


Abstract
Identifying functional elements in promoter sequences is a major goal in computational and experimental genome biology. Here, we describe an algorithm, Local Distribution of Short Sequences for Prokaryotes (LDSS-P), to identify conserved short motifs located at specific positions in the promoters of co-expressed prokaryotic genes. As a test case, we applied this algorithm to a symbiotic nitrogen-fixing bacterium, Sinorhizobium meliloti. The LDSS-P profiles that overlap with the 5′ section of the extracytoplasmic function RNA polymerase sigma factor RpoE2 consensus sequences displayed a sharp peak between -34 and -32 from TSS positions. The corresponding genes overlap significantly with RpoE2 targets identified from previous experiments. We further identified several groups of genes that are co-regulated with characterized marker genes. Our data indicate that in S. meliloti, and possibly in other Rhizobiaceae species, the master cell cycle regulator CtrA may recognize an expanded motif (AACCAT), which is positionally shifted from the previously reported CtrA consensus sequence in Caulobacter crescentus. Bacterial one-hybrid experiments showed that base substitution in the expanded motif either increase or decrease the binding by CtrA. These results show the effectiveness of LDSS-P as a method to delineate functional promoter elements.
INTRODUCTION
Understanding promoter structures and deducing corresponding regulatory networks are major goals in genome biology. Prokaryotic promoters are relatively short (∼50 bp) sequences that provide a recognition site for RNA polymerase in association with a specific sigma factor (1), and thus define the site for transcription initiation. Most bacteria have multiple sigma factors that recognize distinct promoter sequences. A change in sigma factor results in a global change in transcription profile, and this mechanism is used by numerous bacteria as they adapt to changing environments. In addition, promoters are often adjacent to other DNA sequences that serve to bind transcription activators or repressors. These trans-acting proteins add another layer of regulation and may govern further networks of co-expressed genes.
Motif finding is an important step and major challenge to understand and predict promoter functions and regulations. Several approaches are used to predict functional motifs that are recognized by RNA polymerase sigma factors and other DNA binding proteins. Numerous algorithms have been developed during the last two decades. Das and Dai (2) comprehensively reviewed some representative algorithms and categorized into two major categories: (i) probabilistic sequence models where the model parameters are estimated using maximum-likelihood principle or Bayesian inference and (ii) word-based (string-based) approaches that often rely on counting and comparing oligonucleotide frequencies. These algorithms often require pre-grouping or ‘enrichment’ of genes as input. To assemble the set of input sequences, one may use DNA microarray- and RNA sequencing-based transcriptome profiling to reveal genes showing parallel expression, or use chromatin immunoprecipitation and systematic evolution of ligands by exponential enrichment (SELEX) to define DNA sequences with affinity for specific proteins. Such sets of experimentally derived sequences are then analyzed with a probabilistic algorithm.
In some cases, however, there may be little or no pre-existing evidence to characterize all genes, or all expression conditions, especially for non-model systems. In such cases, a consensus-independent method (one that does not require prior knowledge about the targets) may be useful for predicting regulatory networks. Local distribution of short sequences (LDSS) (3) is a de novo motif-finding algorithm based on the presumed presence of conserved short (6–8 mer) sequences in cis to transcription start sites. This algorithm assumes that a functional short sequence, such as an RNA polymerase sigma factor binding site, must be located at a certain defined position in the promoter for its functionality. In the case of eukaryotic promoters, examples are the TATA box and binding sites for NRF-1, SP1, CREB, ATF and E2F (3,4). The original LDSS algorithm has proved to be a simple and powerful method for extracting possible functional short motifs without any knowledge about the genes in a promoter data set, and has been used to analyze and characterize a large number of promoters from plants and animals. A similar algorithm has been included in PEAKS positional footprinting server (5). It supports the analysis of significant motif positional biases up to 6-mer sequences, however, it does not support longer oligomers or combination of 2 or more nucleotides at a position. Also, the PEAKS program is not capable of analyzing large-scale data sets: it is a web-based tool, and it is cumbersome to integrate a systematic data mining process based on the identified candidates using this web program. The original LDSS algorithm has not been tested on prokaryotic promoters. Also, it does not provide a general way to evaluate computationally which genes among the large initial number of mathematically equivalent localized motif peaks are likely co-regulated in a real living system. We have developed an improved algorithm, named LDSS-P, that identifies conserved short motifs that are shared in the promoters of unselected gene sets and in large transcriptome data sets, and is able to evaluate whether these groups of genes are actually co-regulated in the organism. That is, the new algorithm captures both motif-position similarity, and co-expression probability, in a single numerical outcome. This allows one computational approach rather than first identifying all the sets of co-regulated genes, with a subsequent and separate search for shared motifs. A combined spatial and co-expression calculation presents significant challenges for computation time. We made changes to the original LDSS algorithm so we can test all possible 2 to 4 nucleotide combinations that are represented by IUPAC ambiguity codes (6) at each location, thus streamlining the initial comparison processes. Also, LDSS-P employs threading and discretization (modification of variable granularity) to allow parallel computation; this allows efficient calculation using multi-core CPUs and large distributed computing resources such as PC clusters and cloud computing. The LDSS-P program employs a Map-Reduce strategy and the ‘prefix’ parameter function to calculate each unique specified nucleotide space.
In the present paper, we validated the effectiveness of the LDSS (position information) and LDSS-P (position and co-expression) algorithms in bacteria using the plant symbiotic nitrogen-fixing bacterium Sinorhizobium meliloti as the model. We identified several groups of genes that share a short motif at a particular position with respect to mapped 5′ transcription start sites, and that are co-regulated in large scale transcriptome data sets. Our informatics analysis and follow-up experimental data indicate that the cell cycle regulator CtrA may recognize an expanded motif shifted from its previously reported consensus.
