

Abstract
Efavirenz is highly effective at suppressing HIV-1, and the WHO guidelines list it as a component of the first-line antiretroviral (ARV) therapies for treatment-naïve patients. Though the pharmacological basis is unclear, efavirenz is commonly associated with a risk for neuropsychiatric adverse events (NPAEs) when taken at the prescribed dose. In many patients these NPAEs appear to subside after several weeks of treatment, though long-term studies show that in some patients the NPAEs persist. In a recent study focusing on the abuse potential of efavirenz, its receptor psychopharmacology was reported to include interactions with a number of established molecular targets for known drugs of abuse, and it displayed a prevailing behavioral profile in rodents resembling an LSD-like activity. In this report, we discovered interactions with additional serotonergic targets that may be associated with efavirenz-induced NPAEs. The most robust interactions were with 5-HT3A and 5-HT6 receptors, with more modest interactions noted for the 5-HT2B receptor and monoamine oxidase A. From a molecular mechanistic perspective, efavirenz acts as a 5-HT6 receptor inverse agonist of Gs-signaling, 5-HT2A and 5-HT2C antagonist of Gq-signaling, and a blocker of the 5-HT3A receptor currents. Efavirenz also completely or partially blocks agonist stimulation of the M1 and M3 muscarinic receptors, respectively. Schild analysis suggests that efavirenz competes for the same site on the 5-HT2A receptor as two known hallucinogenic partial agonists (±)-DOI and LSD. Prolonged exposure to efavirenz reduces 5-HT2A receptor density and responsiveness to 5-HT. Other ARVs such as zidovudine, nevirapine and emtricitabine did not share the same complex pharmacological profile as efavirenz, though some of them weakly interact with the 5-HT6 receptor or modestly block GABAA currents.
Keywords: Efavirenz, 5-HT2A, LSD, DOI, 5-HT2C, 5-HT6
1. Introduction
Efavirenz [(4S)-6-chloro-4-(2-cyclopropylethynyl)-4-(trifluoromethyl)-2,4-dihydro-1H-3,1-benzoxazin-2-one] is a potent non-nucleoside reverse transcriptase inhibitor (NNRTI) and one of the preferred components of highly active antiretroviral therapy (HAART) [1, 2]. Though very effective at suppressing replication of the virus that causes AIDS, a standard dose of efavirenz is known to carry a significant risk for CNS-mediated NPAEs [3–8]. However, little is known from a mechanistic perspective as to why these NPAEs occur and which CNS off-targets might be involved. The findings of a previous report [9] led us to examine the molecular mechanisms of efavirenz across a broader range of serotonergic targets.
Though efavirenz is considered to have good CNS penetration [10] consistent with its physicochemical properties, only low levels are detected in cerebral spinal fluid (CSF) with a mean concentration across studies of approximately 44 nM that corresponds to CSF levels 0.52% of plasma concentrations [11–13]. Efavirenz has a very high tendency to bind proteins (e.g, approximately 99.8% is bound to plasma proteins) [14, 15], and healthy CSF contains very little protein compared to that in blood plasma (< 1%). Since the concentration of free efavirenz in the aqueous fraction is expected to be low and it binds to protein-rich brain tissue [16], one might expect that 44 nM in CSF, if it accounted for approximately 0.2% (100%−99.8%) of the efavirenz dose, would correspond to an estimated 22 μM (44 nM ÷ 0.002) concentration of efavirenz in brain tissue. A study in rats suggests that efavirenz readily accumulates in the brain to levels that exceed 4.6 times the plasma levels within 1 hr of an i.p. dose of 15 mg/kg [16]. Physiological effects have been reported in rodent studies at efavirenz doses in the same range: 10–30 mg/kg depending upon the behavioral measure examined [9, 17]. High plasma levels of efavirenz (1 – 4 μg/mL) are needed to keep the HIV-1 virus suppressed to clinically meaningful levels [18], but efavirenz also has a narrow therapeutic window [3]: plasma levels less than 1 μg/mL result in virologic failure and while those greater than 2.74 μg/mL result in NPAE [3, 18]. Assuming a similar level of brain accumulation occurs in humans as in rats, then efavirenz plasma levels >2.74 μg/mL would correspond to brain concentrations >40 μM (2.74×10−3 g/L • 1/315.7 g/mole • 4.6). Based upon these types of assumptions and calculations, it would appear that efavirenz can rapidly accumulate in the brain to concentrations in the range of tens of micromolars. Rapid accumulation of relatively high concentrations of efavirenz in the brain and evidence of CNS behavioral effects in animals and humans suggest that this reverse transcriptase inhibitor has CNS off-targets.
