation is not responsible for the observed effects in RNAi,[38,39] the serum expression of several cytokines IL2, IL4, IL5, IL6, IL8, IL10, IL12, TNF-α, IFN-γ and IL-1β was evaluated 48 hours after NPs injection. No changes were observed in cytokines at mRNA levels for either RNAi- or RNAi-M2pep-AuNPs when compared to the sham group (Supplementary Information Figure S14), suggesting the absence of immunostimulatory properties. Since siRNAs can induce nonspecific activation of the immune system through the Toll-like receptors pathway 39, the serum expression of TLR3, TLR7, TLR8 and TLR9 was evaluated 48 hours after NPs injection. No TLR activation was observed for either RNAi- or RNAi-M2pep-AuNPs when compared to the sham group (Supplementary Information Figure S15). These results suggest that both RNAi- and RNAi-M2pep-AuNPs are well tolerated in vivo and that the RNAi silencing is not caused by any innate immune response to a foreign siRNA sequence, but rather by gene-specific silencing.
Since approximately 10% of the NPs also accumulate in the liver (Supplementary Information Figure S7), we evaluated the levels of serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT), 48 hours after NPs injection. A dramatic change in the activity of these two enzymes represents a sign of inflammation and acute liver damage. No liver toxicity or evidence of liver damage was observed for both RNAi- and RNAi-M2pep-AuNPs as the values for AST and ALT activities were within the normal range (Supplementary Information Figure S16).
According to FDA procedures, drugs should be removed via metabolism or excretion processes after they enter the body, to reduce toxicity. Although our in vivo toxicity data demonstrate high RNAi-M2pep-AuNPs' biocompatibility, future studies would focus on long-term accumulation and excretion of the RNAi/peptide nanoparticles to fully translate this nanotherapy to treat patients. However, as NPs' uptake and clearance can be tuned by mediating particles size, charge and surface modification, we chose particles of 14nm diameter that can be excreted through the kidneys and liver. In fact, nanoparticles in the size range of 10-100 nm are generally accepted as efficient delivery agents, determined by in vivo clearance, biodistribution and toxicity.
The contribution of VEGF knockdown in lung TAMs to mice survival was assessed by monitoring survival following two different dosage cohorts (n = 6) of RNAi- or RNAi-M2pep-AuNPs: one with administration of three NP dosages at days 14, 18 and 21 and a second trial following six particle administrations on days 14, 18, 21, 26, 28 and 36. Animals treated with RNAi-M2pep-AuNPs survived significantly longer (P<0.05) in the first trial (Figure 6d) compared to sham and RNAi-AuNPs groups. The trend of prolonged survival following RNAi-M2pep-AuNPs treatment was reproducible (P<0.005) in a second and independent trial that used 6 doses spread over 22 days (Figure 6e).
Our results indicate that the observed survival extension of more than 75% can be attributed to both the silencing of VEGF in TAMs, via the M2pep targeting, and in lung cancer cells, simultaneously. The improved outcome in tumour-bearing mice receiving RNAi-M2pep-AuNPs treatment strongly supports the extraordinary potential of these nanoparticles as adjuvant agents to anticancer therapies.
AuNPs have already found great use as a vehicle for siRNA delivery to lung cancer cells.[40,41] Huschka et al. reported on siRNA delivery to human lung cancer cells (H1299) using light-triggered release via near-infrared (NIR) laser irradiation to silence GFP.[40] Lu et al., also reported the use of NIR light-inducible NF-kappaB downregulation through folate receptor-targeted hollow gold nanospheres carrying siRNA recognizing NF-kappaB p65 subunit. Using micro-positron emission tomography/computed tomography imaging, it was shown that the targeted nanoconstructs exhibited significantly higher tumour uptake in nude mice bearing HeLa cervical cancer xenografts compared to non-targeted nanoparticles following i.v. administration.[41] This work is the first to report on combination therapy that includes gene silencing along with targeted immunomodulation. Our approach enables targeting an angiogenic factor (VEGF) that is produced by inflammatory macrophages and lung cancer cells and thus modulate the recruitment of TAMs and the proliferation of the cancer cells simultaneously.
