

Abstract
Long noncoding RNAs (lncRNAs) were discovered in eukaryotes more than thirty years ago [1]. Recent advances in genomics have led to the discovery that lncRNAs are transcribed pervasively across the genome [2–5]. There are an increasing number of reports that identify lncRNAs whose expression is modulated during cell differentiation or in disease states. However, biological functions for the vast majority of them are yet to be determined. Here, we propose two ways to identify lncRNAs that have biological functions: To identify lncRNAs with dedicated preinitiation complexes (PICs), and to focus on those whose transcription is highly regulated.
Biological roles of long noncoding RNAs (lncRNAs)
Recent advancement in technologies such as next-generation deep sequencing and tiled microarrays has enabled genome-wide analyses of the eukaryotic transcriptome. One of the unexpected findings from these analyses is that a very large number of long non-coding RNAs (lncRNAs) are transcribed from eukaryotic genomes, from budding yeast to humans [2,4–6]. Indeed, current estimation is that more than 80% of eukaryotic genomes are transcribed [5], meaning that thousands of lncRNAs are transcribed in eukaryotic cells. lncRNAs are usually defined as RNA transcripts that are longer than 200 base pairs that do not have the potential to encode protein [7]. High-resolution analyses of lncRNAs revealed that they can arise under a variety of genomic contexts: intergenically or intragenically, and in the sense or antisense orientations. One of the most important questions right now is how many of the thousands of identified lncRNAs play biological roles. Although some researchers believe that the vast majority of lncRNAs are products of stochastic transcription [8], it has become clear that some lncRNAs are capable of possessing regulatory roles in gene expression.
There have been several examples in which transcription of antisense lncRNA leads to down-regulation of its cognate mRNA, and the underlying mechanisms have been reported for many of these cases. For instance, the lncRNA transcript can recruit histone-modifying enzymes to specific genomic loci, thereby creating repressive transcriptional environments. In mammals, the lncRNAs HOTAIR and Xist work in this manner to down-regulate the HOX genes and to inactivate one of the X-chromosomes, respectively [7,9–11]. In the budding yeast, Saccharomyces cerevisiae, transcription of the antisense, lncRNA at the PHO84 gene is coincident with recruitment of the lysine deacetylase (KDAC) Hda1, which suppresses PHO84 mRNA transcription, showing potential evolutionary conservation of lncRNA-mediated gene regulation mechanisms [12,13]. Whether the lncRNA at PHO84 directly recruits Hda1 is still not clear [14]. It should be noted that, in some cases, the act of lncRNA transcription, rather than the lncRNA products, play regulatory roles [15].
These examples in both humans and yeast show that lncRNAs are expressed from an array of contexts across all eukaryotes, and can work through various mechanisms to regulate gene expression, potentially underlying disease pathophysiology. Despite this, functional roles have still not been assigned to the vast majority of lncRNAs. Therefore, establishing a method to systematically identify (or at least enrich for) lncRNAs or lncRNA transcription events that play biological roles would be a very significant step forward. We propose two ways to systematically enrich for lncRNA transcripts or transcription events that likely play biological roles: (1) Identify lncRNAs that have dedicated pre-initiation complexes (PICs). (2) Identify lncRNAs whose transcription is highly regulated.
