


Abstract
Gene-recombinase technologies, such as Cre/loxP-mediated DNA recombination, are important tools in the study of gene function, but have potential side effects due to damaging activity on DNA. Here we show that DNA recombination by Cre instigates a robust antiviral response in mammalian cells, independent of legitimate loxP recombination. This is due to the recruitment of the cytosolic DNA sensor STING, concurrent with Cre-dependent DNA damage and the accumulation of cytoplasmic DNA. Importantly, we establish a direct interplay between this antiviral response and cell–cell interactions, indicating that low cell densities in vitro could be useful to help mitigate these effects of Cre. Taking into account the wide range of interferon stimulated genes that may be induced by the STING pathway, these results have broad implications in fields such as immunology, cancer biology, metabolism and stem cell research. Further, this study sets a precedent in the field of gene-engineering, possibly applicable to other enzymatic-based genome editing technologies.
INTRODUCTION
The adoption of Cre/loxP-based DNA recombination has dominated the field of site-specific gene recombination for the past two decades. This system relies on the bacteriophage P1 Cre recombinase, which promotes the catalytic excision of DNA segments flanked by two palindromic 34-bp loxP sites (1,2). The 34-bp loxP site consists of two 13-bp inverted motifs, and a central 8-bp spacer. Critically, alteration of the central eight nucleotide spacer directly impacts the efficiency of Cre recombination and truncation of this central motif leads to the formation of single stranded nicks in DNA (1).
It became evident more than 15 years ago that sites strongly resembling the 34-bp loxP site (referred to as pseudo-loxP sites) exist in the human and mouse genomes, and can be recombined by Cre (3). Such sites are relatively frequent, with as many as 250 and 300 pseudo-loxP sites predicted in mouse and human genomes, respectively (4). Whether or not Cre activity on such pseudo-loxP sites results in single stranded DNA nicks is ill-defined, but Cre overexpression directly alters the integrity of chromosomes, with increased chromatid breaks, dicentric chromosomes, sister chromatid exchange and aberrant gaps/fragments previously reported (5,6). Such activities are due to the endonuclease activity of Cre on DNA (6), and result in decreased cell proliferation, increased apoptosis and cell accumulation in G2/M phase (4–7). It is noteworthy that these genotoxic effects of Cre are similar to those observed with the topoisomerase I ligase inhibitor camptothecin (5).
The innate immune system is one of the first lines of defense against pathogens such as viruses. It is based on the detection of pathogen associated molecular patterns by specific sensors, which activate a broad response to fight the infection rapidly and recruit the adaptive immune system for further protection. Type I interferons (IFNs) are cytokines that are essential effectors of innate immunity, and their secretion by a few infected cells instigates a global antiviral effect throughout the infected host by inducing more than 2000 genes (8). STING is an intracellular adaptor molecule associated with the endoplasmic reticulum membrane of many immune cells from haematopoietic origin, together with various epithelial cell types (9). In 2008, STING was discovered to play a critical role in detecting pathogen-derived DNA in the cytoplasm (10). Upon transfection of foreign DNA into the cytoplasm, STING is phosphorylated to initiate the transcriptional activation of antiviral genes (including type-I IFN) through the transcription factor IFN regulatory factor 3 (11). In 2013, cyclic-GMP-AMP (cGAMP) synthase (cGAS) was found to operate upstream of STING to directly bind double stranded DNA in the cytoplasm and generate the second messenger cGAMP, which activates STING (12). Recent evidence suggests that DNA damage caused by DNA damaging agents such as camptothecin results in the activation of the cGAS-STING pathway (13). Nonetheless, to our knowledge, the impact of Cre-mediated DNA damage on the innate immune system has not previously been assessed.
Here, while originally aiming at defining the regulatory roles of microRNAs (miRNAs) during viral infections, we observed that Cre activation resulted in the strong induction of an antiviral response, independent of loxP targeting. We demonstrate that Cre-mediated DNA recombination can activate the cytosolic STING pathway, leading to the induction of type-I IFN in mammalian cells.
MATERIALS AND METHODS
Ethics statement
The use of animals and experimental procedures were approved by the Monash Medical Centre Ethics Committee under references MMCA/2008/26/BC, MMCA2012/75BC, MMCA2011/25 and MMCA2012/13.
Animals
C57BL/6 × 129 Dicer1flox/flox(referred to as Dicer1f/f) mice bred to C57BL/6 × 129 R26CreERT mice (14) expressing the Cre-ERT fusion protein (15) (composed of a mutated human estrogen receptor ligand binding domain that selectively binds hydroxy-tamoxifen (OHT), and is sequestered in the cytoplasm in the absence of OHT) from the ROSA26 locus, were previously reported (16). Dicer1f/wt × Cre/ESR1+, Dicer1f/f × Cre/ESR1+, Dicer1wt/wt × Cre/ESR1+ and Dicer1wt/wt × Cre/ESR1− mouse embryonic fibroblasts (MEFs) from day 12–14 embryos were immortalized following transfection of pSG5-SV40-LT-Ag and six successive 1/10 passages. Two different cell lines were generated from two different embryos for both Dicer1f/f × Cre/ESR1+ and Dicer1wt/wt × Cre/ESR1+, and yielded similar results. Primary Sting+/+ and Sting−/− MEFs were used at early passages with no immortalization. For bone marrow derived macrophages (BMDMs), bone marrow was isolated from the femurs of the mice and differentiated in 20% L929-cell-conditioned medium for 6 days at 37°C in a 5% CO2 atmosphere. For in vivo tamoxifen injection, 10-12-week-old mice were injected with tamoxifen (Sigma) (1 mg) diluted in peanut oil by i.p. injection (100 μl) for five consecutive days (days 1–5). Blood mononuclear cells were purified from whole blood on day 12 after euthanasia of the animals with CO2, using adapted Ficoll-Paque plus purification (17) and RNA was purified using the mirVana miRNA isolation kit (Life Technologies). Animal studies were not blinded.
