

Abstract
The ultimate step in chloramphenicol (CAM) biosynthesis is a six-electron oxidation of an aryl-amine precursor (NH2-CAM) to the aryl-nitro group of CAM catalyzed by the non-heme diiron cluster-containing oxygenase CmlI. Upon exposure of the diferrous cluster to O2, CmlI forms a long-lived peroxo intermediate, P, which reacts with NH2-CAM to form CAM. Since P is capable of at most a 2-electron oxidation, the overall reaction must occur in several steps. It is unknown whether P is the oxidant in each step or whether another oxidizing species participates in the reaction. Mass spec product analysis of reactions under 18O2 show that both oxygen atoms in the nitro function of CAM derive from O2. However, when the single turnover reaction between18O2–P and NH2-CAM is carried out in an 16O2 atmosphere, CAM nitro groups contain both 18O and 16O, suggesting that P can be re-formed during the reaction sequence. Such re-formation would require reduction by a pathway intermediate, shown here to be NH(OH)-CAM. Accordingly, the aerobic reaction of NH(OH)-CAM with diferric CmlI yields P and then CAM without an external reductant. A catalytic cycle is proposed in which NH2-CAM reacts with P to form NH(OH)-CAM and diferric CmlI. Then the NH(OH)-CAM re-reduces the enzyme diiron cluster, allowing P to re-form upon O2 binding, while itself being oxidized to NO-CAM. Finally, the re-formed P oxidizes NO-CAM to CAM with incorporation of a second O2-derived oxygen atom. The complete six-electron oxidation requires only two exogenous electrons and could occur in one active site.
INTRODUCTION
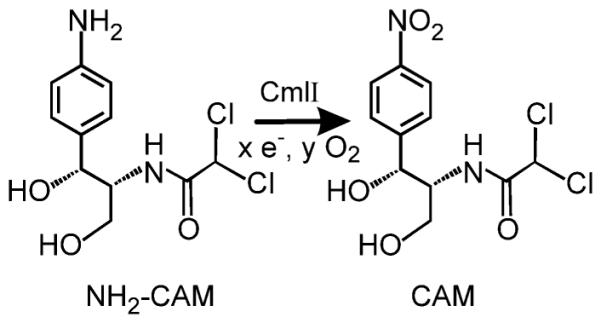
Aryl-nitro-containing molecules serve in many industrial and commercial markets, including materials, dyes, explosives, and pharmaceuticals. The traditional preparation method of such compounds, direct nitration by nitric acid, poses safety concerns and lacks regioselectivity.1 Newer, more selective nitration schemes suffer from similarly harsh reaction conditions.2 Here we turn to biology to examine the production of aryl-nitro-containing natural products from aryl-amine precursors. Not only are such products synthesized under mild, ambient conditions, but they fulfill many important antibiotic and cytostatic functions in nature and have both realized and potential applications to human health.3 One example of such a compound is the antibiotic chloramphenicol (CAM, Scheme 1), which is synthesized in Streptomyces venezuelae by the non-ribosomal peptide synthetase (NRPS) CmlP and associated tailoring enzymes.4 The final tailoring enzyme on the biosynthetic pathway is a non-heme diiron-cluster containing oxygenase (CmlI) that catalyzes the formation of the nitro group via the oxygenation of an aryl-amine precursor (NH2-CAM).
Scheme 1.
Reaction Catalyzed by CmlI
The enzyme-catalyzed six-electron oxidation of aryl-amines to aryl-nitro moieties is a process for which mechanistic consensus has not been reached. There are only three characterized enzymes known to perform this conversion: CmlI and AurF, both of which are non-heme dinuclear iron-cluster-containing enzymes, and PrnD, a Rieske-type non-heme iron oxygenase.5-10 AurF acts on a precursor of the anti-microbial natural product aureothin, catalyzing the oxidation of aminobenzoate to nitrobenzoate.11 Given the structural similarities between AurF and CmlI, including 34% amino acid sequence identity, it is feasible that these two enzymes follow the same N-oxygenation mechanism.5 PrnD catalyzes the final step in the biosynthesis of the antibiotic natural product pyrrolnitrin. It is noteworthy that the cofactors of both of these enzyme classes are only capable of at most 2-electron redox chemistry. Thus, the observed 6-electron aryl-amine (NH2-Ar) oxidation reaction of these enzymes presents a mechanistic puzzle.
The proposals for the mechanism of NH2-Ar to aryl-nitro (NO2-Ar) oxidation by diiron enzymes can be grouped into two general hypotheses. In the sequential three-oxidation hypothesis (Scheme 2, pathway I), the diiron cluster utilizes external electrons to activate O2 in three separate events. Each activated oxygen species, likely a diiron peroxo intermediate (see below), effects a 2-electron oxidation of the substrate or an intermediate. A single turnover thus requires six exogenous electrons. The NH2-Ar substrate is converted to an aryl-hydroxylamine (NH(OH)-Ar) by the first oxidation, to aryl-nitroso (NO-Ar) or aryl-dihydroxylamine (N(OH)2-Ar) by the second oxidation and to the NO2-AR product by the final oxidation. The diiron cluster returns to the diferric resting state at the end of the cycle. This mechanism was originally proposed by Hertweck and Winkler, based on work done on whole cells over-expressing AurF,3 and expanded by Zhao and coworkers based on work on purified AurF and CmlI.6,7 This type of mechanism requires either release of intermediates with subsequent rebinding at another activated metal center or a system to internally transfer the remaining required oxidizing equivalents between active sites in different subunits. The latter possibility seems unlikely given that structural studies show that AurF is only a homodimer, and three sets of oxidizing equivalents, or a homotrimer, would be required to complete the reaction in this case.12
Scheme 2. Two Mechanistic Hypotheses for Aryl-amine Oxygenation Mediated by Diiron Cluster Enzymes.
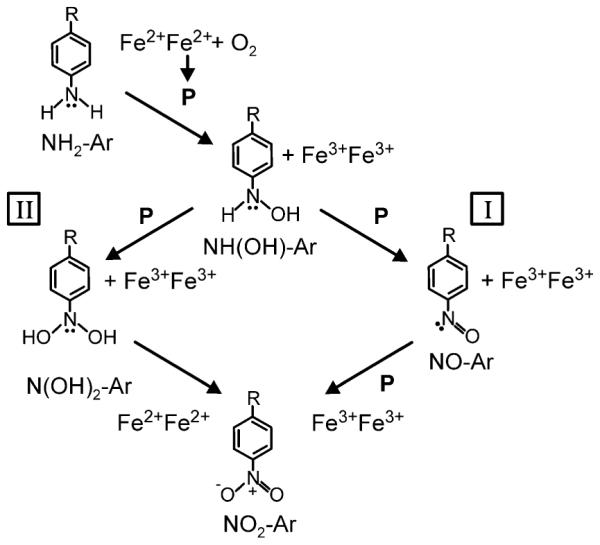
(I) Sequential three-oxidation mechanism requiring six external electrons (2 for each peroxo-intermediate) per turnover and (II) substrate-mediated reduction mechanism requiring a net of two external electrons per turnover because the diferrous cluster is regenerated at the end of the turnover cycle.
The second mechanistic hypothesis, introduced by Bollinger, Krebs and colleagues, invokes a substrate-based reduction step (Scheme 2, pathway II).13 Like the three-step oxidation mechanism, the substrate-based reduction mechanism begins with two 2-electron oxidations to form first NH(OH)-Ar, and then N(OH)2-Ar, intermediates. Using stopped-flow UV-Vis absorbance and Mössbauer spectroscopies, it was reported that the N(OH)2-Ar intermediate can act as a reductant, reducing the diferric diiron cluster to diferrous while itself being oxidized to the final NO2-Ar product. The overall reaction utilizes only two diiron-peroxo species and results in a diferrous cluster at the end of the reaction cycle. As a result, only four exogenous electrons are required for O2 activation in a single turnover, and the final diferrous diiron cluster can be used to activate O2 for the next cycle. Consequently, when considered over multiple cycles, only two external electrons are required per cycle to sustain turnover.
CmlI is an ideal enzyme with which to evaluate these two mechanistic hypotheses for aryl-amine oxidation. We have shown previously that the chemically competent intermediate of the reaction cycle, a diferric peroxo species, CmlI-peroxo (P), can be formed in high yield and has a distinguishable chromophore that can be followed by UV-Vis absorption spectroscopy at 500 nm. P is remarkably stable (t1/2 = 3 h at pH 9 and 4 °C)14 in contrast to the peroxo intermediates of AurF and other diiron oxygenases.15-17 This stability allows the key intermediate to be formed, degassed, transferred to and from an anaerobic chamber, transferred to stopped-flow or freeze-quench instruments, and reacted with the NH2-CAM substrate or one of the other pathway intermediates. Here we show that aryl-amine oxygenation proceeds by a third mechanism in which the reaction cycle ends with the diiron cluster in the diferric state. An N(OH)2-Ar derivative is not a required intermediate. However, diiron cluster reduction by another pathway intermediate serves to increase efficiency and reduce the need for external reducing equivalents in a single turnover. This new mechanism requires only two exogenous electrons, supplied at the beginning of the reaction cycle.