lncRNAs that have dedicated PICs
As far as we know, the vast majority, if not all, of lncRNAs are transcribed by RNA polymerase II (Pol II). Initiation of Pol II transcription absolutely depends on ordered targeting of general transcription factors (GTFs), such as TFIIB and TFIID, to promoters, which leads to the formation of a PIC near the transcriptional initiation sites. Therefore, all protein coding genes that are either actively transcribed, or are poised to be transcribed, have PICs at their promoters. The major source of lncRNA is divergent promoters in both budding yeast [2,4] and humans [16], in which transcription of mRNA and lncRNA initiate from a shared nucleosome depleted region (NDR), where PIC forms (Figure 1). Because NDRs are typically small (less than 300 bp), the resolution afforded by conventional chromatin immuno-precipitation (ChIP) followed by deep sequencing (ChIP-seq) of GTFs cannot determine whether the mRNA and lncRNA share a PIC or they have distinct PICs with high confidence. However, the recent development of ChIP-exo, a super-high resolution ChIP-seq method, enabled genome-wide mapping of PICs at base-pair resolution [17]. The initial report describing the ChIP-exo analyses of GTFs indeed revealed that a significant fraction of divergent promoters on S. cerevisiae genome have two distinct PICs at each end of NDRs, one for mRNA and another for lncRNAs (Figure 1). If a lncRNA has a dedicated PIC, it means that GTFs are targeted in an ordered fashion to assemble the PIC for the lncRNA, and that Pol II is recruited by the PIC only to transcribe the lncRNA. This suggests that the lncRNA represents a discrete transcription unit, and there is a higher likelihood that cells “intend” to transcribe the lncRNA. On the other hand, if a mRNA-lncRNA pair shares a PIC, that suggests that Pol II that transcribe lncRNA may be targeted by the PIC for mRNA transcription. In this case, lncRNA may represent the product of erratic initiation of mRNA, or transcriptional “noise”. Therefore, it is likely that a systematic identification of lncRNAs that have dedicated PICs would enrich for lncRNAs and lncRNA transcription events that play biological roles. ChIP-exo and other super high-resolution method for identifying PIC locations are applicable for metazoan cells [18,19], making this strategy available across eukaryotes.
Figure 1. lncRNAs with a shared PIC and dedicated PIC.
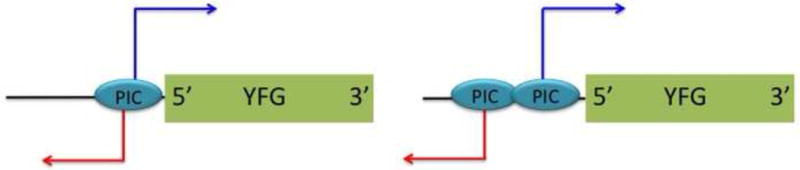
(left) mRNA (blue) and lncRNA (red) share a PIC at the promoter of a protein coding gene (YFG). (right) lncRNA has a dedicated PIC, which is not shared with a mRNA.
lncRNAs whose transcription is highly regulated
Another way we envision to identify lncRNAs that have biological roles is to focus on the transcripts or transcription events that are highly regulated. The rationale is intuitive: There is no reason to regulate transcription of a lncRNA if it is simply a consequence of “noise”. Two different approaches can be taken. One is to identify lncRNAs that are differentially expressed under different circumstances, such as different cell types, growth conditions and disease states. This strategy has been very successful, and a very large number of lncRNAs associated with specific developmental stages or diseases have been identified this way [20][21]. Another approach is to systematically identify lncRNA regulators and their targets. This would involve the identification of lncRNAs that exhibit abnormal transcript levels when the lncRNA regulators are mutated. Of course, these mutations can cause abnormal transcription events, which can produce non-functional lncRNAs. However, most of the lncRNAs that are elevated in these mutants are detectable in wild type cells. In addition, we have started to learn that a lot of cellular resources are used to repress lncRNA transcription as described below. It is therefore difficult to envision that all of these resources are used simply to suppress noise.
The fact that lncRNAs generally initiate from NDRs suggests that their transcription is strongly affected by chromatin structure, much like mRNAs. One way the cell can regulate DNA accessibility is through the use of ATP-dependent chromatin remodeling factors, which can mobilize nucleosomes or alter histone-DNA contacts to regulate DNA accessibility [22]. The first chromatin remodeling factor found to repress lncRNA transcription is Isw2 (Imitation Switch 2) in S. cerevisiae. Isw2 functions to slide nucleosomes along the length of DNA [23,24]. Genome-wide analysis of Isw2 targets revealed that Isw2 functions at NDRs at both the 5′ and 3′ ends of genes [25], and that Isw2 is targeted and represses anti-sense lncRNAs from divergent promoters at the 3′ ends of genes [25,26]. More recently, it was shown that mammalian esBAF, a Swi/Snf family chromatin remodeling factor that is required to maintain embryonic stem cell (ESCs) renewal and pluripotency [27], was found to repress lncRNAs genome-wide [15]. esBAF accomplishes this function by stabilizing nucleosomes at lncRNA initiation sites and maintaining a well-defined NDR [15].