Cell culture
BMDMs, MEFs, Vero cells (ATCC® CCL81™), HEK 293T cells (referred to as HEK throughout the studies) and LentiX™ 293T cells (Clontech) were grown in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% sterile fetal bovine serum (Life Technologies) and 1× antibiotic/antimycotic (Life Technologies) (referred to as complete DMEM). Primary Sting+/+ and Sting−/− MEFs were cultivated in Dulbecco's modified Eagle's medium supplemented with 10% sterile fetal bovine serum, P/S, β-mercaptoethanol and non-essential amino acids. Human A549 cells (ATCC® CCL-185™) were grown in Advanced RPMI medium (Life Technologies) supplemented with 1% sterile fetal bovine serum. HEK Sting and HEK Sting CX43/45DKO cells stably expressing the murine Sting fused to an N-terminal mCherry-tag were previously described (18). MEFs were genotyped regularly for Dicer1 floxed locus using IMR6305 CCTGACAGTGACGGTCCAAAG and IMR6569 CATGACTCTTCAACTCAAACT; Cre/ESR1 locus: IMR1084 GCGGTCTGGCAGTAAAAACTATC and IMR1085 GTGAAACAGCATTGCTGTCACTT. OHT treatment of MEFs was carried out as previously described (16), using 500 nM OHT (Sigma) for 16–24 h, and rinsed the next day with fresh medium. For BMDMs, bone-marrow cells were washed on day 3 of differentiation, with fresh medium supplemented with 20% L929-cell-conditioned medium and 500 nM OHT. The cells were rinsed and collected on day 5, and plated in 24-well plates in 20% L929-cell-conditioned medium, prior to lysis for western blot and RNA analyses at day 6. HEK Sting cells were stimulated for 4 hrs with 15 μl/ml of DMXAA resuspended in DMSO (Sigma).
Western blotting
MEFs were plated at different densities to reach full confluency at 24, 48, 72 or 96 h post OHT treatment (with the exception of Figure 5E). Lysates were separated at 80 V on 10% acrylamide gel for 2 h. Transfer was carried out at 20 V for 1.5 h using Bolt Mini Blot Modules (Life Technologies) to Immobilon-FL (Millipore) membranes. After blocking in Odyssey blocking buffer (LI-COR) for 30 min, the membranes were incubated overnight with 1:1000 mouse monoclonal anti-Viperin (MaP.VIP | #MABF106) (Millipore), rabbit anti-mouse p56 (gift from Ganes Sen, Cleveland Clinic, Cleveland, OH, USA), mouse monoclonal anti-β-tubulin (TU-06 | ab7792) (Abcam) or monoclonal rabbit anti-STING (D2P2F | #13647) (Cell Signaling). Conjugated secondary with Alexa Fluor® 680 dye (Life Technologies) or IRdye800 (Rockland) was subsequently used to image the proteins at 700 or 800 nm with an Odyssey scanner (LI-COR).
Figure 5.

Cre-mediated DNA damage activates the STING pathway. (A) Dicer1wt/wt × Cre/ESR1+MEFs were transfected with 10 nM of the indicated siRNAs overnight, prior to 24 h OHT stimulation. Viperin and p56 levels were analyzed 72 h after stimulation (siNC5 is a non-targeting siRNA and siSting#1Mut is a control siSting#1 sequence mutated to prevent Ago2-mediated mRNA cleavage; Mock is transfection reagent only). Blots are representative of three independent experiments. (B) siRNA-targeted genes in Dicer1wt/wt × Cre/ESR1+MEFs were measured 48 h after transfection of 10 nM of the indicated siRNAs (data are presented as mean of three independent experiments in biological duplicate ± s.e.m. with two-way ANOVA to siSting#1Mut condition). (C) Primary Sting+/+ or Sting−/− MEFs stably expressing Cre or Citrine (Control) were analyzed by RT-qPCR for ISG expression. In each experiment, ISG/18S levels in Cre-expressing cells were normalized to ISG/18S levels in Citrine-expressing cells, for each genotype. Data presented are averaged from three independent experiments ± s.e.m., with unpaired t-tests with Welch's correction. (D) HEK WT, Sting and Sting CX43/45DKO cells expressing an IFN-β-Luciferase reporter were cultured alone (NT and DMXAA conditions), or co-cultured 48 h with Dicer1wt/wt × Cre/ESR1+MEFs pre-treated for 24 h with 10 nM siNC5 or sicGAS#2. DMXAA was used as a known agonist of mouse STING (38). IFN-β-Luciferase expression was reported relative to the NT condition for each cell line (data presented are averaged from two independent experiments in biological triplicate ± s.e.m. with one-way ANOVA to the NT condition shown). (E) 12 000 Dicer1f/f × Cre/ESR1+ MEFs were seeded in 24, 12 or 6-well plates, prior to 24 h treatment with OHT. Viperin and p56 levels were analyzed 72 h after stimulation, the time by which the cells reached high (24-well), intermediate (12-well) or low (6-well) density. Blots are representative of three independent experiments.
Semliki forest virus (SFV) infection
A total of 120 000 MEFs were seeded 3 or 4 days after OHT treatment in 24-well plates, and left to adhere for several hours, prior to infection with semliki forest virus (SFV) in complete DMEM (MOI 2—as determined by plaque forming units in Vero cells) (each condition was carried out in biological triplicate). The cells were rinsed 2 h after infection with fresh medium complemented with 2.5% FCS, and further incubated for 22 h. Virus-containing supernatants were collected at 24 h and series-diluted (10-fold dilutions) on 80% confluent Vero cells in 96-well plates (in 5% FCS complete DMEM). After 48 h, surviving Vero cells were fixed with 10% formalin and stained with 0.1% crystal violet (w/v) in 20% ethanol, before thorough H2O washes.
