


Abstract
Small nucleolar RNAs (snoRNAs) are a class of non-coding RNAs that guide the post-transcriptional processing of other non-coding RNAs (mostly ribosomal RNAs), but have also been implicated in processes ranging from microRNA-dependent gene silencing to alternative splicing. In order to construct an up-to-date catalog of human snoRNAs we have combined data from various databases, de novo prediction and extensive literature review. In total, we list more than 750 curated genomic loci that give rise to snoRNA and snoRNA-like genes. Utilizing small RNA-seq data from the ENCODE project, our study characterizes the plasticity of snoRNA expression identifying both constitutively as well as cell type specific expressed snoRNAs. Especially, the comparison of malignant to non-malignant tissues and cell types shows a dramatic perturbation of the snoRNA expression profile. Finally, we developed a high-throughput variant of the reverse-transcriptase-based method for identifying 2′-O-methyl modifications in RNAs termed RimSeq. Using the data from this and other high-throughput protocols together with previously reported modification sites and state-of-the-art target prediction methods we re-estimate the snoRNA target RNA interaction network. Our current results assign a reliable modification site to 83% of the canonical snoRNAs, leaving only 76 snoRNA sequences as orphan.
INTRODUCTION
SnoRNAs form a specific class of small (60–170 nucleotides, with few exceptions (1)) non-protein coding RNAs that is best known for guiding post-transcriptional modification of other non-protein coding RNAs such as ribosomal and small nuclear RNAs (rRNAs, snRNAs respectively) (2–7). Based on defined sequence motifs and secondary structure elements, snoRNAs are classified as either C/D box or H/ACA box.
C/D box snoRNAs guide 2′-O-methylation and H/ACA snoRNAs pseudouridylation of nucleotides on target molecules. The C box (RUGAUGA, R = A or G) and D box (CUGA) sequence motifs of C/D box snoRNAs, are brought into close proximity when the 5′ and 3′ ends of the molecule fold into a stem structure, forming a kink-turn (8,9). Most C/D box snoRNAs have additional, less conserved, C and D box motifs, the C' and D' boxes, in the central region of the snoRNA. C/D box snoRNAs carry out their function within ribonucleoprotein (RNP) complexes that additionally contain the 15.5K, NOP56, NOP58 and fibrillarin proteins (10,11), the latter catalysing 2′-O-methylation of ribose molecules in the target RNAs (12). Which nucleotide undergoes this modification is determined by the complementarity to the 7 to 21 nucleotides (nt) guide region that is located upstream of the D or D' box: the 5th nucleotide upstream of the D/D' box will undergo the 2′-O-methylation (13–15).
H/ACA box snoRNAs adopt a well-defined secondary structure consisting of two hairpins that are joined by a single-stranded region known as the H box (ANANNA, N = A, C, G or U) and further have an ACA box (AYA, Y = C or U) motif at the 3′ end (16,17). The H/ACA snoRNPs contain the H/ACA snoRNA and a set of four proteins, Dyskerin, Nhp2, Nop10 and Gar1, with Dyskerin acting as the pseudouridine synthase (18). Target recognition by H/ACA box snoRNAs also involves RNA-RNA interactions, of single-stranded regions within interior loops of the two hairpin structures in the snoRNA with the target RNA (19,20).
Canonical snoRNAs accumulate in the nucleolus, the primary site of ribosome synthesis. ScaRNAs (small Cajal body-specific RNAs), are a specific subset of snoRNAs that guide spliceosomal RNA modifications. They are enriched in the Cajal bodies, where the last steps of of spliceosomal RNA biogenesis take place (3). The import of snoRNAs into Cajal bodies is guided by specific sequence motifs, which for H/ACA box snoRNAs are the CAB boxes (UGAG) located in the hairpin loops of the two stem structures (21), whereas C/D box snoRNAs have a long UG dinucleotide repeat element (22). There is evidence that both motifs are recognized by the WDR79 protein, which facilitates the transport to Cajal bodies (22,23). Aside from these snoRNAs that have canonical structures, some long scaRNAs with hybrid structures that are able to function in both methylation and pseudouridylation, have been characterized (1,3). Moreover, the primate-specific Alu repeat elements can give rise to H/ACA box-like snoRNAs; these were coined AluACA RNAs and seem to accumulate in the nucleoplasm (24). The RNA component of the animal (but not of fungal or of other groups of eukaryotes) telomerase RNP (TERC) contains an H/ACA box snoRNA-like domain (25–29), which harbors a CAB box (30) and is essential for telomerase activity (25).
SnoRNAs can guide other types of RNA processing, beyond methylation and pseudouridylation (see ref. (31) for a recent review). For example, SNORD22, SNORD14, SNORD13, SNORD3 and SNORD118 are involved in the processing of ribosomal RNA precursors (32). Even though they have C and D box motifs, these snoRNAs do not seem to undergo the terminal end trimming that is characteristic to C/D box snoRNAs (33). This suggests that additional proteins probably assist these snoRNAs in their function, at the same time preventing the usual C/D box-specific trimming. Some evidences suggest specific functions for snoRNAs encoded in the imprinted 15q11-q13 region: the brain-specific C/D box SNORD115 family regulates the alternative splicing of the serotonin receptor 5-HT(2C) mRNA (34,35), and SNORD116 family members are part of longer RNAs that sequester the Fox family of splicing regulators (36). Many C/D box as well as H/ACA box snoRNAs seem to undergo some kind of processing, yielding smaller fragments whose function remains elusive (33,37). An exception is the H/ACA box snoRNA SCARNA15 whose microRNA-like function has been well documented (38). Whether this function can be more generally carried out by other snoRNAs remains unknown. Recent high-throughput sequencing-based studies identified C/D box-like snoRNAs either as short as 27 nucleotides (33), barely able to host an antisense region. Further there are long non-coding RNAs with snoRNA ends (sno-lncRNAs) described in (36,39). A summary of the currently known structural types of snoRNA is shown in Figure 1.
Figure 1.

Schematic overview of structural types of snoRNAs. (A) Canonical C/D box snoRNAs have a C box and a D box motif located close to the terminal stem, and additional internal C' and D’ boxes. Canonical H/ACA box snoRNAs are composed of two stem loop structures with an internal H box motif and an ACA box motif at the 3′ end. (B) Cajal body-associated snoRNAs additionally have specific localization motifs, which are the CAB box in the case of H/ACA box snoRNAs, and a G/U rich sequence in the case of C/D box snoRNAs. (C) SnoRNAs with hybrid structure that consist of both a C/D box and an H/ACA box domain have been identified. Recent studies have also uncovered extremely short C/D box-like snoRNAs (D) as well as long (several hundred nucleotides) noncoding RNAs with snoRNA ends (E and F).
Despite a few genome-wide surveys, recent studies (22,33) have clearly demonstrated that the catalog of human snoRNA loci is far from complete. The snoRNA data resources (40,41) that used to be standard in the field have either ceased to exist or to be updated, as the focus of the research community has moved towards characterization of snoRNA genes in species other than human (42–46). A recent attempt to improve the accuracy of snoRNA gene annotation (47) clearly demonstrated that a well designed, uniform analysis strategy is needed to expand the catalog of snoRNAs while maintaining annotation accuracy. In this study we have taken a comprehensive approach, combining both: analysis of large-scale data generated by the ENCODE consortium data, as well as developing novel experimental methodology to construct an up-to-date catalog of snoRNA loci in the human genome. Furthermore we characterize their processing patterns, expression profiles across tissues, as well as their potential targets. The data collected in this study is publicly accessible via http://www.bioinf.uni-leipzig.de/publications/supplements/15-065.
MATERIALS AND METHODS
Curation of mature forms of known and novel snoRNA genes
A list of snoRNA genes currently annotated by HGNC was obtained from www.genenames.org (3 March 2014) and the corresponding sequence entries were retrieved from the NCBI Nucleotide database via accession numbers as identifiers. Retrieved sequences were then mapped to the hg19 human genome with BLAT to infer their genomic loci. To annotate the genomic coordinates of mature snoRNA genes, we took advantage of the massive sRNA-seq data produced by the ENCODE Consortium (48). We retrieved the BAM files containing the genomic loci of the reads from 114 sRNA-seq data sets (read length of 101 nt) from the UCSC ENCODE analysis hub (http://genome.ucsc.edu/ENCODE).
To select reads that could support mature snoRNA genes, we used the following criteria: first, we required that either the sRNA-seq read covers at least 75% of a snoRNA gene or the sRNA-seq read was longer than 90 nt, for cases (especially H/ACA box snoRNAs) where the length of the snoRNA gene was presumably too long to be covered in full by the sRNA-seq reads. Second, we required that the first and last genomic positions where the sRNA-seq read mapped were at most 5 nt away from the start and end position of the annotated snoRNA gene to which the read mapped. After thus identifying sRNA-seq reads associated with individual snoRNA genes, we redefined the boundaries of the mature snoRNA forms as the positions where most of the sRNA-reads associated with the locus started or ended, respectively. For snoRNA loci with too few sRNA-seq supporting reads, we manually curated the genomic coordinates of the mature forms based on the sRNA-seq reads profile and inspection of box motifs and secondary structure (see Supplementary Dataset S1). To further validate this procedure, we examined the distance between the 5′ and 3′ ends and the C and D box motifs, respectively. We found that, as shown before (33), the 5′ end of C/D box snoRNA was located 4–5 nt upstream of the C box motif, and the 3′ end at most 5 nt downstream of the D box motif. In turn, we used this information as another indication for curating the 5′ and 3′ end coordinates of the mature snoRNAs for which the sRNA-seq data did not sufficiently or completely cover the loci.