Genetic screens for lncRNA regulators and identification of their targets
Although mutagenesis or manipulation of candidates for lncRNAs regulators to identify highly regulated lncRNAs is feasible, more systematic, unbiased approaches can be taken. So far, large-scale genetic screens for lncRNA regulators have been performed in S. cerevisiae (see figure 2 for schematic drawing of strategies).
Figure 2. Genetic screens to identify regulators of lncRNA expression.
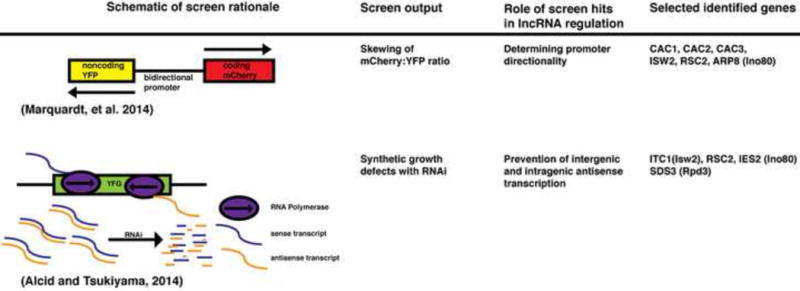
Schematic drawing of the strategies and summary of salient results from two large scale genetic screens for lncRNA regulators. See text for details.
The identification of factors that maintain proper mRNA-lncRNA transcription ratios
Following pioneering work in which a genetic screen was performed to identify genes that repress initiation of intragenic cryptic transcription [28], a genome-wide screen to identify genes that maintain proper relative transcription levels of mRNA and lncRNA pairs at bidirectional promoters was developed [29]. In this screen, a reporter was constructed by using a divergent promoter sequence, fusing a mCherry fluorescent marker on the coding end, and a YPF marker on at the noncoding end, which was introduced to the yeast deletion mutant library collection [30]. This reporter allowed for the systematic identification of mutants in which the relative levels of transcription on the coding and non-coding sides of the divergent promoter is skewed compared to wild type cells. This screen identified many genes involved in chromatin remodeling and chromatin assembly, including subunits of Swr1, Rsc, Ino80, and Isw2 chromatin remodeling factors. They also identified three genes encoding subunits of a histone chaperone the Chromatin Assembly Factors 1 (CAF-1) complex CAC1, CAC2, CAC3. Consistent with the design of the genetic screen, NET-seq [31] analysis on CAF-1 mutants revealed a genome-wide increase in lncRNA transcription relative to mRNA at divergent promoters, making it the first factor identified to have a genome-wide role in dictating transcriptional direction. Interestingly, the elevated lncRNA transcription in CAF-1 mutants is dependent on histone H3 K56 acetylation and Swi/Snf chromatin remodeling complex, demonstrating that multiple chromatin regulators work on lncRNAs for proper control [29]. Although this genetic screen was not specifically designed to identify lncRNAs with biological roles, it is possible that some of the elevated transcripts have biological roles: For example, the increased level of lncRNAs proximal to bidirectional promoters might be a cellular signal of chromatin disruption, which may be required to target chromatin repair factors to the sites of chromatin disruption. For the purpose of identifying functional lncRNA transcripts and transcription events, this genetic screen can also be modified to use different divergent promoters: For example, one can use divergent promoters that form distinct PICs for mRNA and lncRNA, and/or use the promoters that are highly regulated by the environment or growth conditions.