Conventional anti-tumour and gene therapy systems mostly use drugs that lack cellular specificity and suppress inflammation by diminishing protective functions of the immune system. The combination approach reported here for targeting cancer associated immune cells and cancer cells simultaneously, by silencing angiogenic factors that are critical for tumour progression, can effectively shift the tumour microenvironment from pro-oncogenic to anti-tumoral. TAMs are important constituents of the tumour microenvironment, playing active role in promoting tumour progression.[25,26] Hence, a long-lasting and efficient delivery of siRNA to both TAMs and lung cancer cells represents a crucial platform in cancer therapy.
The archetype for cancer treatment has to change from relatively nonspecific cytotoxic agents to selective, mechanism-based therapeutics. The combination of this dual targeted cancer immunotherapy represents a missing link in cancer therapy. Most of the generic siRNA-based nanotherapies are limited by a narrow therapeutic index and significant off-target effects. Our hybrid approach aims to inhibit molecular pathways that are crucial for tumour growth and maintenance via VEGF silencing; whereas in addition to destruction of tumour cells, immune modulation of TAMs endeavours to stimulate a host immune response that results in long-lived tumour destruction, aiming to improve the clinical outcome.
3. Conclusion
We developed a unique nanoformulation that specifically targets murine lung TAMs and delivers siRNA to those cells and to lung cancer cells at the same time. This hybrid approach demonstrates a highly efficacious dual knockdown as a viable, highly potent anticancer immunotherapy. Our hybrid approach represents a paradigm shift from conventional silencing of specific oncogenes in one specific cell population that eradicates an important cell population, TAMs, along with silencing angiogenic factor such as VEGF, which is critical for tumour progression.
Our data prove that passive silencing combined with targeting peptide immunotherapy is able to immuno-modulate TAMs cell-population in the tumour microenvironment, culminating in long-lived tumour eradication along with increased mice survival.
We expect that the results of our work will contribute to the advancement of RNAi technology and will help to develop more efficient siRNA therapeutics for treating lung cancer. A similar targeted delivery strategy could be applied to treat other diseases associated with increased levels of specific markers expressed by resident cells in pathological conditions (other types of cancer, atherosclerosis, fibrosis, asthma).
4. Experimental Section
In vitro screening of siRNAs
Twenty siRNAs (Supplementary Table S1) with 100% homology with the mouse vascular endothelial growth factor (VEGF) gene sequence (NM_001025257.3, GI:160358806) were screened for RNAi potency. For that, mouse BALB/c macrophage J774.2 cell line was transfected with 1 nM (0.01 mg/kg) and 5 nM (0.05 mg/kg) of siRNAs using Lipofectamine 2000 (Invitrogen) according to the manufacture's protocols. VEGF mRNA levels were measured 48 hours after transfection by Real-Time PCR using Taqman probes FAM-MGB (Applied Biosystems) and GAPDH as the reference gene.
Functionalization and characterization of gold nanoparticles (AuNPs)
Bare AuNPs, with an average diameter of ∼15 nm (∼1.64E+12 NPs/mL) and a SPR peak at 520 nm (extinction coefficient 3.67E+08 M-1.cm-1, MW 2.06E+07 g/mol, surface area 7.07E+02 nm2) were purchased from Cytodiagnostics. AuNPs with a 30% saturated PEG layer were prepared by functionalization with α-Mercapto-ω-carboxy PEG (HS-C2H4-CONH-PEG-O-C3H6-COOH, MW. 3500 Da, Sigma) as described elsewhere.[42,43] 30% of saturated PEG layer allows the incorporation of additional thiolated components, such as the thiolated siRNA. See Supplementary Information S2.1 for PEG-AuNPs functionalization and PEG quantification.