Identification of predicted snoRNAs with supporting expression data from the ENCODE project
To uncover additional snoRNA genes that have supporting expression evidence, we first collected predictions of two computational tools, snoSeeker (49) and snoReport (50), that have been specifically designed to predict snoRNA genes. Due to the high computational demand of these tools, we restricted the search space to genomic regions that were supported by at least five reads in the combined set of sRNA-seq samples and extended these loci by 20 nt from the 5′ end and 100 nt from the 3′ end. The predictions of snoSeeker and snoReport were pooled and candidate snoRNA genes overlapping with already annotated snoRNA genes were removed. This step yielded 820,835 putative C/D box snoRNA loci and 316,076 H/ACA box snoRNA loci.
Because the sequence and structure constraints on snoRNAs appear to be weaker compared to, for example, tRNAs, we expect a higher false-positive rate of prediction for snoRNAs compared to tRNAs. Here we used the observation that C/D box snoRNAs undergo precise processing which leaves only 4–5 nt upstream of the C box, and 2–5 nt downstream of the D box (33) to further validate the C/D box snoRNA prediction. Small RNA-seq reads that mapped to C/D box snoRNA loci were considered ‘supportive’ of a snoRNA mature form if the 5′ end of the read was located 4–5 nt upstream of the inferred C box and the 3′ end of the read was located 2–5 nt downstream of the D box. For C/D box snoRNA genes with a predicted length of more than 100 nt, we could only enforce that the 5′ end is processed as expected, but we required that the sRNA-seq reads cover at least 75% of the length of the predicted snoRNA gene or are at least 90 nt in length. For H/ACA box snoRNAs, a read was labelled as supportive if the 5′ end of the read was located ±5 nt around the predicted 5′ end of the snoRNA locus, and the read either covered at least 75% of the length of the snoRNA locus or was at least 90 nt in length. 8,000 predicted C/D box snoRNAs and 7772 predicted H/ACA box snoRNAs had at least one supportive read, but only 121 and 114, respectively, remained when we required at least 1,000 supportive reads (corresponding to 0.087 TPM) in the entire data set. We chose this cut-off because more than 98% of already annotated snoRNAs in HUGO pass this cut-off. In the next step, candidate snoRNA loci were filtered for redundancy and loci overlapping with predictions obtained from deepBase, a survey of the human genome using snoStrip with known vertebrate snoRNAs as query, and GENCODE were removed. Finally, we removed candidates that overlapped with repeat annotation with more than 25% of their length and discarded those that did not have support by uniquely mapped reads. In the end, our de novo prediction yielded 17, 41 and 21 H/ACA box, C/D box and SNORD-like snoRNA loci, respectively. SNORD-like snoRNAs are non-canonical type of C/D box snoRNAs which are shorter than 50 nt in length and hence lack a functional C' and D' box. These putative snoRNAs can be found in Supplementary Dataset S1, filed as ‘de novo’.
In previous work (33), we found that core snoRNP proteins bind snoRNA-like RNAs, that are not reported in snoRNA databases. To capture these cases, we carried out a genome-wide scan for C/D box-like molecules that are supported by sRNA-seq evidence. We started from genomic regions defined by a degenerate C box (TGATGA, TGGTGA, TGATGT, TGATGC or TGTTGA) and a D box (CTGA or ATGA) separated by 10–90 nts. After applying filtering steps as done for canonical C/D box snoRNAs, we obtained 77 C/D-box like candidates that have at least 1,000 supportive reads in the sRNA-seq data, from which 38 are SNORD-like (shorter than 50 nt). These are marked as ‘C/D-box-like’ in Supplementary Dataset S1.
SnoRNA target prediction
SnoRNA target prediction was performed using following data sources: the set of human snoRNA sequences recovered in this study, human ribosomal RNAs sequences (18S (X03205), 28S (U13369 nts 7935–12969), 5.8S (U13369 nts 6623–6779)) (40,41) and sequences of spliceosomal RNAs (U1, U2, U4, U4atac, U6, U6atac, U11, U12) (51) as target RNAs. Experimentally confirmed modification sites were obtained from literature (40,41,52–58), and from a recent high throughput study (59) for pseudouridine sites and from the newly developed RimSeq method for 2′-O-methylation sites.
At first, we predicted putative targets on human rRNAs and snRNAs using RNAsnoop (60) and Plexy (61) for human H/ACA box and human C/D box snoRNAs, respectively. Precomputed RNAup structure profiles of target RNAs (62) were provided to refine interaction predictions with RNAsnoop. Additionally, we used signs of evolutionary conservation as supporting evidence for putative snoRNA–target interactions. To that aim a set of annotated homologous snoRNA sequences and their predicted interactions in deuterostomian species, which were computed with the snoRNA annotation pipeline snoStrip (63), was used. To avoid contamination with repetitive sequences we excluded snoRNA genes overlapping with regions of the UCSC-Repeat-masker track (9 January 2015) from conservation analysis. Subsequently, the Interaction Conservation Index (ICI) (64) was computed for all snoRNA–target RNA interactions.
Information about target sites was gathered with respect to three categories for each snoRNA anti-sense element. First, any previously reported target site (r). Second, the best scoring human target prediction (h1) within the set of human target predictions considering the minimum free energy of the snoRNA–target RNA interaction duplex. And third, the best scoring conserved target prediction (c1) within the set of conserved interactions evaluated by the Interaction Conservation Index. The final assignment of an snoRNA anti-sense element to a target site was based on following rules:
h1, if the best scoring conserved target is the best scoring human target (c1 = h1)
r, if the reported target is the best scoring human target (r = h1)
c1, if the reported target is not the best scoring human target (r ≠ h1); and a human target prediction (hi) exists within the best scoring conserved target predictions (hi = c1)
h1, if no human target prediction exists within the best scoring conserved target predictions (hi ≠ c1).
Selected interactions are accepted, if the interaction is well conserved in deuterostomes with an ICI > 1.0 for box C/D box snoRNAs and an ICI > 0.8 for H/ACA box snoRNAs (see (64) for information on these thresholds). A predicted interaction is classified as highly confident if the resulting modification overlaps a confirmed modified position, that has been identified by a high-throughput approach, or has been reported in literature.
RimSeq library preparation
To identify 2′-O-methylated residues transcriptome-wide we adapted a well-established, low-throughput reverse transcriptase-based protocol (65), which is usually coupled with polyacrylamide gel analysis, and modified it to a high-throughput sequencing protocol. The method is based on the observation that cDNA synthesis is noticeably impaired in the presence of a 2′-O-methyl when deoxynucleotide triphosphate fragments (dNTPs) are limiting (65,66), giving rise to a characteristic pattern of gel banding immediately preceding the 2′-O-methyls, with strong bands at low dNTP concentrations (0.004 mM) (66), becoming weaker with increasing concentrations of dNTPs. These stoppages, which correspond to the position of a 2′-O-methylation site, will generate read ends when RNA fragments are reverse-transcribed under different dNTP concentrations, ligated to adapters and sequenced. 2′-O-methyl positions can be subsequently identified by calculating the ratio of reads starting at given position (5′ ends) to the reads covering it (readthrough reads + 5′ ends) and comparing this ratio to the control. The exact procedure can be found in Supplementary Text S1.
Analysis of the expression profiles of known snoRNA genes and snoRNA-derived fragments based on ENCODE
The expression level of a given snoRNA in a sample was calculated based on the total number of reads (uniquely and multi-mapping) from that sample that overlapped with the snoRNA locus. The normalization of read counts was done relative to the total number of reads obtained in the sample. The ENCODE project generated sRNA-seq samples from a range of cell types, both normal and malignant, as well as from distinct sub-cellular compartments (‘Cell’, ‘Cytosol’, ‘Chromatin’, ‘Nucleus’ and ‘Nucleolus’). Furthermore, to capture various types of small RNAs, the RNA was subjected to various treatments (tobacco acid phosphatase (‘TAP’) to remove cap structures, calf intestinal phosphatase and TAP (‘CIP-TAP’) to further remove 5′ and 3′ phosphates, as well as left untreated ‘No treatment’)). Unsurprisingly, hierarchical clustering of expression levels of snoRNA in the ENCODE samples revealed a strong dependency on the cellular department and the library preparation procedure (Supplementary Figure S1). Consequently, we restricted our analysis of snoRNA expression to samples that were obtained from the cellular compartment ‘cell’ with the TAP-only treatment, as these two factors covered the largest variety of cell types.
SnoRNAs that were more than 80% identical over their entire length to each other were grouped into a snoRNA ‘family’ (see Supplementary Dataset S2 for a list of snoRNAs and their corresponding cluster representatives). The expression level of a cluster representative was defined as the average expression level of all snoRNAs associated with that cluster. When replicates were available, we further averaged expression over replicates. The specificity of expression and the specificity of processing of individual snoRNAs was calculated as follows. We first computed the relative frequency of each snoRNA in the pool of snoRNAs in a given sample. Next, the specificity score S defined as
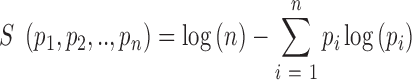 |
was calculated where pi is the normalized frequency of the snoRNA in sample i. The specificity score is maximal when the snoRNA is expressed in a single sample and minimal when the relative frequency of the snoRNA is the same across all samples (Supplementary Figure S2).