The identification of chromatin remodeling complexes that regulate putative functional lncRNAs
Our lab has also developed a genome-wide screen using reconstituted RNA interference (RNAi) as a tool to identify repressors of anti-sense lncRNAs (ASlncRNAs) [32]. The rationale behind the design of the screen is that the global increase in ASlncRNAs in the presence of RNAi would cause growth defects: This would result in a large-scale formation of mRNA:ASlncRNA hybrids, which would be processed by RNAi, causing global destabilization of mRNAs and lncRNAs (Figure 2). This screen identified 408 putative lncRNA repressors, including the chromatin remodeling factors Swr1, Isw2, Rsc, and Ino80, which were all confirmed to be the repressors of ASlncRNAs by RNA-seq. This result suggests that the cell devotes a much larger amount of resources than previously thought to repress lncRNAs. It was shown that Isw2, RSC and Ino80 are targeted to the initiation sites of the ~45% (814) of ASlncRNAs that are repressed by these factors, suggesting that these remodeling factors directly repress transcription of a large number of ASlncRNAs. Most importantly, de-repression of 259 ASlncRNAs in chromatin-remodeling factor mutants is associated with a significant decrease in the level of mRNAs they overlap. This suggests that chromatin-remodeling factors maintain the levels of these 259 mRNAs through repression of transcription of overlapping ASlncRNAs. It is likely that this number represents a gross underestimation of functional ASlncRNA transcription events, as only one growth condition (logarithmic growth in rich medium) was used for RNA analyses. Given the presence of ~400 putative uncharacterized ASlncRNA repressors, it is likely that the mechanism to regulate mRNA levels through ASlncRNA control is utilized far more commonly across the genome than currently appreciated.
Concluding Remarks
Despite the increasing number of reports that identify lncRNAs in the context of development and disease [20,21], functions (if any) of the vast majority of lncRNAs remain to be identified. It is possible that lncRNA products have biological roles: Given that lncRNAs can provide information about the sequence and the location of transcription, they can be the tools for cells to target histone modifying enzymes, chromatin repair factors or other regulators to specific sites. It is also possible that the act of lncRNA transcription itself provide biological functions: It may facilitate co-transcriptional chromatin assembly, chromatin reorganization or interfere with other DNA-dependent processes. By focusing on lncRNAs that have dedicated PICs and those whose expression levels are highly regulated, one would likely be able to systematically enrich for lncRNA transcripts or transcription events that play important biological roles.
Acknowledgments
We thank the members of the Tsukiyama lab for helpful discussions. This work was supported by grant R01 GM058465 (T.T.) and predoctoral fellowship F31 GM101944 (E.A.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vanhe′e-Brossollet C, Vaquero C. Do natural antisense transcripts make sense in eukaryotes? Gene. 1998;211:1–9. doi: 10.1016/s0378-1119(98)00093-6. [DOI] [PubMed] [Google Scholar]
- 2*.Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–1042. doi: 10.1038/nature07747. This manuscript shows that bidirectional promoters are the major source of lncRNAs in budding yeast. [DOI] [PubMed] [Google Scholar]
- 3.Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14:880–893. doi: 10.1038/nrg3594. [DOI] [PubMed] [Google Scholar]
- 4.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. This manuscript describes a high-resolution human lncRNA transcriptome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. This manuscript describes an identification of HOTAIR as a lncRNA that regulates HOX gene transcription. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louro R, Smirnova AS, Verjovski-Almeida S. Long intronic noncoding RNA transcription: Expression noise or expression choice? Genomics. 2009;93:291–298. doi: 10.1016/j.ygeno.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 9*.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. This manuscript shows that a lncRNA HOTAIR plays pivotal roles in cancer metastasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Camblong J, Beyrouthy N, Guffanti E, Schlaepfer G, Steinmetz LM, Stutz F. Transacting antisense RNAs mediate transcriptional gene cosuppression in S. cerevisiae. Genes & development. 