

Abstract
A series of 5’-O-[N-(salicyl)sulfamoyl]-2-aryl-8-aza-3-deazaadenosines were designed to block mycobactin biosynthesis in Mycobacterium tuberculosis (Mtb) through inhibition of the essential adenylating enzyme MbtA. The synthesis of the 2-aryl-8-aza-3-deazaadenosine nucleosides featured sequential copper-free palladium-catalyzed Sonogashira coupling of a precursor 4-cyano-5-iodo-1,2,3-triazolonucleoside with terminal alkynes and Minakawa-Matsuda annulation reaction. These modified nucleosides were shown to inhibit MbtA with apparent Ki values ranging from 6.1 to 25 nM and to inhibit Mtb growth under iron-deficient conditions with minimum inhibitory concentrations ranging from 12.5 to >50 μM.
Keywords: 8-Aza-3-deazaadenosine, Modified Nucleoside, Tuberculosis, Siderophore Biosynthesis, Adenylation Inhibitor
1. Introduction
Tuberculosis (TB) is an infectious disease primarily caused by the bacterium Mycobacterium tuberculosis (Mtb) that recently surpassed HIV as the leading cause of infectious disease mortality [1]. Mtb is easily spread from an actively infected individual by the aerosol route. Upon inhalation Mtb quickly replicates and becomes encased in granulomatous lesions in the lungs. A majority of healthy individuals infected with Mtb are asymptomatic and able to effectively contain, but not clear, the bacteria in these granulomas. Latently infected individuals provide a vast reservoir of the disease and include nearly one-third of the global population. However, individuals – who have an impaired immune system from aging, malnutrition, or co-infection with HIV – are much more likely to develop active TB or reactivate a latent infection. Active TB infections ensue when the granulomas rupture, releasing the bacteria into the sputum leading to the clinical manifestations of hemoptysis and cachexia. Treatment of simple drug-susceptible TB is very challenging, compared to most bacterial infections, requiring 6–9 months of a four-drug regimen comprised of isoniazid, rifampicin, ethambutol, and pyrazinamide [2]. The underlying cause of the persistence and drug tolerance of Mtb in vivo that necessitates this long treatment course is still not fully understood, but is likely multifactorial resulting from variability in the lesion environment and phenotype of individual bacteria [3, 4]. Considering the unique challenges posed by drug susceptible TB, the emergence and dissemination of multidrug resistance TB (MDR-TB) and extensively drug resistant TB (XDR-TB), which are minimally resistant to the two most effective antitubercular agents – isoniazid and rifampicin, is a global health crisis [5]. The European region alone accounts for more than a quarter of the global MDR-TB burden [6]. In order to bring TB back under control, a unified effort will be required to develop improved diagnostics, effective vaccines, and new anti-tubercular agents with novel modes of action.
Mtb requires iron, a trace micronutrient that is highly restricted in a mammalian host, to establish and maintain an infection. To circumvent the nutritional immunity imposed by the host, Mtb synthesizes and secretes a family of small-molecule iron chelators or siderophores known as the mycobactins that extract iron (Fe3+) from host proteins [7]. The Fe3+-mycobactin complex is then imported back into the bacterium through a specialized transport system and the iron is reductively released from the siderophore. The mycobactins share a common biosynthetic pathway for construction of the conserved peptidic core using a mixed nonribosomal peptide synthetase–polyketide synthase (NRPS-PKS) assembly line of six proteins designated MbtA through MbtF [8]. The nucleoside derivative 5’-O-[N-(salicyl)sulfamoyl]adenosine (Sal-AMS, 2, Fig. 1) is a potent nanomolar inhibitor of MbtA, which catalyzes the first committed step in mycobactin synthesis, through the activation of salicylic acid to the mixed anhydride salicyl-AMP intermediate (1) and subsequent loading onto MbtB [9-11]. Sal-AMS blocks synthesis of the mycobactins and correspondingly inhibits growth of Mtb under iron-deficient conditions in vitro and in vivo [12]. Comprehensive SAR studies of 2 demonstrated the N-(salicyl)sulfamoyl moiety is essential for activity and only conservative substitutions to the salicyl ring were permitted [13]. By contrast, the nucleoside is substantially more tolerant to modification with addition of nonpolar aromatic substituents at C-2 exemplified by 2-phenyl-Sal-AMS (3) providing the most potent Sal-AMS derivatives [14]. These studies also showed the N-3 nitrogen atom of adenosine is dispensable for activity [14].
Figure 1.

Structures of Sal-AMP (1), Sal-AMS (2) and 2-phenyl analog of Sal-AMS (3).
To extend the SAR investigation of base-modified Sal-AMS analogues we describe herein the synthesis and evaluation of a series of compounds bearing the isosteric 8-aza-3-deazaadenine nucleobase with various 2-aryl substituents (6a–f, Fig. 2) that uniquely capitalize on the existing SAR. The 8-aza-3-deazaadenine nucleosides have several useful attributes over adenosine and other modified purine nucleosides as elaborated below including stability to cyclonucleoside degradation, lack of intrinsic biological activity, and improved fluorescent properties. Purine nucleoside analogues containing an activated 5’-leaving groups like a sulfamate as found in Sal-AMS are prone to cyclonucleoside formation through attack of the N-3 purine atom onto the C-5’ ribose to afford a 3,5’-cyclonucleoside [15]. This undesired degradation pathway is prevented in 8-aza-3-deazaadenines due to lack of a N-3 atom. As first reported by Franchetti, 8-aza-3-deazaadenosine is devoid of biological activity suggesting it binds poorly to ATP-utilizing proteins, a feature that we exploit to minimize potential off-target toxicity [16].
Figure 2.

Structures of 8-aza-3-deazaadenosine (4), 8-aza-3-deazaguanosine (5), and 2-aryl-8-aza-3-deazaadenine analogs of Sal-AMS (6a–f).
2. Results and discussion
2.1 Chemistry
There are two general synthetic approaches for the preparation of 8-aza-3-deazaadenosine (4) and the homologous guanosine derivative (5) that have been reported in the literature: (1) glycosylation of a protected sugar with a triazolo[4,5-c]pyridine heterocycle and (2) annulation onto a 1H-[1,2,3]triazole-1-β-d-ribofuranoside derivative generated from the glycosylation of the corresponding aglycone, or the 1,3-dipolar cycloaddition of a ribofuranosyl azide to an appropriate dipolarophile, or the Dimroth reaction. The glycosylation strategy suffers from poor selectivity resulting in a mixture of N(1), N(2) and N(3) regiosomeric products [16,17] while the latter procedure involving elaboration of a pre-formed triazole does not allow facile incorporation of C-6 aryl substituents [18-22]. We elected to adapt methodology first reported by Minakawa and Matsuda for the construction of 3-deazapurine nucleosides involving nucleophilic cyclization of 4-carbamoyl- or 4-cyanosubstitued derivatives of 5-ethynyl-1-β-D-ribofuranosylimidazole [23,24]. We were inspired by the successful application of this method for the preparation of related carbocyclic 8-aza-3-deazainosine derivatives reported by Agrofoglio and co-workers [25]. Herein we present the synthesis of protected 5-alkynyl-1-β-D-ribofuranosyl-1H-[1,2,3]triazole-4-carbonitriles and their ring closure leading to the corresponding 2-aryl-8-aza-3-deazaadenosine analogues using a modified Minakawa-Matsuda annulation strategy.
Synthesis of the triazole nucleoside building block 9 was accomplished as shown in Scheme 1 starting from 5-amino-4-carboxamide-1,2,3-triazole nucleoside (7) prepared from 2,3-O-isopropylidene-β-d-ribofuranosyl azide as reported through Dimroth reaction with cyanoacetamide [26-28]. Dehydration of the amide in 7 to the respective nitrile 8 was accomplished by treatment with 4-toluenesulfonyl chloride [28]. Subsequent diazotizationiodination of 8 with diiodomethane and isoamyl nitrite afforded 9 in 75% over two steps, which was superior the inverse reaction sequence via 10 that provided a lower 41% overall yield.
Scheme 1.

Synthesis of compounds 6a–f. Reagents and conditions: (i) Pyr, TsCl, rt; (ii) CH2I2, isoamyl nitrite, 100 °C; (iii) alkyne, K2CO3, (PhCN)2PdCl2, 1,4-dioxane, H2O, 55–65 °C; (iv) NH3–MeOH, 80 °C; (v) NH3–DME, 85 °C; (vi) MeONa–MeOH, 5 °C, then Dowex 50WX8-100; (vii) NH2SO2Cl, MeCN, DMA, rt; (viii) salicylic acid, CDI, MeCN, 60 °C, then substrate, DBU, rt; (ix) 4:1 TFA–H2O, 5 °C.
The Sonogashira coupling of 9 was first optimized with phenylacetylene. We explored a wide variety of conditions, catalysts, and additives (Supplementary data, Table S1) [29]. Optimal conditions were found using bis(benzonitrile)palladium(II) chloride as reported by Minakawa and co-workers [30], but employing K2CO3 as base in 1,4-dioxane with 10 equivalents of H2O at 60 °C to afford 11a in 69% yield. These conditions minimized formation of homocoupled products (not shown) as well as hydrodehalogenated 12 and were used for the other terminal alkynes to afford products 11b–f in 43–77% yield. The requisite terminal alkynes, except phenylacetylene and 1-ethynyl-4-methylbenzene, were prepared by deprotection [31] of their trimethylsilyl precursors obtained using previously reported procedures [31,32] (Supplementary data).
We undertook the key Minakawa–Matsuda annulation of 11a using MeOH saturated with NH3, 80 °C [23,24]. However, we obtained the 4-methoxy-6-phenyl cyclized product 13 (68% yield) due to competitive reaction with methanol. Thus, we next examined non-nucleophilic solvents including 1,4-dioxane, THF, and 1,2-dimethoxyethane (DME). Using 1,4-dioxane saturated with NH3, we obtained at 120 °C an encouraging 29% yield of the desired product 14a along with 61% unreacted 11a. The yield of 14a was further improved to 58% (along with 32% recovered 11a) employing THF saturated with NH3. We attributed the improved yield to the greater solubility of ammonia in THF. Based on this hypothesis, we saturated DME with ammonia at −60 °C then added 11a and heated at 85°C to obtain 14a in an impressive 95% yield. This optimized method was then successfully used for the synthesis of compounds 14b–f.
Methanolysis of compounds 14a–f afforded their corresponding 5’-deacetylated congeners 15a–f, which were then converted to 5’-O-sulfamoyl derivatives 16a–f by treatment with sulfamoyl chloride in MeCN–DMA [33]. These sulfamates 16a–f were coupled to salicylic acid using CDI activation and DBU as a base in MeCN to provide 17a–f [13]. Deprotection of the isopropylidene acetal was accomplished with 80% aqueous TFA and the final products 6a–f were isolated as the triethylammonium salts following silica gel chromatography with 0.5% triethylamine as eluent, which were all greater than 99% pure as determined by HPLC.
We determined the fluorescence properties of compounds 6a–f in MeOH and in aqueous solutions (Table 1). The emission maxima measured for all compounds in MeOH were in the range λmax 409-420 nm (Stokes shifts 95.5-111.5 nm), and in H2O in the range λmax 420-442 nm (Stokes shifts 106.5-137.5 nm). These results are comparable with data given previously for 8-aza-3-deazaadenosine (4) in H2O (λmax 430 nm, Stokes shift 140 nm) [17]. Compounds 6a and 6d–f in MeOH exhibited relatively high values of fluorescence quantum yield (ΦF = 0.32-0.5, compared with ΦF = 1 assumed for 2-aminopurine), while compounds 6b and 6c were less fluorescent (ΦF = 0.06 and 0.11). In contrast, the quantum yield values of 6a–f in H2O were decreased 30- to 250-fold.
Table 1.
Fluorescence properties of compounds 6a–f.
| Compd | Fluorescence emissiona (MeOH) |
Fluorescence emissiona (H2O) |
||
|---|---|---|---|---|
| λmax (nm) | Φ F b | λmax (nm) | Φ F b | |
| 6a | 419 | 0.37 | 433 | 0.0015 |
| 6b | 412 | 0.06 | 433 | 0.0018 |
| 6c | 420 | 0.11 | 442 | 0.002 |
| 6d | 409 | 0.32 | 422 | 0.0019 |
| 6e | 412 | 0.38 | 420 | 0.0025 |
| 6f | 418 | 0.5 | 425 | 0.007 |
Excitation at 305 nm.
Fluorescence quantum yields calculated relative to 2-aminopurine (ΦF = 1).