It has been almost two decades since efavirenz was approved by the FDA and to date, there is only one study that has sought to investigate the receptor neuropharmacology underpinning efavirenz’s CNS targets and this was in the context of its reported recreational use [9]. In that study, a rationalized mechanistic approach was utilized in that receptor targets known to interact with drugs of abuse were selected to narrow the receptor profiling effort leading to a number of CNS receptors being identified as possible targets for efavirenz. At the receptor level, efavirenz was shown to interact with the 5-HT2A, 5-HT2C and GABAA receptors, as well as DAT, SERT, and VMAT2 transporters [9]. Mechanistically, efavirenz potentiated GABA-mediated chloride currents at the GABAA receptor, and functioned as a DAT and SERT blocker [9]. However, in vivo, efavirenz failed to strongly substitute in tests of discrimination for drugs known to have these same functional properties on DAT, SERT, VMAT2 transporters, or the GABAA receptor [9]. In contrast, when efavirenz was used as the discriminative stimulus, LSD partially substituted for efavirenz. Moreover, in rats trained to discriminate LSD from saline, efavirenz partially substituted for LSD and this substitution was blocked by pretreatment with the 5-HT2A receptor selective antagonist MDL100,907 [9]. In a rodent head-twitch assay, efavirenz produced head-twitch responses in wild type but not 5-HT2A-KO mice; though, the response was far weaker than for LSD and initiated much more rapidly [9]. There is a strong positive correlation between compounds that are hallucinogens in humans and those that induce a head-twitch response in rodents [19–21]. Efavirenz also dose-dependently depressed novel open field locomotor activity, similar to LSD in the same strain of mice [9]. For these reasons, it was concluded that efavirenz’s predominate behavioral profile in rodents is most consistent with an LSD-like effect mediated by the 5-HT2A receptor [9].
In this study we report for the first time that efavirenz interacts with the 5-HT3, 5-HT6 receptors and the enzyme MAO-A, and elucidate its molecular mechanisms of action at these targets as well as at the 5-HT2A and 5-HT2C receptors. Within a similar concentration range, efavirenz also blocks agonist responses at the M1 and M3 muscarinic receptors. Schild shift analysis suggests efavirenz acts at the same binding site on the 5-HT2A receptor as the partial agonists LSD and (±)-DOI, but it is unique in that it does not activate the Gq-signaling pathway. Chronic exposure of the 5-HT2A receptor to efavirenz reduces receptor density and responsiveness to 5-HT. We also demonstrate here that efavirenz’s mechanism of action across a range of CNS targets is distinct from other prominent HIV-1 medications like emtricitabine (FTC), nevirapine (NVP) and zidovudine (ZDV).