EXPERIMENTAL PROCEDURES
Reagents
Water used in all experiments was purified with a Millipore Super-Q system. 57Fe (95.5 %) was purchased from Cambridge Isotope Laboratories (Andover, MA). Amino-chloramphenicol(D-threo-1-(4-aminophenyl)-2-dichloroacetylamino-1,3-propanediol hydrochloride) (NH2-CAM) was purchased from Toronto Research Chemicals, Inc. Bicine and other standard reagents used in this study were purchased from Fisher. 18O2 gas (98 atom %) was purchased from Icon Isotopes, Summit, NJ.
CmlI Growth and Purification
CmlI from Streptomyces venezuelae was over-expressed in recombinant E. coli BL21(DE3) and purified as previously described.14 Briefly, cells were grown from 50 ml LB starter cultures in 2 liter shake flasks of M9 minimal medium in the presence of 100 μg/mL ampicillin to an OD ~ 1.0, at which point 50 μM FeCl3 was added, the temperature was lowered to 20 °C, and cells were induced with 150 μM IPTG. After an additional 14-18 h growth, cells were harvested by centrifugation and stored at −80 °C until further use. 57Fe enriched CmlI was prepared from cells grown similarly except for the addition of 57Fe metal dissolved in a minimal volume of aqua regia to a final concentration of 25 μM. To purify CmlI, cells were resuspended in 50 mM potassium phosphate pH 7.4, 300 mM NaCl, 10 mM imidazole, lysed via sonication, and centrifuged. The resulting supernatant was loaded onto a Ni nitrilotriacetic acid column (Qiagen) equilibrated in the same buffer. After loading, the protein was eluted using an imidazole gradient. Protein-containing fractions were pooled and dialyzed against 25 mM Bicine pH 9 and stored at −80 °C until further use. CmlI concentrations were determined by the calculated extinction coefficient checked against denatured protein (ε280 = 50 mM−1 cm−1).
Synthesis and Characterization of NO-CAM and NH(OH)-CAM
The nitroso and hydroxylamine derivatives of chloramphenicol (NO-CAM and NH(OH)-CAM, respectively) were synthesized according to published methods.18 The reported final purification of NO-CAM with Sephadex LH20 was not performed. Instead, normal phase chromatography (SiO2, ethyl acetate) was used as a final purification step for both compounds. The final purity, estimated via integration of the 1H-NMR, was 90% for NO-CAM and 80% for NH(OH)-CAM. Residual proteated solvent (δ 3.31) was the reference compound for 1H-NMR. These products were characterized and found to have the spectral characteristics listed below.
2,2-dichloro-N-[(1R,2R)-1,3-dihydroxy-1-(4-nitroso)propan-2-yl]acetamide (NO-CAM). Green solid: λmax. λmax = 316 nm (aqueous 0.1% formic acid). Rf = 0.40 (SiO2, ethyl acetate). 1H NMR (500 MHz, CD3OD): δH 7.85 (2H, d, J1 = 8 Hz), 7.71 (2H, d, J1 = 8 Hz), 6.24 (1H, s), 5.14 (1H, d, J1 = 3 Hz), 4.18-4.08 (1H, m), 3.82 (1H, dd, J1 = 7 Hz, J2 = 11 Hz), 3.62 (1H, dd, J1 = 6 Hz, J2 = 11 Hz).
2,2-dichloro-N-[(1R,2R)-1,3-dihydroxy-1-(4-hydroxylamine)propan-2-yl]acetamide (NH(OH)-CAM-). λmax = 237 nm (aqueous 0.1% formic acid).Pale yellow gum: 1H NMR (500 MHz, CD3OD): δH 7.25 (2H, d, J1 = 8Hz), 6.94 (2H, d, J1 = 9Hz), 6.930 (1H, s), 4.90 (1H, d, J1 = 4Hz), 4.05-4.02 (1H, m), 3.72 (1H, dd, J1 = 6Hz, J2 = 11Hz), 3.50 (1H, dd, J1 = 6Hz, J2 = 11Hz).
Preparation of CmlI Peroxo Intermediate (P)
CmlI (0.5 – 1.5 mM) was reduced under anaerobic conditions with an excess amount of dithionite in the presence of 0.05 equivalent of methylviologen. Excess reductant and methylviologen were removed using a PD-10 desalting column (G-25, GE Healthcare) in an anaerobic chamber. Aliquots of reduced CmlI were exposed to either 16O2 or 18O2 at ~ 4 °C for several min. P concentration was determined by absorbance at 500 nm (ε = 500 M−1 cm−1) under the assumption of nearly stoichiometric formation of the intermediate.
NH2-CAM Catalytic Walk
All samples were kept at ~4 °C unless otherwise stated and all experiments were performed in 50 mM Bicine buffer pH 9. A sample of P (600 μM) was prepared as described above. A 200 μL aliquot was removed and an absorbance spectrum was taken. This sample was then left on the benchtop to decay; a spectrum was later taken to serve as the P decay control. The remaining P was degassed to remove excess O2, first by purging with Ar gas for 20 min, and then by transferring to the anaerobic chamber and stirring for 10 min in the anaerobic atmosphere. After excess O2 was removed, one equivalent of NH2-CAM (freshly prepared in the anaerobic chamber, 10% MeOH, 90% water) was added and allowed to stir for 5 min. A 100 μL aliquot of this sample was removed and quenched in the anaerobic chamber with 100 μL 5% TFA for HPLC analysis. The remaining enzyme substrate mixture was transferred into a sealable cuvette and brought out of the anaerobic chamber. An absorbance spectrum was taken, after which the sample was exposed to a stream of O2 for 2 min. The spectrum of the resulting sample was taken and an aliquot was removed and quenched in equal volume 5% TFA for HPLC analysis.
NH(OH)-CAM and NO-CAM Catalytic Walks
Diferric resting state CmlI (560 μM) or P (300 μM, prepared as described above) was degassed first by purging with Ar gas for 20 min, and then by transferring to the anaerobic chamber and stirring for 10 min in the anaerobic atmosphere. After excess O2 was removed, one equivalent of NH(OH)-CAM or NO-CAM (freshly prepared in the anaerobic chamber, 10% MeOH, 90% water) was added and allowed to stir for 5 min. A 100 μL aliquot of this sample was removed and quenched in the anaerobic chamber with 100 μL 5% TFA for HPLC analysis. The remaining enzyme substrate mixture was transferred into a sealable cuvette and brought out of the anaerobic chamber. An absorbance spectrum was taken, after which the sample was exposed to a stream of O2 for 2 min. The spectrum of the resulting sample was taken and an aliquot was removed and quenched in equal volume 5% TFA for HPLC analysis.
Oxygen Isotope Incorporation Experiments
P was prepared as above, with the appropriate O2 isotope. For the reactions in H218O, labeled water was introduced when the enzyme was in the diferrous state by three cycles of anaerobic concentration and dilution with 50 mM Bicine buffer, pH 9, prepared in H218O. For the reactions with 18O-P, the vial containing the intermediate was brought into an anaerobic chamber and substrate was added. The sealed vial with P-substrate complex was brought out of the chamber and kept under a positive pressure of 18O2. Reactions were run for 10 min at 4 °C and then acid-quenched. After centrifugation to remove protein precipitate, the supernatant was transferred to an LCMS vial. Reactions of 16O-P were performed similarly expect that the substrate was introduced on the benchtop.
Experiment to Evaluate Exchange of CmlI-peroxo with Atmospheric O2
1 mL of 18O-P (200 μM) was prepared as above and stirred on ice for the duration of the experiment. A stream of 16O2 was applied over the sample. 100 μL aliquots were removed at time points from 1 to 90 min and reacted with 0.3 equivalents of NH2-CAM. Reactions were run for 10 min and then quenched and prepared for LCMS analysis as described above.
Exchange of Products from the Active Site
18O2-P (200 μM) was prepared as above, degassed to remove excess O2, and equilibrated in an anaerobic chamber before 1x NH2-CAM (200 μM) was added. After a 20 s incubation, 1x N16O-CAM (200 μM) was added. The sample was then quickly put under an 18O2 headspace and allowed to react for 10 min. Samples were acid quenched and analyzed by LCMS. Control samples were made by adding buffer instead of NO-CAM.
Spectroscopic and Physical Methods
Mössbauer Sample Preparation and Analysis
Samples for Mössbauer analysis were removed at various points from the catalytic walk experiments described above in which 57Fe-enriched CmlI replaced natural abundance enzyme. To freeze the anaerobic samples, the Mössbauer cup containing the sample was placed in a large Reacti-Vial (Thermo Scientific) while still inside the anaerobic chamber and sealed. After removal from the chamber, the entire vial was promptly frozen in liquid nitrogen. The vial was unsealed for spectral studies.