2.2. Enzyme inhibition and antitubercular activity
Inhibitors 6a–f were evaluated for enzyme inhibition against recombinant MbtA under initial velocity conditions as previously described (see Experimental Section) [34]. The apparent inhibition constants (appKi) were determined by fitting the concentration–response plots to the Morrison equation (eq 1, see Experimental Section) since all compounds exhibited tight-binding behavior. The appKi values ranged from 6.1 to 25 nM (Table 2). The first analogue in the series 6a was 31-fold less potent than the isosteric analogue 2-phenyl-Sal-AMS (3) demonstrating simultaneous deletion of the N-3 atom and introduction of an N-8 atom in 6a was not well tolerated. Nonetheless, 6a is still an exceptionally potent compound and the appKi vastly underestimates the true potency since the assay was performed using supersaturating concentrations of all substrates. Introduction of p-methyl, p-fluoro, and ptrifluoromethyl in 6b, 6c, and 6d, respectively, had a relatively negligible impact on potency and these analogues were only 2–3 fold less potent than 6a. Introduction of the 2-naphthyl substituent in 6e provided the most potent 8-aza-3-deazaadenine analogue with an appKi of 6.1 nM commensurate with 2 while further modification of the naphthyl moiety with a 6-methoxy substituent in 6f was not beneficial leading to a 2-fold loss of potency relative to 6e.
Table 2.
Enzyme inhibition and antimycobacterial activity of 6a–f.

| |||
|---|---|---|---|
| Inhibitor | R | appKi (nM)a | MIC (μM)b |
| 2 | nac | 6.6 ± 1.5d | 0.39d |
| 3 | nac | 0.27 ± 0.07d | 0.049d |
| 6a |
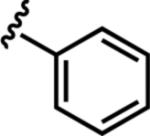
|
8.4 ± 1.0 | 25 |
| 6b |
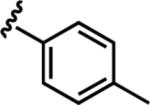
|
16.9 ± 1.9 | 25 |
| 6c |
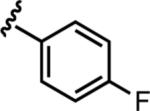
|
13.7 ± 2.3 | 19 |
| 6d |
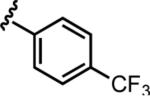
|
25.0 ± 1.9 | >50 |
| 6e |
|
6.1 ± 0.7 | 12.5 |
| 6f |
|
10.5 ± 1.2 | 19 |
Assay performed with 7 nM MbtA, 10 mM ATP, 250 μM salicylic acid, 1 mM PPi.
M. tuberculosis H37Rv grown in glycerol-alanine salts (GAS) medium without ferric ammonium citrate at pH 6.6.
not applicable.
reference 14.
Next, we evaluated these analogs against whole-cell M. tuberculosis H37Rv under iron-deficient and iron-replete conditions as previously described [11]. These antibacterial agents operate by a unique mechanism of action and target iron acquisition by inhibition of siderophore biosynthesis. Under iron-replete conditions, none of the compounds displayed any activity at the maximum concentration evaluated (50 μM) against whole-cell M. tuberculosis H37Rv consistent with their designed mechanism of action. Activity was revealed only under iron-deficient conditions. The minimum inhibitory concentrations (MIC) that resulted in complete inhibition of observable growth under iron-deficient conditions are shown in Table 1. The antitubercular activity of 6a–f excluding 6d ranged between 12.5 and 25 μM, consistent with their relatively flat biochemical SAR. Compared to the lead compound 2, all of the compounds showed considerable loss in whole-cell activity despite displaying similar biochemical potency. These results suggest the nucleoside modification adversely impacted cellular accumulation.
3. Conclusion
The series of 2-aryl-8-aza-3-deazaadenine nucleobase analouges of 5’-O-[N-(salicyl)sulfamoyl]adenosine (2) were synthesized as siderophore inhibitors based on prior SAR studies and evaluated for biochemical activity against MbtA and whole-cell activity under iron-deficient conditions with M. tuberculosis H37Rv. We developed an optimized route to the 2-aryl-8-aza-3-deazaadenosine nucleosides using a modified Minakawa–Matsuda annulation strategy that was highly efficient and enabled facile introduction of C-2 aryl substituents. The SAR studies revealed concurrent introduction of an N-8 atom and deletion of the N-3 atom in the purine base was detrimental to biochemical potency and whole-cell activity. Despite these shortcomings, the compounds showed high chemical stability and promising fluorescent properties that could be useful to study mycobacterial accumulation and localization.
4. Experimental section
4.1. General chemistry methods
Melting points were determined on MEL-TEMP II capillary melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker 400 spectrometer operating at 400.1 MHz and 100.6 MHz, respectively, or on Unity 300 Varian spectrometer operating at 300 MHz and 75.4 MHz, respectively. 19F NMR spectra were recorded on a Bruker 400 spectrometer at 376.4 MHz. The chemical shifts are reported in ppm (δ scale). Mass spectra were recorded using ESI-MS Thermo Q Exactive and Bruker micrOTOF-Q mass spectrometers. UV spectra were measured with Beckman Coulter DU 640 spectrophotometer. Fluorescence spectra were measured on a Shimadzu RF-5301 PC fluorescence spectrophotometer (excitation at 305 nm); quantum yields were calculated relative to 2-aminopurine (ΦF = 1) Microwave heating was performed with Ertec-Poland MW reactor. Thin-layer chromatography (TLC) was carried out on Merck precoated 60 F254 silica gel plates, while column chromatography on Merck silica gel 60H (40-63 μm). Anhydrous 1,4-dioxane, tetrahydrofuran (THF) and 1,2-dimethoxyethane (DME) were prepared by stirring with iron(II) sulfate heptahydrate at room temperature, followed by drying with KOH, distillation with calcium hydride (CaH2) and distillation with sodium/benzophenone. Other anhydrous solvents were prepared as follows: MeOH by treatment with magnesium turnings/iodine and distillation, MeCN and CH2Cl2 by distillation with P2O5, pyridine by drying with KOH and distillation with P2O5, dimethylformamide (DMF) by drying with CaH2 and distillation, triethylamine by distillation with CaH2. Anhydrous dimethylacetamide (DMA) was purchased from Acros. MeOH (Acros) and MeCN (J.T.Baker) used for UV and fluorescence spectra were of HPLC grade.
4.2. Chemical synthesis
4.2.1. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-amino-1H-[1,2,3]triazole-4-carbonitrile (8)
4-Toluenesulfonyl chloride (4.437 g, 22.81 mmol) was added to a solution of 7 [11] (3.975 g, 11.64 mmol) in dry pyridine (55 ml). After being stirred at room temperature for 24 h, the reaction mixture was treated with 5% aq NaHCO3 (100 ml) at 0 °C and with CH2Cl2 (400 ml). The organic layer was separated, while the aqueous one was extracted again with CH2Cl2 (300 ml). Combined organic layers were dried with MgSO4 and evaporated. The residue was chromatographed on a silica gel column with EtOAc-hexane (1:2→2:1) as an eluent to afford solid 8 (3.398 g, 90% yield). UV (MeOH) λmax = 247.5 nm (5900). 1H NMR (DMSO-d6) δ 7.42 (s, 2H, NH2), 6.22 (d, J1’,2’=0.8 Hz, 1H, 1’-H), 5.46 (dd, J2’,1’=0.8 Hz, J2’,3’=5.8 Hz, 1H, 2’-H), 4.96 (dd, J3’,2’=5.8 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.37 (m, 1H, 4’-H), 4.02 (dd, J5’a,5’b=11.6 Hz, J5’a,4’=6.0 Hz, 1H, 5’a-H) 3.85 (dd, J5’b,5’a=11.6 Hz, J5’b,4’=7.2 Hz, 1H, 5’-H), 1.97 (s, 3H, OAc), 1.50 and 1.34 (2×s, 6H, 2×CH3). 13C NMR (DMSO-d6) δ 169.90 (CO), 148.51 (C-5), 113.25 ((CH3)2C), 112.81 (CN), 101.19 (C-4), 88.46 (C-1’), 85.28 (C-4’), 83.33 (C-2’), 81.48 (C-3’), 63.41 (C-5’), 26.61 and 24.94 ((CH3)2C), 20.42 (CO-CH3).
4.2.2. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-iodo-1H-[1,2,3]triazole-4-carbonitrile (9)
To a suspension of 8 (628 mg, 1.94 mmol) in diiodomethane (31.18 g, 116.4 mmol) isoamyl nitrite (797 mg, 6.8 mmol) was added. The resulting mixture was stirred at 100 °C for 2.5 h, then it was applied onto a silica gel column. The column was eluted with CH2Cl2 (in order to collect diiodomethane) followed by CH2Cl2-MeOH (9:1). Isolated crude product was purified on a next silica gel column using EtOAc-hexane (1:2→2:1) to give 9 as a solid (704 mg, 83% yield). UV (MeOH) λmax 239 nm (5400). 1 H NMR (DMSO-d6) δ 6.22 (s, 1H, 1’-H), 5.67 (d, J2’,3’=6.0 Hz, 1H, 2’-H), 5.04 (dd, J3’,2’=5.8 Hz, J3’,4’=1.8 Hz, 1H, 3’-H), 4.49 (m, 1H, 4’-H), 3.92 (dd, J5’a,5’b=12.0 Hz, J5’a,4’=5.2 Hz, 1H, 5’-H), 3.82 (dd, J5’b,5’a=11.8 Hz, J5’b, 4’=7.4 Hz, 1H, 5’-H), 1.91 (s, 3H, OAc), 1.53 and 1.36 (2×s, 6H, 2×CH3). 13C NMR (DMSO-d6) δ 169.72 (CO), 128.46 (C-4), 112.93 ((CH3)2C), 112.04 (CN), 95.35 (C-5), 93.75 (C-1’), 86.23 (C-4’), 83.51 (C-2’), 81.16 (C-3’), 62.86 (C-5’), 26.58 and 24.88 ((CH3)2C), 20.36 (CO-CH3). HRMS [M+H]+ calcd for C13H16IN4O5: 435.0160; found: 435.0156.
4.2.3. General procedure for the coupling of 9 with terminal alkynes
A solution of 9 (dried in vacuo over P2O5 at 50 °C) and bis(benzonitrile)palladium(II) chloride (0.1 equiv) in anhyd 1,4-dioxane (20 ml/mmol) was vigorously deoxygenated with argon for 15 min. To the solution was added K2CO3 (1.0-1.3 equiv), water (10 equiv) and alkyne (3 equiv). The reaction mixture was heated at 55-65 °C overnight, then evaporated. The residue was chromatographed on silica gel column using toluene-EtOAc (98:2→95:5 or 95:5→9:1) to give a crude product. It was subsequently rechromatographed on next silica gel column using hexane-EtOAc (9:1 or 9:1→4:1) to isolate pure 11a–f.
4.2.4. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-phenylethynyl-1H-[1,2,3]triazole-4-carbonitrile (11a)
Solid foam, 69% yield. UV (MeCN): λmax 288.5 nm (14500), 306 nm (12600). 1H NMR (DMSO-d6) δ 7.74 (m, 2H, Ph), 7.62-7.52 (m, 3H, Ph), 6.48 (s, 1H, 1’-H), 5.64 (d, J2’,3’=5.8 Hz, 1H, 2’-H), 5.04 (dd, J3’,2’=5.8 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.56 (m, 1H, 4’-H), 4.00 (m, 2H, 5’-H), 1.90 (s, 3H, OAc), 1.54 and 1.37 (2×s, 6H, 2×CH3). 13C NMR (DMSO-d6) δ 169.78 (CO), 132.12, 131.29 and 129.16 (Ph), 127.65 and 122.64 (C-4, C-5), 118.98 (Ph), 113.05 ((CH3)2C), 110.99 (CN), 105.54 (C≡CPh), 93.01 (C-1’), 86.30 (C-4’), 83.80 (C-2’), 81.14 (C-3’), 70.40 (C≡CPh), 63.13 (C-5’), 26.57 and 24.87 ((CH3)2C), 20.29 (CO-CH3). HRMS [M+H]+ calcd for C21H21N4O5: 409.15120; found: 409.15039.
4.2.5. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-(4-methylphenylethynyl)-1H-[1,2,3]triazole-4-carbonitrile (11b)
Oily material, 77 % yield. UV (MeOH): λmax 296 nm (18500), 312 nm (16100). 1H NMR (DMSO-d6) δ 7.62 (d, J=8.0 Hz, 2H, Ph), 7.36 (d, J=8.0 Hz, 2H, Ph), 6.45 (s, 1H, 1’-H), 5.63 (d, J2’,3’=6.0 Hz, 1H, 2’-H), 5.04 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.56 (m, 1H, 4’-H), 4.02 (dd, J5’a,4’=5.0 Hz, J5’a,5’b=11.8 Hz, 1H, 5’a-H), 3.96 (dd, J5’b,4’=6.8 Hz, J5’b,5’a=12.0 Hz, 1H, 5’b), 2.38 (s, 3H, Ph-CH3), 1.90 (s, 3H, OAc), 1.53 and 1.37 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.77 (CO), 141.62 (C-4Ph), 132.06 (Ph) 129.76 (Ph), 127.81 and 122.45 (C-4, C-5), 115.95 (Ph), 113.04 ((CH3)2C), 111.03 (CN), 106.01 (C≡CPh), 92.89 (C-1’), 86.28 (C-4’), 83.80 (C-2’), 81.14 (C-3’), 69.99 (C≡CPh), 63.12 (C-5’), 26.56 and 24.86 ((CH3)2C), 21.22 (Ph-CH3), 20.28 (CO-CH3). HRMS [M+H]+ calcd for C22H23N4O5: 423.16685; found: 423.16635.