To directly compare snoRNA expression between normal and malignant cells, we averaged the snoRNA expression separately over normal and malignant cell types. The ratio of these quantities gives the fold-change of expression between normal and malignant cells.
Expression profiling of snoRNA-derived fragments
To determine whether processed fragments are generated in a cell type-specific manner, we first separated the reads into those that correspond to the mature snoRNA and to shorter processed products. Because the sRNA-seq samples should in principle contain only full-length RNAs and based on the length distribution of snoRNAs (Supplementary Figure S3), we chose a maximum length of 40 nt for a read to be considered as corresponding to a processed RNA. This is consistent with the length of snoRNA-derived fragments that was reported before (33,37,67,68). Next, we calculated the proportion of processed reads among all reads associated with the snoRNA. Finally, we calculated a specificity score of snoRNA expression or of processing across tissues as described above for the specificity of snoRNA expression (Supplementary Dataset S3 and Supplementary Figure S4).
RESULTS
Curated annotation of the 5′ and 3′ ends of snoRNA genes
In contrast to other types of molecules such as mRNAs and microRNAs, relatively few studies attempted to sequence the full complement of mature human snoRNAs. Thus, the annotation of human snoRNA genes frequently started from computational predictions. Especially in the case of C/D box snoRNAs a consistent procedure for defining the 5′ and 3′ ends of their mature forms is lacking, and different pragmatic definitions such as the longest terminal stem, the longest evolutionarily conserved terminal stem, or the experimentally determined ends were frequently used. However, the data that we obtained in a recent study indicated that C/D box snoRNAs undergo uniform trimming at both the 5′ and the 3′ end (33), irrespective of the length of the terminal stem. Here, we use this observation to provide a complete catalog of curated mature snoRNA 5′ and 3′ ends based on small RNA sequencing data sets.
We first retrieved the 289 C/D box snoRNA, 136 H/ACA box snoRNA and 27 scaRNA genes that were annotated by the HUGO Gene Nomenclature Committee (HGNC) at the time when our study was initiated and mapped them to the human genome (hg19 assembly version from the University of California, Santa Cruz). We further obtained the genomic coordinates of small RNA sequencing reads from 114 data sets that were generated by the ENCODE consortium (48). Intersecting the loci of sequenced small RNAs with those of the HGNC snoRNAs, we identified, for each snoRNA gene, the 5′ and 3′ ends that were most represented among the small RNA sequencing (sRNA-seq) reads (see Materials and Methods for details). We found that these data confirmed the processing pattern that we described previously (33,69), namely that the 5′ end of the mature C/D box snoRNAs is located 4–5 nt upstream of the C box motif and the 3′ end is located up to 5 nt downstream of the D box motif (see Supplementary Figure S5). The curated loci of the mature HGNC snoRNAs are compiled in Supplementary Dataset S1 and Supplementary Dataset S4. For some snoRNAs e.g. SCARNA21, SNORD11B or SNORA58 the sequence inferred from the small RNA sequencing data differed considerably from the sequence defined by the HGNC. Other snoRNAs for which the curated coordinates differed significantly from their known annotation are SNORD81, SNORD49B, SNORD126, SNORD125, SNORD123, SNORD121A, SNORD11B, SNORD127, SNORD58C, SNORD12B, SNORD111B, SNORD105B, SNORD124, SNORD90, SNORD105 and SNORD70. Supplementary Dataset S5 contains visualizations of snoRNA loci including the HGNC sequence, the sRNA-seq read profile along these loci and the 5′ and 3′ ends that were inferred based on the sRNA-seq data. Inspection of sRNA-seq read profiles of three snoRNAs that were annotated in HGNC as H/ACA box snoRNAs (SNORA85, SNORA96, SNORA97) revealed that they are in fact C/D box snoRNAs (named ZL68, ZL5 and ZL6 in (33)) with slightly altered positions. These revised snoRNA sequences are now assigned the gene symbols SNORD142, SNORD143 and SNORD144.
An updated catalog of human snoRNA genes
To further update the human snoRNA catalog, we integrated data from several sources including a de novo genome-wide search (outlined in detail in Figure 2). Specifically, we collected snoRNAs from RFAM-based predictions that were generated by the GENCODE consortium (70), from deepbase (71), and from a snoStrip (63) dataset in deuterostomes (64) (see Materials and Methods for details). Additionally, we performed a genome-wide de novo screen by the workflow summarized in Figure 2. Due to the high computational demand of snoRNA gene finding programs, we restricted our analysis to genomic regions that showed signs of expression in the sRNA-seq data set generated by the ENCODE consortium. Finally, we used the snoReport (50) and snoSeeker (49) software to screen the extracted genomic regions for potential snoRNA genes. Additionally, we implemented a search algorithm that screens for potential C/D box-like snoRNA genes (33) (see Materials and Methods for a detailed description). Due to the vast number of potential snoRNA candidates collected from all these sources, we consolidated these initial candidates to a non-redundant set of putative snoRNA loci and excluded those that overlapped with repeat-annotated genomic regions. Furthermore, we defined a set of strict rules to identify snoRNA candidates whose expression as mature forms was strongly supported by the sRNA-seq data (see Materials and Methods). Finally, we screened and added snoRNAs from recently published literature (24,33,39,72). This analysis yielded more than 160 canonical human snoRNAs that are currently not covered by the human gene annotation (Table 1 and Supplementary Dataset S1). In order to distinguish candidates which have relatively close homologs among the already known snoRNAs, we used the Infernal software and RFAM sequence-structure models (73,74) to assign each snoRNA to the family with the closest homology and a P-value lower than 10−5. Table 1 summarizes the results of these analyses. Finally, we applied at the HGNC for gene names for those snoRNA candidates that showed evolutionary conservation in hominoids and beyond, contained all expected sequence motifs, were found to be expressed as full-length snoRNAs in human and that folded into a canonical structure (H box, ACA box and hairpin-hinge-hairpin-tail structure for H/ACA box snoRNAs, and C box, D box, the typical kink-turn formed by these boxes, and a terminal stem of at least 2 bps for C/D box snoRNAs). For most of the novel snoRNAs, conservation analysis performed with snoStrip (63) could only recover homologs in primates (93 C/D and 8 H/ACA). For 13 C/D box snoRNAs and 7 H/ACA box snoRNAs no homologs at all could be retrieved. Reliably determining if these snoRNAs are indeed evolutionary new inventions, specific to human and primates is beyond the current methodology.
Figure 2.

Outline of the snoRNA annotation strategy used in this study. We combined de novo search on ENCODE sRNA-seq expressed regions with snoRNA genes and predictions from various databases. All predicted candidate sequences were checked for a supportive sRNA-seq read pattern to identify high confidence, currently not annotated snoRNA genes. Finally, snoRNAs from all sources were merged and filtered for redundancy to establish a comprehensive map of human snoRNA loci.
Table 1. Overview of known and novel snoRNAs analyzed in this study.
| Known snoRNAs | Novel snoRNAs | ||||||
|---|---|---|---|---|---|---|---|
| Type of snoRNA | Total count | Currently recognized by the HGNC | Changes/Additions requested at the HGNC | Not sufficiently supported to be added to HGNC | Total count | Additions requested at the HGNC | Not sufficiently supported to be added to HGNC |
| H/ACA box | 179 | 136 | +39/-3 | 4 | 11 | 9 | 2 |
| AluACA | 348 | 0 | 0 | 348 | 6 | 0 | 6 |
| C/D box | 376 | 295 | 48 (+4) | 29 | 41 | 14 (+21) | 6 |
| C/D-box like | 18 | 0 | 0 (+8) | 10 | 98 | 0 (+98) | 0 |
| scaRNAs | 29 | 27 | 2 | 0 | 0 | 0 | 0 |
| Telomerase | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| sno-lnc RNAs | 11 | 0 | 0 | 11 | 0 | 0 | 0 |
‘Known snoRNAs’ are either annotated by the HGNC or extracted from recently published literature (24,33,39,64,72), and/or the public databases GENCODE and deepBase. ‘Novel snoRNAs’ are those genes that do not overlap any of the known ones, naturally. In brackets, we provide counts of putative snoRNAs that give at least rise to processed transcripts, but only partially fulfil our criteria for applying at the HNGC for gene names (new HGNC prefix pending).