Mössbauer spectra were recorded with home-built spectrometers using Janis Research Super-Varitemp dewars, which allowed studies in the temperature range from 1.5 to 200 K and applied magnetic fields up to 8.0 T. Mössbauer spectral simulations were performed using the WMOSS software package (SEE Co., Edina, MN). Isomer shifts are quoted relative to Fe metal at 298 K. All Mössbauer figures were prepared using SpinCount software.19
UV-Vis and Stopped-Flow
Absorbance spectra were taken on an Agilent Cary 60 UV-Vis spectrophotometer at room temperature. Stopped-flow experiments were performed using an Applied Photophysics model SX.18MV stopped-flow device. In reactions of P with O2 and NH2-CAM or NH(OH)-CAM, P (~260 μM) was prepared as above and loaded into one syringe. The second syringe contained 130 μM NH2-CAM or 260 µM NH(OH)-CAM in O2 saturated buffer (~1.8 mM O2). In the reactions of the CmlIred with O2 and NO-CAM, CmlIred (100 μM) was prepared as above and combined with NO-CAM (100 μM) in the anaerobic chamber. The ES complex is nearly stoichiometrically formed at the concentrations employed. The ES complex was loaded into a syringe in the anaerobic chamber. The second syringe contained O2-saturated buffer. In these reactions, the stopped-flow device had been previously scrubbed of O2 by flushing with dithionite solution and then anaerobic buffer. The kinetic data were analyzed to extract reciprocal relaxation times using the nonlinear regression function of the Applied Photophysics ProData Viewer program. Each reaction time course was fit to a summed exponential expression, Eq. 1, which is appropriate for a series of first order or pseudo-first order reactions.20 In this equation, Abst is the observed absorbance at time t, Ampi is the observed amplitude for exponential phase i, τi is the relaxation time for phase i, and Absinf is the final absorbance at the end of the reaction. For a linear series of n reaction steps, n phases are required for the fit. Fitting statistics were reported by the fitting program, and each reaction was repeated at least 10 times to determine the average fitting parameters and errors.
| (Eq. 1) |
Analytical Methods
Metabolite Analysis
To analyze products in the reaction of CmlI with substrate, 150 μL aliquots of P diluted to 100-300 μM were promptly transferred to pre-cooled Reacti-Vials with stir bars at 4 °C. The indicated equivalents of substrate were added to the stirring P. (Substrate stocks were 2-20 mM in 10% MeOH, 90% water, kept at 4 °C. Normal substrate addition was 1-15 μL, so the amount of MeOH added to the reaction was minimal). Substrate equivalents were calculated vs. concentration of P unless otherwise stated. The reactions were run for 10 min unless otherwise indicated, at which point they were quenched with 150 μL 5% TFA or 75 μL 10 M HCl/50 μL 7.5 M HCOONa/50 μL 10 M NaOH. The reaction mixture was centrifuged at 4 °C at maximum speed for 20 min to pellet the protein precipitate, and then the supernatant was transferred to a clean Eppendorf microcentrifuge tube and either stored at −80 °C or analyzed by HPLC immediately.
Under the highly acidic conditions of the quench procedure, NH(OH)-CAM converts to NO-CAM which may subsequently dimerize.3 To control for this non-enzymatic conversion, some reactions were spin-quenched instead of acid-quenched by centrifuging an aliquot for 20 min in an Amicon Ultra-0.5 mL 10K (Millipore), washing the retentate with 150 μL of buffer, and centrifuging again for another 20 min. Some experimental procedures required an acid quench. After surveying several acid quench procedures, the 5.0% TFA acid quench was found to be the least damaging, with retention of >95% of CAM product and ~70% of the NO-CAM product.
HPLC
HPLC analysis was performed using a Waters 1525 binary HPLC pump and Waters 2487 dual λ absorbance detector with an Agilent Zorbax SB-C18 column. Products were monitored in dual wavelength mode at 280 nm and 316 nm. The method was isocratic 75% buffer A (water with 0.1% v/v formic acid) and 25% buffer B (MeOH with 0.1% v/v formic acid) for 15 min followed by a 5 min wash with buffer B at a flow rate of 2.5 mL/min. By this method, NH2-CAM elutes at 1.2 min, NO-CAM at 11.2 min and CAM at 10 min.
UPLC/MSe and Analysis
Instrumentation for UPLC/MSe analysis was a Waters Acquity UPLC with a Waters HSS T3 C18 2.1 mm × 100 mm column (1.7 μm diameter particles) coupled to a Waters Synapt G2 HDMS quadrupole orthogonal acceleration time of flight mass spectrometer (Waters Corp., Milford, MA USA). A 15 min linear gradient separation was run at a flow rate of 0.400 mL/min at 35 °C using A: water containing 0.1% v/v formic acid and B: acetonitrile containing 0.1% v/v formic acid: 3% B, 0 min to 1 min; 3% B to 97% B, 1 min to 9 min; 97% B, 9 min to 11 min; 97% B to 3% B, 11 min to 12 min; 3% B 12 min to 15 min. Spectra were collected in negative mode.
Determination of 18O incorporation into product was complicated by the fact that the substrate and product both contain two Cl atoms, for which two common isotopes, 35Cl and 37Cl, are two mass units apart just as 16O and 18O. To establish a baseline for the isotope patterns, the reaction of P and NH2-CAM was performed under an 16O2 atmosphere in quadruplicate and the natural abundance at each m/z was noted. These were loaded into an excel spreadsheet to be iteratively subtracted from the value obtained from reactions with 18O2. Under the these conditions, the retention time for CAM and NO-CAM was 4.5 min and the retention time for NH2-CAM was 1.3 min. The m/z values for NH2-CAM are 291.03, 293.03 and 295.03, for NO-CAM are 305.01, 307.01, 309.01 and 311.01, and for CAM are 321.01, 323.01, 325.01, 327.01 and 329.01.
RESULTS
Two Oxygen Atoms from O2 are Incorporated into Chloramphenicol
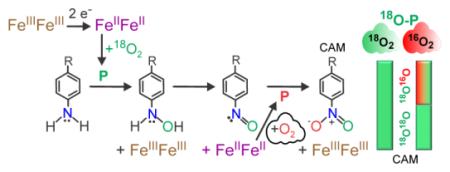
To determine the source of the oxygen atoms in the nitro-function of CAM, P was formed under either an 16O2 or 18O2 atmosphere and then reacted with NH2-CAM. Reactions were carried out at 4 °C with 0.3 equivalent of substrate, allowed to run for 10 min and then acid quenched as described in Experimental Procedures. Equivalents of substrate here and in future experiments are calculated vs. concentration of P unless otherwise stated. When 18O2-P was reacted with NH2-CAM in an 18O2 environment, the resulting CAM product was 93 ± 2% doubly labeled with 18O (Table 1). NO-CAM, observed as a minor product, was >95% singly labeled with 18O. Based on the isotopic purity of the 18O2 source, we could expect a maximum of 98% incorporation. Additional 16O incorporated during the reaction is likely to be due to contamination from atmospheric O2 during experimental manipulations. Thus, it appears that both of the O atoms of the CAM product derive from P (and therefore from O2), and not from H2O. To confirm this, reduced CmlI (CmlIred) was exchanged into H218O buffer and then supplied with 16O2 to form 16O2-P. Reaction with NH2-CAM yielded only 16O-containing product (Table 1).
Table 1.
Oxygen Incorporation into CAM Products in the Reaction of P with NH2-CAMa
| oxygen isotope: atmosphere / P / solvent | ||||
|---|---|---|---|---|
|
|
||||
| product | oxygen incorporation |
18O2/18O2 /H216O % |
16O2/18O2/H216O % |
16O2/16O2 /H218O % |
| CAM |
16O, 16O 16O, 18O 18O 18O |
0.4 ± 0.2 7 ± 2 93 ± 2 |
10 ± 1 44 ± 1 47 ± 2 |
100 0 0 |
|
| ||||
| NO-CAM |
16O 18O |
4 ± 1 96 ± 1 |
8 ± 6 92 ± 6 |
98 ± 2 3 ± 2 |
16O2-P or 18O2-P was reacted with 0.3 equivalents NH2-CAM under different atmosphere and solvent conditions shown. Resultant products were analyzed by LCMS as described in the Experimental Procedures. All reactions done in buffer: 50 mM Bicine, pH 9, 4 °C.
When the reaction of 18O2-P with NH2-CAM was carried out in an 16O2 atmosphere, approximately 50% of the CAM produced was doubly labeled, while the remainder was either singly labeled (~40%) or had no 18O incorporation (10%) (Table 1). NO-CAM product labeling fell slightly, but remained >90%. No significant difference in oxygen incorporation was observed when 1.0 equivalent of substrate was used instead of 0.3.