4.2.6. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-(4-fluorophenylethynyl)-1H-[1,2,3]triazole-4-carbonitrile (11c)
Oily material, 43% yield. UV (MeOH): λmax 290 nm (16900), 306.5 nm (14200). 1 H NMR (DMSO-d6) δ 7.82 (m, 2H, Ph), 7.40 (m, 2H, Ph), 6.49 (s, 1H, 1’-H), 5.63 (d, J2’,3’=5.6 Hz, 1H, 2’-H), 5.04 (dd, J3’,2’=5.8 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.56 (m, 1H, 4’-H), 3.99 (m, 2H, 5’-H), 1.90 (s, 3H, OAc), 1.53 and 1.37 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.79 (CO), 163.45 (d, JC,F=251.84 Hz, C-4Ph), 134.92 (d, JC,F=8.83 Hz, C-2Ph, C-6Ph), 127.56 and 122.63 (C-4, C-5), 116.61 (d, JC,F=22.74 Hz, C-3Ph, C-5Ph), 115.51 (C-1Ph), 113.07 ((CH3)2C), 110.97 (CN), 104.57 (C≡CPh), 92.99 (C-1’), 86.32 (C-4’), 83.83 (C-2’), 81.15 (C-3’), 70.26 (C≡CPh), 63.14 (C-5’), 26.57 and 24.88 ((CH3)2C), 20.29 (CO-CH3). 19F NMR (DMSO d6) δ −30.69. HRMS [M+H]+ calcd for C21H20N4O5F: 427.14177; found: 427.14114.
4.2.7. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-(4-trifluoromethylphenylethynyl)-1H-[1,2,3]triazole-4-carbonitrile (11d)
Oily material, 69 % yield. UV (MeOH): λmax 287.5 nm (22600), 306 nm (17800). 1H NMR (DMSO-d6) δ 7.97 (d, J=8.4 Hz, 2H, Ph) ,7.90 (d, J=8.4 Hz, 2H, Ph), 6.54 (s, 1H, 1’-H), 5.65 (d, J2’,3’=5.8 Hz, 1H, 2’-H), 5.05 (dd, J3’,2’=5.6 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.57 (m, 1H, 4’-H), 4.00 (m, 2H, 5’-H), 1.89 (s, 3H, OAc), 1.54 and 1.37 (2×s, 6H, C(CH3)2. 13C NMR (DMSO-d6) δ 169.79 (CO), 133.02 (Ph), 130.73 (q, JC,F=32.4 Hz, C-4Ph), 127.09, 123.31 and 123.10 (C-4, C-5, Ph), 125.99 (q, JC,F=3.7 Hz, C-3Ph, C-5Ph), 123.66 (q, JC,F=272.8 Hz, CF3), 113.06 ((CH3)2C), 110.85 (CN), 103.52 (C≡CPh), 93.20 (C-1’), 86.39 (C-4’), 83.87 (C-2’), 81.15 (C-3’), 72.39 (C≡CPh), 63.15 (C-5’), 26.56 and 24.86 ((CH3)2C), 20.28 (CO-CH3). 19F NMR (DMSO d6) δ 14.02. HRMS [M+H]+ calcd for C22H20N4O5F3: 477.13858; found: 477.13531.
4.2.8. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-(naphth-2-ylethynyl)-1H-[1,2,3]triazole-4-carbonitrile (11e)
From fractions containing homogenous 11e white crystalline material was separated, while evaporation of the filtrate resulted in the oily residue (57% total yield). UV (MeCN): λmax 266 nm (25900), 276.5 nm (35700), 312 nm (26700). 1H NMR (DMSO-d6) δ 8.44 (d, J=0.4Hz, 1H, naphthyl), 8.07 (m, 2H, naphthyl), 8.02 (m, 1H, naphthyl), 7.73 (dd, J=1.4 Hz, J=8.6 Hz, 1H, naphthyl), 7.66 (m, 2H, naphthyl), 6.54 (s, 1H, 1’-H), 5.66 (d, J2’,3’=6.0 Hz, 1H, 2’-H), 5.06 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.58 (m, 1H, 4’-H), 4.01 (m, 2H, 5’-H), 1.90 (s, 3H, OAc), 1.56 and 1.38 (2×s, 6H, 2×CH3). 13C NMR (DMSO-d6) δ 169.80 (CO), 133.46, 133.16, 132.28, 128.86, 128.40, 128.29, 127.88, 127.69, 127.47, 127.38, 122.66 and 116.23 (naphthyl, C-4, C-5), 113.06 ((CH3)2C), 111.05 (CN), 105.98 (C≡C-naphthyl), 92.99 (C-1’), 86.36 (C-4’), 83.86 (C-2’), 81.16 (C-3’), 70.65 (C≡C-naphthyl), 63.15 (C-5’), 26.58 and 24.87 ((CH3)2C), 20.30 (CO-CH3). HRMS [M+H]+ calcd for C25H23N4O5: 459.16685; found: 459.16596.
4.2.9. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-(6-methoxynaphth-2-ylethynyl)-1H-[1,2,3]triazole-4-carbonitrile (11f)
For rechromatography CH2Cl2 was used to give 11f as solid foam in 60% yield. UV (MeCN): λmax 227 nm (21600), 281.5 nm (11800), 331.5 nm (11800). 1H NMR (DMSO-d6) δ 8.34 (d, J=0.8 Hz, 1H, naphthyl), 7.96 (m, 2H, naphthyl), 7.68 (dd, J=1.8 Hz, J=8.6 Hz, 1H, naphthyl), 7.43 (d, J=2.4 Hz, 1H, naphthyl), 7.28 (dd, J=2.6 Hz, J=9.0 Hz, 1H, naphthyl), 6.51 (s, 1H, 1’-H), 5.65 (d, J2’,3’=6.8 Hz, 1H, 2’-H), 5.06 (m, 1H, 3’-H), 4.57 (m, 1H, 4’-H), 4.00 (m, 2H, 5’-H), 3.92 (s, 3H, OCH3), 1.90 (s, 3H, OAc), 1.55 and 1.38 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.80 (CO), 159.12 (C-6naphthyl), 135.27, 132.96, 129.96, 128.08, 127.88, 127.72, 127.64, 122.42 and 119.96 (naphthyl, C-4, C-5), 113.51 and 113.05 (naphthyl, (CH3)2C), 111.10 (CN), 106.58 and 106.27 (naphthyl, C≡C-naphthyl), 92.88 (C-1’), 86.32 (C-4’), 83.83 (C-2’), 81.16 (C-3’), 70.12 (C≡C-naphthyl), 63.15 (C-5’), 55.44 (OCH3), 26.59 and 24.87 ((CH3)2C), 20.30 (CO-CH3). HRMS [M+H]+ calcd for C26H25N4O6: 489.17741; found: 489.17714.
4.2.10 General procedure for cyclization reactions
To a solution of 1-(5-O-acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-5-alkynyl-1H-[1,2,3]triazole-4-carbonitrile (11a–f; dried under vacuum at room temperature) in DME (10 ml/mmol) was added ca 10 M NH3/DME (30 ml/mmol) and the resulting mixture was heated in a Parr reactor at 85 °C for 40 h, then it was evaporated.
4.2.11. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-4-amino-6-phenyl-1H-[1,2,3]triazole[4,5-c]pyridine (14a)
Chromatography on silica gel column with hexane-EtOAc (2:1→1:1) resulted in 14a as solid foam (95% yield). UV (MeOH): λmax 249 nm (22900), 315 nm (9900). 1H NMR (DMSO-d6) δ 8.10 (m, 2H, Ph), 7.63 (s, 1H, 7-H), 7.47 (m, 2H, Ph), 7.40 (m, 1H, Ph), 7.30 (s, 2H, NH2), 6.70 (d, J1’,2’=1.6 Hz, 1H, 1’-H), 5.72 (dd, J2’,1’=1.6 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.10 (dd, J3’,2’=6.0 Hz, J3’,4’=2.4 Hz, 1H, 3’-H), 4.48 (m, 1H, 4’-H), 4.06 (dd, J5’a,5’b=11.8 Hz, J5’a,4’=5.4 Hz, 1H, 5’a-H), 3.87 (dd, J5’b,5’a=11.8 Hz, J5’b,4’=6.6 Hz, 1H, 5’b-H), 1.86 (s, 3H, OAc), 1.58 and 1.39 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.84 (CO), 152.65 and 151.41 (C-4, C-6), 139.49, 138.98, 130.71, 128.71, 128.39 and 126.87 (Ph, C-3a, C-7a), 113.05 ((CH3)2C), 90.85 and 90.48 (C-1’, C-7), 84.90 (C-4’), 83.44 (C-2’), 81.51 (C-3’), 63.41 (C-5’), 26.69 and 25.01 ((CH3)2C), 20.31 (CO-CH3). HRMS [M+H]+ calcd for C21H24N5O5: 426.1772; found: 426.1787.
4.2.12. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-4-amino-6-(4-methylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (14b)
Solid (95% yield). UV (MeOH): λmax 256.5 nm (31000), 316.5 nm (13500). 1H NMR (DMSO-d6) δ 8.01 (d, J=8.0 Hz, 2H, Ph), 7.60 (s, 1H, 7-H), 7.27 (m, 4H, Ph, NH2), 6.69 (s, 1H, 1’-H), 5.72 (d, J2’,3’=5.6 Hz, 1H, 2’-H), 5.10 (dd, J3’,2’=5.8 Hz, J3’,4’=1.4 Hz, 1H, 3’-H), 4.48 (m, 1H, 4’-H), 4.06 (dd, J5’a,5’b=11.8 Hz, J5’a,4’=5.4 Hz, 1H, 5’a-H), 3.86 (dd, J5’b,5’a=11.8 Hz, J5’b,4’=6.6 Hz, 1H, 5’b-H), 2.35 (s, 3H, CH3), 1.87 (s, 3H, OAc), 1.57 and 1.39 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.84 (CO), 152.64 and 151.34 (C-4, C-6), 139.55, 138.18, 136.19, 130.61, 129.00 and 126.77 (Ph, C-3a, C-7a), 113.04 ((CH3)2C), 90.80 (C-1’), 89.92 (C-7), 84.89 (C-4’), 83.42 (C-2’), 81.52 (C-3’), 63.41 (C-5’), 26.69 and 25.01 ((CH3)2C), 20.78 (Ph-CH3), 20.31 (CO-CH ). HRMS [M+H]+ 3 calcd for C22H26N5O5: 440.1934; found: 440.1925.
4.2.13. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-4-amino-6-(4-fluorophenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (14c)
Chromatography on silica gel column with hexane-EtOAc (2:1→1:2) resulted in 14c as solid foam (80% yield). UV (MeOH): λmax 248.5 nm (37000), 318 nm (15500). 1H NMR (DMSO-d6) δ 8.15 (m, 2H, Ph), 7.63 (s, 1H, 7-H), 7.30 (m, 4H, Ph, NH2), 6.69 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.72 (dd, J2’,1’=1.2 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.10 (dd, J3’,2’=6.0 Hz, J3’,4’=2.4 Hz, 1H, 3’-H), 4.48 (m, 1H, 4’-H), 4.07 (dd, J5’a,5’b=11.6 Hz, J5’a,4’=5.2 Hz, 1H, 5’a-H), 3.87 (dd, J5’b,5’a=11.6 Hz, J5’b,4’=6.8 Hz, 1H, 5’b-H), 1.87 (s, 3H, OAc), 1.57 and 1.39 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.84 (CO), 162.62 (d, JC,F=245.88 Hz, C-4Ph), 151.58 and 151.41 (C-4, C-6), 139.54, 135.44 and 130.63 (C-1Ph, C-3a, C-7a), 128.93 (d, JC,F=8.76 Hz, C-2Ph, C-6Ph), 115.22 (d, JC,F=21.22 Hz, C-3Ph, C-5Ph), 113.05 ((CH3)2C), 90.83 (C-1’), 90.28 (C-7), 84.94 (C-4’), 83.45 (C-2’), 81.53 (C-3’), 63.41 (C-5’), 26.68 and 25.00 ((CH3)2C), 20.30 (CO-CH3). 19F NMR (DMSO d6) δ −37.96. HRMS [M+H]+ calcd for C21H23N5O5F: 444.16832; found: 444.16778.