An updated catalog of human snoRNA target interactions
The primary function of snoRNAs is to guide the modification of specific sites in ribosomal and spliceosomal RNAs. There are, however, some well documented snoRNAs with non-canonical function, like SNORD115 that has been reported to regulate alternative splicing (75) or SNORD116 that forms the ends of longer non-coding RNAs (36). To provide an up-to-date annotation of the targets in our snoRNA catalog, we here combined target predictions based on state-of-the-art computational methods (31) with experimental data on snoRNA-guided RNA modifications. The computational target prediction follows three main steps. First, RNAsnoop (60) and Plexy (61) are used to predict human targets based on primary sequence features, secondary structure of the snoRNA, the accessibility of the target region, and the predicted minimum free energy of the snoRNA–target duplex. In a second step the evolutionary conservation of the predicted interaction within vertebrates is evaluated using the Interaction Conservation Index (ICI) (64)). In brief, the ICI combines stability of an individual interaction between snoRNA and target RNA within a single species with the range of conservation of the equivalent interaction among species for which a homologous snoRNA exists. Roughly, an ICI score > 1 can be interpreted as the specific interaction being better than alternative predictions in all species where a snoRNA homolog is present. We also used a coarse-grained encoding of the conservation termed ‘levelC’ that indicates the depth of conservation in the phylogenetic tree of eukaryotes. Lastly, we identified the highest-confidence interactions among the predicted interactions as those interactions, for which a corresponding snoRNA-guided RNA modification has also been reported in human. Data on snoRNA-guided modifications was gathered from snoRNAbase and available literature, or from very recently conducted experiments that were designed to identify RNA modifications in high-throughput. In particular, we obtained data on pseudouridine modifications to validate predicted interactions of H/ACA box snoRNAs from two studies (59,76). For 2′-O-methylation sites, however, no such high-throughput data exists. To fill this gap, we developed a novel method termed RimSeq, which ports the principles of 2′-O-methylation site identification used in primer extension assays (77–79) to a high-throughput approach using next generation sequencing. A detailed description and evaluation of the RimSeq procedure is outlined in Supplementary Text S1 with inferred modifications sites being displayed in Supplementary Dataset S6. Using the computational predictions and the data obtained from high-throughput experiments and modifications reported in literature we could identify ten novel high confidence interactions between snoRNAs and target molecules. For two target sites whose methylation has been reported to be guided by a known snoRNA we predicted an additional guiding snoRNA: the D'-box ASE of SNORD136 for 18S-683, and snoID_0337 for 18S-1326. Additionally, the methylations that were experimentally identified at 18S-1606 and 18S-1410 could be assigned to previously considered orphan snoRNAs SNORD73A/B and to novel snoRNA snoID_0340, respectively. Guiding H/ACA box snoRNAs could be assigned to two previously mapped pseudouridylation sites, 18S-681 and 28S-4266. Concerning the pseudouridylation sites that emerged from high-throughput data, we could predict guiding snoRNAs in three (18S-1046, 18S-1232, and 28S-2619) out of the four cases; we could not identify a guiding snoRNA for the pseudouridine at position 1177 in human 18S rRNA reported by Carlilie et al. (59). Details of this analysis are summarized in Table 2 (see Supplementary Dataset S6 for a full listing).
Table 2. List of predicted interactions between nucleotides whose modification has been confirmed experimentally and the corresponding guide snoRNAs.
| Modified nucleotide | Currently assigned guide | Support by HTP | Newly predicted guide | Location of ASE | ICI | Conservation level of the snoRNA |
|---|---|---|---|---|---|---|
| 18S-683 | SNORD19 | - | SNORD136 | D' | 1.22 | Eutherians |
| 18S-1326 | SNORD33 | + | snoID_0337 | D' | 1.84 | Primates |
| 18S-1410 | NA | + | snoID_0340 | D | 1.33 | Primates |
| 18S-1606 | NA | + | SNORD73A/B | D' | 1.17 | Tetrapodes and Teleostes |
| 18S-681 | unknown | + | SNORA14A/B | 5′ stem | 0.84 | Amniotes |
| 18S-681 | unknown | + | SNORA55 | 3′ stem | 1.2 | Tetrapodes |
| 28S-4266 | unknown | - | SNORA78 | 5′ stem | 0.92 | Tetrapodes and Teleostes |
| 18S-1046 | NA | + | SNORA57 | 3′ stem | 0.9 | Deuterostomes |
| 18S-1232 | NA | + | SNORA70A/B/E/11/14 | 5′ stem | 1.18 | Vertebrates |
| 28S-2619 | NA | + | SNORA38A/B | 5′-stem | 0.84 | Therians |
The modification data originated either from snoRNAbase (https://www-snorna.biotoul.fr/), in which case guide snoRNAs were sometimes already assigned, or from the high-throughput (HTP) approaches, in which case the guiding snoRNAs were not known so far. We further provide the location of the ASE which is predicted to take part in the interaction, the Interaction Conservation Index (ICI) of the interaction and the conservation level of the predicted snoRNA guide.
For C/D box snoRNA target prediction we excluded the SNORD3 and SNORD13 snoRNA families that have established non-canonical functions in pre-rRNA cleavage (80,81). Hence, we obtained a total of 393 snoRNA sequences, of which 275 are canonical C/D box snoRNAs and 118 are members of the multi-copy (mc) gene families SNORD113, SNORD114, SNORD115, and SNORD116. For the majority (∼83%) of these sequences we could annotate both a D and a D' box sequence motif (Table 3). In contrast, only a few (∼21%) of the C/D box-like snoRNAs appear to possess both D and D' boxes. In many cases the D' box could not be reliably annotated either due to the short length of these snoRNA like genes or the lack of evolutionary conservation or sequence motif signals.
Table 3. Overview of snoRNA considered for target prediction.
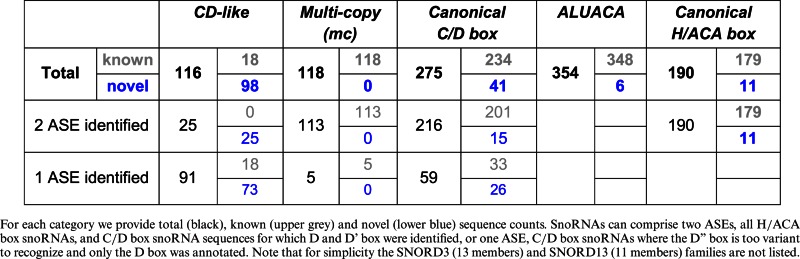
|
In total, we applied target prediction methods to 863 = (25 + 113 + 216) × 2 + (91 + 5 + 59) anti-sense elements (ASEs) covering all cataloged C/D box and C/D box-like snoRNAs. The snoRNA target prediction results are listed in detail in Supplementary Dataset S1. Table 2 depicts high-confidence interactions, for which additional experimental evidence of RNA modification is available. Summarizing results obtained from target prediction and reported interactions, we can currently associate more than two thirds (∼70%) of the C/D box snoRNAs with a specific rRNA or snRNA target. However, 118 C/D box snoRNA genes remain classified as orphan. Interestingly, the vast majority of these (91) were found to have two ASEs. Here, the question remains if these snoRNAs interact with their target RNAs in a way that fails to be recognized by our computational target prediction methods or if these snoRNAs execute entirely biologically different functions than guiding modifications. Excluding the multi-copy (mc) snoRNA genes, 48 C/D box snoRNAs with canonical features remain without a predicted or known target in rRNA or snRNA (Figure 3A). Of these, SNORD97 is reported as enriched in chromatin-associated RNAs (caRNAs) (82). Because a detailed analysis of the mc snoRNA families did not reveal convincing target predictions, we excluded these families from further analyses. Although most of the known and novel C/D box snoRNAs have both D and D' boxes, only a minority of those indeed interact with targets at both anti-sense elements (Figure 3A).
Figure 3.

Distribution of orphans, single guides (sg), and double guides (dg) among known and novel snoRNAs based on our target predictions. (A) Of the 275 canonical C/D box snoRNAs, 48 are orphan, 38 are double guides and 189 are single guides. Of the latter, 93 (56 D'+D box, +37 no D' box) have a functional ASE adjacent to the D-box and 96 adjacent to the D'-box. (B) Of 190 canonical H/ACA box snoRNA sequences 30 remain orphan (of which SNORA73A/B have a non-canonical role in 18S rRNA maturation (83)), 89 are double guides and 71 are single guides. Of the latter 45 have a functional ASE in the 5′ stem, and 26 in the 3′ stem.
For H/ACA box snoRNA target prediction, we only used canonical genes and excluded those sequences encoded within Alu repeats (ALUACAs), for which evolutionary conservation information cannot be reliably obtained. A canonical H/ACA box snoRNA forms a double stem-loop structure each possessing a respective ASE in its interior loop. Therefore, a total of 380 (2 x 190) ASEs (Table 3) were considered for target prediction. In total, our analysis of reported and predicted targets associated ∼85% of the H/ACA box snoRNA sequences with at least one target Uracil in an rRNA or snRNA. About 55% of the guiding snoRNAs have targets for both ASEs, while the remaining appear to guide pseudouridylation at one site only. In the majority of these cases, the target site is predicted to interact with the 5′ stem (Figure 3B). Among the 30 snoRNAs for which no canonical targets were reported or predicted is SNORA73A/B. This is in agreement with the reported non-canonical interaction of the yeast homolog snR30 with the 18S RNA (83). The conserved potential for base-pairing of these molecules suggests that the mechanism is well conserved to vertebrates. Furthermore, there is evidence that SNORA73A functions as a putative regulator of chromatin function (82).
Finally, we summarized the evidence and features used to infer snoRNA–target RNA interaction sites as heatmaps depicted in Figure 4 (see Supplementary Figure S6 for a high resolution version). Blue and red colors indicate low and high evidence for the interaction, respectively. It is apparent that after our analysis only a small fraction of snoRNAs remains orphan, which is indicated by the blue color in the column ‘reported’ and by the low value of the Interaction Conservation Index (ICI, see Materials and Methods) for both ASEs. Several interactions, mainly for the newly identified snoRNAs, seem to be primate specific (column ‘levelC’: blue and column ‘ICI’: white/red). Interestingly, C/D box snoRNAs seem to have a single-guide tendency (column ‘ICI’ is white/red for either D or D’ box, but relatively rarely for both). For the 59 snoRNAs for which we could not identify a D' box, the classification as single-, double-guide or orphan snoRNA remains preliminary (grey cells on D' box side). Although the majority of C/D box snoRNAs encode both a D and a D’ box and have associated ASEs, for only ∼17% we predicted high-scoring interactions for both ASEs. Among single-guide C/D box snoRNAs, the predicted interaction preferentially involves the D’ box-associated ASE (96 cases vs. 56 with guiding at the D box-associated ASE). This is in strong contrast to the pattern of evolutionary conservation, since the D box generally shows stronger conservation. H/ACA box snoRNAs, however, are predominantly (56%) double guiding. For those with one guiding ASE, the ASE is preferentially located in the 5′ stem (45 of cases compared to 26 that have the single guiding ASE in the 3′ stem).