MATERIALS AND METHODS
Preparation of a promoter and 5′ untranslated region (UTR) data set
Approximately 37.8 million Raw reads from empirically defined 5′ transcription start sites (7) were mapped onto the reference genomic sequences of S. meliloti 1021 (GenBank accession numbers AL591688, AE006469 and AL591985 for chromosome, pSymA and pSymB, respectively) using the Mosaik program (8). The 5′ TSS-enriched library was created from RNA samples representing 16 different growth and stress conditions (Supplementary Table S1); this set did not include samples from nitrogen-fixing nodules. It is anticipated that because many genes are co-transcribed in operons, the number of TSS will be substantially lower than the number of annotated genes (9). A matrix that contains the count of mapped TSS reads at each position was created for each of the 3 replicons. Locations with fewer than 25 mapped TSS sites were removed from the matrix. Using the refined matrices, we employed Savitsky–Golay's smoothing differentiation with 8-point convolution (10) to identify significant TSS peaks by removing random spikes. Although some S. meliloti genes have long 5′ untranslated regions, a TSS peak was assigned to a gene only when an identified peak located within 150 bp from the 5′ end nucleotide of a coding sequence (CDS) to be sure a TSS is not mistakenly assigned to an unrelated CDS. In the event two or more TSS peaks were found within the 150 bp, all TSS peaks were included for later analyses, except for those with a read count lower than 20% and/or those located within a 50 bp radius of the highest peak. In the present study, we focused on the most accurate TSSs; these were assigned to 2217 out of 6235 annotated CDS. The non-redundant promoter and 5′-UTR data set (‘promoter data set’ hereafter) was then created by extracting the 100 bp upstream and 50 bp downstream sequences from each TSS.
LDSS-P analysis
The LDSS-P approach includes two components: LDSS (position) and co-expression analyses. All the programs described here are available through our website (http://cmgm.stanford.edu/biology/long/files/ichida2016/LDSS-P_dist.zip).
LDSS calculation
LDSS analysis was carried out with a combination of C++ programs incorporating the algorithm described in the previous paper (3). All programs were compiled using Intel C++ compiler version 11.1.059 (Intel, Santa Clara, CA, USA) and were run on a Linux PC cluster (Fujitsu Primergy RX200 S5, Intel Xeon X5570 2.93 GHz processors, 12 GB memory, CentOS 5.4, 48 nodes, 384 cores in total). All possible hexamer sequences were searched for possible placement in each sequence position in the promoter data set (described above). In addition to the standard four nucleotides, combinations of all 2 and 3 nucleotide possibilities were included for positions 2–5 within the hexamer. The possible number of ambiguous nucleotides for any one site is not limited in the present analysis, thus exact matches and corresponding ambiguous hexamers are all counted. For example, we established a set of all occurrences of the hexamer ATGCAT. We also formed a set of all sequences AWGCAT and AHGCAT, where W can be either T or A, and H can be T, A, or C (or ‘not-G’), and so on (6) for all 2 and 3 nucleotide combinations in positions 2–5. In order to assure the effective motif length and specificity at the border, ambiguity codes were not permitted at position 1 and 6. This allowed incorporation of ambiguity in the first pass of the search. The occurrence of the hexamer sequences was counted for each nucleotide position relative to the TSS. A running moving average was computed with a bin of 5 bp, and used to identify localized motif peaks, which would indicate a sequence position where a motif is significantly localized. The average count (height) divided by standard deviation of counts at all possible genome positions within the start-site database was designated as ‘fold standard deviation’ (FSD). The left and right borders of a peak were adjusted to maximize FSD within the range of 5 bp upstream and downstream from that sequence position. All localized motif peaks were loaded into a data set that included for each entry: the sequence; left- and right-border positions; peak height; and average height within and outside of the peak. For motifs found by incorporating ambiguity, the best corresponding ATGC-only peak (that with the highest FSD) was annotated as the representative ATGC-only motif.
Co-expression analysis
A gene expression vector was created from 261 SymbiosisChip transcriptome results from 24 projects (described in Results and Supplementary Table S1). Hybridization signals were normalized for each project using the RMA method (11). The normalized values were combined for each gene to create global expression vectors. Distance between two genes X and Y was defined as (1 - Pearson's linear correlation coefficient), and is expressed by following equation:
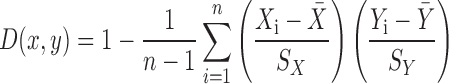 |
Where sX and sY denotes standard deviation of all hybridization signals from gene X and Y, respectively. Distances between pairwise combinations of all 6091 annotated S. meliloti genes on the SymbiosisChip were calculated.
To identify localized motif peaks corresponding to genes with analogous (the direction and level of increase/decrease is similar) transcription patterns, we calculated the distances from all possible pairwise combinations of genes in that peak (a peak reflects a group of genes having the motif at one particular position from TSS). As a control, the same number of genes was randomly chosen from the entire database set, and from these a distance population was constructed. A Mann–Whitney U test was used to determine whether the pairwise distance between the genes in a peak was significantly different from pairwise distances for a randomly chosen population. The test was repeated 1000 times with different random populations. For these data, the P-value represents the percentage of trials that were not significant.