4.2.14. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-4-amino-6-(4-trifluoromethylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (14d)
Chromatography on silica gel column with hexane-EtOAc (2:1) gave 14d as crystalline material (80% yield), mp 154-157 °C. UV (MeOH): λmax 248 nm (25600), 322.5 nm (10900). 1H NMR (DMSO-d6) δ 8.32 (d, J=8.0 Hz, 2H, Ph), 7.84 (d, J=8.4 Hz, 2H, Ph), 7.79 (s, 1H, 7-H), 7.42 (s, 2H, NH2), 6.72 (d, J1’,2’=0.8 Hz, 1H, 1’-H), 5.73 (dd, J2’,1’=1.2 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.11 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.49 (m, 1H, 4’-H), 4.06 (dd, J5’a,5’b=11.8 Hz, J5’a,4’=5.4 Hz, 1H, 5’a-H), 3.87 (dd, J5’b,5’a=11.6 Hz, J5’b,4’=6.8 Hz, 1H, 5’b-H), 1.86 (s, 3H, OAc), 1.58 and 1.39 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.83 (CO), 151.60 and 150.81 (C-4, C-6), 142.86 (Ph), 139.43 and 131.06 (C-3a, C-7a), 128.80 (q, JC,F=31.70 Hz, C-4Ph), 127.47 (Ph), 125.35 (q, JC,F=3.51 Hz, C-3Ph, C-5Ph), 124.34 (q, JC,F=272.20 Hz, CF3), 113.04 ((CH3)2C), 91.69 (C-7), 90.92 (C-1’), 85.05 (C-4’), 83.48 (C-2’), 81.54 (C-3’), 63.39 (C-5’), 26.68 and 24.99 ((CH3)2C), 20.29 (CO-CH3). 19F NMR (DMSO-d6) δ 14.75. HRMS [M+H]+ calcd for C22H23N5O5F3: 494.16513; found: 494.16504.
4.2.15. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-4-amino-6-(naphth-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine (14e)
Chromatography on silica gel column with hexane-EtOAc (2:1→1:1) afforded 14e as solid (95% yield). UV (MeOH): λmax 252.5 nm (59200), 325 nm (18100). 1H NMR (DMSO-d6) δ 8.67 (s, 1H, naphthyl), 8.27 (dd, J=1.6 Hz, J=8.8 Hz, 1H, naphthyl), 8.02-7.94 (m, 3H, naphthyl), 7.82 (s, 1H, 7-H), 7.55 (m, 2H, naphthyl), 7.38 (s, 2H, NH2), 6.74 (s, 1H, 1‘-H), 5.76 (d, J2’,3’=6.0 Hz, 1H, 2’-H), 5.12 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.50 (m, 1H, 4’-H), 4.08 (dd, J5’a,5’b=11.6 Hz, J5’a,4’=5.6 Hz, 1H, 5’a-H), 3.88 (dd, J5’b,5’a=12.0 Hz, J5’b,4’=6.8 Hz, 1H, 5’b-H), 1.87 (s, 3H, OAc), 1.59 and 1.40 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 169.85 (CO), 152.41 and 151.50 (C-4, C-6), 139.60, 136.40, 133.10, 132.95, 130.82, 128.41, 127.83, 127.52, 126.50, 126.44, 125.99 and 124.82 (naphthyl, C-3a, C-7a), 113.04 ((CH3)2C), 90.95 and 90.88 (C-1’, C-7), 84.97 (C-4’), 83.44 (C-2’), 81.55 (C-3’), 63.41 (C-5’), 26.71 and 25.03 ((CH3)2C), 20.32 (CO-CH3). HRMS [M+H]+ calcd for C25H26N5O5: 476.19340; found: 476.19348.
4.2.16. 1-(5-O-Acetyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-4-amino-6-(6-methoxynaphth-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine (14f)
Solid (88% yield). UV (MeOH): λmax 260 nm (39600), 297 nm (15200), 327.5 (18600). 1H NMR (DMSO-d6) δ 8.59 (s, 1H, naphthyl), 8.22 (d, J=8.8 Hz, 1H, naphthyl), 7.90 (dd, J=3.4 Hz, J=8.6 Hz, 2H, naphthyl), 7.75 (s, 1H, 7-H), 7.34 (m, 3H, naphthyl, NH2), 7.19 (m, 1H, naphthyl), 6.72 (s, 1H, 1’-H), 5.75 (d, J2’,3’=6.0 Hz, 1H, 2’-H), 5.11 (m, J3’,2’=6.0 Hz, J3’,4’=1.6 Hz., 1H, 3’-H), 4.50 (m, 1H, 4’-H), 4.07 (dd, J5’a,5’b=11.8 Hz, J5’a,4’=5.4 Hz, 1H, 5’a-H), 3.88 (m, 4H, 5’b-H, OCH3), 1.87 (s, 3H, OAc), 1.59 and 1.40 (2×s, 6H, C(CH3)2) 13C NMR (DMSO-d6) δ 169.86 (CO), 157.78 (C-6naphthyl), 152.68 (C-6), 151.44 (C-4), 139.64, 134.51, 134.17, 130.70, 129.98, 128.34, 126.72, 125.90, 125.29 and 118.98 (naphthyl, C-3a, C-7a), 113.06 ((CH3)2C), 105.86 (naphthyl), 90.87 (C-1’), 90.36 (C-7), 84.94 (C-4’), 83.43 (C-2’), 81.56 (C-3’), 63.42 (C-5’), 55.23 (OCH3), 26.72 and 25.03 ((CH3)2C), 20.32 (CO-CH3). HRMS [M+H]+ calcd for C26H28N5O6: 506.20396; found 506.20301.
4.2.17. General procedure for deacetylation reactions
To a cooled to 0 °C solution of compound 14a–f in anhyd 1,4-dioxane/MeOH (1:1; 20 ml/mmol) or in anhyd CH2Cl2/MeOH (1:1; 20 ml/mmol) was added 0.25 M MeONa in anhydrous MeOH (0.08 equiv). The mixture was allowed to react under argon at 5 °C for 20 h. It was again cooled to 0 °C and neutralized with Dowex 50WX8-100 ion exchange resin, which was then separated by filtration. The filtrate was evaporated and the residue was subjected to chromatography on silica gel column with CH2Cl2-MeOH (99:1→95:5) to afford purified product 15a–f as solid or solid foam in 79-96% yield.
4.2.18. 4-Amino-1-(2,3-O-isopropylidene-β-D-ribofuranosyl)-6-phenyl-1H-[1,2,3]triazole[4,5-c]pyridine (15a)
Crude material after chromatography was crystallized from MeOH to give white crystals, mp 105 °C. 1H NMR (DMSO-d6) δ 8.10 (m, 2H, Ph), 7.68 (s, 1H, 7-H), 7.46 (m, 2H, Ph), 7.40 (m, 1H, Ph), 7.34 (s, 2H, NH2), 6.63 (d, J1’,2’=1.6 Hz, 1H, 1’-H), 5.65 (dd, J2’,1’=1.8 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.05 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 5.00 (t, J5’OH,5’H=5.6 Hz, 1H, 5’-OH), 4.23 (m, 1H, 4’-H), 3.30 (m, 2H, 5’-H), 1.56 and 1.38 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 152.53 (C-6), 151.47 (C-4), 139.44, 139.05, 130.86, 128.77, 128.48 and 126.93 (Ph, C-3a, C-7a), 112.91 ((CH3)2C), 91.00 (C-1’), 90.73 (C-7), 87.86 (C-4’), 83.25 (C-2’), 81.72 (C-3’), 61.02 (C-5’), 26.80 and 25.05 ((CH3)2C). HRMS [M+H]+ calcd for C19H22N5O4: 384.16718; found 384.16706.
4.2.19. 4-Amino-1-(2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(4-methylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (15b)
1H NMR (DMSO-d6) δ 8.00 (d, J=8.0 Hz, 2H, Ph), 7.63 (s, 1H, 7-H), 7.32 (s, 2H, NH2), 7.27 (d, J=8.0 Hz, 2H, Ph), 6.61 (d, J1’,2’=1.6 Hz, 1H, 1’-H), 5.64 (dd, J2’,1’=1.2 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.05 (dd, J3’,2’=6.0 Hz, J3’,4’=1.6 Hz, 1H, 3’-H), 4.96 (brs, 1H, 5’-OH), 4.24 (m, 1H, 4’-H), 3.31 (m, 2H, 5’-H), 2.35 (s, 3H, CH3), 1.57 and 1.38 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 152.22 (C-6), 151.25 (C-4), 139.43, 138.23, 135.98, 130.69, 129.01 and 126.79 (Ph, C-3a, C-7a), 112.86 ((CH3)2C), 90.99 (C-1’), 90.28 (C-7), 87.79 (C-4’), 83.17 (C-2’), 81.66 (C-3’), 60.98 (C-5’), 26.75 and 25.01 ((CH3)2C), 20.79 (Ph-CH3). HRMS [M+H]+ calcd for C20H24N5O4: 398.18283; found: 398.18213.
4.2.20. 4-Amino-1-(2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(4-fluorophenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (15c)
1H NMR (DMSO-d6) δ 8.15 (dd, J=5.8 Hz, J=8.6 Hz, 2H, Ph), 7.66 (s, 1H, 7-H), 7.31-7.23 (m, 4H, Ph, NH2), 6.60 (s, 1H, 1’-H), 5.63 (d, J2’,3’=6.0 Hz, 1H, 2’-H), 5.05 (dd, J3’,2’= 6.0 Hz, J3’,4’=1.6 Hz, 1H, 3’-H), 4.96 (t, J5’OH,5’H=5.4 Hz, 1H, 5’-OH), 4.24 (m, 1H, 4’-H), 3.31 (m, 2H, 5’-H), 1.57 i 1.38 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 162.58 (d, JC,F=246.14 Hz, C-4Ph), 151.38 (C-4, C-6), 139.37 (C-7a), 135.46 (d, JC,F=2.97 Hz, C-1Ph), 130.71 (C-3a), 128.90 (d, JC,F=8.42 Hz, C-2Ph, C-6Ph), 115.18 (d, JC,F=21.26 Hz, C-3Ph, C-5Ph), 112.87 ((CH3)2C), 91.03 (C-1’), 90.50 (C-7), 87.76 (C-4’), 83.17 (C-2’), 81.62 (C-3’), 60.97 (C-5’), 26.74 and 25.00 ((CH3)2) 19F NMR (DMSO d6) δ −38.02. HRMS [M+H]+ calcd for C19H21N5O4F: 402.15776; found: 402.15771.
4.2.21. 4-Amino-1-(2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(4-trifluoromethylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (15d)
1H NMR (DMSO-d6) δ 8.32 (d, J=8.0 Hz, 2H, Ph), 7.83 (m, 3H, Ph, 7-H), 7.41 (s, 2H, NH2), 6.64 (d, J1’,2’=1.6 Hz, 1H, 1’-H), 5.65 (dd, J2’,1’=1.6 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.06 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.97 (t, J5’OH,5’H=5.4 Hz, 1H, 5’-OH), 4.25 (m, 1H, 4’-H), 3.31 (m, 2H, 5’-H), 1.57 and 1.38 (2×s, 6H, C(CH3)2) 13C NMR (DMSO-d6) δ 151.59 and 150.66 (C-4, C-6), 142.90 (Ph), 139.27 (C-7a), 131.16 (C-3a), 128.76 (q, JC,F=30.72 Hz, C-4Ph), 127.47 (Ph), 125.34 (q, JC,F=3.08 Hz, C-3Ph, C-5Ph), 124.35 (q, JC,F=271.51 Hz, CF3), 112.87 ((CH3)2C), 91.95 and 91.13 (C-1’, C-7), 87.89 (C-4’), 83.24 (C-2’), 81.64 (C-3’), 60.97 (C-5’), 26.74 and 25.00 ((CH3)2). 19F NMR (DMSO-d6): 14.79. HRMS [M+H]+ calcd for C20H21N5O4F3: 452.15456; found: 452.15393.