Three possible explanations were considered for why the aerobic reactions of 18O2-P with NH2-CAM incorporate a significant amount of 16O: (1) P exchanges with water, converting 18O2-P to 18O/16O-P and/or 16O2-P, (2) P exchanges with the atmospheric O2, converting 18O2-P to 16O2-P, (3) P re-forms with 16O2 from the aerobic atmosphere after the P-decay species, diferric CmlI (CmlIox), is re-reduced by a pathway intermediate.
It was shown above that the reaction of 16O2-P in H218O yields only 16O product, indicating that neither the peroxo species nor products exchange with water. This finding rules out the first possible explanation. To test the second possibility and determine whether P can exchange with atmospheric O2, a stock of 18O2-P was made and then placed under a steady stream of 16O2. At time points between 1 and 100 min, an aliquot of P was removed and reacted with NH2-CAM for 10 minutes. No time dependent change in the 18O incorporation pattern was observed (Figure S1), showing that P does not exchange with ambient O2. In the absence of alternative explanations, the reduction of the enzyme by a substrate-based intermediate to allow re-formation of P with atmospheric 16O2 as part of the reactive cycle is strongly supported. The fact that >90% of the NO-CAM remained 18O-labeled also supports the hypothesis that the first oxygen atom is added by an 18O2-P and the second by a 16O2-P. The reducing agent required to re-form P is identified below.
Although the fraction of CAM with a mixed oxygen-labeled nitro group clearly increases in an 16O2 atmosphere, a significant portion of the product remains doubly labeled with 18O. A possible explanation for this observation is that a pathway intermediate prior to CAM escapes the active site and subsequently reacts with unreacted 18O2-P. This possibility is explored below.
NO-CAM Reacts with P
The detection of NO-CAM by LCMS during turnover of NH2-CAM suggests the possibility that NO-CAM is an intermediate in the reaction pathway. To test this possibility, NO-CAM was synthesized from CAM using a zinc reduction method previously reported.18 An anaerobic solution containing 1 eq. of NO-CAM and diferrous CmlI (CmlIred) was rapidly mixed with O2-saturated buffer at 4 °C (Scheme 3 and Figure 1A) to yield Sample A.
Scheme 3.

Figure 1.

Reaction of CmlIred with NO-CAM. (A) 100 μM NO-CAM was added anaerobically to 100 μM CmlIred, forming the nearly stoichiometric ES complex CmlIred-[NO-CAM] (black trace) that was rapidly mixed with O2 saturated buffer (~1.8 mM) to yield Sample A (blue trace). Inset: HPLC analysis of CmlIred-[NO-CAM] yielded mostly NO-CAM (black HPLC trace) as expected, while analysis of Sample A shows that the NO-CAM was converted into CAM product (blue HPLC trace). (B) Stopped-flow time course of the reaction described in (A). The time course (black) fits to a triple summed exponential (red, residuals in blue). 1/τ1 = 24 ± 1 s−1, 1/τ2 = 0.6 ± 0.1 s−1 and 1/τ3 = 0.05 ± 0.01 s−1. Buffer: 50 mM Bicine, pH 9, 4 °C.
The time course of the reaction monitored at 500 nm by stopped-flow spectroscopy is fit well by the sum of three exponential expressions with reciprocal relaxation times of 24 ± 1, 0.6 ± 0.1 and 0.05 ± 0.01 s−1, respectively (Figure 1B). Given the previously observed rapid, irreversible formation of P, and the direction and magnitude of the spectral change,14 it is likely that the fastest of the values correlates with the rate constant for P formation. However, it remains unclear whether the slower two phases arise from a two-step reaction or two parallel pathways leading to loss of P. In either case, these reciprocal relaxation times imply that the reaction steps occur with rate constants 100-1000 fold faster than the rate constant for P autodecay (~0.0006 s−1). It is evident that the increase in rate constant for P decay was caused by a reaction(s) that leads to formation of CAM because analysis of Sample A by HPLC showed only this product (Figure 1, inset). The yield of CAM was ~50% in these experiments. Less than stoichiometric yields were consistently observed for reactions utilizing NH2-CAM and, to a lesser extent, NO-CAM as substrates. The low yields may result from uncoupling of the reaction at the high enzyme and substrate concentrations used in single turnover experiments (see Discussion). A control reaction of diferric CmlI and NO-CAM yielded no CAM product or spectral changes either in the presence or absence of O2, demonstrating that NO-CAM reacts exclusively with the P form of CmlI.
NH(OH)-CAM Reacts with P and CmlIox
NH(OH)-CAM was synthesized from NO-CAM by reaction with reduced glutathione.18 NH(OH)-CAM was reacted separately with P and CmlIox under aerobic conditions, followed by acid quench. Remarkably, both reactions yielded CAM as the primary product, with a small amount of NO-CAM as either a product or a side product of acid-mediated decay of NH(OH)-CAM. NH(OH)-CAM is a relatively unstable compound which could potentially yield CAM in a non-specific reaction. To explore this possibility, it was acid quenched in buffer. This control yielded only NO-CAM product, likely resulting from the acid-mediated decay of NH(OH)-CAM in the quench procedure. These results indicate that the observed CAM production was mediated by CmlI (Figure 2).
Figure 2.
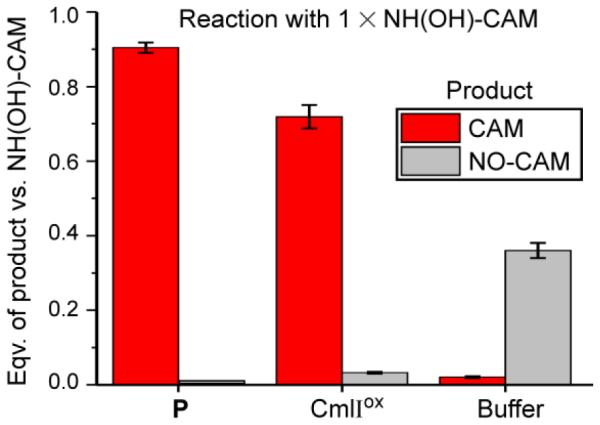
Reaction with one equivalent NH(OH)-CAM aerobically for 10 min. The reaction of 300 μM P or CmlIox with NH(OH)-CAM yields primarily CAM product. Control incubation with buffer (50 mM Bicine, pH 9) yields primarily NO-CAM, formed during the acid quench of the reaction. The apparent yield of NO-CAM is decreased about 50% by dimerization of the NO-CAM species into azoxy-CAM.3,21,22 Buffer: 50 mM Bicine, pH 9, 4 °C.
When reacted aerobically with a 10-fold excess of NH(OH)-CAM, P and CmlIox produced 5.9 ± 0.6 and 5.7 ± 0.1 equivalents of CAM product, respectively, under the conditions of Figure 2. The remaining NH(OH)-CAM was converted to NO-CAM in the acid quench. Thus, both reactions are catalytic with no requirement for external reducing equivalents. This result suggests that NH(OH)-CAM can provide the reducing equivalents required to reduce CmlIox and prime it to produce P in the presence of O2.
The reaction between CmlIox and NH(OH)-CAM was followed by UV-Vis spectroscopy to characterize pathway intermediates. The addition of NH(OH)-CAM directly to CmlIox (Scheme 4 and Figure 3) in an anaerobic environment resulted in the formation of a diferrous CmlI Sample B. An aliquot of Sample B removed and quenched anaerobically yielded an HPLC peak with the same retention time as NO-CAM. Oxygen was then added to sample B, forming primarily CmlIox, Sample C. HPLC analysis of Sample C showed that the NO-CAM species had disappeared and CAM was formed in its place. Total CAM yield was ~70% versus initial P concentration.
Scheme 4.
Figure 3.
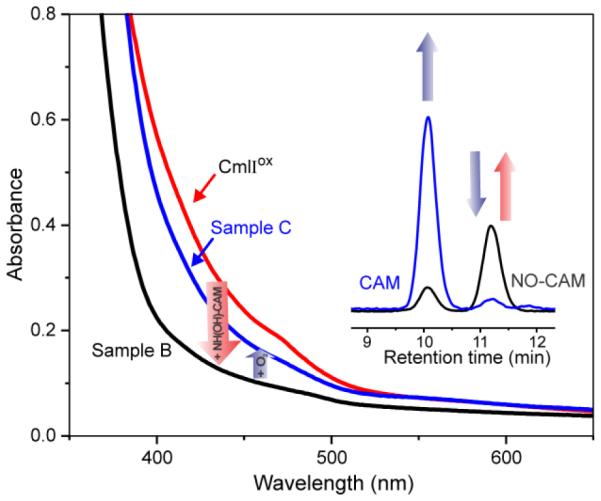
Optical absorbance spectra and product analysis in the reaction of 560 μM NH(OH)-CAM with 560 μM anaerobic CmlIox (red spectrum), which yields Sample B (black spectrum) which contains NO-CAM product (black HPLC trace). Addition of O2 yields Sample C (blue spectrum) which contains primarily CmlIox and CAM product (blue HPLC trace). Buffer: 50 mM Bicine, pH 9, 4 °C.