Pathway representation and matrix-based motif discovery using MEME to identify additional conserved motifs
The annotations of S. meliloti 1021 genes were obtained from the EnsemblBacteria database Release 21 (12). Genes were mapped to the reference pathways using a public KEGG automatic annotation server version 1.6a (http://www.genome.jp/tools/kaas/). Each LDSS-P positive peak was subjected to statistical representation analysis against each one of the pathways using the ‘representation’ program. Briefly, the representation factor was defined as the number of overlapping genes divided by the expected number of overlapping genes drawn from an LDSS-P positive peak and all 2650 genes in the promoter data set. The representation factor and the hypergeometric probability was calculated according to a previous report (13). LDSS-P positive peaks with the highest representation factor were subjected to a search with MEME analysis to find extended conserved motifs in the promoter. The ‘nr_prom’ is a program to create the input promoter sequences from a LDSS-P positive peak. MEME Suite software Version 4.9.1 patch1 (compiled from source code; (14)) was used to identify conserved motifs within a set of promoters with options (settings: -dna -maxsize 60 000 -mod zoops -nmotifs 3 -minw 6 -maxw 50 -revcomp).
Bacterial one-hybrid assay
The omega-based bacterial one-hybrid (B1H) assay was carried out as described (15). The plasmids pB1H2w2-zif268 (Plasmid #18045) and pH3U3-zif268 (Plasmid #18046) were obtained from Addgene (Cambridge, MA, USA). An attR1-ChroramphenicolR-ccdB-attR2 cassette (Gateway vector conversion system, Invitrogen, Carlsbad, CA, USA) was introduced in frame between KpnI and XbaI sites of pB1H2w2-zif268 to create a Gateway-compatible derivative, pHiS577. The coding sequence of ctrA (SMc00654) and the flanking two attP sites were amplified from the Gateway ORFeome library (16) with primers HI571 (TCGCGTTAACGCTAGCATGGATCTC) and HI572, and subjected to a Gateway LR reaction. A D51E mutation was introduced to ctrA by site-directed mutagenesis with primers HI939 (GGTTCAGTTCGAGGAGAATGATGTCG) and HI940 (TCCTCGAACTGAACCTGCCGGACAT) using a CloneEZ PCR cloning kit (GenScript, Piscataway, NJ, USA). The resulting plasmid was designated pHiS636. To construct reporter fusions, a 45 bp sequence containing the CtrA binding motif in the upstream region of ctrA-P2 promoter (17) was used as the scaffold. Two oligonucleotides (TACACCCGGGCGGCC TCGATACACCTTGCCAGAGTGAATCAGAATTTGTTAACCATTTGGCGAATTCTTTACACTTT and its complementary sequence; the underline denotes the TTAACCMT motif identified by LDSS-P and the first and last 15 bp are homologous to pH3U3-zif268 for CloneEZ reaction) were synthesized, dissolved at 100 μM in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) containing 50 mM sodium chloride, and heat denatured at 95°C for 5 min, then gradually cooled down to room temperature. The resulting double-stranded fragment was cloned between NotI and EcoRI sites of pH3U3-zif268 by CloneEZ. Plasmids with a base substitution on and adjacent to the TTAACCMT motif were created in similar way.
To test the interaction between RpoZ-CtrA(D51E) fusion and the CtrA binding motifs, Escherichia coli US0 (hisB− pyrF− rpoZ−; Addgene #18049) was transformed with pHiS577 and pHiS636 or its mutated derivatives. The overnight culture was washed twice with 10 mM magnesium sulfate and spotted on M9 plates supplemented with 0.2% D-glucose, 2.5 μg/ml thiamine hydrochloride, 50 μg/ml ampicillin and 25 μg/ml kanamycin, and incubated at 30°C for 2 days. LB plates containing the same antibiotics were used as control. Digital images of the plates were taken using a ChemiDoc MP (Bio-rad) under epi-white illumination.
RESULTS AND DISCUSSION
Improved LDSS analysis identifies sequence motifs in bacteria
Previously, hexamer and octamer sequences were shown to work well for LDSS analysis in eukaryotes (4). We used hexamer searches as a starting point for LDSS-P analysis in prokaryotes. For each hexamer sequence, a distribution profile in relation to distance from the TSS was analyzed at a global scale in S. meliloti. This analysis produced 614 656 (42 [combinations of A, C, G, and T at position 1 and 6] × 144 [the 4 nucleotides and the ambiguity codes representing the mixture of 2 (6 codes) and 3 (4 codes) nucleotides at positions 2–5]) distribution profiles with 2 797 994 possible localized motif peaks. Of all possible hexamer motifs, there were 17 970 (0.64%) localized motif peaks containing motifs with no ambiguity codes. The distribution of localized motif peaks is shown in Figure 1. Localized motif peaks were observed most often at 4 major regions: around −95, −40, −15 and +4 from TSS. The regions near −40 and −15 likely correspond to the conserved −35 and −10 regions recognized by prokaryotic sigma factors. The number of genes within the peak (integration) ranged from 2 to 1735 (126.50 ± 115.12; average ± standard deviation). The FSD (see Materials and Methods) ranged from 0.30 to 23.25 (2.49 ± 0.88; average ± standard deviation). Sequence distribution peaks that showed a higher FSD were more symmetrical and well separated from background (data not shown). Localized motif peaks with ambiguity nucleotide codes were assigned to sub-groups based on the peak locations and FSD in their corresponding ATGC-only motif sequences. For example, a grouping process for the AGRACA (R indicates A or G) peak compares the FSD values of corresponding peaks in AGAACA and AGGACA, and designates a unique group identifier consisting of the ATGC-only motif sequence and the left and right border positions of the peak (e.g. AGGACA_−59_−50). This grouping is particularly useful since introducing ambiguity to a motif increases the combination of nucleotides by an order of magnitude and thus would make it more difficult to find the most representative groups from the large number of peaks with similar nucleotide sequences. This grouping information makes it easier to identify the most significant localized motifs from a large variety of similar motifs with ambiguity.