2. Materials and Methods
2.1. Chemicals
Radiochemicals were from Perkin Elmer (Saint Louis, MO): 4-(2″-Methoxy)-phenyl-1-[2″-(N-2-pyridinyl)-p-fluorobenzamido]ethyl-piperazine ([3H]MPPF, NET-1109, 80 Ci/mmol); [3H]methylspiperone ([3H]MSP, NET-856, 84 Ci/mmol); [3H]Lysergic acid diethylamide ([3H]LSD, NET638, 70 Ci/mmol); [3H]Mesulergine (NET1148, 80 Ci/mmol); [3H]BRL-43694 (granisetron) (NET1030, 65 Ci/mmol); [3H]GR113808 (NET1152, 85 Ci/mmol). Efavirenz, [(4S)-6-chloro-4-(2-cyclopropylethynyl)-4-(trifluoromethyl)-2,4-dihydro-1H-3,1-benzoxazin-2-one], and the other antiretroviral drugs (emtricitabine (FTC), zidovudine (ZDV) and nevirapine (NVP)) were purchased from Sequoia Research Products Limited (Pangbourne, UK). MDL100,907 was synthesized and kindly provided by Dr. Kenner C. Rice (NIDA/NIAAA). Unless otherwise noted, all other drugs and reagents were purchased from Tocris Biosciences (via R&D Systems, Inc. in Minneapolis, MN) or Sigma-Aldrich (St. Louis, MO). All ARV drugs were solubilized in DMSO at concentrations ranging from 10–100 mM and diluted at least 1:1000 v/v in the final assay solution.
2.2. Profiling Serotonin Receptors by Radioligand Binding
The interaction of efavirenz (10 μM) with serotonin receptors was measured by its ability to displace specifically bound radioligands from the metabotropic 5-HT1A, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT4, 5-HT5A, 5-HT6 and 5-HT7 receptors, and an ionotropic 5-HT3A receptor using the conditions outlined in Table 1. With the exception of the 5-HT4 receptor, which was sourced from Duncan Hartley guinea pig striatal tissue, all other serotonin receptors were cloned receptors heterologously expressed in mammalian cell lines lacking the receptor subtypes of interest. Radioligand, purified membranes expressing individual serotonin receptors and a fixed concentration of 10 μM efavirenz in a total volume of 1 mL binding buffer were allowed to equilibrate. Receptors were then isolated by rapid filtration through glass fiber filters pretreated for 10 min with 0.5% polyethyleneimine (Sigma-Aldrich) and three rapid washes with 3 mL of ice-cold wash buffer (50 mM Tris, pH 7.4 at 0–2°C). GF/C filters were used for membranes derived from cell cultures and GF/B filters for membranes derived from brain tissue (Brandel, Gaithersburg, MD). Dried filters were cut into individual scintillation vials, filled with 3.5 ml of scintillation fluid, mixed, and the radioactivity bound to the filters was quantified via scintillation spectroscopy. Membrane protein concentrations varied from 0.02–0.06 mg/mL. All data for each experiment was measured in triplicate. Each experiment was then repeated two or three times and the mean values of these experiments were reported with their associated SEM. A one-way ANOVA followed by a Bonferroni post-hoc analysis set to high stringency (P < 0.001) was used to determine significant displacement of the radioligand.
Table 1.
Conditions for measuring efavirenz’s ability to displace radioligands specifically bound to the serotonin receptor subtypes. Cloned receptors expressed in cell lines served as the source of all subtypes, except for the 5-HT4 receptor. The letter(s) in front of the receptor subtype designate the species: h = human, m = mouse, r = rat, gp = guinea pig.