The oxidation state of the diiron cluster at each stage of the reaction was directly determined in a parallel Mössbauer experiment as shown in Table 2 (spectra and parameters show in Supporting Information Figures S2-S5). The anaerobic addition of approximately one equivalent of NH(OH)-CAM to CmlIox generates Sample B, the major component, representing 65% of the total iron in the sample, of which was assigned to a diferrous cluster (Figures 4 and S2). The remaining species were assigned to diferric clusters (~30%) and a small amount of unassigned mononuclear ferric material (~7%). Addition of O2 to this sample formed Sample C, which by UV-Vis appeared as primarily CmlIox. Accordingly, it exhibited the Mössbauer parameters of the diferric enzyme, which represents ~ 90% of the total iron in the sample. The different Mössbauer parameters of the CmlIox before and after the reaction with NH(OH)-CAM (See Figures S2 and S5 in the Supporting Information) could be due to the perturbation of the diiron centers by substrate/product binding or a change in the pH of the cluster environment; further studies are needed to resolve the detailed mechanisms resulting in such a difference.
Table 2.
Mössbauer Quantification of CmlI Forms during Single-Turnover Reactions
| Samplea | ||||||
|---|---|---|---|---|---|---|
| Speciesb (%) | B | C | Dc | E | Fd | G |
| CmlIox | 30 ± 3 | 90 ± 3 | 17 ± 3 | 85 ± 3 | 35 ± 3 | 75 ± 5 |
| CmlIred | 65 ± 3 | 0 | 68 ± 2 | 0 | 30 ± 3 | 0 |
| Unassigned mononuclear ferric species |
7 ± 3 | 9 ± 3 | 14 ± 3 | 14 ± 3 | 30 ± 5 | 23 ± 5 |
Mössbauer spectra and parameters of the species found in each sample are presented in Supporting Information Figures S2-S5.
Species are identified by the oxidation state of the diiron cluster. They may also have intermediate products bound in some cases. An ~10% conversion to diferric is expected in the Mössbauer samples vs the optical time course experiments due to autodecay of P during addition time required to prepare these high concentration samples. The presented quantifications do not include P, see Supporting Information for details.
Starting sample of P prior to degassing contained 72% P, 24% CmlIox, and 4% unassigned ferric species.
Starting sample of P prior to degassing contained 60% P, 36% CmlIox, and 4% unassigned ferric species.
Figure 4.

4.2 K Mössbauer spectra of a sample after anaerobically reacting with 1 eq. NH(OH)-CAM with CmlIox (A), and the same sample after further addition of O2 (B). The red lines are experimental data, the black lines are spectral simulations (See the Supporting Information for detailed analysis). The blue line represents the CmlIred simulation in A.
Addition of approximately one equivalent of NH(OH)-CAM to P resulted in Sample D (Scheme 5), which has a UV-Vis spectrum that reflects a mixture of diferric and diferrous species (Figure 5, black trace). Analysis of Sample D by HPLC shows generation of CAM product in ~45% yield vs. initial NH(OH)-CAM concentration (Figure 5, inset, black). Addition of O2 to Sample D produced Sample E (Figure 5, red trace) and an increase in CAM product, presumably arising from unreacted NH(OH)-CAM (Figure 5, inset, red). The spectrum of Sample E is best fit by summing the spectra of approximately 80% CmlIox and 20% P.
Scheme 5.
Figure 5.
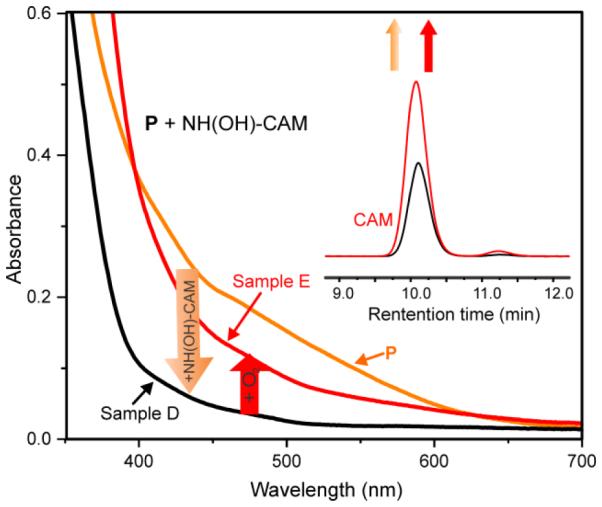
Absorbance spectra and product analysis in the anaerobic reaction of 300 μM NH(OH)-CAM with 300 μM P (orange trace), which yields Sample D (black spectrum). Addition of O2 forms Sample E (red trace). Inset: HPLC analysis of CAM and NO-CAM products from Sample D (black trace) and Sample E (red trace). Buffer: 50 mM Bicine, pH 9, 4 °C.
Mössbauer analysis of parallel samples (Figure S3) is generally consistent with the results observed by UV-Vis spectroscopy. In the Mössbauer experiment, P was formed in 72% yield and then degassed, leaving ~ 60% P and ~36% diferric CmlI (plus 4% mono-ferric material). After anaerobic addition of one equivalent of NH(OH)-CAM versus total active site concentration and a 5 minute incubation, the resulting Sample D was ~68% CmlIred and ~17% diferric clusters (Table 2). The observation of CmlIred and CAM (Figure 5) suggests that this reaction proceeds at least in part by the route proposed by Bollinger, Krebs and coworkers for reaction of NH(OH)-Ar with the peroxo intermediate of AurF. The latter reaction yields NO2-Ar and diferrous AurF (see Scheme 2, pathway II). 13 The generation of 68% CmlIred was accompanied in this experiment by production of 62% CAM (product yield differences were observed between the optical and Mössbauer experiments, but the trends remained the same). These values support the reduction of CmlIox by N(OH)2-CAM to yield roughly equal amounts of CAM and CmlIred. A small amount of NO-CAM is also observed suggesting that the CmlIox fraction also reacts with NH(OH)-CAM to yield CmlIred and NO-CAM. Consequently, only part of the observed CmlIred derives from pathway II, thereby slightly decreasing the relative yield of CAM.
Addition of excess O2 to Sample D formed Sample E, which Mössbauer spectra show to contain ~85% CmlIox. This value is similar to the approximately 80% CmlIox and 20% P observed by UV-Vis spectroscopy. The difference in the percentages derived from UV-Vis and Mössbauer data can be attributed to some P decay during the extended Mössbauer sample preparation process. Formation of Sample E is accompanied by formation of an additional 10% CAM product, which probably arises from the re-formation of P and its reaction with residual NH(OH)-CAM. It is unclear why the remaining unreacted CmlIred is converted to CmlIox rather than P but we demonstrate above that the process of forming P is less than stoichiometric, resulting in a significant amount of adventitious CmlIox formation.
Reaction of CmlIred, O2, and NH2-CAM Confirms NH(OH)-CAM and NO-CAM as Intermediates
It is shown above that P reacts individually with NH2-CAM, NH(OH)-CAM and NO-CAM to produce chloramphenicol; however it is also important to demonstrate the reaction of NH2-CAM beginning with CmlIred also proceeds through the NH(OH)-CAM and NO-CAM intermediates. Accordingly, the reaction between NH2-CAM, Cmlred, and O2 was monitored in a stepwise fashion using UV-Vis spectroscopy (Scheme 6 and Figure 6). The addition of O2 to CmlIred formed P, which was made anaerobic and then reacted with one equivalent of NH2-CAM. The resulting species, Sample F, lacks the strong absorbance band at 375 nm that is characteristic of CmlIox and the band at 500 nm characteristic of P. Sample F looks similar to CmlIred save for an additional shoulder at ~ 400 nm, which may be attributable to to the presence of a substrate or product. NO-CAM has significant absorbance at 400 nm, and so a sample of CmlIred combined with NO-CAM was prepared for comparison. Sample F exhibits a spectrum similar to that of the comparison sample, supporting its assignment as CmlIred bound to one equivalent of NO-CAM. Differences between Sample F and authentic diferrous CmlI-NO-CAM can be attributed to an estimated 10% O2 contamination introduced during handling of the P sample, leading to oxidation of the diferrous cluster after the CmlIred-NO-CAM complex is formed. When oxygen was added to Sample F, a species which exhibits a spectrum that overlays with an independently prepared sample of decayed P (i.e. CmlIox) was observed (Sample G).
Scheme 6.
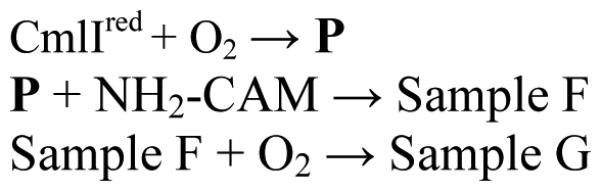
Figure 6.