Figure 1.

Distribution of localized motif peak positions. Values along the vertical axis represent cumulative occurrence of LDSS (×) and LDSS-P (•) positive pentamer and hexamer peaks at each position along the horizontal axis. The criteria to identify localized motif (by LDSS) and co-expressed (LDSS-P) peaks are described in the text. Localized motifs identified by LDSS and LDSS-P positive peaks lay in specific regions: for LDSS, peaks tended to occur at around −95, −40, −15, and +4 from TSS; for LDSS-P, peaks typically occurred at around −40 and −15.
We tested whether the improved LDSS algorithm can identify known −35 and −10 motifs with reasonable sensitivity. RpoE2 is an RNA polymerase ECF sigma factor that is responsible for activating the transcription of general stress response genes in S. meliloti. The target genes of RpoE2 have a consensus sequence of GGAAC-N15/16-gcgTTt (lowercase characters indicate less conservation) in their promoters (18). The LDSS profiles of GGAACA, GGAACC, GGAACG and GGAACT, which partly overlaps the −35 motif of the RpoE2 promoter consensus, is shown in Figure 2. The LDSS-P algorithm detected major peaks between −36 and −32 (consisted from 37 genes, FSD 7.621), −36 and −28 (27 genes, FSD 3.376) and −35 and −32 (20 genes, FSD 5.323) from GGAAA, GGAACC and GGAACT, respectively. No significant localized peak was detected from GGAACG. The total number of unique genes in these 3 peaks was 84. We compared these 84 genes with the significantly induced genes by RpoE2 overexpression identified by transcriptome analysis using microarrays (7). Of the 87 genes that were increased more than 2-fold by RpoE2 overexpression and with at least 1 TSS assigned in our promoter data set, 54 were found in the GGAAC(A/C/T) peaks; this represented 62.1% of the gene sets. The transcriptome results should include both direct and indirect targets of RpoE2-polymerase. Our results showed strong matching with the RpoE2-overexpression data, and indicate that almost 2/3 of the implied target genes are directly controlled by RpoE2. This analysis showed that LDSS can identify conserved functional motifs from a set of unclassified bacterial promoters without an instruction set or prior knowledge of targets.
Figure 2.

Distribution profiles of GGAACN, RpoE2 target consensus motifs. Examples of hexamer analysis for GGAACN, which partly overlaps with the RpoE2 consensus motif. The vertical axis indicates the total number of promoters with the motif. The horizontal axis indicates relative position from the transcription start site (position at 0 corresponds to the TSS). Crosses (×) and filled circles (•) indicate raw count and running average, respectively, within a 5 bp window.
Global LDSS-P analysis of S. meliloti
We used a large set of global transcription data for S. meliloti to identify localized motif peaks associated with coordinated transcription. These transcriptomic data were generated using the SymbiosisChip, a custom Affymetrix microarray platform (19), and are all based on comparison between appropriate wild-type and/or empty vector controls and the test construct or condition. These data sets include tests for function of numerous regulatory proteins (Supplementary Table S1).
We created a gene distance matrix by calculating the distance (1 - Pearson's linear correlation coefficient) of normalized signal intensity between all possible pairwise combinations of S. meliloti genes from every test condition. We calculated the distances of all possible pairwise combinations of genes within a peak; treated it as a population; and statistically asked (using a non-parametric Mann–Whitney U test) if a set of genes grouped by LDSS has a significantly closer distance population than that of a randomly chosen group of the same number of sequences (P < 0.05). For each peak, we repeated this test 1000 times with different sets of randomly selected genes to evaluate the robustness of the grouping and calculated a functional P-value, which is the percentage of trials in which the two distance populations showed no significant statistical difference between the LDSS-grouped peak and randomly selected genes. We focused on LDSS peaks with 5 or more genes to obtain a reasonable number of groups. We selected 241 918 peaks (8.65% of all peaks in the previous step) which had an uncorrected functional P-value less than 0.05: these peaks were categorized as LDSS-P positive. The genes corresponding to these LDSS-P positive peaks not only share a hexamer motif at a specific position in promoters but also show similar expression patterns (increase and decrease). Figure 1 shows the distribution of LDSS-P positive motifs that are shared within a group of genes expressed in parallel (note filled circles). We observed two major peaks centered at −39 and −15. Almost all of the peaks upstream of −50 and downstream of −10, including the two peaks near −95 and +4 in the previous step, were rejected after expression pattern-based screening. We implemented a program (‘extract_best_peak_by_distance’) to pick the best LDSS-P positive peaks, namely those with the shortest average distance among each ATGC-only motif group (described above). The process efficiently removed multiple similar ambiguous motifs which might be derived from the same ATGC-only functional element (data not shown). This process selected 4905 most representative peaks from the 103 558 LDSS-P positive peaks, and these peaks are subjected to the pathway representation analysis.