Statistical analyses
Statistical analyses were carried out using Prism 6 (GraphPad Software Inc.). Two-tailed unpaired non-parametric Mann–Whitney U tests or two-tailed unpaired t-tests with Welch's correction (to compare pairs of conditions), one-way ANOVA with Tukey's multiple comparisons tests (to compare more than two conditions) or two-way ANOVA with Sidak's multiple comparisons tests (to compare groups of conditions) are shown when appropriate. Symbols used: *P≤ 0.05, **P≤ 0.01, ***P≤ 0.001, ****P≤ 0.0001.
RESULTS
Cre-ERT activation induces a strong antiviral response in mouse embryonic fibroblasts (MEFs) and animals
We have recently reported an important role for microRNAs in the regulation of inflammation through the control of nuclear factor kappa B (NF-κB) signaling in MEFs (19). To define the impact of miRNAs on viral infections following global miRNA depletion, we infected immortalized MEFs with SFV, 4 days after OHT-induced Cre-ERT-mediated deletion of Dicer1 (16). We chose this duration based on our previous observation that miRNAs are decreased by more than 50–60% at that point in time in this model (16,20). Cre-ERT-mediated deletion of Dicer1 resulted in a marked decrease in viral titers (Figure 1A), with concurrent induction of the key antiviral proteins p56 (21) (encoded by Ifit1) and Viperin (22) (encoded by Rsad2) (Figure 1B). Unexpectedly, this effect was observed in both Dicer1f/f × Cre/ESR1+ and heterozygous Dicer1f/wt × Cre/ESR1+ cells. Accordingly, both Ifit1 and Rsad2 were induced in vivo in peripheral blood mononuclear cells from Dicer1f/f × Cre/ESR1+ and Dicer1f/wt × Cre/ESR1+ animals treated with tamoxifen, 7 days after the last injection (Figure 1C). Because in Dicer1f/wt × Cre/ESR1+cells the levels of most miRNAs were not changed (Gantier, M., unpublished observation), we investigated the potential contribution of the Cre-ERT system to this antiviral activity, independent of Dicer1 deletion and miRNA biogenesis. Remarkably, transient Cre-ERT activation alone resulted in the induction of p56 and Viperin in Dicer1wt/wt × Cre/ESR1+ MEFs, while being sufficiently robust to strongly inhibit SFV replication, independent of the OHT treatment itself (Figure 1D and E).
Figure 1.

Cre-ERT-mediated activation of an antiviral response. (A) MEFs treated for 24 h with OHT were expanded for 3 days, prior to 24 h infection in biological triplicate with SFV (MOI 2). Viral titers were assayed with log10 fold dilutions on confluent Vero cells as shown. NI: not infected (uninfected cells were stained with crystal violet). (B) Time course showing the activity of 24 h OHT treatment of Dicer1f/f x Cre/ESR1+ and Dicer1f/wt Cre/ESR1 + MEFs on the levels of Viperin and p56. (A, B) Data shown are representative of a minimum of two independent experiments. NT: non-treated. (C) Peripheral blood mononuclear cells from mice injected with oil or tamoxifen for five consecutive days, and culled on day 12, were analyzed by RT-qPCR for ISG expression (each point represents one mouse; mean ± s.e.m. and unpaired Mann-Whitney U tests are shown). Data shown relative to the mean ISG/18S expression of the oil control samples. nd: non-detected in three out of four mice. (D) MEFs treated for 24 h with OHT were expanded for two days, prior to 24 h infection in biological triplicate with SFV (MOI 2) and viral titers on confluent Vero cells as in (A). (E) Time course showing the activity of 24 h OHT treatment of Dicer1wt/wt x Cre/ESR1+ and Cre/ESR1− MEFs on the levels of Viperin and p56. (C-E) Data shown are representative of three independent experiments or mice.
Dose-dependent activation of type-I IFN gene signatures in MEFs by Cre-ERT
Having shown a strong antiviral effect of Cre-ERT activation in the absence of loxP site recombination, we decided to investigate the potential mechanism at play. Speculating the recruitment of type-I IFNs, genome-wide microarray analyses of three different Cre/ESR1+ MEF cell lines was carried out (Dicer1f/f × Cre/ESR1+, Dicer1f/wt × Cre/ESR1+ and Dicer1wt/wt × Cre/ESR1+) (Figure 2A). This analysis confirmed that OHT stimulation of Cre-ERT, and not the recombination of targeted loxP sites, resulted in the induction of a classical type I IFN-stimulated gene (ISG) signature—including the antiviral genes Rsad2, Ifit1, Ifih1, Ddx58 and Ifnb1 (Figure 2A). Of note, this induction was delayed and exhibited only 48–72 h post initial OHT treatment (Figure 2B and Supplementary Figure S1A). In addition, the expression of many antiviral genes was basally higher in all Cre/ESR1+ lines compared to wild-type MEFs, indicative of leaky Cre-ERT activity, consistent with a previous report (5)(Figure 2A and B). We also noted significant variations in the basal levels of certain ISGs between different Cre/ESR1+ lines, with the example of Rsad2 which was >20-fold higher in Dicer1wt/wt × Cre/ESR1+ compared to Dicer1f/wt × Cre/ESR1+, possibly explaining the earlier/higher induction of Viperin levels seen in Figure 1 (Figure 2B, Supplementary Figure S1A and not shown). Given that Cre-ERT was found to lack off-target effects with lower OHT doses, we next assessed the induction of p56 and Viperin with different doses of OHT. Although clearly dose-dependent, induction of the antiviral proteins was still apparent at 10 nM OHT, a dose previously thought to avoid Cre off-target effects (5) (Figure 2C).
Figure 2.