2009;23:1534–1545. doi: 10.1101/gad.522509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell. 2007;131:706–717. doi: 10.1016/j.cell.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 14.Castelnuovo M, Rahman S, Guffanti E, Infantino V, Stutz F, Zenklusen D. Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nature structural & molecular biology. 2013;20:851–858. doi: 10.1038/nsmb.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15*.Hainer SJ, Gu W, Carone BR, Landry BD, Rando OJ, Mello CC, Fazzio TG. Suppression of pervasive noncoding transcription in embryonic stem cells by esBAF. Genes & Development. 2015;29:362–378. doi: 10.1101/gad.253534.114. This manuscript reports that a mammalian chromatin remodeling factor esBAF represses lncRNA transcription by stabilizing nucleosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Core LJ, Waterfall J, Lis J. Nascent RNA Sequencing Reveals Widespread Pausing and Divergent Initiation at Human Promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. doi: 10.1038/nature10799. This manuscript describes the development of ChIP-exo, a super high-resolution method to determine protein binding sites on chromatin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang GS, Chen XA, Park B, Rhee HS, Li P, Han KH, Mishra T, Chan-Salis KY, Li Y, Hardison RC, et al. A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep. 2014;8:514–527. doi: 10.1016/j.celrep.2014.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skene PJ, Henikoff S. A simple method for generating high-resolution maps of genome-wide protein binding. Elife. 2015;4:e09225. doi: 10.7554/eLife.09225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulitsky I, Bartel David P. lincRNAs: Genomics, Evolution, and Mechanisms. Cell. 154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flynn RA, Chang HY. Long Noncoding RNAs in Cell-Fate Programming and Reprogramming. Cell stem cell. 2014;14:752–761. doi: 10.1016/j.stem.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartholomew B. Regulating the Chromatin Landscape: Structural and Mechanistic Perspectives. Annual Review of Biochemistry. 2014;83:671–696. doi: 10.1146/annurev-biochem-051810-093157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fazzio TG, Tsukiyama T. Chromatin Remodeling In Vivo: Evidence for a Nucleosome Sliding Mechanism. Molecular Cell. 2003;12:1333–1340. doi: 10.1016/s1097-2765(03)00436-2. [DOI] [PubMed] [Google Scholar]
- 24.Whitehouse I, Tsukiyama T. Antagonistic forces that position nucleosomes in vivo. Nat Struct Mol Biol. 2006;13:633–640. doi: 10.1038/nsmb1111. [DOI] [PubMed] [Google Scholar]
- 25**.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450:1031–1035. doi: 10.1038/nature06391. This manuscript reveales that Isw2 chromatin remodeling factor represses antisense lncRNA transcription. [DOI] [PubMed] [Google Scholar]
- 26.Yadon AN, Van de Mark D, Basom R, Delrow J, Whitehouse I, Tsukiyama T. Chromatin remodeling around nucleosome-free regions leads to repression of noncoding RNA transcription. Molecular and cellular biology. 2010;30:5110–5122. doi: 10.1128/MCB.00602-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, Lessard J, Nesvizhskii AI, Ranish J, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proceedings of the National Academy of Sciences. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, Winston F. Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS biology. 2008;6:e277. doi: 10.1371/journal.pbio.0060277. This manuscript describes a large-scale genetic screen for repressors of cryptic intragenic lncRNAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29**.Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S. A chromatin-based mechanism for limiting divergent noncoding transcription. Cell. 2014;157:1712–1723. doi: 10.1016/j.cell.2014.04.036. This manuscript describes a genetic screen for factors involved in determining transcriptional direction from divergent promoters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 31.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Alcid EA, Tsukiyama T. ATP-dependent chromatin remodeling shapes the long noncoding RNA landscape. Genes Dev. 2014;28:2348–2360. doi: 10.1101/gad.250902.114. This manuscript describes a genetic screen for repressors of antisense lncRNAs, and showed that regulation of mRNA transcription through anti-sense lncRNA is likely far more common than previously thought. [DOI] [PMC free article] [PubMed] [Google Scholar]