It is a challenging and labor intensive task to narrow down and identify motifs that are shared within a particular group of genes from a large number of LDSS-P positive peaks, even after the grouping process described above. As a test case for the identification of functional hexamer motifs corresponding to a particular groups of genes, we statistically analyzed the correlation between LDSS-P positive peaks and metabolic and signaling pathways. We chose KEGG pathway as the reference because it is one of the most comprehensive, well structured, and generally applicable pathway information data sets (20). We applied a statistical analysis which uses the representation factor (defined as the number of detected overlapping genes divided by the expected number of overlapping genes drawn from two independent peaks, and its hypergeometric probability; (13)) to identify LDSS-P positive motifs which are significantly over-represented in the pathway of interest. The strongest LDSS-P positive peaks (see above) were searched for representation against each of the 148 KEGG pathways, and motifs with hypergeometric probability of less than 0.05 were considered to be significant. Table 1 shows examples of the most significant LDSS-P positive peaks corresponding to KEGG pathways. For each of these, there were four or fewer potential motif sequences (as determined in the previous calculation) so as to avoid too much ambiguity.
Table 1. The most significant LDSS-P positive peaks corresponding to KEGG pathways.
| Motif position | ||||||
|---|---|---|---|---|---|---|
| KEGG ID | Pathway | Motif | from | to | Number of genes | Rep. factora |
| 00010 | Glycolysis / Gluconeogenesis | ARATCG | −89 | −78 | 33 | 6.67 |
| 00030 | Pentose phosphate pathway | GSCRCC | −88 | −80 | 70 | 13.16 |
| 00040 | Pentose and glucuronate interconversions | AASYGC | −55 | −46 | 55 | 11.11 |
| 00051 | Fructose and mannose metabolism | GCHATC | −9 | 2 | 48 | 10.71 |
| 00052 | Galactose metabolism | GKCWAG | −24 | −13 | 55 | 15.00 |
| 00061 | Fatty acid biosynthesis | AWRCCA | −94 | −83 | 29 | 14.29 |
| 00190 | Oxidative phosphorylation | CDTGTT | −44 | −36 | 34 | 4.94 |
| 00240 | Pyrimidine metabolism | AAACBT | −101 | −93 | 17 | 6.12 |
| 00250 | Alanine, aspartate and glutamate metabolism | GCDTGC | −46 | −34 | 64 | 9.30 |
| 00270 | Cysteine and methionine metabolism | GACYSG | −39 | −30 | 42 | 13.79 |
| 00290 | Valine, leucine and isoleucine biosynthesis | TYCCKC | −83 | −75 | 50 | 13.04 |
| 00330 | Arginine and proline metabolism | TTGCSC | −77 | −67 | 38 | 5.80 |
| 00480 | Glutathione metabolism | CWTGYC | −47 | −36 | 96 | 13.79 |
| 00500 | Starch and sucrose metabolism | TRCGCG | −74 | −65 | 33 | 10.00 |
| 00520 | Amino sugar and nucleotide sugar metabolism | AAMARG | 2 | 12 | 72 | 10.26 |
| 00620 | Pyruvate metabolism | ARATCG | −89 | −78 | 33 | 6.38 |
| 00680 | Methane metabolism | TCAHCC | −101 | −93 | 19 | 6.12 |
| 00710 | Carbon fixation in photosynthetic organisms | CCKCCG | −72 | −63 | 47 | 11.11 |
| 00770 | Pantothenate and CoA biosynthesis | AVCGGA | 1 | 12 | 55 | 12.00 |
| 00860 | Porphyrin and chlorophyll metabolism | CWTRCA | −39 | −33 | 44 | 7.89 |
| 00970 | Aminoacyl-tRNA biosynthesis | CCKSAA | −99 | −91 | 52 | 10.71 |
| 01200 | Carbon metabolism | TCAHCC | −101 | −93 | 19 | 2.78 |
| 01210 | 2-Oxocarboxylic acid metabolism | TYCCKC | −83 | −75 | 50 | 9.09 |
| 01212 | Fatty acid metabolism | AWRCCA | −94 | −83 | 29 | 9.09 |
| 02010 | ABC transporters | CKMTTG | −46 | −38 | 117 | 2.06 |
| 02020 | Two-component system | TTWAYC | −40 | −27 | 66 | 6.96 |
| 02040 | Flagellar assembly | TAACSA | −34 | −26 | 22 | 9.68 |
| 03010 | Ribosome | AWAMGC | −15 | −7 | 46 | 9.09 |
| 03018 | RNA degradation | GSKTGC | −81 | −69 | 73 | 22.22 |
| 03030 | DNA replication | CARSGG | −97 | −86 | 53 | 25.00 |
| 03060 | Protein export | TCRMTT | −51 | −37 | 65 | 18.75 |
| 03070 | Bacterial secretion system | TGGDAT | −59 | −52 | 14 | 10.34 |
| 03430 | Mismatch repair | CARSGG | −97 | −86 | 53 | 13.64 |
| 04112 | Cell cycle - Caulobacter | TTWAMC | −36 | −27 | 45 | 10.00 |
| 05134 | Legionellosis | TAACSA | −34 | −26 | 22 | 20.00 |
| M00001 | Glycolysis (Embden-Meyerhof pathway) | THCCGC | −83 | −75 | 39 | 18.75 |
| M00003 | Gluconeogenesis | THCCGC | −83 | −75 | 39 | 27.27 |
| M00004 | Pentose phosphate cycle | CATHTT | −50 | −41 | 33 | 17.65 |
| M00048 | Inosine monophosphate biosynthesis | GCHATC | −9 | 2 | 48 | 33.33 |
| M00144 | NADH: quinone oxidoreductase, prokaryotes | TCMKCT | −98 | −91 | 24 | 10.71 |
| M00178 | Ribosome, bacteria | AWAMGC | −15 | −7 | 46 | 9.09 |
| M00237 | Branched-chain amino acid transport system | TCMTKT | −43 | −34 | 48 | 12.00 |
| M00359 | Aminoacyl-tRNA biosynthesis, eukaryotes | CCSTYG | −47 | −38 | 85 | 16.67 |
aRepresentation factor.