Cre-ERT-dependent activation of type-I IFN gene signatures. (A) Microarray analysis of MEFs (48 h post-initial OHT treatment for Dicerwt/wt × Cre/ESR1+ and Cre/ESR1− cells and 96 h for Dicer f/wt × Cre/ESR1+and Dicer f/f cells), restricted to 23 ISGs. NT: non-treated. (B) RT-qPCR analysis of ISGs in MEFs after 24 h OHT treatment. Data are shown relative to non-treated Cre/ESR1− cells (data are presented as mean of three independent experiments in biological duplicate ± s.e.m.). (C) Dose-response study of varying concentrations of OHT on MEFs, on the levels of Viperin and p56, 72 h after stimulation. (B and C) Data shown are representative of three independent experiments.
Cre induces antiviral genes in mouse macrophages and human epithelial cells
To define whether the antiviral signature observed was restricted to MEFs, primary BMDMs from Dicer1f/wt × Cre/ESR1+, Dicer1wt/wt × Cre/ESR1+ and the control Dicer1wt/wt × Cre/ESR1− mice were treated with OHT. Similar to MEFs, induction of p56 and Viperin by OHT in BMDMs was also Cre-ERT dependent (Figure 3A and Supplementary Figure S1B). Expression analysis of eight known ISGs in OHT-treated Dicer1wt/wt × Cre/ESR1+ BMDMs confirmed the hallmark of a type-I IFN response in these cells (Figure 3B). Furthermore, stable overexpression of Cre in human A549 cells was also able to induce ISGs, confirming that these observations were not restricted to the murine Cre-ERT model (Figure 3C). Critically, the Cre-mediated ISG induction relied on the endonuclease activity of Cre on DNA since stable overexpression of an enzymatic point mutant (Cre-R173K (5)) was not able to induce IFIT2, while expressing as much Cre-R173K mRNA as the native gene (Figure 3D). Collectively, these results demonstrate that the activation of a type-I IFN antiviral response by Cre, in both mouse and human cells, depends on the catalytic activity of Cre.
Figure 3.
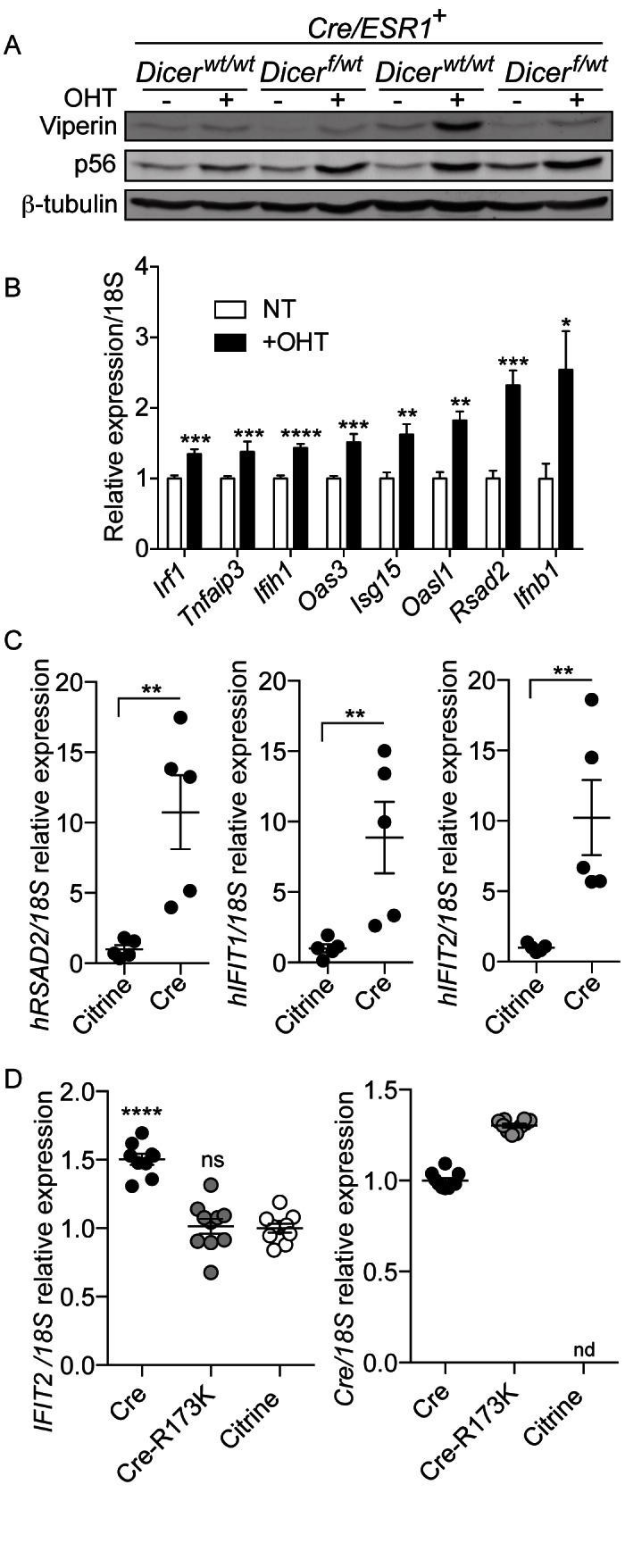
Cre catalytic activity on DNA induces antiviral genes in human and mouse cells. (A) Viperin and p56 levels in BMDMs from Dicer1f/wt × Cre/ESR1+ and Dicer1wt/wt × Cre/ESR1+ animals treated with OHT. (B) BMDMs from Dicer1wt/wt × Cre/ESR1+mice were analyzed for expression of the indicated ISGs by RT-qPCR analysis (data are presented as mean of three independent experiments in biological duplicate ± s.e.m., with unpaired Mann–Whitney U tests to the non-treated condition). (A and B) Data shown are representative of three independent experiments or mice. (C) Human A549 cells stably expressing Cre or Citrine (control) were analyzed by RT-qPCR for ISG expression. Data presented are from five independent cell lines ± s.e.m., reported relative to the mean ISG/18S expression of the Citrine samples, with unpaired Mann–Whitney U tests to the control condition. (D) Left panel: human A549 cells stably expressing Cre, Cre-R173K or Citrine (control) were analyzed by RT-qPCR for IFIT2/18S expression, reported relative to the Citrine condition. Right panel: RT-qPCR of Cre/18S expression reported relative to the Cre condition (nd: not detected). Data presented are from two independent experiments, with five biological replicates per experiment ± s.e.m., with one-way ANOVA to the Citrine condition.