The M2 macrophage-binding peptide (M2pep) [12], with the sequence YEQDPWGVKWWY (with no modifications at C- and N-terminals, AnaSpec), was coupled to PEG-AuNPs by a carbodiimide chemistry assisted by N-hydroxisuccinimide (EDC/NHS coupling reaction) between the carboxylated PEG terminal and the primary amine groups of the peptide. See Supplementary Information S2.2 for M2pep coupling and quantification. A peptide with the sequence KKKGRKKRRQRRR (with no modifications at C- and N-terminals, AnaSpec) was used as a non-targeting peptide (control). The most efficient siRNA duplex for mouse VEGF with the sequence 5′: CCCACAUACACACAUAUAUUU (sense) and 3′: UUGGGUGUAUGUGUGUAUAUA (antisense) was selected from the in vitro screening described above and was used for subsequent nanoparticle functionalization and in vitro and in vivo testing. Thiolated anti-VEGF siRNA labelled with Alexa Fluor® 488 (Qiagen) for mouse VEGF was dissolved in 1 ml of 0.1 M DTT, extracted three times with ethyl acetate, and further purified through a desalting Illustra™ NAP™-5 column Sephadex G-25 DNA grade (GE Healthcare) according to the manufacturer's instructions. The purified thiolated siRNAs, were incubated at a concentration of 10 μM, with RNase-free solution of the M2pep-PEG-AuNPs (10 nM) containing 0.08% SDS. Subsequently, the salt concentration was increased from 0.05 to 0.3 M NaCl with brief ultrasonication following each addition to increase the coverage of oligonucleotides on the nanoparticle surface. After functionalization during 16 hours at 4°C, the particles were purified by centrifugation (20.000×g, 20 mins, 4°C), and re-suspended in diethyl pyrocarbonate (DEPC)-water. This procedure was repeated 3 times. The number of siRNA per nanoparticle was determined by using Quant-It Ribogreen RNA assay (Invitrogen).
For NP size determination, samples were analysed through Dynamic Light Scattering (DLS) with a nanoparticle analyser from Wyatt Dyna Pro Plate Reader at an angle of 90° and at 25°C in water. Zeta potential measurements of all NPs were performed in water and 0.1 M KCl at 25°C using a Zeta PALS (Brookhaven Instruments) analyser. The morphology and size of the NPs was examined in a field emission gun transmission electron microscope (TEM, JEOL 2100F, 120 kV) using 2% phosphotungstic acid (1:1 v/v) for negative staining. The NP suspension was dripped onto carbon grids (Ted Pella, Inc.) and allowed to air dry before TEM observation.
In vitro nanoparticles' delivery
A549-luciferase-C8 human lung adenocarcinoma cells (Bioware® Cell Line P/N 119266) were grown in F-12K medium (Kaighn's modification of Ham's F-12 medium) with L-glutamine (ATCC®), 10% heat inactivated fetal bovine serum (FBS, Invitrogen), 100 U/ml penicillin and 100 μg/mL streptomycin (Invitrogen), and maintained at 37°C in 5% CO2. Cells were seeded in 24-well plates at a density of 1E+05 cells/well and grown for 24 hours prior to incubation with NPs. On the day of incubation, 0.05 mg/kg of siRNA conjugated on M2pep-AuNPs and siRNA-AuNPs only were added to cells at approximately 50% confluence in F-12K medium supplemented only with 10% heat inactivated fetal bovine serum. After 24 hours, cells were fixed with 4% paraformaldehyde for 15 min at 37°C on cover slips. The cover slips were washed twice in PBS. After DAPI staining, one drop of aqueous mounting medium was added on the cover slip and inverted carefully on a glass slide. The images were acquired in a Nikon A1R Ultra-Fast Spectral Scanning confocal microscope. To visualize the cellular uptake of NPs via TEM, A549-luciferase-C8 human lung adenocarcinoma cells were incubated with all NPs for 48 hours and fixed in 1.5% glutaraldehyde in PBS for 1 hour and then in 1% osmium tetroxide in PBS for an additional hour. After dehydration in increasing concentrations of ethanol, slides were embedding in 100% pure epoxy resin (1:1) and imaged in a JEOL 2100F TEM (120 kV).