4.2.22. 4-Amino-1-(2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(naphth-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine (15e)
Crystalline material, mp 167-170 °C. 1H NMR (DMSO-d6) δ 8.68 (s, 1H, naphthyl), 8.28 (dd, J=1.8 Hz, J=8.6 Hz, 1H, naphthyl), 8.00 (d, J=8.8 Hz, 2H, naphthyl) ,7.95 (m, 1H, naphthyl), 7.85 (s, 1H, 7-H), 7.55 (m, 2H, naftyl-H), 7.36 (s, 2H, NH2), 6.66 (d, J1’,2’ =1.6 Hz, 1H, 1’-H), 5.68 (dd, J2’,1’=1.6 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.07 (dd, J3’,2’=6.4 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.99 (t, J5’OH,5’H=5.4 Hz, 1H, 5’-OH), 4.26 (m, 1H, 4’-H), 3.33 (m, 2H, 5’-H), 1.58 and 1.39 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 152.27 (C-6), 151.49 (C-4), 139.48 (C-7a), 136.43, 133.09 and 132.96 (naphthyl), 130.93 (C-3a), 128.43, 127.81, 127.51, 126.47, 126.41, 125.99 and 124.84 (naphthyl), 112.87 ((CH3)2C), 91.18 (C-7), 91.04 (C-1’), 87.87 (C-4’), 83.21 (C-2’), 81.68 (C-3’), 60.99 (C-5’), 26.77 and 25.02 ((CH3)2C). HRMS [M+H]+ calcd for C23H24N5H4: 434.18283; found: 434.18195.
4.2.23. 4-Amino-1-(2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(6-methoxynaphth-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine (15f)
Crude material after chromatography was crystallized from hexane-EtOAc to give white crystals, mp 140 °C. 1H NMR (DMSO-d6) δ 8.59 (s, 1H, naphthyl), 8.22 (dd, J=1.6 Hz, J=8.8 Hz, 1H, naphthyl), 7.89 (dd, J=5.6 Hz, J=8.8 Hz, 2H, naphthyl), 7.78 (s, 1H, 7-H), 7.36 (d, J=2.0 Hz, 1H, naphthyl), 7.30 (s, 2H, NH2), 7.20 (dd, J=2.4 Hz, J=8.8 Hz, 1H, naphthyl), 6.64 (d, J1’,2’=1.6 Hz, 1H, 1’-H), 5.67 (dd, J2’,1’=1.4 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.07 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.98 (t, J5’OH,5’H=5.4 Hz, 1H, 5’-OH), 4.26 (m, 1H, 4’-H), 3.90 (s, 3H, OCH3), 3.32 (m, 2H, 5’-H), 1.58 and 1.39 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 157.75 (C-6naphthyl), 152.50 (C-6), 151,39 (C-4), 139.48 (C-7a), 130.78 (C-3a), 134.47, 134.18, 129.96, 128.33, 126.68, 125.85, 125.28 and 118.92 (naphthyl), 112.86 ((CH3)2C), 105.84 (naphthyl), 91.02 (C-1’), 90.56 (C-7), 87.79 (C-4’), 83.16 (C-2’), 81.65 (C-3’), 60.97 (C-5’), 55.21 (OCH3), 26.75 and 25.01 ((CH3)2C). HRMS [M+H]+ calcd for C24H26N5O5: 464.19340; found: 464.19342.
4.2.24. General procedure for sulfamoylation reactions
To a stirred solution of compound 15a–f (dried in vacuo over P2O5 at room temperature) in MeCN (15 ml/mmol) containing DMA (50 equiv) was added 1.33 M sulfamoyl chloride [35] in MeCN (2.5 equiv) at 0 °C. The mixture was stirred on ice-cooling for 15 min, then at room temperature overnight. It was evaporated and the resulting residue was chromatographed on silica gel column with CH2Cl2-MeOH (98:2→95:5) to obtain product 16a–f contaminated with DMA as solid foam (yields 51-71% calculated from 1H NMR spectra).
4.2.25. 4-Amino-6-phenyl-1-(5-O-sulfamoyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-1H-[1,2,3]triazole[4,5-c]pyridine (16a)
1H NMR (DMSO-d6) δ 8.11 (m, 2H, Ph), 7.68 (s, 1H, 7-H), 7.55 (brs, 2H, SO3NH2), 7.43 (m, 3H, Ph), 7.31 (brs, 2H, NH2), 6.77 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.65 (dd, J2’,1’=1.2 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.13 (dd, 1H, J3’2’=6.0 Hz, J3’,4’=2.4 Hz, 1H, 3’-H), 4.48 (dt, J4’,3’=2.3 Hz, J4’,5’= 6.5 Hz, 1H, 4’-H), 4.06 and 3.91 (2×dd, J5’,4’=6.6 Hz, J5’a,5’b=10.6 Hz, 2H, 5’-H), 1.58 and 1.40 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 152.83 and 151.44 (C-4, C-6), 139.56 (C-7a), 138.94 (Ph), 130.66 (C-3a), 128.76, 128.41 and 126.91 (Ph), 113.29 ((CH3)2C), 90.38 and 90.31 (C-1’, C-7), 84.80 (C-4’), 83.51 (C-2’), 81.50 (C-3’), 67.63 (C-5’), 26.68 and 25.00 ((CH3)2C). HRMS [M+H]+ calcd for C19H23N6O6S: 463.1394; found: 463.1413.
4.2.26. 4-Amino-6-(4-methylphenyl)-1-(5-O-sulfamoyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-1H-[1,2,3]triazole[4,5-c]pyridine (16b)
1H NMR (DMSO-d6) δ 8.02 (d, J=8.0 Hz, 2H, Ph), 7.65 (s, 1H, 7-H), 7.55 (s, 2H, SO3NH2), 7.27 (m, 4H, NH2, Ph), 6.75 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.65 (dd, J2’,1’=1.4 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.13 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.48 (dt, J4’,3’=2.1 Hz, J4’,5’=6.6 Hz, 1H, 4’-H), 4.06 and 3.91 (2×dd, J5’,4’=6.7 Hz, J5’a,5’b=10.7 Hz, 2H, 5’-H), 2.35 (s, 3H, CH3), 1.58 and 1.40 (2×s, 6H, C(CH3)2. 13C NMR (DMSO-d6) δ 152.80 and 151.35 (C-4, C-6), 139.58 (C-7a), 138.21 and 136.13 (Ph), 130.53 (C-3a), 128.99 and 126.80 (Ph), 113.26 ((CH3)2C), 90.33 and 89.73 (C-1’, C-7), 84.76 (C-4’), 83.48 (C-2’), 81.49 (C-3’), 67.61 (C-5’), 26.67 and 24.99 ((CH3)2C), 20.78 (CH3). HRMS [M+H]+ calcd for C20H25N6O6S: 477.1556; found: 477.1563.
4.2.27. 4-Amino-6-(4-fluorophenyl)-1-(5-O-sulfamoyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-1H-[1,2,3]triazole[4,5-c]pyridine (16c)
1H NMR (DMSO-d6) δ 8.16 (m, 2H, Ph), 7.68 (s, 1H, 7-H), 7.56 (s, 2H, SO3NH2), 7.31 (m, 4H, NH2, Ph), 6.75 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.65 (dd, J2’,1’=1.2 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.13 (dd, J3’,2’=6.0 Hz, J3’,4’=2.4 Hz, 1H, 3’-H), 4.48 (dt, J4’,3’=2.1 Hz, J4’,5’=6.6 Hz, 1H, 4’-H), 4.07 and 3.92 (2×dd, J5’,4’=6.5 Hz, J5’a,5’b=10.5 Hz, 2H, 5’-H), 1.58 and 1.40 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 162.66 (d, JC,F=245.6 Hz, C-4Ph), 151.73 and 151.44 (C-4, C-6), 139.58 (C-7a), 135.41 (Ph), 130.57 (C-3a), 128.97 (d, JC,F=8.22 Hz, C-2Ph, C-6Ph), 115.24 (d, JC,F=21.47 Hz, C-3Ph, C-5Ph), 113.30 ((CH3)2C), 90.40 and 90.12 (C-1’, C-7), 84.81 (C-4’), 83.51 (C-2’), 81.48 (C-3’), 67.63 (C-5’), 26.67 and 24.99 ((CH3)2C). 19F NMR (DMSO-d6) δ: -37.94. HRMS [M+H]+ calcd for C19H22N6O6SF: 481.1300; found: 481.1308.
4.2.28. 4-Amino-1-(5-O-sulfamoyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-6-(4-trifluoromethylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (16d)
1H NMR (DMSO-d6) δ 8.32 (d, J=8.0 Hz, 2H, Ph), 7.84 (s and d, 3H, Ph, 7-H), 7.54 (brs, 2H, SO3NH2), 7.44 (s, 2H, NH2), 6.78 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.66 (dd, J2’,1’=1.1 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.14 (dd, J3’,2’=6.0 Hz, J3’,4’=2.4 Hz, 1H, 3’-H), 4.49 (dt, J4’,3’=2.0 Hz, J4’,5’=6.4 Hz, 1H, 4’-H), 4.07 and 3.92 (2×dd, J5’,4’=6.5 Hz, J5’a,5’b=10.5 Hz, 2H, 5’-H), 1.58 and 1.40 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 151.61 and 150.94 (C-4, C-6), 142.80 (Ph), 139.42 (C-7a), 130.98 (C-3a), 128.80 (q, JC,F=31.70 Hz, C-4Ph), 127.49 (Ph), 125.34 (q, JC,F=3.53 Hz, C-3Ph, C-5Ph), 124.34 (q, JC,F=272.05, CF3), 113.28 ((CH3)2C), 91.52 (C-7), 90.48 (C-1’), 84.85 (C-4’), 83.52 (C-2’), 81.43 (C-3’), 67.60 (C-5’), 26.67 and 24.99 ((CH3)2C). 19F NMR (DMSO-d6) δ14.78. HRMS [M+H]+ calcd for C20H22N6O6SF3: 531.1268; found: 531.1288.
4.2.29. 4-Amino-6-(naphth-2-yl)-1-(5-O-sulfamoyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-1H-[1,2,3]triazole[4,5-c]pyridine (16e)
1H NMR (DMSO-d6) δ 8.68 (s, 1H, naphthyl), 8.28 (dd, J=1.6 Hz, J=8.8 Hz, 1H, naphthyl), 7.98 (m, 3H, naphthyl), 7.87 (s, 1H, 7-H), 7.55 (m, 4H, naphthyl, SO3NH2), 7.39 (s, 2H, NH2), 6.80 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.69 (dd, J2’,1’=1.2 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.15 (dd, J3’,2’=6.2 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.51 (dt, J4’,3’=2.3 Hz, J4’,5’=6.6 Hz, 1H, 4’-H), 4.08 and 3.93 (2×dd, J5’,4’=6.6 Hz, J5’a,5’b=10.6 Hz, 2×1H, 5’-H), 1.60 and 1.41 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 152.58 and 151.51 (C-4, C-6), 139.63 (C-7a), 136.35, 133.12 and 132.95 (naphthyl), 130.75 (C-3a), 128.43, 127.83, 127.52, 126.52, 126.43, 126.04 and 124.85 (naphthyl), 113.28 ((CH3)2C), 90.78 and 90.43 (C-1’, C-7), 84.84 (C-4’), 83.50 (C-2’), 81.51 (C-3’), 67.62 (C-5’), 26.69 and 25.00 ((CH3)2C). HRMS [M+H]+ calcd for C23H25N6O6S: 513.1556; found: 513.1550.
4.2.30. 4-Amino-6-(6-methoxynaphth-2-yl)-1-(5-O-sulfamoyl-2,3-O-isopropylidene-β-D-ribofuranosyl)-1H-[1,2,3]triazole[4,5-c]pyridine (16f)
1H NMR (DMSO-d6) δ 8.60 (s, 1H, naphthyl), 8.23 (dd, J=1.8 Hz, J=8.6 Hz, 1H, naphthyl), 7.90 (m, 2H, naphthyl), 7.80 (s, 1H, 7-H), 7.56 (s, 2H, SO3NH2), 7.35 (m, 3H, naphthyl, NH2), 7.20 (dd, J=2.4 Hz, J=8.8 Hz, 1H, naphthyl), 6.78 (d, J1’,2’=1.2 Hz, 1H, 1’-H), 5.68 (dd, J2’,1’=1.4 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.15 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.50 (dt, J4’,3’=2.1 Hz, J4’,5’=6.6 Hz, 1H, 4’-H), 4.08 (dd, J5a’,4’=6.8 Hz, J5’a,5’b=10.8 Hz, 2H, 5’a-H), 3.93 (m, 4H, 5’b-H, OCH3), 1.60 and 1.41 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 157.79 (C-6naphthyl), 152.84 and 151.45 (C-4, C-6), 139.66 (C-7a), 134.54, 134.13, 130.60, 130.00, 128.34, 126.72, 125.93, 125.32 and 118.98 (naphthyl, C-3a), 113.29 ((CH3)2C), 105.86 (naphthyl), 90.42 and 90.18 (C-1’, C-7), 84.79 (C-4’), 83.48 (C-2’), 81.51 (C-3’), 67.61 (C-5’), 55.24 (OCH3), 26.69 and 25.00 ((CH3)2C). HRMS [M+H]+ calcd for C24H27N6O7S: 543.1656; found: 543.1668.