Cre-ERT activation induces DNA damage and accumulation of cytoplasmic DNA products
Given the previous reports that Cre overexpression promotes DNA damage, leading to decreased cell proliferation (4–6), we next assessed DNA damage following OHT treatment of Cre/ESR1+MEFs. In accordance with these previous reports, the number of γ-H2A.X-positive cells was significantly increased after OHT treatment of Cre/ESR1+ cells (Figure 4A and B). Critically, part of the γ-H2A.X staining seen in OHT-treated Cre/ESR1+ cells was often present in foci outside of the nucleus (Figure 4A). Such cytoplasmic localization was recently shown for histone H3 upon DNA damage with the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA), concurrent with leakage of DNA products to the cytoplasm (13). Speculating that the DNA damage promoted by Cre could also lead to such cytoplasmic DNA leakage, we next analyzed OHT-treated Cre/ESR1+ cells for DNA staining using a pan anti-DNA antibody. This analysis revealed increased levels of cytoplasmic DNA foci upon OHT stimulation of Cre/ESR1+ cells, but not the control Cre/ESR1− cells, clearly pointing to an accumulation of cytoplasmic DNA upon Cre activation (Figure 4C and D). This led us to speculate that the type-I IFN response observed with Cre was instigated by the activation of a cytoplasmic DNA sensor.
Figure 4.
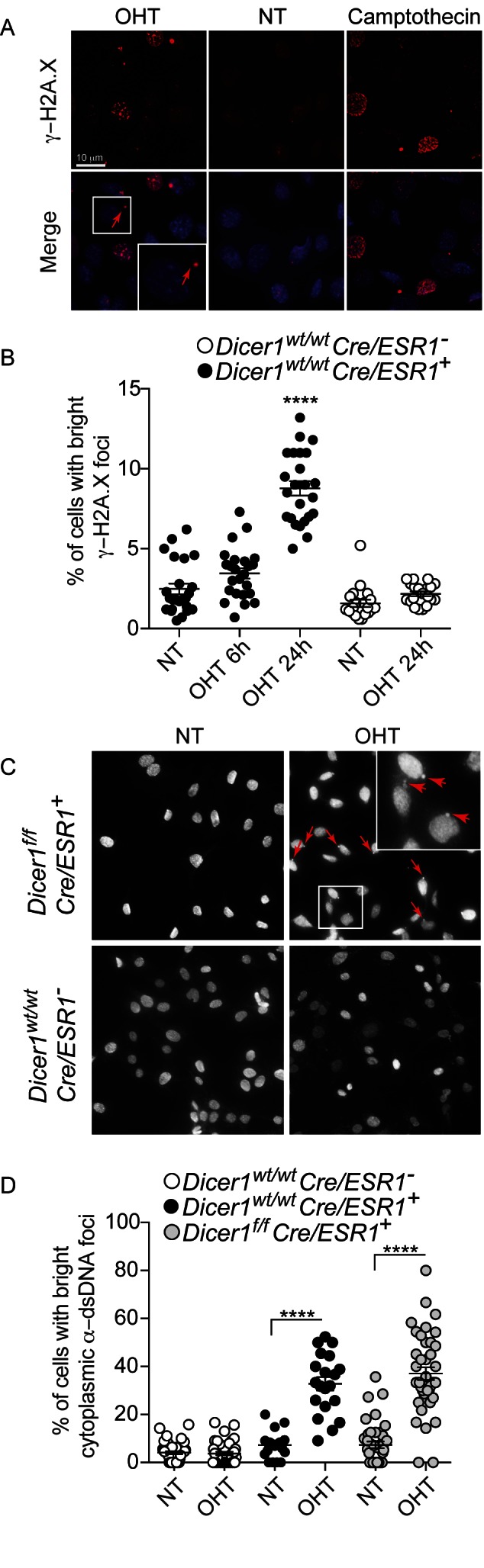
Cre-ERT activation increases DNA damage and cytoplasmic DNA. (A) Immunofluorescence of γ-H2A.X staining (red) of Dicer1wt/wt × Cre/ESR1+ MEFs after 72 h OHT (red arrows point to a cytoplasmic bright foci) or camptothecin treatment (5 μM for 2 h—positive control), and (B) quantification of cells showing bright foci per field (each point represents one field; data are averaged from two experiments for Cre/ESR1+ MEFs ± s.e.m. with one-way ANOVA to Cre/ESR1+ NT condition shown). (C) Immunofluorescence of anti-DNA staining in MEFs after 72 h OHT treatment (red arrows point to cytoplasmic foci) and (D) quantification of cells showing bright cytoplasmic foci (each point represents one field; data are averaged from two independent experiments ± s.e.m. with one-way ANOVA to NT conditions shown). NT: non-treated.