In vivo targeting of RNAi-M2pep-AuNPs
The lung cancer orthotopic murine model was achieved by inducing BALB/c nude mice (initial weight ∼22 g; 6 weeks age) via intratracheal instillation with A549-luciferase-C8 human lung adenocarcinoma cells (2E+06 cells), to establish human non-small cell lung carcinoma tumour growth model. Six animals in each group were induced with lung tumours and, 14 days after tumour growth, RNAi- and RNAi-M2pep-AuNPs were intratracheal administered on days 14, 18 and 21 with additional particles' administration on days 14, 18, 21, 26, 28 and 36. Mice were monitored for tumour burden via luminescence along with measuring tumour size. All animal experiments were authorized by the Research Animal Care Committees, in agreement with local state and federal regulations. All in vivo studies were performed with 0.05 mg/kg of siRNA conjugated on the surface of the nanoparticles. All animals were anesthetised by intraperitoneal injection of a mixture of xylazine (10 mg/kg) and ketamine (100 mg/kg) in 0.9% sterile PBS. The animals were then intubated by a nonsurgical technique. Using a 1 mL subcutaneous (sub-Q) syringe with a 30 gauge, 5/16-inch needle, a suspension containing 0.05 mg/kg of siRNA conjugated on NPs in 50 μl sterile water was instilled, holding the syringe bevel side up and parallel with the trachea, and injected the full volume of instillate into the trachea. The animals were recovered on a heating pad and monitored until reactive and then receive a dose of buprenex 0.05-0.1 mg/kg every 8 hours for 24 hours if signs of pain or distress were observed. All experiments used 6 mice per treatment group unless noted otherwise. Animals were euthanized with CO2 inhalation.
Preparation of BAL cells/fluid
Bronchoalveolar lavage (BAL) cells were used to follow NPs internalization in immune cells (especially macrophages) derived from BALB/c nude mice. BAL was performed by instilling and immediately retrieving three 0.5 mL aliquots of normal saline (PBS) via a tracheal cannula. Cell counts on cytospin (cytology method designed to concentrate cells that are found in small numbers) preparations were determined on unprocessed lavage fluids using a haematocytometer and the differences between macrophages, lymphocytes and neutrophils were determined. Cells were fixed in dry freshly poured high quality methanol at 4°C for 10 minutes. The cells were imaged by light microscopy. For lysosome co-localization studies, the lysosomal dye LysoTracker® Red DND-99 (Invitrogen) was included at a final concentration of 100 nM. Cells were then fixed with 4% paraformaldehyde in 1× PBS for 15 min at 37 °C. The cells were imaged by confocal microscopy. Quantification of the colocalization was achieved by measuring RGB intensity for red, green and yellow (merged=colocalization) colours from confocal images. The intensity level of the three colours was measured by the RGB code. In the RGB colour model a secondary colour is formed by the sum of two primary colours (red, green or blue) of equal intensity where yellow (secondary colour) is red+green. Every secondary colour is the complement of one primary colour.
Biodistribution studies: quantification of NPs accumulation in the tumour and other organs
Biodistribution of the functionalized NPs in tissues associated with clearance (liver, spleen, and kidney) as well as lung (tumour), intestine and heart was measured by ICP-MS. Briefly, 7 days after NPs instillation, mice were killed to collect major organs, which were rinsed with ethanol three times and then air-dried into clean vials for acid digestion (aqua-regia 3HCl:1HNO3). After 1 day of strong acid digestion, the samples were analysed by ICP-MS.
Flow cytometry
Mice were sacrificed and perfused with sterile PBS. Cell suspension from blood (drawn from the tail vein into heparin-containing PBS) and lung tissues (digested with DNAse I and collagenase D for 30 minutes at 37°C, as described elsewhere [42]) were incubated with antibodies against CD11b (Abcam, ab8878, 5 μg/mL) and F4/80hi (Abcam, ab6640, 10 μg/ml) and probed with PE/Cy5 Streptavidin (Biolegend, 1.25 μg/mL) and Streptavidin-Allophycocyanin-Cy7 (BD Biosciences, 0.6 μg/mL) and to identify and sort tumour-associated macrophages (CD11b+F4/80hi TAMs). Only the macrophages with double staining for CD11b and F4/80hi were sorted and used for further analysis. Alexa Fluor® 488-labelled siRNA-AuNPs were identified in the FITC channel. Data were acquired on FACS Canto II HTS-1 (BD Biosciences) flow cytometer.