Spectroscopic snapshots of the CmlI catalytic cycle and HPLC analysis of associated products. The reaction began with 600 μM CmlIred (grey trace), to which O2 was added to form P (orange trace). Anaerobic addition of 600 μM NH2-CAM yields Sample F (solid black trace), proposed to be CmlIred-NO-CAM by comparison to independently prepared 600 μM CmlIred-NO-CAM (dotted black trace). The addition of O2 to Sample F yields Sample G (solid blue trace), which nearly overlays with an independently prepared 600 μM decayed P sample (dotted blue trace). Inset: HPLC analysis of products in Sample F (black trace) and Sample G (blue trace). Sample F yields NO-CAM as the primary product and Sample G shows that NO-CAM has been consumed and CAM produced. Buffer: 50 mM Bicine, pH 9, 4 °C
Aliquots of Sample F and G were analyzed by HPLC. Sample F was found to contain primarily NO-CAM in an acid quench experiment. Repeating this experiment using a spin quench method gave the same results, eliminating the possibility that the sample contained NH(OH)-CAM, which was then converted to NO-CAM under acidic conditions. A small amount of CAM was also present corroborating the 10% O2 contamination during handling of P proposed above. An aliquot of the reaction end point Sample G contained no NO-CAM, presumably because it had been converted to CAM, which was detected as the sole product. These results suggest that CmlI is re-reduced concomitant with the production of NO-CAM, and that the addition of O2 converts the new CmlIred to CmlIox (presumably with P as an intermediate) while NO-CAM is converted to CAM. The yield of CAM from a single turnover reaction using NH2-CAM as the substrate is ≤ 30%, in contrast to the nearly stoichiometric yields observed when using NH(OH)-CAM as the substrate in a single turnover experiment.
Corroborating evidence for the proposed oxidation states was provided by analyzing a parallel experiment by Mössbauer spectroscopy (Table 2, Figure S4). P was formed in ~60% yield as CmlIred reacted with O2. The remaining iron was 36% unreacted CmlIox and 4% of a paramagnetic component that can be attributed to adventitious mononuclear iron sites in the enzyme. Formation of Sample F by addition of NH2-CAM to P in the absence of excess O2 causes the disappearance of >90% of P and the emergence of CmlIred. Interestingly, only ~55% of P decays to CmlIred in Sample F; the rest decays to one or more unidentified mononuclear ferric species, representing ~ 30% of the total iron in the sample. Addition of O2 to yield sample G regenerates ~75% diferric enzyme in the sample, the rest of the sample is still the mononuclear ferric species (~ 23%). Therefore, the Mössbauer results suggest that P is prone to degradation by an unknown mechanism when reacting with NH2-CAM under the experimental conditions we applied, causing significant uncoupling of the reaction. Based on the amount of CmlIred generated (35%) in the anaerobic reaction, it is understandable that the yield of CAM from a single turnover reaction using NH2-CAM as the substrate is sub-stoichiometric as observed in HPLC analysis.
CmlI is in the Oxidized State After a Single Turnover
It has been reported that the AurF single turnover ends with the enzyme in the diferrous state (Scheme 2, pathway II).10 Under conditions of stoichiometric NH(OH)-Ar substrate and excess O2, the AurF diferric peroxo species re-formed and was detected by its characteristic Mössbauer spectrum and absorbance at 500 nm.10 It is shown here that the reactions of P with NH2-CAM and NO-CAM end with CAM production and CmlI in the diferric state. In addition, the reaction of P with NH(OH)-CAM yields an intermediate diferrous state and CAM prior to O2 addition. When assessed by its optical spectrum, a fraction of this sample is found as P at its completion. The N(OH)2 reduction mechanism proposed by Bollinger and coworkers suggests that the reaction of P with a substoichiometric amount of NH2-Ar substrate should result in the decay and partial reformation of P. The conclusion that CmlI does not end up in the reduced state after a single turnover of the native substrate is confirmed by the time course of a reaction of P with a substoichiometric concentration of NH2-CAM shown in Figure 7, red trace. Rapidly mixing P with 0.25 or 0.5 equivalents of NH2-CAM in an oxygenated buffer at 4 °C led to only a rapid decay of P to CmlIox. In contrast, the reaction with 1.0 equivalent NH(OH)-CAM showed decay and then partial reformation of P within two seconds (Figure 7, blue trace). The observation of re-formed P suggests that the reaction of P with NH(OH)-CAM leads to substantial formation of the diferrous cluster, as reported for AurF-peroxo and NH(OH)-benzoate.13 Rates of P decay and reformation are sufficiently similar such that only a small percentage of decay is observed before the reformation process begins. Thus, using equivalents of NH(OH)-CAM below or above the stoichiometric amount masks the reformation process.
Figure 7.
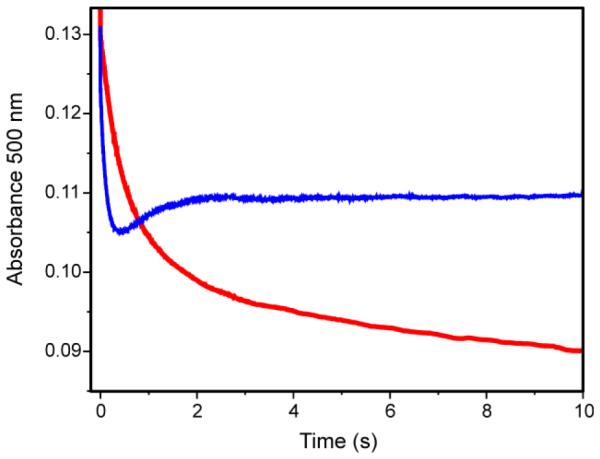
Single turnover reaction of P with 0.5 equiv NH2-CAM (red trace) and 1.0 equiv NH(OH)-CAM (blue trace). Single wavelength (500 nm) stopped flow traces for the reaction of 260 μM P and 130 μM NH2-CAM or 260 µM NH(OH)-CAM (post-mix concentrations) in the presence of excess O2. No P reformation is observed in the reaction with the native substrate NH2-CAM, suggesting that the in-vitro reaction ends in the diferric state. The reactions using the same concentrations of NH2-CAM and NH(OH)-CAM give the same results but the reformation of P in the case of the latter substrate is masked due to the rates of the decay and re-formation reactions. Buffer: 50 mM Bicine, pH 9, 4 °C.
Exchange of NO-CAM from the CmlI Active Site
In order to test whether NO-CAM can leave and reenter the active site during the reaction cycle, 18O2-P was degassed to remove excess O2, and equilibrated in an anaerobic chamber before 1 equiv. NH2-CAM was added. After a 20-s incubation, 1 equiv. N16O-CAM was added. The sample was then quickly put under an 18O2 headspace and allowed to react for 10 min. Samples were acid quenched and analyzed by LCMS. Control samples were made by adding buffer instead of NO-CAM. If the NO-CAM made during the reaction cannot leave the active site, then only CAM containing 18O in the nitro group should be made. If, however, NO-CAM can leave and subsequently re-bind in the active site after 16O-P has formed, then there should be some CAM formed with both 18O and 16O in the nitro group. As shown in Figure 8 and Table S2, approximately 20% more 16O,18O CAM is formed compared to the control, suggesting that at least 20% of the NO-CAM dissociates during the course of the experiment. Unfortunately, the short lifetime of the NH(OH)-CAM intermediate prevented a similar evaluation of dissociation from the active site.
Figure 8.
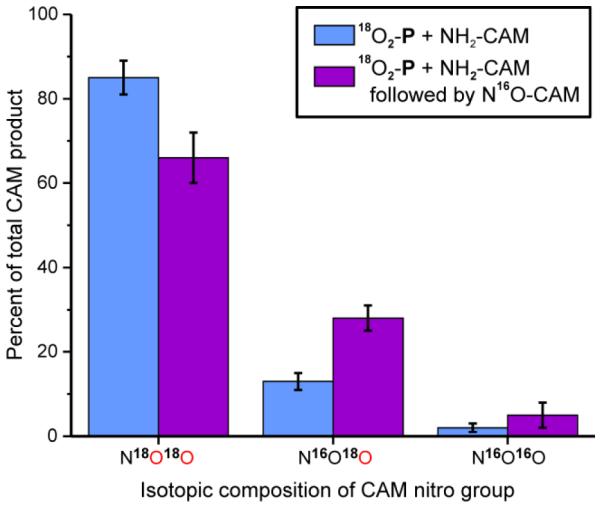
Reaction of 200 μM of NH2-CAM with 200 μM 18O2-P, followed 20 s later by addition of buffer or NO-CAM under an 18O2 atmosphere. Presence of mixed labeled CAM in the reaction in which NO-CAM was added shows that NO-CAM can exchange from the active site given a 20 s incubation period. Buffer: 50 mM Bicine, pH 9, 4 °C.