STING-dependent activation of antiviral genes upon Cre-ERT activation or Cre overexpression
Induction of ISG responses to several DNA-damaging agents was recently shown to result in cytoplasmic DNA leakage, sensed by the cytosolic cGAS-STING innate immune pathway (13,23). We posited that a similar mechanism could be at play with Cre. Accordingly, we examined the involvement of the cGAS-STING pathway in Cre-dependent ISG induction. Small interfering RNA (siRNA)-mediated knockdown of STING or cGAS in Dicer1f/f × Cre/ESR1+ and Dicer1wt/wt × Cre/ESR1+ MEFs, before OHT-driven Cre-ERT activation, strongly inhibited p56 and Viperin induction in both cell types (Figure 5A and B; Supplementary Figure S2A and B). To confirm these results, we next assessed the requirement for STING in the induction of antiviral Rsad2 and Ifit1 genes upon stable expression of Cre in primary MEFs from wild-type and Sting-deficient mice. Similarly to what we observed upon downregulation of Sting in Cre/ESR1+ MEFs, genetic ablation of STING abolished the induction of ISGs upon stable Cre overexpression in primary MEFs (Figure 5C). Collectively, these results establish the requirement for STING in inducing antiviral genes by Cre overexpression or Cre-ERT activation.
Cell-density-dependent amplification of Cre-mediated ISG induction
cGAMP (2′-5′), the product of cGAS and the effector of the cGAS-STING pathway, can be transmitted between cells through connexins that form gap junctions to confer bystander immunity in adjacent cells (18). In line with our microarray analysis (Figure 2) that indicated a basal engagement of cGAS in Cre/ESR1+MEFs, we performed co-culture studies of Dicer1f/f × Cre/ESR1+ or Dicer1wt/wt × Cre/ESR1+ MEFs with HEK cells expressing the murine Sting and an IFN-β-Luciferase reporter (18). In these experiments, we observed significantly upregulated IFN-β expression (see siNC5 condition; Figure 5D and Supplementary Figure S2C). This effect was not seen in co-cultures with WT HEK cells, which are not naturally responsive to cGAMP (Figure 5D) (18). This effect was also absent in co-cultures with HEK cells double-deficient in connexin 43 (CX43) and 45 (CX45), but which expressed Sting (18); nor was it seen when downregulation of cGAS was carried out in Cre/ESR1+ MEFs prior to co-culture (Figure 5D and Supplementary Figure S2C). Collectively, these results support the existence of a basal and functional cGAMP-like activity in Cre/ESR1+ MEFs, since cGAS, CX43/45 and STING are necessary for activation of a bystander effect in HEK cells, initiated from Cre/ESR1+ cells. However, this cGAMP-like activity was not significantly induced in Cre/ESR1+ MEFs treated with OHT using this co-culture system, possibly relating to high basal cGAMP-like activities (not shown). Nevertheless, in line with an amplification of STING engagement driven by Cre-ERT activation, OHT-dependent induction of p56 and Viperin in Cre/ESR1+ MEFs was directly dependent on cell:cell interactions, and was virtually absent in cells grown at low density (Figure 5E and Supplementary Figure S2D).
DISCUSSION
The diversity of inducible and/or tissue-specific Cre-expressing transgenic lines created to date suggest that the Cre/loxP-based system is likely to remain an essential and highly efficacious technology for the foreseeable future, despite the advent of new gene-editing technologies such as TALENs and CRISPR-Cas9. Importantly, the non-specific effects of Cre are well documented in the literature, in vitro (in cell types including: HEK 293 cells, HeLa cells, NIH3T3 cells, COS-7 cells, CV-1 cells, U2OS cells and primary MEFs (5–7)), but also in vivo (for example in the striatum or hippocampus (7), in spermatids (24), in hematopoietic cells (25), in the heart (26), in c-Myc-transformed B cell lymphomas (27) and the central nervous system (28)). Consequently, the use of Cre-only controls or self-deleting Cre has been recommended in several instance to help mitigate the non-specific effects of Cre (5–7,27–29).
In the present work we aimed to define the overall contribution of miRNAs to the modulation of antiviral responses. Unexpectedly, we noted that partial deletion of Dicer1, in Dicer1f/wt × Cre/ESR1+ MEFs, resulted in a similar antiviral response to that observed upon complete Dicer1 deletion (Figure 1). Given prior reports of a non-specific activity of Cre-ERT activation in MEFs, we investigated the potential contribution of Cre in the antiviral effects observed. Relying on (i) the downregulation of Sting expression in Cre/ESR1+ MEFs through two different siRNAs, and (ii) Sting-deficient primary MEFs overexpressing Cre, we establish that Cre-ERT activation and Cre overexpression activate a type-I IFN response, through recruitment of cytosolic STING.
Given both the prior demonstrations that Cre overexpression or Cre-ERT activation promote DNA damage (4–6), as well as our own observations (Figure 4), we propose that Cre-dependent STING activation is mediated by DNA products leaked to the cytoplasm. This is consistent with our observation of cytoplasmic DNA staining upon Cre-ERT activation, and two independent studies establishing STING activation upon DNA damage by camptothecin and several other DNA-damaging agents (DMBA, cisplatin, etoposide and γ-irradiation) (13,23). Importantly, deficiency of the DNaseIII TREX1, involved in the degradation of cytoplasmic DNA, was found to exacerbate the accumulation of cytoplasmic DNA and histone H3 upon induction of DNA damage, while leading to an increased STING-mediated ISG induction (13). These findings clearly point to a critical role for cytoplasmic DNA of endogenous origin leaked from the nucleus, in STING activation by DNA damage. Although mechanistic differences between Cre-mediated DNA damage and that of agents such as camptothecin are likely, the kinetics of ISG induction upon camptothecin or DMBA treatment is in line with our observations of Cre-ERT activation by OHT, seen around 48–72 h (13). This is in agreement with a build up of DNA damage (seen with increased γ-H2A.X staining at 24 h and beyond), and an amplification of the response between cells.