Macrophages in vitro toxicity assays
For macrophages viability tests, a regular MTT assay (Invitrogen) was performed in FACS-sorted lung macrophages (CD11b+F4/80hi TAMs), isolated after intratracheal instillation, and subjected to a dose of 5 nM of siRNA on NPs (equivalent to 0.05 mg/kg of siRNA in vivo) during 14 days. For co-localization of nanoparticles with lysosomes, TAMs were recovered and sorted from treated mice, maintained in low serum medium and imaged immediately in order to avoid cell death under low serum conditions.[44,45]
Quantitative PCR
Total RNA from BALB/c macrophage J774.2 cell line, A549-luciferase-C8 human lung adenocarcinoma cells and from BALB/c nude mice tissues induced with lung cancer was extracted using RNeasy Plus Mini Kit (Qiagen) according to the manufacter's protocol. cDNA was produced using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) using 500 ng of total RNA. qRT-PCR was performed with Taqman probes FAM-MGB for VEGF and GAPDH (Applied Biosystems). GAPDH was used as a reference gene. The reactions were processed using Light Cycler 480 II Real-time PCR machine (Roche) using TaqMan® Gene Expression Master Mix (Applied Biosystems) under the following cycling steps: 2 min at 50 °C for UNG activation; 10 min at 95 °C; 40 cycles at 95 °C for 15 s; 60 °C for 60 s. At least three independent repeats for each experiment were carried out. Gene expression was determined as a difference in fold after normalizing to the housekeeping gene GAPDH expression.
Histology and immunohistochemistry
After mice have been sacrificed and perfused with sterile PBS, lungs were snap-frozen in optimum cutting temperature compound (Tissue-Tek®) and cut on a cryostat microtome and air-dried overnight. Frozen sections were thawed and stained for hematoxylin and eosin (H&E) using standard procedures. Stained tissue sections were then imaged by light microscopy. For immunolocalisation, the lung cancer tissues were embedded and sliced into a 3 μm section. After fixation with 4% paraformaldehyde in PBS, lung tissue sections were treated with blocking solution containing 4% rabbit serum in PBS. The tissue slide was incubated with Anti-VEGF antibody (1:1000, Abcam) in PBS containing 1% BSA for 60 min, and was washed three times in PBS. Alexa Fluor® 568 Goat Anti-Mouse IgG (Invitrogen) was used as secondary antibody (1:200; Exc/Emi: 578/603nm). Nuclei were DAPI stained before slide mounting with Fluoromount™ Aqueous Mounting Medium (Sigma). Stained tissue sections were then imaged by fluorescence microscopy.
Analysis of tumour growth and animal survival
Non-invasive longitudinal monitoring of tumour progression was followed by scanning mice with the IVIS Spectrum-bioluminescent and fluorescent imaging system (Xenogen XPM-2 Corporation) from mice bearing tumours from A549-luciferase-C8 human lung adenocarcinoma cells (n = 6 animals per treated group). 15 minutes before imaging, mice were intraperitoneally injected with 150 μL of D-luciferin (30 mg/mL, Perkin Elmer) in DPBS. Whole-animal imaging was performed at the indicated time points for two cohorts of mice, 3 (on days 14, 18 and 21) and 6 doses (on days 14, 18, 21, 26, 28 and 36) of NPs. Kaplan-Meier survival curves were designed by evaluating mice survival follow up of the two cohorts. In order to compare the survival distributions of mice treated with RNAi- and RNAi-M2pep-AuNPs (compared to non-treated mice) log-rank tests were performed.