DISCUSSION
It is shown here that the CmlI reaction cycle intermediate P reacts with the native substrate NH2-CAM as well as the reaction pathway intermediates NH(OH)-CAM and NO-CAM to yield CAM. Moreover, NH(OH)-CAM is found to reduce CmlIox to yield CmlIred, which can subsequently form P in the presence of O2. These observations provide the basis for the new single turnover mechanistic cycle shown in Scheme 7 in which re-reduction of CmlIox by NH(OH)-CAM, formed during the reaction of P with NH2-CAM, allows formation of CAM with the input of only two non-substrate-derived electrons (supplied in vivo by an as yet unidentified reductase). This cycle is conceptually similar to a recent model proposed for AurF.13 However, it differs in both the pathway intermediate that serves as the reductant and the state of the enzyme diiron cluster at the conclusion of a single turnover cycle. The mechanistic significance of these observations will be discussed here.
Scheme 7.
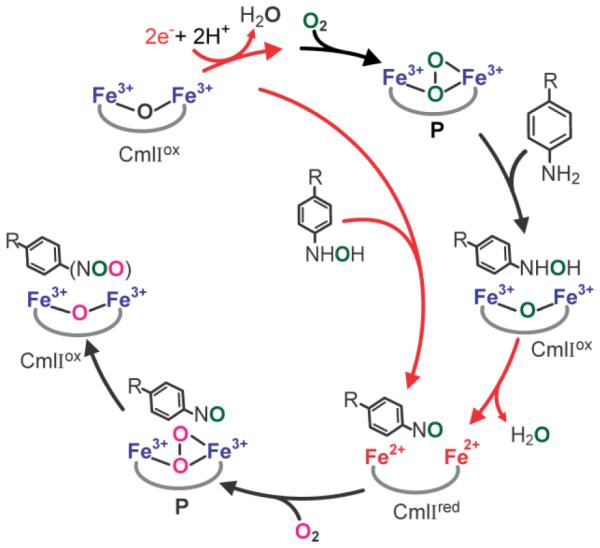
Proposed Single Turnover Reaction Pathway of CmlI. Reduction steps are shown in red.
Comparison to a Model Requiring Three Successive Reactions with P
As shown in Scheme 2, pathway I, the original model for the 6-electron oxidation of NH2-Ar substrates envisioned three similar successive oxidation reactions, each utilizing a P-like intermediate (or a high valent species derived from it). This model was supported by the observation that a titration of P with NH2-CAM, or the equivalent titration for the AurF system, had approximately a 3:1 stoichiometry.3,14,17,23 The current results show that the reaction of NH(OH)-CAM with either CmlIox or P approaches 1:1 stoichiometry and yields CAM as the major product. These results suggest that the original mechanistic proposal is not correct and that at least one step of the reaction involves oxidation of a pathway intermediate coupled to reduction of CmlIox.
Based on the stepwise study of the reaction shown in Figure 6, the reaction of NH2-CAM with P (i.e. CmlIred + O2) to produce CAM also proceeds by utilizing a pathway intermediate to re-reduce the diiron cluster. The reductive pathway intermediate is again NH(OH)-CAM. However, the overall yield of CAM from this reaction is typically 33% or less, due presumably to uncoupling. We believe that this fortuitous fractional loss of yield led to the observed roughly 3:1 P to NH2-CAM ratio that supported the original mechanistic proposal.
Comparison of the Mechanistic Models Employing a Pathway Intermediate as an Electron Source
The observation by Bollinger, Krebs, and coworkers that the reaction of NH(OH)-Ar with the AurF-peroxo intermediate led to NO2-Ar product plus diferrous AurF under anaerobic conditions initiated a new way to think about the amine oxygenase chemistry.13 It was proposed that N(OH)2-Ar is formed as an intermediate and then acts as the reducing agent for diferric AurF as it itself is oxidized to NO2-Ar. However, despite the structural similarity and common reaction type of AurF and CmlI, we do not believe that CmlI follows this mechanistic pathway during its native cycle. Scheme 8, pathways II and III, respectively, compare an AurF-type mechanism to that proposed here for CmlI for CAM formation. We believe that pathway III better describes the native reaction for CmlI for five reasons: (1) NH(OH)-CAM is shown to act as an efficient reducing agent for CmlIox, and it is the consensus first intermediate in both pathways II and III. (2) In general, hydroxylamines are efficient reducing agents for iron. The reduction of ferric to ferrous ion is well documented in the organic synthetic literature, and it is an essential step of colorimetric assays to determine iron concentration.18,24,25 In contrast, N(OH)2-CAM is more likely to hydrolyze quickly to the NO-CAM intermediate rather than act as a reducing agent. (3) The single turnover reaction of stoichiometric NH2-CAM and P proceeds through NO-CAM, and NO-CAM is independently shown to react with P to form CAM. (4) After a single turnover of sub-stoichiometric NH2-CAM and P under aerobic conditions, CmlIox rather than CmlIred or P is observed. This requires that the pathway reducing agent not act in the final step of the pathway because doing so would generate a reduced diiron cluster after the final step. (5) As shown in Scheme 8, a single turnover reaction of 18O-P in an 16O2 atmosphere and H216O would be expected to give mixed-labeled product by pathway III but not by pathway II. Mixed-labeled CAM product was, in fact, observed for the CmlI reaction (Table 1).
Scheme 8. Comparison of Oxygen Incorporation during CAM Formation via Different Pathways.
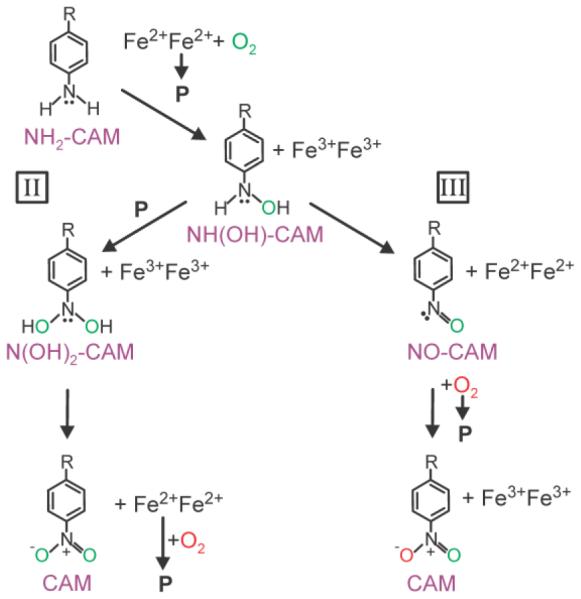
Although pathway III better describes the native CmlI pathway, the off-cycle or artificial reaction of P with NH(OH)-CAM was carried out to make a direct comparison to the analogous reaction in the AurF cycle. In accord with the results reported for the anaerobic reaction of the AurF-peroxo intermediate reaction with NH(OH)-Ar,13 we find that CmlIred and CAM are produced when P is reacted with NH(OH)-CAM. This reaction would not be on-pathway for Scheme 8, pathway III, as only CmlIox and NH(OH)-CAM are found together in the active site, thereby allowing the redox reaction to occur. Consequently, the off-pathway reaction for CmlI might occur as proposed for AurF (Scheme 8, pathway II). However, the subsequent addition of O2 was found to yield additional CAM and CmlI finishes in the mostly oxidized rather than reduced or P states expected based on the reported AurF reaction. 13 Additional kinetic and spectroscopic studies will be required to fully characterize the off-pathway reaction of P with NH(OH)-CAM.