4.2.31. General procedure for the preparation of compounds 17a–f
To a solution of compound 16a–f (contaminated with DMA) in MeCN (20 ml/mmol) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (1.5 equiv) and the mixture was stirred at room temperature for 15 min. In a separate flask 1,1'-carbonyldiimidazole (1.4 equiv) and salicylic acid (1.4 equiv) were dissolved in MeCN (20 ml/mmol of substrate) and the resulting solution was stirred at 60 °C for 2 h, then it was cooled to room temperature. Both solutions were combined and stirred at room temperature overnight. After evaporation the residue was chromatographed on silica gel column with CH2Cl2-MeOH (98:2→9:1). Crude product obtained was rechromatographed on silica gel column using EtOAc to give pure 17a–f as solid foam in 56-86% yield.
4.2.32. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-2,3-O-isopropylidene-β-D-ribofuranosyl}-6-phenyl-1H-[1,2,3]triazole[4,5-c]pyridine (17a)
UV (MeOH): λmax 246 nm (32500), 309.5 nm (13700). 1H NMR (DMSO-d6) δ 13.49 (s, 1H, OH), 8.12 (d, J=7.2 Hz, 2H, Ph), 7.77 (dd, J=1.8 Hz, J=8.2 Hz, 1H, sal), 7.66 (s, 1H, 7-H), 7.43 (m, 3H, Ph), 7.25 (m, 3H, NH2, sal), 6.71 (m, 3H, sal, 1’-H), 5.59 (dd, J2’,1’=1.6 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.16 (dd, J3’,2’=6.2 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.47 (dt, J4’,3’=2.1 Hz, J4’,5’=6.5 Hz, 1H, 4’-H), 4.07 (dd, J5’a,4’=7.0 Hz, J5’a,5’b=11.0 Hz, 1H, 5’a-H), 3.91 (dd, J5’b,4’=6.0 Hz, J5’b,5’a=10.8 Hz, 1H, 5’b-H), 1.56 and 1.36 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 171.11 (CO), 160.69 (C-OHsal), 152.76 and 151.43 (C-4, C-6), 139.41 (C-7a), 138.96 (Ph), 132.57 (sal), 130.67 (C-3a), 129.81 (sal), 128.65, 128.35 and 126.96 (Ph), 119.72, 117.50 and 116.52 (sal), 113.17 ((CH3)2C), 90.66 and 90.36 (C-1’, C-7), 84.94 (C-4’), 83.31 (C-2’), 81.61 (C-3’), 67.18 (C-5’), 26.69 and 24.96 ((CH3)2C). HRMS [M+H]+ calcd for C26H27N6O8S: 583.1606; found: 583.1607.
4.2.33. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-2,3-O-isopropylidene-β-D-ribofuranosyl}-6-(4-methylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (17b)
UV (MeOH): λmax 249.5 nm (31400), 309.5 nm (15900). 1H NMR (DMSO-d6) δ 13.49 (s, 1H, OH), 8.03 (d, J=8.4 Hz, 2H, Ph), 7.77 (dd, J=2.0 Hz, J=8.0 Hz, 1H, sal), 7.62 (s, 1H, 7-H), 7.24 (m, 5H, NH2, sal, Ph), 6.72 (m, 2H, sal), 6.67 (d, J1’,2’=2.0 Hz, 1H, 1’-H), 5.58 (dd, J2’,1’=1.8 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.16 (dd, J3’,2’=6.2 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.46 (m, 1H, 4’-H), 4.06 (dd, J5’a,4’=6.8 Hz, J5’a,5’b=10.8 Hz, 1H, 5’a-H), 3.91 (dd, J5’b,4’=6.4 Hz, J5’b,5’a=10.8 Hz, 1H, 5’b-H), 2.35 (s, 3H, CH3), 1.56 and 1.36 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 171.19 (CO), 160.72 (C-OHsal), 152.80 and 151.38 (C-4, C-6), 139.49 (C-7a), 138.16 and 136.18 (Ph), 132.65 (sal), 130.60 (C-3a), 129.85 (sal), 129.01 and 126.89 (Ph), 119.72, 117.56 and 116.57 (sal), 113.21 ((CH3)2C), 90.67 and 89.84 (C-1’, C-7), 84.94 (C-4’), 83.32 (C-2’), 81.63 (C-3’), 67.24 (C-5’), 26.71 and 24.98 ((CH3)2C), 20.82 (CH3). HRMS [M+H]+ calcd for C27H29N6O8S: 597.1762; found: 597.1760.
4.2.34. 4-Amino-6-(4-fluorophenyl)-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-2,3-O-isopropylidene-β-D-ribofuranosyl}-1H-[1,2,3]triazole[4,5-c]pyridine (17c)
UV (MeOH): λmax 246.5 nm (33300), 311.5 nm (14400). 1H NMR (DMSO-d6) δ 13.47 (s, 1H, OH), 8.17 (m, 2H, Ph), 7.77 (dd, J=1.8 Hz, J=8.2 Hz, 1H, sal), 7.65 (s, 1H, 7-H), 7.27 (m, 5H, NH2, sal, Ph), 6.72 (m, 2H, sal), 6.66 (d, J1’,2’=1.6 Hz, 1H, 1’-H), 5.58 (dd, J2’,1’=1.6 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.16 (dd, J3’,2’=6.2 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.47 (dt, J4’,3’=2.0 Hz, J4’,5’=6.2 Hz, 1H, 4’-H), 4.07 (dd, J5’a,4’=6.6 Hz, J5’a,5’b=10.6 Hz, 1H, 5’a-H), 3.92 (dd, J5’b,4’=6.0 Hz, J5’b,5’a=10.8 Hz, 1H, 5’b-H), 1.56 and 1.36 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 171.09 (CO), 162.60 (d, JC,F=244.93 Hz, C-4Ph), 160.68 (C-OHsal), 151.65 and 151.42 (C-4, C-6), 139.38 (C-7a), 135.40 (d, JC,F=2.18 Hz, C-1Ph), 132.57 (sal), 130.60 (C-3a), 129.79 (sal), 129.04 (d, JC,F=8.27 Hz, C-2Ph, C-6Ph), 119.71, 117.49 and 116.51 (sal), 115.14 (d, JC,F=21.31 Hz, C-3Ph, C-5Ph), 113.20 ((CH3)2C), 90.75 and 90.18 (C-1’, C-7), 84.87 (C-4’), 83.25 (C-2’), 81.52 (C-3’), 67.18 (C-5’), 26.69 and 24.95 ((CH3)2C). 19F NMR (DMSO-d6) δ −38.08. HRMS [M+H]+ calcd for C26H26N6O8SF: 601.1511; found: 601.1518.
4.2.35. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-2,3-O-isopropylidene-β-D-ribofuranosyl}-6-(4-trifluoromethylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine (17d)
UV (MeOH): λmax 243.5 nm (45100), 312 nm (17200). 1H NMR (DMSO-d6) δ 13.47 (s, 1H, OH), 8.34 (d, J=8.4 Hz, 2H, Ph), 7.79 (m, 4H, Ph, 7-H, sal), 7.41 (s, 2H, NH2), 7.24 (m, 1H, sal), 6.71 (m, 3H, sal, 1’-H), 5.57 (dd, J2’,1’=2.0 Hz, J2’,3’=6.0 Hz, 1H, 2’-H), 5.17 (dd, J3’,2’=6.2 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.49 (dt, J4’,3’=2.1 Hz, J4’,5’=6.2 Hz, 1H, 4’-H), 4.09 and 3.95 (2×dd, J5’,4’=6.3 Hz, J5’a,5’b=10.9 Hz, 2H, 5’-H), 1.57 and 1.36 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 171.15 (CO), 160.69 (C-OHsal), 151.62 and 150.91 (C-4, C-6), 142.81 (Ph), 139.20 (C-7a), 132.60 (sal), 131.08 (C-3a), 129.80 (sal), 128.74 (q, JC,F=31.80 Hz, C-4Ph), 127.60 (Ph), 125.28 (q, JC,F=3.77 Hz, C-3Ph, C-5Ph), 124.37 (q, JC,F=272.19 Hz, CF3), 119.70, 117.52 and 116.53 (sal), 113.27 ((CH3)2C), 91.65 and 90.97 (C-1’, C-7), 84.87 (C-4’), 83.27 (C-2’), 81.44 (C-3’), 67.22 (C-5’), 26.71 and 24.96 ((CH3)2C). 19F NMR (DMSO-d6) δ 14.78. HRMS [M+H]+ calcd for C27H26N6O8SF3: 651.1479; found: 651.1479.
4.2.36. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-2,3-O-isopropylidene-β-D-ribofuranosyl}-6-(naphth-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine (17e)
UV (MeOH): λmax 251 nm (29600), 317.5 nm (9400). 1H NMR (DMSO-d6) δ 13.50 (s, 1H, OH), 8.70 (s, 1H, naphthyl), 8.29 (dd, J=1.6 Hz, J2=8.4 Hz, 1H, naphthyl), 7.98 (m, 3H, naphthyl), 7.84 (s, 1H, 7-H), 7.77 (m, 1H, sal), 7.54 (m, 2H, naphthyl), 7.36 (s, 2H, NH2), 7.24 (m, 1H, sal), 6.72 (m, 3H, sal, 1’-H), 5.62 (dd, J2’,1’=1.6 Hz, J2’,3’=6.4 Hz, 1H, 2’-H), 5.18 (dd, J3’,2’=6.2 Hz, J3’,4’=2.2 Hz, 1H, 3’-H), 4.50 (m, 1H, 4’-H), 4.09 (dd, J5’a,4’=6.8 Hz, J5’a,5’b=10.8 Hz, 1H, 5’a-H), 3.94 (dd, J5’b,4’=6.0 Hz, J5’b,5’a=10.8 Hz, 1H, 5’b-H), 1.58 and 1.37 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 171.14 (CO), 160.70 (C-OHsal), 152.54 and 151.51 (C-4, C-6), 139.47 (C-7a), 136.39, 133.10 and 132.98 (naphthyl), 132.58 (sal), 130.79 (C-3a), 129.81 (sal), 128.50, 127.76, 127.47, 126.43, 126.33, 126.08 and 124.96 (naphthyl) 119.72, 117.50 and 116.52 (sal), 113.19 ((CH3)2C), 90.85 and 90.78 (C-1’, C-7), 84.94 (C-4’), 83.27 (C-2’), 81.59 (C-3’), 67.19 (C-5’), 26.71 and 24.97 ((CH3)2C). HRMS [M+H]+ for C30H29N6O8S: 633.1762; found: 633.1767.
4.2.37. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-2,3-O-isopropylidene-β-D-ribofuranosyl}-6-(6-methoxynaphth-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine (17f)
UV (MeOH): λmax 247 nm (58700), 323.5 nm (27300). 1H NMR (DMSO-d6) δ 13.48 (s, 1H, OH), 8.61 (s, 1H, naphthyl), 8.25 (dd, J=1.8 Hz, J=8.6 Hz, 1H, naphthyl), 7.90 (dd, 2H, naphthyl), 7.77 (m, 2H, 7-H, sal), 7.33 (m, 3H, NH2, naphthyl), 7.25 (m, 1H, sal), 7.19 (dd, J=2.6 Hz, 1H, naphthyl), 6.71 (m, 3H, sal, 1’-H), 5.61 (dd, J2’,1’=1.8 Hz, J2’,3’=6.2 Hz, 1H, 2’-H), 5.18 (dd, J3’,2’=6.0 Hz, J3’,4’=2.0 Hz, 1H, 3’-H), 4.49 (dt, J4’,3’=2.3 Hz, J4’,5’=6.3 Hz, 1H, 4’-H), 4.09 (dd, J5’a,4’=6.8 Hz, J5’a,5’b=10.8 Hz, 1H, 5’a-H), 3.93 (m, 4H, 5’b-H, OCH3), 1.58 and 1.37 (2×s, 6H, C(CH3)2). 13C NMR (DMSO-d6) δ 171.17 (CO), 160.71 (C-OHsal), 157.76 (C-6naphthyl), 152.78 and 151.45 (C-4, C-6), 139.51 (C-7a), 134.52 and 134.14 (naphthyl), 132.62 (sal), 130.68 (C-3a), 130.08, 129.84, 128.38, 126.69, 125.99, 125.44, 119.72, 118.89, 117.54 and 116.54 (sal, naphthyl), 113.22 ((CH3)2C), 105.83 (naphthyl), 90.80 and 90.28 (C-1’, C-7), 84.89 (C-4’), 83.25 (C-2’), 81.59 (C-3’), 67.22 (C-5’), 55.24 (OCH3), 26.73 and 24.98 ((CH3)2C). HRMS [M+H]+ calcd for C31H31N6O9S: 663.1868; found: 663.1870.