AACCAT may represent an extended CtrA binding motif
As shown in Table 1, a TTWAMC (W and M indicate A/T and A/C, respectively) motif was significantly over-represented at between −36 and −27 from TSS among the cell-cycle regulation pathway genes (ccr04112). We chose this pathway as a model because it is well characterized, and the mechanism and genes for cell cycle regulation are highly conserved in α-proteobacteria. The pathway was drawn based on the findings in Caulobacter crescentus. The KAAS program identified 30 orthologous genes in S. meliloti, and these genes include the essential master cell cycle transcriptional regulators dnaA, gcrA and ctrA and an essential cell-cycle regulated DNA methyltransferase ccrM (21). There were 48 genes in this LDSS-P positive peak in our promoter data set (Supplementary Table S2). We used the promoter sequence of these 48 genes and MEME program to identify the conserved nucleotide motifs that spanned outside the identified TTWAMC motif. The sequence logo (22), which visualizes nucleotide conservation at each position by MEME analysis, is shown in Figure 3.
Figure 3.

Conserved nucleotides in TTWAMC group. The sequence conservation for the TTWAMC motif found at between −37 and −25 from TSS in the S. meliloti database. The nucleotide representation logo was generated using MEME as described in Materials and Methods. The height of each nucleotide in the logo represents the conservation of the nucleotide at each position.
The analysis revealed that there were additional highly conserved nucleotides in two places upstream and downstream of the identified TTWAMC motif: an adenine and thymine (AT) were immediately adjacent to the 3′ end of TTWAMC, plus 5 nt upstream we observed a second motif, GTTAACC. The TTAACC part of the GTTAACC motif was marked positive by the proposed LDSS-P criterion (22 genes; an uncorrected functional P-value of 0.004 for TTAACC between −80 and −77). In contrast, the GTTAAC was not captured due to dissimilar expression patterns (0.129 for GTTAAC between −84 and −78). It is most likely that because LDSS-P relies on the localization of relatively short k-mers, some of such sequences are too common among the promoters or genomic sequences due to the organisms GC bias and other nucleotide preferences. In such a case, it would not be possible to distinguish significant localizations from such background noise.
We found the overall conserved motif (GTTAACC-N5-TTWAMCAT) shows overlap with the reported consensus sequence of CtrA binding motif (TTAA-N7-TTAA; (23)). CtrA is an essential transcriptional regulator that coordinates DNA replication, cell division and polar morphogenesis, and is considered the master cell cycle regulator in α-proteobacteria (24). In S. meliloti, phosphorylated CtrA binds to a consensus sequence TAA-N7-TTAAC based on the analysis in the promoter region of ctrA (17). We hypothesized that the AACCAT is a part of an extended CtrA binding motif.
We tested this hypothesis by asking whether a point mutation in the AACCAT motif changes interaction with CtrA, using a bacterial one-hybrid assay as a reporter for protein–DNA binding. A translational fusion with an RNA polymerase ω subunit, RpoZ, and a constitutively active D51E mutant of S. meliloti CtrA was created and used to determine the interaction between CtrA (D51E) and its binding motifs (with the extended AACCAT sequence). We created a set of pH3U3-derivative plasmids, containing wild-type or mutagenized CtrA binding motifs upstream of ura and pyrF genes driven by a weak minimal promoter elements. In this system, histidine and uracil auxotrophy in the host E. coli strain US0 is (25–29) complemented only when the CtrA (D51E) protein fused with RpoZ binds to the motif. This allows binding of an RNA polymerase holoenzyme to the −35/−10 region of the weak promoter, so transcription can initiate. The degree of complementation (ura and pyrF expression) reflects the stability of interaction between the protein and DNA. We confirmed that the D51E mutation results in enhanced binding to the CtrA binding motif (data not shown). The interactions of CtrA (D51E) with wild-type versus 12 different mutagenized CtrA binding motifs, and with zif268 (the binding sequence of a well characterized zinc-finger transcriptional factor, Zif268, as a negative control; (30)) are shown in Figure 4. The T2C (the second thymine in the TTAACCAT motif was substituted to cytosine), A4G, C5T, C6G, A7G and T8C mutations caused significant loss of CtrA binding, whereas T1C, A3G and C6A did not alter the binding. The C6T mutation caused better binding of CtrA, suggesting that the C6 position is an important determinant for CtrA binding affinity. Unexpectedly, one of the mutations at the flanks of the motif, a thymine to cytosine transversion at 3 bp upstream of the motif (Mut1), resulted in better binding, comparable to that of the C6T (Mut7) mutation. These results reveal that the expanded CAT and the upstream regions in addition to the previously identified TTAAC motif influence the binding of CtrA. Here, LDSS-P analysis was effective in identifying novel functional motifs in promoters in S. meliloti, and we propose it can be used similarly for other bacteria.
Figure 4.

Bacterial one-hybrid assay of CtrA (D51E) and mutated CtrA binding motifs. At left, sequences for mutants 1–12 are shown underneath the newly proposed CtrA binding consensus. Degree of binding is summarized to the right of each sequence (from − for no binding to +++ for the strongest binding). At right, growth of cells on M9 minimal medium or LB rich medium due to successful binding of CtrA to the motif for each mutant.
The combination of LDSS-P plus other matrix-based motif finding tools is very powerful because LDSS-P is tolerant to unrelated promoters that may be present in the input promoter set, thanks to its principle that focuses on the occurrence of short k-mers, and can increase the sensitivity to find less conserved motifs in the subsequent analysis.