In vivo toxicity
Assessment of mice body weight was performed on all the animal groups during 46 days after NP exposure (dose of 0.05 mg/kg siRNA in six doses treatment cohort). To study the immunostimulation of NPs, the serum expression of cytokines IL2, IL4, IL5, IL6, IL8, IL10, IL12, TNF-α, IFN-γ and IL-1β, as well as the expression of Toll-like receptors TLR3, TLR7, TLR8 and TLR9 was evaluated via qRT-PCR analysis as described in Quantitative PCR methods section, 48 hours after NPs administration. Heparinized blood from treated mice was layered using Ficoll-Histopaque (Histopaque®-1077 sterile-filtered, density: 1.077 g/mL, Sigma) density gradient medium and fresh Eagle's MEM supplemented with fetal bovine serum (FBS, Invitrogen). The mixture was centrifuged (800×g, 20 min) at RT, and pure lymphocytes were collected by gentle removal of the lymphocyte layer at the Ficoll-MEM interface. To evaluate liver toxicity, serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were quantified 48 hours after NPs administration using commercially available diagnostic kits (AST and ALT Activity Assay Kits, Sigma). AST is involved in the conversion of aspartate to glutamate in liver and other organs and ALT is involved in alanine synthesis in the liver.
Statistics
Differences between groups were examined using Student's paired t test through SPSS statistical package (version 17, SPSS Inc.). All error bars used in this report are mean ± SD of at 3 to 6 independent experiments. Statistically significant P values were indicated in Figures and/or captions as ***, P < 0.005; **, P < 0.01; *, P < 0.05. All in vivo experiments used 6 mice per treatment group unless noted otherwise.
Supplementary Material
Acknowledgments
This work was funded in part by Science Foundation Ireland under Grant Number 11/PI/08, the National Key Basic Research Program (973 Project) (No. 2011CB933101 and 2015CB931802), National Natural Scientific Fund (No.81225010 and 81327002), 863 project of China (2012AA022703 and 2014AA020700), Shanghai Science and Technology Fund (No.13NM1401500). ERE was supported in part by NIH R01 GM49039. JC acknowledges Marie Curie International Outgoing Fellowship (FP7-PEOPLE-2013-IOF, Project no. 626386) and FT for Marie Curie grant Agreement (PIEF-GA-2012-332-332462). The authors also thank to the Peterson Nanotechnology Materials Core Facility and the Swanson Biotechnology Center at the Koch Institute (MIT).
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Quail DF, Joyce JA. Nature Medicine. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yokota J. Carcinogenesis. 2000;21:497–503. doi: 10.1093/carcin/21.3.497. [DOI] [PubMed] [Google Scholar]
- 3.Pollard JW. Nature Reviews Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 4.Quatromoni JG, Eruslanov E. American Journal of Translational Research. 2012;4:376–389. [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Cook J, Hagemann T. Current Opinion in Pharmacology. 2013;13:595–601. doi: 10.1016/j.coph.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 7.Tang XQ, Mo CF, Wang YS, Wei DD, Xiao HY. Immunology. 2013;138:93–104. doi: 10.1111/imm.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Getts DR, et al. Science Translational Medicine. 2014:6. doi: 10.1126/scitranslmed.3007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Journal of Experimental Medicine. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu SS, et al. Molecular Pharmaceutics. 2013;10:975–987. doi: 10.1021/mp300434e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Low PS, Henne WA, Doorneweerd DD. Accounts of Chemical Research. 2008;41:120–129. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- 12.Cieslewicz M, Tang JJ, Yu JL, Cao H, Zavaljevski M, Motoyama K, Lieber A, Raines EW, Pun SH. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:15919–15924. doi: 10.1073/pnas.1312197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidson BL, Mccray PB. Nature Reviews Genetics. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimm D. Advanced Drug Delivery Reviews. 2009;61:672–703. doi: 10.1016/j.addr.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Whitehead KA, Langer R, Anderson DG. Nature Reviews Drug Discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanasty R, Dorkin JR, Vegas A, Anderson D. Nature Materials. 2013;12:967–977. doi: 10.1038/nmat3765. [DOI] [PubMed] [Google Scholar]