4.2.38. General procedure for the preparation of compounds6a–f
Compound 17a–f was treated with cooled 80% aq trifluoroacetic acid (10 ml/mmol) and, if necessary to dissolve the starting material, MeOH was added. The resulting mixture was stirred at 5 °C until deprotection was complete. Then it was evaporated and co-evaporated several times with MeOH. The residue was chromatographed on silica gel column with EtOAc-MeOH-Et3N (90:10:0.5→70:30:0.5) to obtain product 6a–f as triethylammonium salt (solid foam, 65-75% yield).
4.2.39. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-β-D-ribofuranosyl}-6-phenyl-1H-[1,2,3]triazole[4,5-c]pyridine triethylammonium salt (6a)
UV (MeOH): λmax 245.5 nm (37700), 309 nm (16200). UV (H2O): λmax 245 nm (30800), 303.5 nm (12700). Fluorescence emission in MeOH: λmax 419 nm, ΦF = 0.37; in H2O: λmax 433 nm, ΦF = 0.0015. 1H NMR (DMSO-d6) δ 13.57 (s, 1H, OH), 9.09 (brs, 1H, Et3NH+), 8.16 (m, 2H, Ph), 7.79 (dd, J=2.0 Hz, J=7.6 Hz, 1H, sal), 7.57 (s, 1H, 7-H), 7.39 (m, 3H, Ph), 7.25 (m, 3H, NH2, sal), 6.72 (m, 2H, sal), 6.27 (d, J1’,2’=5.2 Hz, 1H, 1’-H), 5.62 (d, J2’-OH,2’-H=6.0 Hz, 1H, 2’-OH), 5.46 (d, J3’-OH,3’-H=5.2 Hz, 1H, 3’-OH), 4.71 (m, 1H, 2’-H), 4.36 (m, 1H, 3’-H), 4.25 (m, 2H, 4’-H, 5’a-H), 4.16 (m, 1H, 5’b-H), 3.04 (brs, 6H, (CH3CH2)3NH+), 1.15 (t, J=7.4 Hz, 9H, (CH3CH2)3NH+). 13C NMR (DMSO-d6) δ 170.92 (CO), 160.67 (C-OHsal), 152.47 and 151.45 (C-4, C-6), 139.05 (C-7a), 138.88 (Ph), 132.57 (sal), 130.89 (C-3a), 129.83 (sal), 128.58, 128.33 and 127.07 (Ph), 119.85, 117.51 and 116.55 (sal), 90.48 and 90.33 (C-1’, C-7), 82.97 (C-4’), 73.03 (C-2’), 70.55 (C-3’), 68.66 (C-5’), 45.71 ((CH3CH2)3NH+), 8.62 ((CH3CH2NH+). HRMS [M-H]− calcd for C23H21N6O8S: 541.1147; found: 541.1141.
4.2.40. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-β-D-ribofuranosyl}-6-(4-methylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine triethylammonium salt (6b)
UV (MeOH): λmax 250 nm (33700), 311.5 nm (16400). UV (H2O): λmax 249.5 nm (27400), 306.5 nm (14000). Fluorescence emission in MeOH: λmax 412 nm, ΦF = 0.06; in H2O: λmax 433 nm, ΦF = 0.0018. 1H NMR (DMSO-d6) δ 13.56 (s, 1H, OH), 8.06 (d, J=8.0 Hz, 2H, Ph), 7.80 (dd, J=1.8 Hz, J=7.8 Hz, 1H, sal), 7.52 (s, 1H, 7-H), 7.24 (m, 5H, Ph, sal, NH2), 6.72 (m, 2H, sal), 6.25 (d, J1’,2’=5.6 Hz, 1H, 1’-H), 5.61 (d, J2’OH,2’H=6.0 Hz, 1H, 2’-OH), 5.46 (d, J3’OH,3’H=5.2 Hz, 1H, 3’-OH), 4.70 (m, 1H, 2’-H), 4.35 (m, 1H, 3’-H), 4.24 (m, 2H, 4’-H, 5’a-H), 4.17 (m, 1H, 5’b-H), 3.02 (m, 6H, (CH3CH2)3NH+), 2.32 (s, 3H, CH3), 1.15 (t, J=7.2 Hz, 9H, (CH3CH2)3NH+). 13C NMR (DMSO-d6) δ 171.37 (CO), 160.64 (C-OHsal), 152.15 and 151.27 (C-4, C-6), 139.02 (C-7a), 138.01 and 135.81 (Ph), 132.48 (sal), 130.74 (C-3a), 129.79 (sal), 128.93 and 126.93 (Ph), 119.84, 117.43 and 116.48 (sal), 90.34 and 90.06 (C-1’, C-7), 82.96 (C-4’), 72.97 (C-2’), 70.51 (C-3’), 68.63 (C-5’), 45.50 ((CH3CH2)3NH+), 20.73 (CH3), 8.46 ((CH3CH2)3NH+). HRMS [M-H]− calcd for C24H23N6O8S: 555.1304; found: 555.1294.
4.2.41. 4-Amino-6-(4-fluorophenyl)-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-β-D-ribofuranosyl}-1H-[1,2,3]triazole[4,5-c]pyridine triethylammonium salt (6c)
UV (MeOH): λmax 246 nm (39800), 308.5 nm (16000). UV (H2O): λmax 245 nm (30000), 304.5 nm (13400). Fluorescence emission in MeOH: λmax 420 nm, ΦF = 0.11; in H2O: λmax 442 nm, ΦF = 0.002. 1H NMR (DMSO-d6) δ 13.56 (s, 1H, OH), 8.21 (m, 2H, Ph), 7.79 (m, 1H, sal), 7.56 (s, 1H, 7-H), 7.23 (m, 5H, NH2, Ph, sal), 6.72 (m, 2H, sal), 6.26 (d, J1’,2’=5.6 Hz, 1H, 1’-H), 5.61 (d, J2’-OH,2’H=6.4 Hz, 1H, 2’-OH), 5.46 (d, J3’-OH,3’-H=4.8 Hz, 1H, 3’-OH), 4.70 (m, 1H, 2’-H), 4.36 (m, 1H, 3’-H), 4.24 (m, 2H, 4’-H, 5’a-H), 4.18 (m, 1H, 5’b-H), 3.08 (m, 6H, (CH3CH2)3NH+), 1.17 (t, J=7.4 Hz, 9H, (CH3CH2)3NH+). 13C NMR (DMSO-d6) δ 170.90 (CO), 162.56 (d, JC,F=244.88 Hz, C-4Ph), 160.65 (C-OHsal), 151.45 and 151.35 (C-4, C-6), 138.93 (C-7a), 135.33 (Ph), 132.57 (sal), 130.85 (C-3a), 129.81 (sal), 129.18 (d, JC,F=8.42 Hz, C-2Ph, C-6Ph), 119.82, 117.50 and 116.54 (sal), 115.06 (d, JC,F=21.47 Hz, C-3Ph, C-5Ph), 90.43 and 90.37 (C-1’, C-7), 83.07 (C-4’), 72.92 (C-2’), 70.49 (C-3’), 68.65 (C-5’), 45.68 ((CH3CH2)3NH+), 8.60 ((CH3CH2)3NH+). 19F NMR (DMSO-d6) δ −38.30. HRMS [M-H]− calcd for C23H20N6O8SF: 559.1053; found: 559.1051.
4.2.42. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-β-D-ribofuranosyl}-6-(4-trifluoromethylphenyl)-1H-[1,2,3]triazole[4,5-c]pyridine triethylammonium salt (6d)
UV (MeOH): λmax 244 nm (46900), 310 nm (18700). UV (H2O): λmax 244 nm (46900), 309 nm (18000). Fluorescence emission in MeOH: λmax 409 nm, ΦF = 0.32; in H2O: λmax 422 nm, ΦF = 0.0019. 1H NMR (DMSO-d6) δ 13.54 (s, 1H, OH), 8.39 (d, J=8.4 Hz, 2H, Ph), 7.76 (m, 4H, Ph, sal, 7-H), 7.37 (s, 2H, NH2), 7.24 (m, 1H, sal), 6.71 (m, 2H, sal), 6.28 (d, J1’,2’=6.0 Hz, 1H, 1’-H), 5.62 (d, J2’-OH,2’-H=6.4 Hz, 1H, 2’-OH), 5.47 (d, J3’-OH,3’-H=5.2 Hz, 1H, 3’-OH), 4.69 (m, 1H, 2’-H), 4.36 (m, 1H, 3’-H), 4.24 (m, 3H, 4’-H, 5’-H), 3.09 (m, 6H, (CH3CH2)3NH+), 1.17 (t, J=7.2 Hz, 9H, (CH3CH2)3NH+). 13C NMR (DMSO-d6) δ 171.00 (CO),160.66 (C-OHsal), 151.64 and 150.57 (C-4, C-6), 142.67 (Ph), 138.62 (C-7a), 132.55 (sal), 131.35 (C-3a), 129.78 (sal), 128.62 (q, JC,F=32.12 Hz, C-4Ph), 127.70 (Ph), 125.17 (q, JC,F=3.85 Hz, C-3Ph, C-5Ph), 124.39 (q, JC,F=276.08 Hz, CF3), 119.77, 117.46 and 116.52 (sal), 91.84 and 90.67 (C-1’, C-7), 83.27 (C-4’), 72.92 (C-2’), 70.43 (C-3’), 68.65 (C-5’), 45.68 ((CH3CH2NH+), 8.60 ((CH3CH2)3NH+). 19F NMR (DMSO-d6) δ 14.74. HRMS [M-H]− calcd for C24H20N6O8SF3: 609.1021; found: 609.1013.
4.2.43. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-β-D-ribofuranosyl}-6-(napht-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine triethylammonium salt (6e)
UV (MeOH): λmax 251 nm (46300), 315 nm (14800). UV (H2O): λmax 250 nm (48200), 311.5 nm (13600). Fluorescence emission in MeOH: λmax 412 nm, ΦF = 0.38; in H2O: λmax 420 nm, ΦF = 0.0025. 1H NMR (DMSO-d6) δ 13.58 (s, 1H, OH), 8.73 (s, 1H, naphthyl), 8.34 (dd, J=1.8 Hz, J=8.6 Hz, 1H, naphthyl), 8.06 (m, 1H, naphthyl), 7.92 (m, 2H, naphthyl), 7.80 (dd, J=1.8 Hz, J=7.8 Hz, 1H, sal), 7.74 (s, 1H, 7-H), 7.51 (m, 2H, naphthyl), 7.32 (s, 2H, NH2), 7.24 (m, 1H, sal), 6.72 (m, 2H, sal), 6.30 (d, J1’,2’=6.0 Hz, 1H, 1’-H), 5.64 (d, J2’OH,2’H=6.0 Hz, 1H, 2’-OH), 5.48 (d, J3’OH,3’H=5.2 Hz, 1H, 3’-OH), 4.75 (m, 1H, 2’-H), 4.38 (m, 1H, 3’-H), 4.28 (m, 2H, 4’-H, 5’a-H), 4.20 (m, 1H, 5’b-H), 3.07 (brs, 6H, (CH3CH2)3NH+), 1.16 (t, J=7.2 Hz, 9H, (CH3CH2)3NH+). 13C NMR (DMSO-d6) δ 170.98 (CO), 160.68 (C-OHsal), 152.22 and 151.51 (C4, C6), 139.02 (C-7a), 136.29, 133.05 and 133.01 (naphthyl), 132.52 (sal), 131.01 (C-3a), 129.83 (sal), 128.65, 127.68, 127.33, 126.34, 126.14, 126.05 and 125.11 (naphthyl), 119.82, 117.46 and 116.51 (sal), 90.92 and 90.40 (C-1’, C-7), 83.05 (C-4’), 72.92 (C-2’), 70.53 (C-3’), 68.63 (C-5’), 45.65 ((CH3CH2)3NH+), 8.62 ((CH3CH2)3NH+). HRMS [M-H]− calcd for C27H23N6O8S: 591.1304; found: 591.1295.