Annotation of motifs in the S. meliloti genome with LDSS-P
We noted some groups of genes within LDSS-P peaks that empirically showed similarity in their gene expression patterns. Among the short nucleotide motifs that LDSS found at certain defined distances from TSS, there were two groups: (i) motifs determining transcriptional specificity, which are LDSS-P positive peaks near −39 and −15, consistent with previous reports in rhizobia (7,31) and with the canonical model for the σ70 promoter sequence (a hexamer at −35 separated by 15–21 nucleotide from the −10 hexamer in E. coli; (32)); and (ii) sequences corresponding to peaks at −95 and +4 from the TSS. We infer that these peaks at −95 and +4 are not primarily involved in transcriptional control, at least in the conditions covered by our transcriptome data sets, because these groups of genes did not show significant co-expression in the transcriptional data sets, thus rejected by LDSS-P analysis. We asked whether the peak at +4 is due to nucleotide bias from start codons which are located within 6 nucleotides of the TSS, however, we found only 12 of such genes in our promoter data set. Therefore, it is unlikely that these peaks are the bias from start codons located near TSS. Another possible explanation for the +4 peak would be a result of nucleotide preference in transcriptional initiation by RNA polymerase. In considering the peak at −90, we note the report that in E. coli, a flexibly tethered alpha subunit C-terminal domain, which recognizes the upstream sequence of the −35 element, can bind non-specifically to more distant DNA sequences, making contacts up to −90 from TSS (33). It is also possible that −95 and +4 peaks influence binding of other regulators, and/or affect other levels of gene expression control.
Recent advances in genome sequencing technology have enabled rapid acquisition of bacterial whole genome sequences. Since orthologous DNA binding proteins and their target nucleotide sequences are expected to be well conserved between phylogenetically related organisms, we propose that LDSS-P positive peaks in a model organism are likely to be functional in other related species. Indeed, these short motifs may be better conserved than the TSS themselves, due to the selective pressure from motif functionality in protein binding. Therefore, the transcriptional regulatory networks identified in a model organism can be efficiently transferred into non-model species by converting raw TSS locations into LDSS-P positive motifs.
We annotated the S. meliloti 1021 genome with the most significant 4606 LDSS-P positive motifs relative to the corresponding TSS locations. This produced more than 88 000 annotations for the promoter regions of previously annotated coding sequences: each annotation indicates the corresponding LDSS-positive motif; relative distance from TSS; the representing ATGC-only motif peak; and statistical evaluations. The data were stored in standard tab-delimited GFF3 format, and are freely available from our website (http://cmgm.stanford.edu/biology/long/files/ichida2016/). These files can be easily imported into any of the many platforms that support GFF3, including GBrowse and NCBI Graphical Sequence Viewer (http://www.ncbi.nlm.nih.gov/tools/sviewer/), one of the most widely used web-based genome browsers (Supplementary Figure S1).
Supplementary Material
Acknowledgments
The authors thank Melanie Barnett, Robert Fisher, Claus Lang, and Alisa Lehman (ordered alphabetically) for providing unpublished SymbiosisChip results. We also thank Melanie Barnett, Claus Lang, and Lucinda Smith for critical reading and valuable suggestions. HI was supported by Research Fellowship for Young Scientists and Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science (JSPS).
FUNDING
Japan Society for the Promotion of Science [KAKENHI to H.I.]; National Institutes of Health (NIH) [R01GM093628 to S.R.L.]. Funding for open access charge: Stanford University research account from NIH [R01GM093628 to S.R.L.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Browning D.F., Busby S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 2.Das M.K., Dai H.K. A survey of DNA motif finding algorithms. BMC Bioinformatics. 2007;8(Suppl. 7):S21. doi: 10.1186/1471-2105-8-S7-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto Y.Y., Ichida H., Matsui M., Obokata J., Sakurai T., Satou M., Seki M., Shinozaki K., Abe T. Identification of plant promoter constituents by analysis of local distribution of short sequences. BMC Genomics. 2007;8:67. doi: 10.1186/1471-2164-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamamoto Y.Y., Ichida H., Abe T., Suzuki Y., Sugano S., Obokata J. Differentiation of core promoter architecture between plants and mammals revealed by LDSS analysis. Nucleic Acids Res. 2007;35:6219–6226. doi: 10.1093/nar/gkm685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellora N., Farre D., Mar Alba M. PEAKS: identification of regulatory motifs by their position in DNA sequences. Bioinformatics. 2007;23:243–244. doi: 10.1093/bioinformatics/btl568. [DOI] [PubMed] [Google Scholar]
- 6.Nomenclature Committee of the International Union of Biochemistry. Nomenclature for incompletely specified bases in nucleic acid sequences. Recommendations 1984. Eur. J. Biochem. 1985;150:1–5. doi: 10.1111/j.1432-1033.1985.tb08977.x. [DOI] [PubMed] [Google Scholar]