- 17.Conde J, Edelman ER, Artzi N. Adv Drug Deliv Rev. 2015;81:169–183. doi: 10.1016/j.addr.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg MS, Xing DY, Ren Y, Orsulic S, Bhatia SN, Sharp PA. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:745–750. doi: 10.1073/pnas.1016538108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conde J, et al. ACS Nano. 2012;6:8316–8324. doi: 10.1021/nn3030223. [DOI] [PubMed] [Google Scholar]
- 20.Conde J, et al. Biomaterials. 2013;34:7744–7753. doi: 10.1016/j.biomaterials.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 21.Dahlman JE, et al. Nature Nanotechnology. 2014;9:648–655. doi: 10.1038/nnano.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng D, Giljohann DA, Chen DL, Massich MD, Wang XQ, Iordanov H, Mirkin CA, Paller AS. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:11975–11980. doi: 10.1073/pnas.1118425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinstein S, Peer D. Nanotechnology. 2010:21. doi: 10.1088/0957-4484/21/23/232001. [DOI] [PubMed] [Google Scholar]
- 25.Colegio OR, et al. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balkwill F. Nature Reviews Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 27.Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Chemistry & Biology. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 28.De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJAM, Geertsma RE. Biomaterials. 2008;29:1912–1919. doi: 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 29.Heron M, Grutters JC, ten Dam-Molenkamp KM, Hijdra D, van Heugten-Roeling A, Claessen AME, Ruven HJT, van den Bosch JMM, van Velzen-Blad H. Clinical and Experimental Immunology. 2012;167:523–531. doi: 10.1111/j.1365-2249.2011.04529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cursiefen C, et al. Journal of Clinical Investigation. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE, Han SH, Alitalo K, Koh GY. Blood. 2009;113:5650–5659. doi: 10.1182/blood-2008-09-176776. [DOI] [PubMed] [Google Scholar]
- 32.Leuschner F, et al. Nature Biotechnology. 2011;29:1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh YW, Park CS, Yoon DH, Suh C, Huh J. Plos One. 2014:9. doi: 10.1371/journal.pone.0087066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gulubova M, Ananiev J, Yovchev Y, Julianov A, Karashmalakov A, Vlaykova T. Journal of Molecular Histology. 2013;44:679–692. doi: 10.1007/s10735-013-9520-9. [DOI] [PubMed] [Google Scholar]
- 35.Shi C, Pamer EG. Nature Reviews Immunology. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegel R, Ma JM, Zou ZH, Jemal A. Ca-A Cancer Journal for Clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 37.Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Nature Biotechnology. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- 38.Kleinman ME, et al. Nature. 2008;452:591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whitehead KA, Dahlman JE, Langer RS, Anderson DG. Annual Review of Chemical and Biomolecular Engineering. 2011;2:77–96. doi: 10.1146/annurev-chembioeng-061010-114133. [DOI] [PubMed] [Google Scholar]
- 40.Huschka R, Barhoumi A, Liu Q, Roth JA, Ji L, Halas NJ. ACS Nano. 2012;6:7681–7691. doi: 10.1021/nn301135w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu W, Zhang G, Zhang R, Flores LG, Huang Q, Gelovani JG, Li C. Cancer Res. 2010;70:3177–88. doi: 10.1158/0008-5472.CAN-09-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conde J, Bao C, Cui D, Baptista PV, Tian F. J Control Release. 2014;183:87–93. doi: 10.1016/j.jconrel.2014.03.045. [DOI] [PubMed] [Google Scholar]
- 43.Conde J, Rosa J, de la Fuente JM, Baptista PV. Biomaterials. 2013;34:2516–2523. doi: 10.1016/j.biomaterials.2012.12.015. [DOI] [PubMed] [Google Scholar]
- 44.Kim JA, Salvati A, Åberg C, Dawson KA. Nanoscale. 2014;6:14180–14184. doi: 10.1039/c4nr04970e. [DOI] [PubMed] [Google Scholar]
- 45.Monopoli MP, Aberg C, Salvati A, Dawson KA. Nat Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.