Figure 4.

Visualization of predicted snoRNA–target RNA interactions. Each row displays binding properties of one snoRNA sequence. The columns represent: ICI: Interaction Conservation Index of selected target (scaled to [−1,1]); rank: rank of selected interaction within set of predictions for the snoRNA ASE in the human genome (1: respective interaction is best for this ASE, else:1/(log of rank) (scaled)); levelC: level of conservation of the interaction among deuterostomes (1: primates, 2: eutherians, 3: therians, 4: mammals, 5: amniotes, 6: tetrapodes, 7: tetrapodes and teleosts, 8: vertebrates, 9: deuterostomes, scaled to [−1,1], gray denotes human-specific); MFE: minimum free energy (MFE) of the interaction (scaled to [-1,1]); reported: target reported in the literature (1: yes, −1: no). For each cell, the value is also illustrated by the position of the vertical black line relative to the 0-value line, located in the middle of the cells. Supplementary Figure S6 provides the heatmaps in higher resolution with snoRNA names next to the rows. (A) C/D box snoRNAs (not including multi-copy and C/D-like snoRNAs). The left set of columns refer to the ASE upstream of D box; the right set of columns the ASE adjacent to D' box (a grey line if the D' box and the particular ASE could not be annotated). (B) H/ACA box snoRNAs (not including the ALUACA class). The left set of columns refer to the 5′ stem-associated ASE, whereas the right set of columns refer to the 3′ stem-associated ASE.
The human scaRNAs can be grouped into tandem C/D box (4), tandem H/ACA box (1), hybrids of C/D and H/ACA boxes (5), canonical C/D box (2) and canonical H/ACA box (17). Thus, the pool of scaRNAs can potentially interact with target RNAs at 78 = (4 + 1 + 5) × 4 + (2 + 17) × 2 sites. Due to their intricate structure, we could not reliably annotate all potential ASEs for six scaRNAs, leaving a total of 71 ASEs that were subjected to further analysis. An evolutionarily conserved target could be recovered for 43 cases including seven sites that are newly predicted. In particular, the elongated isoform of SCARNA21, an H/ACA box snoRNA embedded in a C/D box snoRNA, was found to harbor three additional functional ASEs (Figure 5 and Supplementary Text S2). Most intriguingly, the snRNA U12 residue targeted by the 5′ ASE of the H/ACA domain of this scaRNA is directly adjacent to the newly predicted target of D' box-associated ASE. The 3′ ASE of the H/ACA part is predicted to guide a modification in the U6atac snRNA, which is part of the minor spliceosome, as is the U12 snRNA. Thus, our results suggest an important role of SCARNA21 in the maturation of snRNAs of the minor spliceosome.
Figure 5.

Structure of the elongated SCARNA21. The snoRNA-characteristic sequence motifs are enclosed in a black frame. The C/D box domain folds into the characteristic terminal stem and the obligatory kink-turn motif. The H/ACA domain forms the typical double-hairpin structure. Predicted target sites for the ASEs are displayed in the grey boxes. From 5′ to 3′, the predicted functions are: guide1: U12–17, guide2l&r: U12–18 (102), guide3l&r: U6atac-83, guide4: 28S-4426. See Supplementary Text S2 for details about these interactions. The figure was produced with R2R (103).
Expression profiling of human snoRNAs
The plasticity of snoRNA expression across cell types has been relatively poorly studied, although changes in snoRNA expression have been observed in cancers (84). Due to the diverse set of both normal and malignant cell types profiled by the ENCODE consortium, this data set constitutes an excellent source to study cell type specific expression of snoRNAs in detail. Our analysis revealed that the pool of both H/ACA box and the C/D box snoRNAs is dominated by a few abundantly expressed snoRNAs (Figure 6A). As an illustration, 21 C/D box and 18 H/ACA box snoRNAs account for more than 80% of sRNA-seq reads captured for the respective snoRNA class. Of these abundantly expressed snoRNAs, only two of the C/D box family (SNORD83A and SNORD64) and only two of the H/ACA family (SNORA11 and SNORA51) lack well confirmed target sites on ribosomal RNAs and snRNAs. However, we previously predicted that SNORD83A targets 18S-468 (64), a site also known to be modified, whereas here we further predicted that SNORD64 targets U1-53. A conserved interaction between SNORA51 and 28S-1849 was predicted in (64), and SNORA11 appears to target 18S-1350. (see also Supplementary Dataset S1). The abundantly expressed snoRNAs thus appear to target largely rRNAs, and may therefore be essential for ribosome biogenesis. Consistently, these snoRNAs also show little variation in expression across cell types (Figure 6B denoted by red stars; high resolution versions of these figures including gene names can be found in Supplementary Figure S2). Some snoRNAs do exhibit cell type-specific, particularly neuronal expression. Among them are the neuron-specific orphan SNORD115 and SNORD116 families (34,75,85) as well as snoRNAs with canonical ribosomal targets such as SNORD100 and SNORD33. The H/ACA box SNORA35 (86), which has the strongest cell type specificity among the H/ACA box snoRNAs, is predicted to target 18S-566 through the 5′ ASE and U7-7 through the 3′ ASE. A comprehensive list of snoRNAs that show cell type specific expression can be found in Table 4.
Figure 6.

Expression profiling of snoRNA genes in ENCODE sRNA-seq data. (A) The pool of human snoRNA genes is dominated by a few abundantly expressed snoRNA genes. (B) Evaluation of tissue specific expression of snoRNA genes. The top panel shows values for C/D box snoRNAs, while the bottom panel does for H/ACA box snoRNAs. The higher the specificity score is the more biased is the expression to a specific tissue or cell type. MFOCP is an acronym for melanocytes, fibroblasts, osteoblasts, chondrocytes and placental tissue. The red stars mark the 20 most highly expressed C/D box (upper panel) and H/ACA box (lower panel) snoRNAs in the entire data set (further details in Supplementary Figure S2).
Table 4. Summary of snoRNAs with a highly cell type-specific expression (specificity score > 0.6, see Materials and Methods).
| SnoRNA name | Sub-class of cells that show strongly biased expression | Associated ENCODE samples |
|---|---|---|
| SNORD115 family, SNORD116 family, SNORD100, SNORD109, SNORD107, SNORD29 | Neurons | H1_neurons |
| SNORD33, SNORD81, SNORD105, SNORD68, SNORD11, SNORD36A, SNORD102, SNORD111, SNORD12B, SNORD30, SNORD69, SNORD32A (2), SNORD12, SNORD22, SNORD50A, SNORD11B, SNORD55, SNORD105B | Neurons and lymphoblastoid cells | H1_neurons, GM12878 |
| SNORD11B | Neurons and pericytes | H1_neurons, HPC_PL |
| SNORD112 | MFOCP | HCH |
| SNORD113-8 (7) | MFOCP | hMSC-BM |
| SNORD114-22 (28) | MFOCP | HPIEpC |
| SNORD7 | Neurons and Endothelial cells | H1_neurons, HAoEC |
| SNORD46, SNORD42A | Mammary gland and lymphoblastoid cells | HMEpC and GM12878 |
| SNORD125, SNORD85, SNORD91A | hematopoietic, neurons and lymphoblastoid cells | CD34+, H1_neuron, GM12878 |
| SNORA35, SNORA36B (3) | Neurons | H1_neurons |
| SNORA54, SNORA22, SNORA16A (2), SNORA48, SNORA63, SNORA14B(2), SNORA5A | Neurons and lymphoblastoid cells | H1_neurons, GM12878 |
| SNORA47 | Neurons, hematopoietic and lymphoblastoid cells | H1_neurons, CD34+, GM12878 |
| SNORA55 | Neurons and pericytes | H1_neurons, HPC_PL |
MFOCP stands for melanocytes, fibroblasts, osteoblasts, chondrocytes and placental tissue.
Hierarchical clustering of a subset of sRNA-seq samples that have been generated from decapped (tobacco acid phosphatase (TAP)-treated) RNAs isolated from whole cells (Supplementary Figure S7), revealed a striking separation of normal and malignant cell lines. Several snoRNAs seem to be differentially expressed in all cancer cell lines compared to cells of non-malignant origin, consistent with the results of prior studies that identified snoRNAs as putative cancer biomarkers (87–91). Our results also parallel a recent finding of increased expression of a specific set of tRNAs in cancers, with possible consequences on translation in these cells (92). As an entry point into investigations into cancer-associated snoRNAs we compiled the list of snoRNAs with the most significant differential expression in cancer cell lines (see Supplementary Tables S1, S2A and S2B).
Among non-malignant cells and tissues, we found that cells of neuronal origin form one cluster, due to a relatively large number of neuron-specific snoRNAs. Other cell types show more similar profiles, although the mammary gland and hematopoietic cell types tend to cluster closer together, as do the muscle and adipose tissue. The remaining cell types (melanocytes, fibroblasts, osteoblasts, chondrocytes and placental tissue) form one big cluster with no clear boundaries (Supplementary Figures S7 and S8).