Critically, we found that cell density was essential in the detection of an ISG response in OHT-treated Cre/ESR1+ MEFs (Figure 5). Given that Cre-mediated DNA damage is unlikely to be dependent on cell confluency, this finding supports the concept of an amplification of the ISG response between cells. Furthermore, the requirement for cell:cell interactions suggest that such amplification predominantly relies on an IFN-independent mediator. Our observation that Cre-ERT-driven ISG induction is entirely blunted by two different siRNAs that downregulate cGas expression in Cre/ESR1+ MEFs supports the concept that cGAS is involved in detecting cytoplasmic DNA products generated by Cre. cGAS downregulation was also found to decrease ISG induction by camptothecin and DMBA in Trex1-deficient MEFs (13). Given that cGAMP, the product of cGAS, is diffusible between cells that form gap junctions, we reasoned that it was most likely involved in the amplification of the ISG response between confluent cells.
We showed that Dicer1wt/wt × Cre/ESR1+ and Dicer1fl/fl × Cre/ESR1+ MEFs exhibited basal levels of cGAMP-like activity, dependent on cGAS expression in MEFs, and CX43/45/STING expression in adjacent recipient cells. This basal cGAMP-like activity was presumably involved in Viperin/p56 induction seen at 96 h in highly confluent, non-stimulated Dicer1wt/wt × Cre/ESR1+ and Dicer1fl/fl × Cre/ESR1+MEFs (Figure 1). Nonetheless, we were unable to detect an increased cGAMP-like activity upon OHT treatment of Cre/ESR1+ MEFs, relying on the same biological assay. Importantly, treatment of wild-type immortalized MEFs with camptothecin, only led to a ∼1.5–2-fold increase of cGAMP-like activity (Pépin et al., in preparation). Given that Cre-ERT-mediated DNA damage is not expected to be as strong as that of camptothecin, as it does not equally affect every cell, it is likely that the sensitivity of our cGAMP biological assay was not great enough to differentiate increases of cGAMP activity from basal cGAMP levels in a small proportion of cells. However, it is noteworthy that the activity of STING can be modulated independently of cGAS/cGAMP by other DNA-binding proteins (9), or by membrane fusions (30). As such, we cannot rule out that the activation of STING following Cre-ERT activation partially relies on another DNA sensor than cGAS, whose function could be controlled by cGAS and basal cGAMP levels (31), but not mediated by cGAMP.
We found that the catalytic activity of Cre was essential for its ability to induce an IFN-response in human cells. Similar to its effects on DNA damage and cell proliferation (4,6), this indicates that the DNA cleavage by Cre is required for STING activation. Given that this activity of Cre was independent of loxP sites, it is reasonable to assume that it pertains to its activity on pseudo-loxP sites (27). Whether or not Cre activity on genomic pseudo-loxP sites results in single stranded DNA nicks and cytoplasmic DNA products remains to be defined, but it would be consistent with its reported nickase activity when binding to ‘mutated’ loxP sites (1).
In summary, we have shown here that DNA damage resulting from legitimate and illegitimate Cre enzymatic activity can result in the activation of the STING antiviral pathway. Given the vast array of genes regulated by the IFN pathway (∼2000 genes (8)), and the wide range of their auto- and paracrine functions, these findings demonstrate the need for caution in the interpretation of data from Cre/loxP-based studies that do not include appropriate Cre-only controls (reviewed in (28)). Beyond the previously known effects of Cre on DNA damage, the additional involvement of IFNs by Cre recombination discovered in this work has functional consequences for many pathways and tissues, through modulation of several key transcription factors/modulators, including the IRF/STAT, SMAD, KLF, HDAC and ETS families (32). As such, in addition to having implications for studies of the immune system, the IFN induction reported here has broad consequences for many other fields such as cancer biology, metabolism, stem cell research and neurology (9,33–35).
Although the STING pathway described in this work may not be functional in every cell type, our studies indicate that in vitro experiments relying on Cre/ESR1+ cell models should be performed at low densities, to limit the risk of amplification and minimize the potential bias introduced by the IFN pathway. In addition, we propose that measuring the expression of select ISGs could be useful, in order to rule out potential off-target effects of Cre technologies related to other aspects of DNA damage, with specific relevance to hematopoietic tissues where STING is functional. Finally, our findings suggest that gene-editing technologies relying on DNA nickases (such as Cas9 nickase mutant (36)) may also be under the innate immune radar, and that similar caution and appropriate controls should be used in experiments employing these technologies. Conversely, novel gene editing technologies that do not rely on DNA cleavage, such as CRISPR/Cas9 cytidine deaminase (37), should help avoid innate immune recognition by the pathways described here.
Supplementary Material
Acknowledgments
We thank A.T. Irving for help with SFV infections, D.N. Watkins for important discussions, M. Bilandzic for providing cell lines, and F.A. Cribbin and R.E. Smith for help with the redaction of this paper.