| Receptor Subtype | Cell type for expression | Radioligand | Drug for defining non-specific binding | Binding buffer and conditions |
|---|---|---|---|---|
| h5-HT1A | HEK293 | [3H]MPPF | 5 μM NAN-190 | Buffer A; 90 minutes at 25°C |
| h5-HT2A | HEK293 | [3H]MSP | 5 μM mianserin | Buffer A; 90 minutes at 25°C |
| h5-HT2B | HEK293 | [3H]LSD | 30 μM mianserin | Buffer C; 90 minutes at 25°C |
| h5-HT2C | HEK293 | [3H]mesulergine | 5 μM mianserin | Buffer A; 90 minutes at 25°C |
| m5-HT3 | HEK293 | [3H]BRL-43694 | 10 μM mianserin | Buffer A; 90 minutes at 25°C |
| gp5-HT4 | Striatum | [3H]GR113808 | 30 μM serotonin | Buffer A; 30 minutes at 25°C |
| h5-HT5A | CHO-K1 | [3H]LSD | 100 μM serotonin | Buffer B; 60 minutes at 37°C |
| r5-HT6 | HeLa | [3H]LSD | 30 μM chlorpromazine | Buffer C; 60 minutes at 37°C |
| h5-HT7 | CHO | [3H]LSD | 10 μM serotonin | Buffer B; 120 minutes at 25°C |
Binding Buffer A = 50 mM Tris, pH = 7.4
Binding Buffer B = 10 mM MgSO4, 0.5 mM EDTA, 50 mM Tris, pH 7.4
Binding Buffer C = 5 mM MgCl2, 250 μM sodium ascorbate, 50 mM Tris, pH = 7.4
2.3. Measuring the affinity of efavirenz for selected serotonin receptor subtypes using competition radioligand binding
Selected receptors were examined further in competition-type concentration-response curves to measure their affinity for efavirenz. The binding conditions and procedure were the same as those described above and in Table 1 except that increasing concentrations, instead of a fixed concentration, of efavirenz was used in order to generate concentration-response curves and extract IC50 values. Equilibrium inhibition constants (Ki), representing binding affinities, were calculated from the IC50 values using the Cheng-Prusoff equation: Ki = IC50/(1 + [ligand]/KD). Concentration-response curves were fitted with a four parameter logistic equation that included a variable slope using a 95% confidence interval for all curve-fits using Graphpad Prism version 4.0.
2.4. Intracellular Calcium and IP-One Assay: measurement of Gq-coupled response
Individual cloned human 5-HT2A, 5-HT2B or 5-HT2c receptors were stably expressed in HEK293 cells. Since an endogenous human M3 muscarinic receptor [22–24] is expressed in HEK293 cells, untransfected HEK293 cells served as the source of the M3 receptor. The cloned human M1 muscarinic receptor was stably expressed in CHO cells. HEK293 cells were grown in Dulbecco’s modified eagles medium (DMEM) complete which is DMEM supplemented with 50 units/mL streptomycin, 50 μg/mL penicillin, 1 mM sodium pyruvate, and 10% fetal bovine serum. The media of transfected cells additionally contained 0.1 mg/mL G418 to maintain selection. In the case of the 5-HT2c receptor, dialyzed serum (Innovative Research Inc., Novi, MI) was employed as no signal could be obtained when using undialyzed FBS (ThermoFisher Scientific, Waltham, MA); however, undialyzed FBS was used in the case of the 5-HT2A receptor since virtually identical responses were observed regardless of the serum type. CHO cells stably expressing the cloned human M1 muscarinic receptor were grown in Ham’s F12 supplemented with 50 units/mL streptomycin, 50 μg/mL penicillin, 1 mM sodium pyruvate, and 10% fetal bovine serum, and 0.1 mg/mL G418 to maintain selection.
Cells expressing the receptor of interest were seeded into Poly-L-Lysine coated, black-walled 96 well plates at a density of 120,000 cells per well. The following day, media was removed and cells were loaded for three hours in the dark with the FLIPR Calcium-6 QF dye (Molecular Devices, Sunnyvale CA) dissolved in Hank’s buffered saline supplemented with 20 mM HEPES pH = 7.4. In cases where drug pretreatments were needed (as in the case of antagonist blocking an agonist response), the pretreatment drug or vehicle was added to the loading dye solution. Following dye loading and any pretreatments, changes in intracellular calcium were detected as rapid changes in fluorescent signal measured at 485 nm excitation, 525 nm emission with a cutoff filter set at 515 nm using a Flexstation 3 (Molecular Devices, Sunnyvale, CA). The baseline calcium signal was measured for 120 seconds followed by injection of the test drug and reading of any change in the calcium signal for an additional 780 seconds. At the termination of the experiment all signals were baseline subtracted and the change in intracellular calcium was quantified as the area under the curve, measured by integration (Prism version 4.0). All data were normalized to maximal functional response defined as the asymptote of the sigmoidal curve.