Limited evidence of tissue-specificity of snoRNA-derived fragments
Several studies described snoRNA-derived fragments and suggested that, with some exceptions, the pattern of processing is conserved across snoRNAs and tissues (37,67). Furthermore, various groups proposed that snoRNA-derived fragments may have non-canonical functions (37,38,68,75,93–97). We asked whether the relative proportion of short (less than 40 nt) snoRNA-derived fragments differs between snoRNAs and whether it differs across cell types (see Materials and Methods) for a given snoRNA. We found that the majority of C/D box snoRNAs (75%) are found predominantly as mature forms in the data. That is, the proportion of processing products is <50% of the reads associated with the snoRNA. The cumulative distribution of this proportion is shown in Supplementary Figure S9. Furthermore, we found only minor differences in this proportion across the tissues where the snoRNAs are expressed. Notable exceptions are the SNORD115, 116, 113 and 114 families. A group of snoRNA comprising SNORD50, SNORD19, SNORD32B, SNORD123, SNORD111, SNORD72, SNORD93, SNORD23 and SNORD85 gives rise to over 90% of the processed fragments. However, we did not find evidence that the frequency of shorter forms is cell type-specific (Supplementary Dataset S3 and Supplementary Figure S4).
DISCUSSION
Among the small RNAs, snoRNAs have a relatively long history, going back to the late 1960s (98), and several hundred snoRNA genes have been catalogued. snoRNA-LBME-db (https://www-snorna.biotoul.fr/) is an outstanding resource in this domain (41), providing detailed information to more than 361 snoRNA genes and their target RNAs. This database has, however, not been updated lately and is missing out on the technological advances of deep sequencing. Indeed, the wide availability of deep sequencing technologies has prompted thorough investigations into the processing and expression patterns of all types of RNA molecules including snoRNAs (33,69), and the improved understanding of the biogenesis of these molecules, in turn, allows to build more accurate identification protocols when scanning large-scale data sets. A recent controversy concerning the criteria that were used in identifying novel snoRNA genes (47), demonstrates again that only a thorough, well defined strategy can be used to map snoRNA genes on a genome-wide scale. To that aim, we here combined known sequence and structure properties of snoRNAs, as well as recently described characteristic patterns of processing and expression evidence to generate an updated catalog of human C/D box and H/ACA box snoRNAs.
Our analysis suggests that although many genomic regions may give rise to potential RNA molecules that are processed by the snoRNA-processing machinery and may even be bound by the core proteins of the snoRNP complex (33,69), it is only about 700 snoRNAs that are expressed at a significant level. Even more challenging than the identification of novel snoRNA genes is the task of finding the target RNAs. The main reason is that the interaction typically involves only a short region making it necessary to take additional signs of evidence such as evolutionary conservation into consideration. On the other hand, making this strategy fall short on species-specific modifications that have been reported as well (99,100). In this study, we extended the snoRNA interaction network in human being able to suggest functions for many of the novel snoRNAs as well as assign snoRNA guides to three previously reported ‘orphan’ modifications and five modifications identified by high-throughput methods during this study. In total, we were able to reduce the percentage of reported orphan snoRNAs from ∼40% to ∼20% compared to data currently listed in snoRNA-LBME-db. Our thorough target prediction strategy could, however, not identify reliable targets on ribosomal RNAs and snRNAs for the multicopy snoRNA families SNORD113, SNORD114, SNORD115 and SNORD116, which once more supports the hypothesis that these snoRNAs act in a non-canonical manner. Among canonical, evolutionarily conserved snoRNAs we currently annotate still 76 as orphan. Clearly, high throughput protocols such as RimSeq and Ψ-seq applied to RNA extracted from various tissues have the potential to uncover not yet recognized modification sites and further reduce the list of orphan snoRNAs. How many of the orphan snoRNAs are to execute non-canonical functions remains difficult to answer and will in most cases require detailed experiments for each snoRNA in question.
The C-D'-C'-D box architecture of C/D box snoRNAs seems to be crucial for correct formation and function of the snoRNP complex (101) equipping each C/D box snoRNA with two potential guide regions for modification of other RNAs. Our analysis clearly revealed that C/D box snoRNAs that have a predicted or reported guide for both ASEs are only a minority constituting about 15% of all catalogued snoRNAs. Among the snoRNAs with a single target, the D' box ASE is surprisingly preferred over the ASE located at the generally more conserved D box. The underlying reason for this observation is not clear, but it might be that the ASE at the D' box is catalytically more active. In contrast to C/D box snoRNAs most H/ACA box snoRNAs function as double guides. This in accordance with higher constraints on the sequences through the need of structure formation, resulting in higher evolutionary conservation of the sequences.
Finally, our analysis paints a new picture of the plasticity of cell or tissue specific expression of snoRNAs. Although it has been long known that neurons specifically express a large number of snoRNAs, we were also able to identify several snoRNAs that show specific expression in cells other than neurons. Especially, there is a striking difference in snoRNA expression between normal and malignant cells. The big question here is if the changes in snoRNA expression are reflected in the processing of the target molecules such as rRNAs and whether this has a consequence for mRNA translation. Our study facilitates new avenues into this direction by providing a carefully curated catalog of snoRNAs and their associated snRNA and rRNA modifications that serves as a basis for any study on this topic.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available here.
FUNDING
Swiss National Science Foundation (SNF) [31003A_147013 to H.J., M.Z.]; DFG-funded Collaborative Research Center ‘Obesity Mechanisms’ [CRC1052 to S.K.]; Marie Curie Initial Training Network, RNPnet [289007 to R.G.] from the European Commission. Funding for open access charge: SNF.
Conflict of interest statement. None declared.
REFERENCES
- 1.Marz M., Gruber A.R., Höner Zu Siederdissen C., Amman F., Badelt S., Bartschat S., Bernhart S.H., Beyer W., Kehr S., Lorenz R., et al. Animal snoRNAs and scaRNAs with exceptional structures. RNA Biol. 2011;8:938–946. doi: 10.4161/rna.8.6.16603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Decatur W.A., Fournier M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002;27:344–351. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- 3.Darzacq X., Jády B.E., Verheggen C., Kiss A.M., Bertrand E., Kiss T. Cajal body-specific small nuclear RNAs: a novel class of 2′-O-methylation and pseudouridylation guide RNAs. EMBO J. 2002;21:2746–2756. doi: 10.1093/emboj/21.11.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.d'Orval B.C., Bortolin M.-L., Gaspin C., Bachellerie J.-P. Box C/D RNA guides for the ribose methylation of archaeal tRNAs. The tRNATrp intron guides the formation of two ribose-methylated nucleosides in the mature tRNATrp. Nucleic Acids Res. 2001;29:4518–4529. doi: 10.1093/nar/29.22.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109:145–148. doi: 10.1016/s0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]
- 6.Matera A.G., Terns R.M., Terns M.P. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 7.Bratkovič T., Rogelj B. Biology and applications of small nucleolar RNAs. Cell. Mol. Life Sci. 2011;68:3843–3851. doi: 10.1007/s00018-011-0762-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tollervey D., Kiss T. Function and synthesis of small nucleolar RNAs. Curr. Opin. Cell Biol. 1997;9:337–342. doi: 10.1016/s0955-0674(97)80005-1. [DOI] [PubMed] [Google Scholar]
- 9.Darzacq X., Kiss T. Processing of intron-encoded box C/D small nucleolar RNAs lacking a 5′, 3′-terminal stem structure. Mol. Cell. Biol. 2000;20:4522–4531. doi: 10.1128/mcb.20.13.4522-4531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiss T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001;20:3617–3622. doi: 10.1093/emboj/20.14.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKeegan K.S., Debieux C.M., Boulon S., Bertrand E., Watkins N.J. A Dynamic Scaffold of Pre-snoRNP Factors Facilitates Human Box C/D snoRNP Assembly. Mol. Cell. Biol. 2007;27:6782–6793. doi: 10.1128/MCB.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tollervey D., Lehtonen H., Jansen R., Kern H., Hurt E.C. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell. 1993;72:443–457. doi: 10.1016/0092-8674(93)90120-f. [DOI] [PubMed] [Google Scholar]
- 13.Nicoloso M., Qu L.H., Michot B., Bachellerie J.P. Intron-encoded, antisense small nucleolar RNAs: the characterization of nine novel species points to their direct role as guides for the 2′-O-ribose methylation of rRNAs. J. Mol. Biol. 1996;260:178–195. doi: 10.1006/jmbi.1996.0391. [DOI] [PubMed] [Google Scholar]
- 14.Kiss-László Z., Henry Y., Bachellerie J.P., Caizergues-Ferrer M., Kiss T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell. 1996;85:1077–1088. doi: 10.1016/s0092-8674(00)81308-2. [DOI] [PubMed] [Google Scholar]