Although this data set strongly supports the reaction pictured in Scheme 7 and Scheme 8, pathway III as the best fit for the CmlI cycle, it also reveals the complexity of the reaction. For example, in the reaction of NH2-CAM with 18O-P in an 16O2 atmosphere and H216O, doubly 18O-labled product was observed in addition to the mixed-labeled CAM product predicted by Scheme 8, pathway III. Two ways in which this might come about are: (1) partial dissociation of NO-CAM and subsequent reaction with unreacted 18O-P, or (2) partial dissociation of NH(OH)-CAM and subsequent reaction with 18O-P through pathway II. We view the latter possibility as unlikely because CmlIox and NH(OH)-CAM are formed simultaneously in the active site where the observed redox reaction to form Cmlred and NO-CAM would be expected to occur efficiently. Approximately 50% of the NH(OH)-CAM would have to dissociate and react through pathway II in order to account for the amount of mixed-label CAM observed. However, very little CAM and no NH(OH)-CAM are observed in the stepwise reaction of NH2-CAM with P illustrated in Figure 6 prior to addition of O2. In contrast, generation of mixed-label CAM via the partial dissociation of NO-CAM is possible based on the exchange experiment illustrated in Figure 8. It is possible that these alternative pathways as well as the lower than stoichiometric yields in some cases described above result from performing the reaction under single turnover conditions without the native reductant. Indeed, protein interactions are very important throughout the diiron oxygenase family but cannot be explored in the current case because the native reductase has not been identified for any amine oxygenase.26-28
Mechanistic Significance
The reactivity of the various activated forms of O2 created by diiron clusters is of great current interest.29,30 In the past, there has been significant focus on the high-valent species that can transfer oxygen into very stable C-H bonds, such as in the reaction of sMMOH compound Q with methane.31-33 The potential reactivity of peroxo or superoxo intermediates has, by contrast, come to light only recently.30,34,35 The activity, novel structure, and long lifetime of the CmlI peroxo intermediate P offer an opportunity to explore new aspects of the formation and reaction of peroxo species in diiron-cluster containing enzymes. Indeed, we find that P, proposed to have a µ-η1η2 peroxo core,14 is quite reactive with both aromatic amine and aromatic nitroso substrates. The 18O2 incorporation studies reported here show that both of the nitro oxygens in the final product derive from O2, but it is clear that they are added in a stepwise fashion from different O2 molecules (via two successively formed P intermediates), because a mixed-isotope product is obtained when 18O-P is reacted with NH2-CAM in an 16O atmosphere. Thus, CmlI is a monooxygenase where the reactive oxygen species is formed prior to O-O bond cleavage, quite distinct from the high-valent strategy used by sMMOH-type diiron monooxygenases.31 The use of a diferric peroxo species may allow the enzyme to catalyze a wide range of N-oxygenation reactions. Over the course of the amine to nitro conversion, the substrate becomes increasingly electron deficient. Thus P must be ambiphilic, able to shift from acting as an electrophilic species for the oxygenation of NH2-CAM to a more nucleophilic oxidant for oxygenation of NO-CAM. It is significant that, despite the monooxygenase reactivity of CmlI, it cannot oxygenate C-H bonds of aromatic or aliphatic hydrocarbons.14 While this may be attributed to several causes including the relative bond stability of these substrates, it may also reflect the unique structure of the CmlI P intermediate and its environment. For example, this environment may restrict access to the protons thought to play a role in the generation of the high-valent oxo or bis µ-oxo intermediates of hydrocarbon monooxygenases.15,33
Conclusion
Aromatic amine oxygenation deviates from the paradigm of diiron oxygenation chemistry in several ways. While cluster reduction and formation of a peroxo intermediate is shared among oxygen-activating diiron oxygenases, the structure of the peroxo intermediate P of the CmlI aromatic amine oxygenase is unique.14 This specialized peroxo intermediate is highly reactive with NH2-CAM and the other amine oxidation pathway intermediates, while the peroxo intermediates of other oxygenases serve primarily as unreactive stepping stones to reactive high valent intermediates. Another remarkable aspect of the diiron cluster of the aromatic amine oxygenases CmlI and AurF is their ability to use intermediate products as mid-pathway diiron cluster reductants in place of the reductase used by all other diiron oxygenases. This means that a 6-electron oxidation of the NH2-Ar substrate could occur without dissociation of the intermediate products, thereby avoiding release of hydroxylamino- and nitroso-intermediates that might be damaging to the cell. While the results presented here show that dissociation is possible at the NO-CAM stage, the time scale for release versus the rate of normal catalysis appears to be slow. The versatility of the P intermediate allows it to react with intermediate products that it would not normally encounter in the native pathway to CAM formation. In particular, the P reaction with NH(OH)-CAM would not normally occur because they are not present at the same time in the cycle. Nevertheless, this reaction can occur with high efficiency when these reactants are separately prepared and mixed, and it leads to the same CAM product, albeit through an alternative reaction pathway.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the financial support of this work from grants NIH GM100943 and NIH GM118030 (to J.D.L.), NIH GM38767 (to L.Q.) and graduate traineeship NIH GM08700 (to A.J.K.). Y.G. acknowledges financial support from Carnegie Mellon University. We thank Joseph Dalluge for assistance in mass spectral analysis.
Footnotes
Supporting Information
Detailed experimental procedures for quantification and fitting of Mössbauer spectra Figures S1-S5 and Tables S1-S2. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Yan G, Yang M. Org. Biomol. Chem. 2013;11:2554. doi: 10.1039/c3ob27354g. [DOI] [PubMed] [Google Scholar]
- (2).McPake CB, Murray CB, Sandford G. ChemSusChem. 2012;5:312. doi: 10.1002/cssc.201100423. [DOI] [PubMed] [Google Scholar]
- (3).Winkler R, Hertweck C. Angew. Chem. Int. Ed. 2005;44:4083. doi: 10.1002/anie.200500365. [DOI] [PubMed] [Google Scholar]
- (4).He J, Magarvey N, Piraee M, Vining LC. Microbiology. 2001;147:2817. doi: 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- (5).Lu HG, Chanco E, Zhao HM. Tetrahedron. 2012;68:7651. doi: 10.1016/j.tet.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Simurdiak M, Lee J, Zhao H. ChemBioChem. 2006;7:1169. doi: 10.1002/cbic.200600136. [DOI] [PubMed] [Google Scholar]
- (7).Lee J, Simurdiak M, Zhao H. J. Biol. Chem. 2005;280:36719. doi: 10.1074/jbc.M505334200. [DOI] [PubMed] [Google Scholar]
- (8).Lee J, Zhao H. Angew. Chem. Int. Ed. 2006;45:622. doi: 10.1002/anie.200502903. [DOI] [PubMed] [Google Scholar]
- (9).Zocher G, Winkler R, Hertweck C, Schulz GE. J. Mol. Biol. 2007;373:65. doi: 10.1016/j.jmb.2007.06.014. [DOI] [PubMed] [Google Scholar]
- (10).Platter E, Lawson M, Marsh C, Sazinsky MH. Arch. Biochem. Biophys. 2011;508:39. doi: 10.1016/j.abb.2011.01.010. [DOI] [PubMed] [Google Scholar]
- (11).He J, Hertweck C. J. Am. Chem. Soc. 2004;126:3694. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- (12).Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc. Natl. Acad. Sci. USA. 2008;105:6858. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li N, Korboukh VK, Krebs C, Bollinger JM., Jr. Proc. Natl. Acad. Sci. USA. 2010;107:15722. doi: 10.1073/pnas.1002785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Makris TM, Vu VV, Meier KK, Komor AJ, Rivard BS, Münck E, Que L, Jr., Lipscomb JD. J. Am. Chem. Soc. 2015;137:1608. doi: 10.1021/ja511649n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- (16).Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- (17).Korboukh VK, Li N, Barr EW, Bollinger JM, Jr., Krebs C. J. Am. Chem. Soc. 2009;131:13608. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Eyer P, Schneller M. Biochem. Pharmacol. 1983;32:1029. doi: 10.1016/0006-2952(83)90621-4. [DOI] [PubMed] [Google Scholar]
- (19).Petasis DT, Hendrich MP. Methods Enzymol. 2015;563:171. doi: 10.1016/bs.mie.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Groce SL, Miller-Rodeberg MA, Lipscomb JD. Biochemistry. 2004;43:15141. doi: 10.1021/bi048690x. [DOI] [PubMed] [Google Scholar]
- (21).Entwistle ID, Gilkerson T, Johnstone RAW, Telford RP. Tetrahedron. 1978;34:213. [Google Scholar]
- (22).Spain JC. Biodegradation of Nitroaromatic Compounds. Springer US; New York: 1995. [DOI] [PubMed] [Google Scholar]
- (23).Winkler R, Zocher G, Richter I, Friedrich T, Schulz GE, Hertweck C. Angew. Chem. Int. Ed. 2007;46:8605. doi: 10.1002/anie.200703089. [DOI] [PubMed] [Google Scholar]
- (24).Saywell LG, Cunningham BB. Ind. Eng. Chem. Anal. Ed. 1937;9:67. [Google Scholar]
- (25).Rao GG, Somidevamma G. Fresen J. Anal. Chem. 165:432. [Google Scholar]
- (26).Wallar BJ, Lipscomb JD. Biochemistry. 2001;40:2220. doi: 10.1021/bi002298b. [DOI] [PubMed] [Google Scholar]
- (27).Liang AD, Lippard SJ. Biochemistry. 2014;53:7368. doi: 10.1021/bi500892n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bailey LJ, Acheson JF, McCoy JG, Elsen NL, Phillips GN, Jr., Fox BG. Biochemistry. 2012;51:1101. doi: 10.1021/bi2018333. [DOI] [PubMed] [Google Scholar]
- (29).Bollinger JM, Jr., Diao Y, Matthews ML, Xing G, Krebs C. Dalton Trans. 2009:905. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tinberg CE, Lippard SJ. Biochemistry. 2010;49:7902. doi: 10.1021/bi1009375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wallar BJ, Lipscomb JD. Chem. Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- (32).Banerjee R, Proshlyakov Y, Lipscomb JD, Proshlyakov DA. Nature. 2015;518:431. doi: 10.1038/nature14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tinberg CE, Lippard SJ. Acc. Chem. Res. 2011;44:280. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Beauvais LG, Lippard SJ. J. Am. Chem. Soc. 2005;127:7370. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- (35).Xing G, Barr EW, Diao Y, Hoffart LM, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr. Biochemistry. 2006;45:5402. doi: 10.1021/bi0526276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.