FUNDING
Australian National Health and Medical Research Council [1022144 to M.P.G., 1062683 to M.P.G., B.R.G.W., in part]; Australian National Health and Medical Research Council Career Development Fellowship [1063914 to D.J.G.]; Australian Research Council Future Fellowship [140100594 to M.P.G.]; Victorian Government's Operational Infrastructure Support Program. Funding for open access charge: Hudson Institute of Medical Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Abremski K., Wierzbicki A., Frommer B., Hoess R.H. Bacteriophage P1 Cre-loxP site-specific recombination. Site-specific DNA topoisomerase activity of the Cre recombination protein. J. Biol. Chem. 1986;261:391–396. [PubMed] [Google Scholar]
- 2.Sauer B., Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. U.S.A. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thyagarajan B., Guimaraes M.J., Groth A.C., Calos M.P. Mammalian genomes contain active recombinase recognition sites. Gene. 2000;244:47–54. doi: 10.1016/s0378-1119(00)00008-1. [DOI] [PubMed] [Google Scholar]
- 4.Karimova M., Abi-Ghanem J., Berger N., Surendranath V., Pisabarro M.T., Buchholz F. Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucleic Acids Res. 2013;41:e37. doi: 10.1093/nar/gks1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loonstra A., Vooijs M., Beverloo H.B., Allak B.A., van Drunen E., Kanaar R., Berns A., Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9209–9214. doi: 10.1073/pnas.161269798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silver D.P., Livingston D.M. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol. Cell. 2001;8:233–243. doi: 10.1016/s1097-2765(01)00295-7. [DOI] [PubMed] [Google Scholar]
- 7.Pfeifer A., Brandon E.P., Kootstra N., Gage F.H., Verma I.M. Delivery of the Cre recombinase by a self-deleting lentiviral vector: efficient gene targeting in vivo. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11450–11455. doi: 10.1073/pnas.201415498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rusinova I., Forster S., Yu S., Kannan A., Masse M., Cumming H., Chapman R., Hertzog P.J. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41:D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barber G.N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y.T., Grishin N.V., et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 12.Wu J., Sun L., Chen X., Du F., Shi H., Chen C., Chen Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn J., Xia T., Konno H., Konno K., Ruiz P., Barber G.N. Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun. 2014;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badea T.C., Wang Y., Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J. Neurosci. 2003;23:2314–2322. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feil R., Brocard J., Mascrez B., LeMeur M., Metzger D., Chambon P. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10887–10890. doi: 10.1073/pnas.93.20.10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gantier M.P., McCoy C.E., Rusinova I., Saulep D., Wang D., Xu D., Irving A.T., Behlke M.A., Hertzog P.J., Mackay F., et al. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res. 2011;39:5692–5703. doi: 10.1093/nar/gkr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gantier M.P., Williams B.R. Monitoring innate immune recruitment by siRNAs in mammalian cells. Methods Mol. Biol. 2010;623:21–33. doi: 10.1007/978-1-60761-588-0_2. [DOI] [PubMed] [Google Scholar]
- 18.Ablasser A., Schmid-Burgk J.L., Hemmerling I., Horvath G.L., Schmidt T., Latz E., Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gantier M.P., Stunden H.J., McCoy C.E., Behlke M.A., Wang D., Kaparakis-Liaskos M., Sarvestani S.T., Yang Y.H., Xu D., Corr S.C., et al. A miR-19 regulon that controls NF-kappaB signaling. Nucleic Acids Res. 2012;40:8048–8058. doi: 10.1093/nar/gks521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu D., Hu Y., Tong S., Williams B.R., Smyth G.K., Gantier M.P. The use of miRNA microarrays for the analysis of cancer samples with global miRNA decrease. RNA. 2013;19:876–888. doi: 10.1261/rna.035055.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fensterl V., Sen G.C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011;31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzgerald K.A. The interferon inducible gene: Viperin. J. Interferon Cytokine Res. 2011;31:131–135. doi: 10.1089/jir.2010.0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartlova A., Erttmann S.F., Raffi F.A., Schmalz A.M., Resch U., Anugula S., Lienenklaus S., Nilsson L.M., Kroger A., Nilsson J.A., et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity. 2015;42:332–343. doi: 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt E.E., Taylor D.S., Prigge J.R., Barnett S., Capecchi M.R. Illegitimate Cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13702–13707. doi: 10.1073/pnas.240471297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higashi A.Y., Ikawa T., Muramatsu M., Economides A.N., Niwa A., Okuda T., Murphy A.J., Rojas J., Heike T., Nakahata T., et al. Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J. Immunol. 2009;182:5633–5640. doi: 10.4049/jimmunol.0802413. [DOI] [PubMed] [Google Scholar]
- 26.Buerger A., Rozhitskaya O., Sherwood M.C., Dorfman A.L., Bisping E., Abel E.D., Pu W.T., Izumo S., Jay P.Y. Dilated cardiomyopathy resulting from high-level myocardial expression of Cre-recombinase. J. Card. Fail. 2006;12:392–398. doi: 10.1016/j.cardfail.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt-Supprian M., Rajewsky K. Vagaries of conditional gene targeting. Nat. Immunol. 2007;8:665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- 28.Harno E., Cottrell E.C., White A. Metabolic pitfalls of CNS Cre-based technology. Cell Metab. 2013;18:21–28. doi: 10.1016/j.cmet.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 29.Toxic alert. Nature. 2007;449:378. doi: 10.1038/449378a. [DOI] [PubMed] [Google Scholar]
- 30.Holm C.K., Rahbek S.H., Gad H.H., Bak R.O., Jakobsen M.R., Jiang Z., Hansen A.L., Jensen S.K., Sun C., Thomsen M.K., et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016;7:10680. doi: 10.1038/ncomms10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoggins J.W., MacDuff D.A., Imanaka N., Gainey M.D., Shrestha B., Eitson J.L., Mar K.B., Richardson R.B., Ratushny A.V., Litvak V., et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hertzog P., Forster S., Samarajiwa S. Systems biology of interferon responses. J. Interferon Cytokine Res. 2011;31:5–11. doi: 10.1089/jir.2010.0126. [DOI] [PubMed] [Google Scholar]
- 33.Essers M.A., Offner S., Blanco-Bose W.E., Waibler Z., Kalinke U., Duchosal M.A., Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 34.York A.G., Williams K.J., Argus J.P., Zhou Q.D., Brar G., Vergnes L., Gray E.E., Zhen A., Wu N.C., Yamada D.H., et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell. 2015;163:1716–1729. doi: 10.1016/j.cell.2015.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGlasson S., Jury A., Jackson A., Hunt D. Type I interferon dysregulation and neurological disease. Nat. Rev. Neurol. 2015;11:515–523. doi: 10.1038/nrneurol.2015.143. [DOI] [PubMed] [Google Scholar]
- 36.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016 doi: 10.1038/nature17946. doi:10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conlon J., Burdette D.L., Sharma S., Bhat N., Thompson M., Jiang Z., Rathinam V.A., Monks B., Jin T., Xiao T.S., et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J. Immunol. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.