- 15.Cavaillé J., Nicoloso M., Bachellerie J.P. Targeted ribose methylation of RNA in vivo directed by tailored antisense RNA guides. Nature. 1996;383:732–735. doi: 10.1038/383732a0. [DOI] [PubMed] [Google Scholar]
- 16.Balakin A.G., Smith L., Fournier M.J. The RNA world of the nucleolus: two major families of small RNAs defined by different box elements with related functions. Cell. 1996;86:823–834. doi: 10.1016/s0092-8674(00)80156-7. [DOI] [PubMed] [Google Scholar]
- 17.Ganot P., Caizergues-Ferrer M., Kiss T. The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev. 1997;11:941–956. doi: 10.1101/gad.11.7.941. [DOI] [PubMed] [Google Scholar]
- 18.Lafontaine D.L., Bousquet-Antonelli C., Henry Y., Caizergues-Ferrer M., Tollervey D. The box H+ ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 1998;12:527–537. doi: 10.1101/gad.12.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganot P., Bortolin M.L., Kiss T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell. 1997;89:799–809. doi: 10.1016/s0092-8674(00)80263-9. [DOI] [PubMed] [Google Scholar]
- 20.Bortolin M.L., Ganot P., Kiss T. Elements essential for accumulation and function of small nucleolar RNAs directing site-specific pseudouridylation of ribosomal RNAs. EMBO J. 1999;18:457–469. doi: 10.1093/emboj/18.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richard P., Darzacq X., Bertrand E., Jády B.E., Verheggen C., Kiss T. A common sequence motif determines the Cajal body-specific localization of box H/ACA scaRNAs. EMBO J. 2003;22:4283–4293. doi: 10.1093/emboj/cdg394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marnef A., Richard P., Pinzón N., Kiss T. Targeting vertebrate intron-encoded box C/D 2′-O-methylation guide RNAs into the Cajal body. Nucleic Acids Res. 2014;42:6616–6629. doi: 10.1093/nar/gku287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tycowski K.T., Shu M.-D., Kukoyi A., Steitz J.A. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol. Cell. 2009;34:47–57. doi: 10.1016/j.molcel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jády B.E., Ketele A., Kiss T. Human intron-encoded Alu RNAs are processed and packaged into Wdr79-associated nucleoplasmic box H/ACA RNPs. Genes Dev. 2012;26:1897–1910. doi: 10.1101/gad.197467.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell J.R., Cheng J., Collins K. A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol. 1999;19:567–576. doi: 10.1128/mcb.19.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q., Kim N.-K., Feigon J. Architecture of human telomerase RNA. Proc. Natl. Acad. Sci. U.S.A. 2011;108:20325–20332. doi: 10.1073/pnas.1100279108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Podlevsky J.D., Marz M., Qi X., Hoffmann S., Stadler P.F., Chen J.J.-L. Identification of purple sea urchin telomerase RNA using a next-generation sequencing based approach. RNA. 2013;19:852–860. doi: 10.1261/rna.039131.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi X., Li Y., Honda S., Hoffmann S., Marz M., Mosig A., Podlevsky J.D., Stadler P.F., Selker E.U., Chen J.J.-L. The common ancestral core of vertebrate and fungal telomerase RNAs. Nucleic Acids Res. 2013;41:450–462. doi: 10.1093/nar/gks980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ulyanov N.B., Shefer K., James T.L., Tzfati Y. Pseudoknot structures with conserved base triples in telomerase RNAs of ciliates. Nucleic Acids Res. 2007;35:6150–6160. doi: 10.1093/nar/gkm660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jády B.E., Bertrand E., Kiss T. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body–specific localization signal. J. Cell Biol. 2004;164:647–652. doi: 10.1083/jcb.200310138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bratkovič T., Rogelj B. The many faces of small nucleolar RNAs. Biochim. Biophys. Acta. 2014;1839:438–443. doi: 10.1016/j.bbagrm.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Lafontaine D.L., Tollervey D. Birth of the snoRNPs: the evolution of the modification-guide snoRNAs. Trends Biochem. Sci. 1998;23:383–388. doi: 10.1016/s0968-0004(98)01260-2. [DOI] [PubMed] [Google Scholar]
- 33.Kishore S., Gruber A.R., Jedlinski D.J., Syed A.P., Jorjani H., Zavolan M. Insights into snoRNA biogenesis and processing from PAR-CLIP of snoRNA core proteins and small RNA sequencing. Genome Biol. 2013;14:R45. doi: 10.1186/gb-2013-14-5-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kishore S., Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 35.Doe C.M., Relkovic D., Garfield A.S., Dalley J.W., Theobald D.E.H., Humby T., Wilkinson L.S., Isles A.R. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum. Mol. Genet. 2009;18:2140–2148. doi: 10.1093/hmg/ddp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin Q.-F., Yang L., Zhang Y., Xiang J.-F., Wu Y.-W., Carmichael G.G., Chen L.-L. Long noncoding RNAs with snoRNA ends. Mol. Cell. 2012;48:219–30. doi: 10.1016/j.molcel.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 37.Scott M.S., Ono M., Yamada K., Endo A., Barton G.J., Lamond A.I. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Res. 2012;40:3676–3688. doi: 10.1093/nar/gkr1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ender C., Krek A., Friedländer M.R., Beitzinger M., Weinmann L., Chen W., Pfeffer S., Rajewsky N., Meister G. A human snoRNA with microRNA-like functions. Mol. Cell. 2008;32:519–528. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X.-O., Yin Q.-F., Wang H.-B., Zhang Y., Chen T., Zheng P., Lu X., Chen L.-L., Yang L. Species-specific alternative splicing leads to unique expression of sno-lncRNAs. BMC Genomics. 2014;15:287. doi: 10.1186/1471-2164-15-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie J., Zhang M., Zhou T., Hua X., Tang L., Wu W. Sno/scaRNAbase: a curated database for small nucleolar RNAs and cajal body-specific RNAs. Nucleic Acids Res. 2007;35:D183–D187. doi: 10.1093/nar/gkl873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lestrade L., Weber M.J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006;34:D158–D162. doi: 10.1093/nar/gkj002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis J.C., Brown D.D., Brown J.W. The small nucleolar ribonucleoprotein (snoRNP) database. RNA. 2010;16:664–666. doi: 10.1261/rna.1871310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y., Liu J., Jia C., Li T., Wu R., Wang J., Chen Y., Zou X., Chen R., Wang X.-J., et al. Systematic identification and evolutionary features of rhesus monkey small nucleolar RNAs. BMC Genomics. 2010;11:61. doi: 10.1186/1471-2164-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu T.-T., Zhu D., Chen W., Deng W., He H., He G., Bai B., Qi Y., Chen R., Deng X.W. A global identification and analysis of small nucleolar RNAs and possible intermediate-sized non-coding RNAs in Oryza sativa. Mol. Plant. 2013;6:830–846. doi: 10.1093/mp/sss087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner P.P., Bateman A., Poole A.M. SnoPatrol: how many snoRNA genes are there? J. Biol. 2010;9:4. doi: 10.1186/jbiol211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaur D., Gupta A.K., Kumari V., Sharma R., Bhattacharya A., Bhattacharya S. Computational prediction and validation of C/D, H/ACA and Eh_U3 snoRNAs of Entamoeba histolytica. BMC Genomics. 2012;13:390. doi: 10.1186/1471-2164-13-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Makarova J.A., Kramerov D.A. SNOntology: Myriads of novel snoRNAs or just a mirage? BMC Genomics. 2011;12:543. doi: 10.1186/1471-2164-12-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J.-H., Zhang X.-C., Huang Z.-P., Zhou H., Huang M.-B., Zhang S., Chen Y.-Q., Qu L.-H. snoSeeker: an advanced computational package for screening of guide and orphan snoRNA genes in the human genome. Nucleic Acids Res. 2006;34:5112–5123. doi: 10.1093/nar/gkl672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hertel J., Hofacker I.L., Stadler P.F. SnoReport: computational identification of snoRNAs with unknown targets. Bioinformatics. 2008;24:158–164. doi: 10.1093/bioinformatics/btm464. [DOI] [PubMed] [Google Scholar]
- 51.Marz M., Kirsten T., Stadler P.F. Evolution of spliceosomal snRNA genes in metazoan animals. J. Mol. Evol. 2008;67:594–607. doi: 10.1007/s00239-008-9149-6. [DOI] [PubMed] [Google Scholar]
- 52.Maden T. Ribosomal RNA. Click here for methylation. Nature. 1996;383:675–676. doi: 10.1038/383675a0. [DOI] [PubMed] [Google Scholar]
- 53.Maden B.E. Identification of the locations of the methyl groups in 18 S ribosomal RNA from Xenopus laevis and man. J. Mol. Biol. 1986;189:681–699. doi: 10.1016/0022-2836(86)90498-5. [DOI] [PubMed] [Google Scholar]
- 54.Ofengand J., Bakin A. Mapping to nucleotide resolution of pseudouridine residues in large subunit ribosomal RNAs from representative eukaryotes, prokaryotes, archaebacteria, mitochondria and chloroplasts. J. Mol. Biol. 1997;266:246–268. doi: 10.1006/jmbi.1996.0737. [DOI] [PubMed] [Google Scholar]
- 55.Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A.P., Fabris D., Agris P.F. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCloskey J.A., Rozenski J. The Small Subunit rRNA Modification Database. Nucleic Acids Res. 2005;33:D135–D138. doi: 10.1093/nar/gki015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dönmez G., Hartmuth K., Lührmann R. Modified nucleotides at the 5′ end of human U2 snRNA are required for spliceosomal E-complex formation. RNA. 2004;10:1925–1933. doi: 10.1261/rna.7186504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu Y.T., Shu M.D., Steitz J.A. Modifications of U2 snRNA are required for snRNP assembly and pre-mRNA splicing. EMBO J. 1998;17:5783–5795. doi: 10.1093/emboj/17.19.5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tafer H., Kehr S., Hertel J., Hofacker I.L., Stadler P.F. RNAsnoop: efficient target prediction for H/ACA snoRNAs. Bioinformatics. 2010;26:610–616. doi: 10.1093/bioinformatics/btp680. [DOI] [PubMed] [Google Scholar]