- 7.Schlüter J.P., Reinkensmeier J., Barnett M.J., Lang C., Krol E., Giegerich R., Long S.R., Becker A. Global mapping of transcription start sites and promoter motifs in the symbiotic alpha-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics. 2013;14:156. doi: 10.1186/1471-2164-14-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee W.P., Stromberg M.P., Ward A., Stewart C., Garrison E.P., Marth G.T. MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One. 2014;9:e90581. doi: 10.1371/journal.pone.0090581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becker A., Berges H., Krol E., Bruand C., Ruberg S., Capela D., Lauber E., Meilhoc E., Ampe F., de Bruijn F.J., et al. Global changes in gene expression in Sinorhizobium meliloti 1021 under microoxic and symbiotic conditions. Mol. Plant Microbe Interact. 2004;17:292–303. doi: 10.1094/MPMI.2004.17.3.292. [DOI] [PubMed] [Google Scholar]
- 10.Savitzky A., Golay M.J.E. Smoothing and fifferentiation of fata by simplified least squares procedures. Anal. Chem. 1964;36:1627–1639. [Google Scholar]
- 11.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 12.Kersey P.J., Allen J.E., Christensen M., Davis P., Falin L.J., Grabmueller C., Hughes D.S., Humphrey J., Kerhornou A., Khobova J., et al. Ensembl Genomes 2013: scaling up access to genome-wide data. Nucleic Acids Res. 2014;42:D546–D552. doi: 10.1093/nar/gkt979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S.K., Lund J., Kiraly M., Duke K., Jiang M., Stuart J.M., Eizinger A., Wylie B.N., Davidson G.S. A gene expression map for Caenorhabditis elegans. Science. 2001;293:2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- 14.Bailey T.L., Williams N., Misleh C., Li W.W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noyes M.B., Meng X., Wakabayashi A., Sinha S., Brodsky M.H., Wolfe S.A. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res. 2008;36:2547–2560. doi: 10.1093/nar/gkn048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroeder B.K., House B.L., Mortimer M.W., Yurgel S.N., Maloney S.C., Ward K.L., Kahn M.L. Development of a functional genomics platform for Sinorhizobium meliloti: construction of an ORFeome. Appl. Environ. Microbiol. 2005;71:5858–5864. doi: 10.1128/AEM.71.10.5858-5864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnett M.J., Hung D.Y., Reisenauer A., Shapiro L., Long S.R. A homolog of the CtrA cell cycle regulator is present and essential in Sinorhizobium meliloti. J. Bacteriol. 2001;183:3204–3210. doi: 10.1128/JB.183.10.3204-3210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauviac L., Philippe H., Phok K., Bruand C. An extracytoplasmic function sigma factor acts as a general stress response regulator in Sinorhizobium meliloti. J. Bacteriol. 2007;189:4204–4216. doi: 10.1128/JB.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnett M.J., Toman C.J., Fisher R.F., Long S.R. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16636–16641. doi: 10.1073/pnas.0407269101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Nisco N.J., Abo R.P., Wu C.M., Penterman J., Walker G.C. Global analysis of cell cycle gene expression of the legume symbiont Sinorhizobium meliloti. Proc. Natl. Acad. Sci. U.S.A. 2014;111:3217–3224. doi: 10.1073/pnas.1400421111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider T.D., Stephens R.M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quon K.C., Marczynski G.T., Shapiro L. Cell cycle control by an essential bacterial two-component signal transduction protein. Cell. 1996;84:83–93. doi: 10.1016/s0092-8674(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 24.Pini F., De Nisco N.J., Ferri L., Penterman J., Fioravanti A., Brilli M., Mengoni A., Bazzicalupo M., Viollier P.H., Walker G.C., et al. Cell cycle control by the master regulator CtrA in Sinorhizobium meliloti. PLoS Genet. 2015;11:e1005232. doi: 10.1371/journal.pgen.1005232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugawara M., Epstein B., Badgley B., Unno T., Xu L., Reese J., Gyaneshwar P., Denny R., Mudge J., Bharti A.K., et al. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol. 2013;14:R17. doi: 10.1186/gb-2013-14-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneiker-Bekel S., Wibberg D., Bekel T., Blom J., Linke B., Neuweger H., Stiens M., Vorholter F.J., Weidner S., Goesmann A., et al. The complete genome sequence of the dominant Sinorhizobium meliloti field isolate SM11 extends the S.meliloti pan-genome. J. Biotechnol. 2011;155:20–33. doi: 10.1016/j.jbiotec.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 27.Reeve W., Chain P., O'Hara G., Ardley J., Nandesena K., Brau L., Tiwari R., Malfatti S., Kiss H., Lapidus A., et al. Complete genome sequence of the Medicago microsymbiont Ensifer(Sinorhizobium)medicae strain WSM419. Stand Genomic Sci. 2010;2:77–86. doi: 10.4056/sigs.43526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Margaret I., Becker A., Blom J., Bonilla I., Goesmann A., Gottfert M., Lloret J., Mittard-Runte V., Ruckert C., Ruiz-Sainz J.E., et al. Symbiotic properties and first analyses of the genomic sequence of the fast growing model strain Sinorhizobium fredii HH103 nodulating soybean. J. Biotechnol. 2011;155:11–19. doi: 10.1016/j.jbiotec.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 29.Schuldes J., Rodriguez Orbegoso M., Schmeisser C., Krishnan H.B., Daniel R., Streit W.R. Complete genome sequence of the broad-host-range strain Sinorhizobium fredii USDA257. J. Bacteriol. 2012;194:4483. doi: 10.1128/JB.00966-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christensen R.G., Gupta A., Zuo Z., Schriefer L.A., Wolfe S.A., Stormo G.D. A modified bacterial one-hybrid system yields improved quantitative models of transcription factor specificity. Nucleic Acids Res. 2011;39:e83. doi: 10.1093/nar/gkr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLellan S.R., MacLean A.M., Finan T.M. Promoter prediction in the rhizobia. Microbiology. 2006;152:1751–1763. doi: 10.1099/mic.0.28743-0. [DOI] [PubMed] [Google Scholar]
- 32.Gruber T.M., Gross C.A. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- 33.Saecker R.M., Record M.T., Jr, Dehaseth P.L. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011;412:754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.