4.2.44. 4-Amino-1-{5-O-[N-(2-hydroxybenzoyl)sulfamoyl]-β-D-ribofuranosyl}-6-(6-methoxynapht-2-yl)-1H-[1,2,3]triazole[4,5-c]pyridine triethylammonium salt (6f)
UV (MeOH): λmax 247.5 nm (47000), 322.5 nm (21200). UV (H2O): λmax 243 nm (26600), 318.5 nm (12600). Fluorescence emission in MeOH: λmax 418 nm, ΦF = 0.5; in H2O: λmax 425 nm, ΦF = 0.007. 1H NMR (DMSO-d6) δ 13.57 (s, 1H, OH), 8.65 (d, J=1.6 Hz, 1H, naphthyl), 8.30 (dd, J=1.8 Hz, J=8.6 Hz, 1H, naphthyl), 7.96 (d, J=8.8 Hz, 1H, naphthyl), 7.82 (m, 2H, naphthyl, sal), 7.68 (s, 1H, 7-H), 7.27 (m, 4H, naphthyl, sal, NH2), 7.15 (dd, J=2.6 Hz, J=9.0 Hz, 1H, naphthyl), 6.72 (m, 2H, sal), 6.29 (d, J1’,2’=6.0 Hz, 1H, 1’-H), 5.64 (d, J2’OH,2’H=6.0 Hz, 1H, 2’-OH), 5.48 (d, J3’OH,3’H=5.2 Hz, 1H, 3’-OH), 4.74 (m, 1H, 2’-H), 4.38 (m, 1H, 3’-H), 4.24 (m, 3H, 4’-H, 5’-H), 3.89 (s, 3H, OCH3), 3.04 (q, J=7.2 Hz, 6H, (CH3 CH2)3NH+), 1.16 (t, J=7.4 Hz, 9H, (CH3CH2)3NH+). 13C NMR (DMSO-d6) δ 171.01 (CO), 160.69 (COHsal), 157.68 (C-6naphthyl), 152.47 and 151.45 (C-4, C-6), 139.05 (C-7a), 134.46 and 134.08 (naphthyl), 132.56 (sal), 130.90 (C-3a), 130.23 (naphthyl), 129.85 (sal), 128.42, 126.61, 125.94 and 125.59 (naphthyl), 119.83 (sal), 118.69 (naphthyl), 117.49 and 116.53 (sal), 105.70 (naphthyl), 90.41 and 90.35 (C-1’, C-7), 83.05 (C-4’), 72.89 (C-2’), 70.53 (C-3’), 68.66 (C-5’), 55.20 (OCH3), 45.62 ((CH3CH2)3NH+), 8.71 ((CH3CH2)3NH+). HRMS [M-H]− calcd for C28H25N6O9S: 621.1409; found: 621.1402.
4.3. MbtA Enzyme Assay
MbtA was expressed in E. coli and purified as described. MbtA concentration was determined by active site titration with 2 as described [34]. The inhibition assays were performed as reported in duplicate under initial velocity conditions. In brief the reaction was initiated by adding 10 μL [32P]PPi with 7 nM MbtA in 90 μL reaction buffer (278 μM salicylic acid, 11.1 mM ATP, 1.11 mM PPi, 83.3 mM Tris-HCl, pH 7.5, 11.1 mM MgCl2, 2.22 mM DTT) at 37 °C in the presence of eight different concentrations of the inhibitor (1.5-fold dilution from 100 nM down to 8.78 nM and a 3-fold dilution to 2.93 nM). The reaction was terminated at 20 min by the addition of 200 μL of quenching buffer (350 mM HClO4, 100 mM PPi, 1.8 % w/v activated charcoal). The charcoal was pelleted by centrifugation and washed once with 500 μL H2O and analyzed by liquid scintillation counting as described. Fractional initial velocities were fit by nonlinear regression analysis to the Morrison equation (eq 1) using GraphPad Prism 5.0
| (eq 1) |
where, I represents the concentration of inhibitor, is the apparent inhibition constant, E is the active enzyme concentration (determined by active-site titration), vi is the initial rate at each [I], and v0 is the initial rate of the DMSO control. A fitted curve for compound 6e is shown (Fig. 3) as an example.
Figure 3.

Dose-response of fractional initial velocity of [32P]-ATP formation catalyzed by MbtA as a function of inhibitor 6e concentration.
4.4. M. tuberculosis H37Rv MIC Assay
All compounds minimum inhibitory concentrations (MICs) were experimentally determined as previously described. MICs were determined in triplicate in iron-deficient GAST according to the broth microdilution method using compounds from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. Isoniazid was used as a positive control while DMSO was employed as a negative control. All measurements reported herein used an initial cell density of 104–105 cells/assay and growth monitored at 10–14 days, with the untreated and DMSO-treated control cultures reaching an OD620 0.2–0.3. Plates were incubated at 37 °C (100 μL/well) and growth was recorded by measurement of optical density at 620 nm.
Supplementary Material
Acknowledgments
This work was supported by the Polish Ministry of Science and Higher Education grant no. N N405 2516 33 (J.Z.), under statutory financing, and under the KNOW program. It was also supported by a grant from the NIH (AI070219 to C.C.A.) and the Intramural Research Program of the NIAID, NIH (C.E.B.). We wish to thank Dr. Lukasz Marczak and Dr. Barbara Swarcewicz for MS spectra measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found in the online version at
References
- 1.Global Tuberculosis Report 2015. World Health Organization; Geneva: 2015. ( http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf?ua=1) [Google Scholar]
- 2.Horsburgh CR, Jr., Barry CE, 3rd, Lange C. Treatment of tuberculosis. N. Engl. J. Med. 2015;373:2149–2160. doi: 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- 3.Lenaerts A, Barry CE, 3rd, Dartois V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol. Rev. 2015;264:288–307. doi: 10.1111/imr.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Companion handbook to the WHO guidelines for the programmatic management of drug-resistant tuberculosis. World Health Organization; Geneva: 2014. [PubMed] [Google Scholar]
- 6.Tuberculosis in the WHO European Region 2015. World Health Organization; Geneva: 2015. ( http://www.euro.who.int/__data/assets/pdf_file/0010/273169/WTBD_2015_FS_Final_ENG.pdf) [Google Scholar]
- 7.Fang Z, Sampson SL, Warren RM, van Pittius NCG, Newton-Foot M. Iron acquisition strategies in mycobacteria. Tuberculosis. 2015;95:123–130. doi: 10.1016/j.tube.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 8.McMahon MD, Rush JS, Thomas MG. Analysis of MbtB, MbtE, and MbtF suggest revisions to the mycobactin biosynthesis pathway in Mycobacterium tuberculosis. J. Bacteriol. 2012;194:2809–2818. doi: 10.1128/JB.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 10.Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006;273:409–419. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- 11.Somu RV, Boshoff H, Qiao C, Bennett EM, Barry CE, 3rd, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 12.Lun S, Guo H, Adamson J, Cisar JS, Davis TD, Chavadi SS, Warren JD, Quadri LE, Tan DS, Bishai WR. Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother. 2013;57:5138–5140. doi: 10.1128/AAC.00918-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiao C, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, III, Aldrich CC. 5’-O-[(N-Acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: an adenylation enzyme required for siderophore biosynthesis of the mycobactins. J. Med. Chem. 2007;50:6080–6094. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008;51:5349–5370. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi C, Tiwari D, Wilson DJ, Seiler CL, Schnappinger D, Aldrich CC. Bisubstrate inhibitors of biotin protein ligase in Mycobacterium tuberculosis resistant to cyclonucleoside formation. ACS Med. Chem. Lett. 2013;4:1213–1217. doi: 10.1021/ml400328a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franchetti P, Messini L, Cappellacci L, Grifantini M, Nocentini G, Guarracino P, Marongiu ME, La Colla P. 8-Aza derivatives of 3-deazapurine nucleosides. Synthesis and in vitro evaluation of antiviral and antitumor activity. Antiviral Chem. Chemother. 1993;4:341–352. [Google Scholar]
- 17.May JA, Jr., Townsend LB. Synthesis of v-triazolo[4,5-c]pyridine nucleosides and 4-(β-D-ribofuranosyl)amino-1,2,3-thiadiazolo[5,4-b]pyridine via a rearrangement. J. Org. Chem. 1976;41:1449–1456. doi: 10.1021/jo00870a035. [DOI] [PubMed] [Google Scholar]
- 18.Meyer RB, Jr., Revankar GR, Dan Cook P, Ehler KW, Schweizer MP, Robins RK. Synthesis of 6-amino-1,2,3-triazolo[4,5-c]pyridin-4(5H)one (8-aza-3-deazaguanine) and 6-amino-1-(β-D-ribofuranosyl)-1,2,3-triazolo[4,5-c]pyridin-4(5H)one (8-aza-3-deazaguanosine) via novel ring closure procedures. J. Heterocyclic Chem. 1980;17:159–169. [Google Scholar]
- 19.Earl RA, Townsend LB. The synthesis of 8-aza-3-deazaguanosine [6-amino-1-(β-D-ribofuranosyl)-v-triazolo[4,5-c]pyridin-4-one] via a novel 1,3-dipolar cycloaddition reaction. Can. J. Chem. 1980;58:2550–2561. [Google Scholar]
- 20.Stimac A, Townsend LB, Kobe J. The synthesis of v-triazolo[4,5-c]pyridine nucleosides. Nucl. Nucl. 1991;10:727–728. [Google Scholar]
- 21.Stimac A, Leban I, Kobe J. An efficient stereospecific method for the synthesis of 8-aza-3-deazaguanine nucleosides from glycosyl azides. Synlett. 1999:1069–1073. [Google Scholar]
- 22.Jeselnik M, Jaksa S, Kobe J. Synthesis of 8-aza-3-deazaisoguanosine by a novel ring closure of dinitriles by sodium alkoxides. Croat. Chem. Acta. 2004;77:153–160. [Google Scholar]
- 23.Minakawa N, Matsuda A. Nucleosides and nucleotides. 114. A convenient method for the synthesis of 3-deazapurine nucleosides from AICA-riboside. Tetrahedron Lett. 1993;34:661–664. [Google Scholar]
- 24.Minakawa N, Matsuda A. Nucleosides and nucleotides. 116. Convenient syntheses of 3-deazaadenosine, 3-deazaguanosine, and 3-deazainosine via ring closure of 5-ethynyl-1-β-D-ribofuranosylimidazole-4-carboxamide or -carbonitrile. Tetrahedron. 1993;49:557–570. [Google Scholar]
- 25.Joubert N, Schinazi RF, Agrofoglio LA. Efficient Pd(0)-catalyzed synthesis of 1,2,3-triazolo-3’-deoxycarbanucleosides and their analogues. Tetrahedron. 2005;61:11744–11750. [Google Scholar]
- 26.Hutzenlaub W, Tolman RL, Robins RK. Azapurine nucleosides. 1. Synthesis and antitumor activity of certain 3-β-d-ribofuranosyl- and 2′deoxy-d-ribofuranosyl-v-triazolo[4,5-d]pyrimidines. J. Med. Chem. 1972;15:879–883. doi: 10.1021/jm00279a001. [DOI] [PubMed] [Google Scholar]
- 27.Biagi G, Giorgi I, Livi O, Scartoni V. Synthesis of new 2-substituted 9-β-d-ribofuranosyl-8-azahypoxanthines. Il Farmaco. 1992;47:525–536. [Google Scholar]
- 28.Ostrowski T, Januszczyk P, Cieslak M, Kazmierczak-Baranska J, Nawrot B, Bartoszak-Adamska E, Zeidler J. 5-Ethynyl-1-β-d-ribofuranosyl-1H-[1,2,3]triazole-4-carboxylic acid amide (ETCAR) and its analogues: synthesis and cytotoxic properties. Bioorg. Med. Chem. 2011;19:4386–4398. doi: 10.1016/j.bmc.2011.05.050. [DOI] [PubMed] [Google Scholar]
- 29.Negishi E, Anastasia L. Palladium-catalyzed alkynylation. Chem. Rev. 2003;103:1979–2017. doi: 10.1021/cr020377i. and ref. therein. [DOI] [PubMed] [Google Scholar]
- 30.Minakawa N, Takayuki T, Sasaki T, Matsuda A, Ueda T. Nucleosides and nucleotides. 96. Synthesis and antitumor activity of 5-ethynyl-1-β-d-ribofuranosylimidazole-4-carboxamide (EICAR) and its derivatives. J. Med. Chem. 1991;34:778–786. doi: 10.1021/jm00106a045. [DOI] [PubMed] [Google Scholar]
- 31.Benanti TL, Saejueng P, Venkataraman D. Segregated assemblies in bridged electron-rich and electron-poor π-conjugated moieties. Chem. Commun. 2007:692–694. doi: 10.1039/b610565c. [DOI] [PubMed] [Google Scholar]
- 32.Crisp GT, Flynn BL. Palladium-catalyzed coupling of terminal alkynes with 5-(trifluoromethanesulfonyloxy)pyrimidine nucleosides. J. Org. Chem. 1993;58:6614–6619. [Google Scholar]
- 33.Okada M, Iwashita S, Koizumi N. Efficient general method for sulfamoylation of a hydroxyl group. Tetrahedron Lett. 2000;41:7047–7051. [Google Scholar]
- 34.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, 3rd, Aldrich CC. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J. Med. Chem. 2006;49:7623–7635. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of prolyl adenylate analogs. Bioorg. Chem. 1996;24:273–289. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.