- 61.Kehr S., Bartschat S., Stadler P.F., Tafer H. PLEXY: efficient target prediction for box C/D snoRNAs. Bioinformatics. 2011;27:279–280. doi: 10.1093/bioinformatics/btq642. [DOI] [PubMed] [Google Scholar]
- 62.Mückstein U., Tafer H., Hackermüller J., Bernhart S.H., Stadler P.F., Hofacker I.L. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–1182. doi: 10.1093/bioinformatics/btl024. [DOI] [PubMed] [Google Scholar]
- 63.Bartschat S., Kehr S., Tafer H., Stadler P.F., Hertel J. snoStrip: a snoRNA annotation pipeline. Bioinformatics. 2014;30:115–116. doi: 10.1093/bioinformatics/btt604. [DOI] [PubMed] [Google Scholar]
- 64.Kehr S., Bartschat S., Tafer H., Stadler P.F., Hertel J. Matching of Soulmates: coevolution of snoRNAs and their targets. Mol. Biol. Evol. 2014;31:455–467. doi: 10.1093/molbev/mst209. [DOI] [PubMed] [Google Scholar]
- 65.Maden B.E., Corbett M.E., Heeney P.A., Pugh K., Ajuh P.M. Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie. 1995;77:22–29. doi: 10.1016/0300-9084(96)88100-4. [DOI] [PubMed] [Google Scholar]
- 66.Maden B.E. Mapping 2′-O-methyl groups in ribosomal RNA. Methods. 2001;25:374–382. doi: 10.1006/meth.2001.1250. [DOI] [PubMed] [Google Scholar]
- 67.Taft R.J., Glazov E.A., Lassmann T., Hayashizaki Y., Carninci P., Mattick J.S. Small RNAs derived from snoRNAs. RNA. 2009;15:1233–1240. doi: 10.1261/rna.1528909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Falaleeva M., Stamm S. Processing of snoRNAs as a new source of regulatory non-coding RNAs: snoRNA fragments form a new class of functional RNAs. Bioessays. 2013;35:46–54. doi: 10.1002/bies.201200117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deschamps-Francoeur G., Garneau D., Dupuis-Sandoval F., Roy A., Frappier M., Catala M., Couture S., Barbe-Marcoux M., Abou-Elela S., Scott M.S. Identification of discrete classes of small nucleolar RNA featuring different ends and RNA binding protein dependency. Nucleic Acids Res. 2014;42:10073–10085. doi: 10.1093/nar/gku664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G., et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang J.-H., Shao P., Zhou H., Chen Y.-Q., Qu L.-H. deepBase: a database for deeply annotating and mining deep sequencing data. Nucleic Acids Res. 2010;38:D123–D130. doi: 10.1093/nar/gkp943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Machyna M., Kehr S., Straube K., Kappei D., Buchholz F., Butter F., Ule J., Hertel J., Stadler P.F., Neugebauer K.M. The coilin interactome identifies hundreds of small noncoding RNAs that traffic through Cajal bodies. Mol. Cell. 2014;56:389–399. doi: 10.1016/j.molcel.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 73.Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kishore S., Khanna A., Zhang Z., Hui J., Balwierz P.J., Stefan M., Beach C., Nicholls R.D., Zavolan M., Stamm S. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum. Mol. Genet. 2010;19:1153–1164. doi: 10.1093/hmg/ddp585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., León-Ricardo B.X., Engreitz J.M., Guttman M., Satija R., Lander E.S., et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carey M.F., Peterson C.L., Smale S.T. The primer extension assay. Cold Spring Harb. Protoc. 2013;2013:164–173. doi: 10.1101/pdb.prot071902. [DOI] [PubMed] [Google Scholar]
- 78.Boorstein W.R., Craig E.A. Primer extension analysis of RNA. Methods Enzymol. 1989;180:347–369. doi: 10.1016/0076-6879(89)80111-9. [DOI] [PubMed] [Google Scholar]
- 79.Raymond C.K., Roberts B.S., Garrett-Engele P., Lim L.P., Johnson J.M. Simple, quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. RNA. 2005;11:1737–1744. doi: 10.1261/rna.2148705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cavaillé J., Hadjiolov A.A., Bachellerie J.P. Processing of mammalian rRNA precursors at the 3′ end of 18S rRNA. Identification of cis-acting signals suggests the involvement of U13 small nucleolar RNA. Eur. J. Biochem. 1996;242:206–213. doi: 10.1111/j.1432-1033.1996.0206r.x. [DOI] [PubMed] [Google Scholar]
- 81.Kass S. The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell. 1990;60:897–908. doi: 10.1016/0092-8674(90)90338-f. [DOI] [PubMed] [Google Scholar]
- 82.Dupuis-Sandoval F., Poirier M., Scott M.S. The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip. Rev. RNA. 2015;6:381–397. doi: 10.1002/wrna.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fayet-Lebaron E., Atzorn V., Henry Y., Kiss T. 18S rRNA processing requires base pairings of snR30 H/ACA snoRNA to eukaryote-specific 18S sequences. EMBO J. 2009;28:1260–1270. doi: 10.1038/emboj.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mannoor K., Liao J., Jiang F. Small nucleolar RNAs in cancer. Biochim. Biophys. Acta. 2012;1826:121–128. doi: 10.1016/j.bbcan.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bortolin-Cavaillé M.-L., Cavaillé J. The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader–Willi locus generate canonical box C/D snoRNAs. Nucleic Acids Res. 2012;40:6800–6807. doi: 10.1093/nar/gks321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cavaillé J., Buiting K., Kiefmann M., Lalande M., Brannan C.I., Horsthemke B., Bachellerie J.P., Brosius J., Hüttenhofer A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14311–14316. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mannoor K., Shen J., Liao J., Liu Z., Jiang F. Small nucleolar RNA signatures of lung tumor-initiating cells. Mol. Cancer. 2014;13:104. doi: 10.1186/1476-4598-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin Y., Li Z., Ozsolak F., Kim S.W., Arango-Argoty G., Liu T.T., Tenenbaum S.A., Bailey T., Monaghan A.P., Milos P.M., et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012;40:8460–8471. doi: 10.1093/nar/gks637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gao L., Ma J., Mannoor K., Guarnera M.A., Shetty A., Zhan M., Xing L., Stass S.A., Jiang F. Genome-wide small nucleolar RNA expression analysis of lung cancer by next-generation deep sequencing. Int. J. Cancer. 2014 doi: 10.1002/ijc.29169. doi:10.1002/ijc.29169. [DOI] [PubMed] [Google Scholar]
- 90.Ronchetti D., Mosca L., Cutrona G., Tuana G., Gentile M., Fabris S., Agnelli L., Ciceri G., Matis S., Massucco C., et al. Small nucleolar RNAs as new biomarkers in chronic lymphocytic leukemia. BMC Med. Genomics. 2013;6:27. doi: 10.1186/1755-8794-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ronchetti D., Todoerti K., Tuana G., Agnelli L., Mosca L., Lionetti M., Fabris S., Colapietro P., Miozzo M., Ferrarini M., et al. The expression pattern of small nucleolar and small Cajal body-specific RNAs characterizes distinct molecular subtypes of multiple myeloma. Blood Cancer J. 2012;2:e96. doi: 10.1038/bcj.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gingold H., Tehler D., Christoffersen N.R., Nielsen M.M., Asmar F., Kooistra S.M., Christophersen N.S., Christensen L.L., Borre M., Sørensen K.D., et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell. 2014;158:1281–1292. doi: 10.1016/j.cell.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 93.Abel Y., Clerget G., Bourguignon-Igel V., Salone V., Rederstorff M. [Beyond usual functions of snoRNAs] Med. Sci. 2014;30:297–302. doi: 10.1051/medsci/20143003018. [DOI] [PubMed] [Google Scholar]
- 94.Scott M.S., Avolio F., Ono M., Lamond A.I., Barton G.J. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput. Biol. 2009;5:e1000507. doi: 10.1371/journal.pcbi.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ono M., Scott M.S., Yamada K., Avolio F., Barton G.J., Lamond A.I. Identification of human miRNA precursors that resemble box C/D snoRNAs. Nucleic Acids Res. 2011;39:3879–3891. doi: 10.1093/nar/gkq1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brameier M., Herwig A., Reinhardt R., Walter L., Gruber J. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res. 2011;39:675–686. doi: 10.1093/nar/gkq776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Röther S., Meister G. Small RNAs derived from longer non-coding RNAs. Biochimie. 2011;93:1905–1915. doi: 10.1016/j.biochi.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 98.Maxwell E.S., Fournier M.J. The small nucleolar RNAs. Annu. Rev. Biochem. 1995;64:897–934. doi: 10.1146/annurev.bi.64.070195.004341. [DOI] [PubMed] [Google Scholar]
- 99.Makarova J.A., Kramerov D.A. Analysis of C/D box snoRNA genes in vertebrates: The number of copies decreases in placental mammals. Genomics. 2009;94:11–19. doi: 10.1016/j.ygeno.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 100.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M., et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bleichert F., Gagnon K.T., Brown B.A. 2nd, Maxwell E.S., Leschziner A.E., Unger V.M., Baserga S.J. A dimeric structure for archaeal box C/D small ribonucleoproteins. Science. 2009;325:1384–1387. doi: 10.1126/science.1176099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schattner P., Barberan-Soler S., Lowe T.M.P. A computational screen for mammalian pseudouridylation guide H/ACA RNAs. RNA. 2006;12:15–25. doi: 10.1261/rna.2210406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weinberg Z., Zasha W., Breaker R.R. R2R - software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinformatics. 2011;12:3. doi: 10.1186/1471-2105-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.