IP-One levels were measured in HEK293 cells stably expressing the 5-HT2A receptor. Cells were seeded into Poly-L-Lysine coated, black walled 96 well plates at a density of 20,000 cells per well. IP-One levels were measured using the IP-One HTRF® assay kit (cisbio Assays, Bedford, MA) per manufacturer’s protocol.
2.5. Effect of prolonged exposure to efavirenz on receptor density and Gq-coupled activation of the 5-HT2A receptor
HEK293 cells stably expressing the 5-HT2A receptor were split into four T-25 flasks to 20% confluency and were allowed 24 hrs to attach. Media was then removed by aspiration and replaced with either vehicle (1:1000 v/v DMSO), 30 μM 5-HT, or 30 μM efavirenz. Two flasks were designated for the vehicle treatment as one of them would be used later for a very short (15 min) exposure to efavirenz. For three days, the media was replaced with fresh media containing the same treatments every 12 hrs. At the end of the third day, one of the vehicle treated flasks was also treated with 30 μM efavirenz for only 15 min. This group served as a control to ensure all efavirenz could be removed with our washing procedure prior to measuring the change in intracellular Ca2+ or receptor density (Bmax). For the washing procedure, media was removed from all four flasks and cells were washed twice by incubating for 30 min in 10 mL DMEM complete. Following the second wash, cells were harvested for either the calcium assay as described above with no more than 8 hrs total between replating cells and performing the assay, or for radioligand binding to estimate receptor density (Bmax). For the radioligand binding assay, membranes were prepared as described above for each of the four treated groups. The 5-HT2A receptor density was then estimated using 0.5 nM [3H]MSP followed by the calculation of Bmax at saturation using a rectangular hyperbola model Bmax = (Y •(KD+X))/X, where Y is [specific binding] and X = [radioligand concentration]. The KD for [3H]MSP at the cloned human 5-HT2A receptor had been previously determined to be 246 pM [25]. The protein concentration for each sample was determined using the BCA assays as described in section 2.8.
2.6. Cyclic adenosine monophosphate (cAMP) Assay: measurement of Gs-coupled responses
Changes in intracellular cAMP levels were measured in HeLa cells stably expressing the rat 5-HT6 receptor using a luminescent cAMP-Glo kit (Promega, Madison, WI) terminal assay as per the manufacturer’s protocol with the following modifications. A total of 10,000 cells were seeded per well in a 96 well plate and grown in DMEM complete (see above). The following day, media was removed and cells were pretreated for 30 min in DMEM supplemented with 500 μM of the phosphodiesterase inhibitors 3-Isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich, St. Louis, MO) and 100 μM Ro 20-1724 (Santa Cruz Biotechnology, Inc., Dallas, Texas). Next, cells were treated either alone or with a combination of vehicle, efavirenz or 5-HT for 30 min and then lysed in 20 μL lysis buffer for an additional 30 min with vigorous shaking (600 rpm on an IKA MTS 2/4 digital microplate shaker). A total of 10 μL of this lysate was transferred to a half-area plate containing 20 μL cAMP-Glo™ detection solution and the plate was mixed for 1 minute at 600 rpm and incubated at room temperature for 20 min. Finally, 40 μL of kinase glo® reagent was added to each well, mixed for 1 minute at 600 rpm and incubated at room temperature for 10 min. Luminescence measurements were performed using the Flexstation 3 set to luminescence mode with 100 millisecond integration time. All data were normalized to maximal stimulatory concentration of forskolin (10 μM).
2.7. Inhibition of monoamine oxidase A (MAO-A) activity
Recombinant human MAO-A was overexpressed in insect Hi5 cells and lysates of these cells containing approximately 4 μg/mL of enzyme were washed in 100 mM potassium phosphate, pH = 7.4, then pretreated for 15 min at 37°C with vehicle or 10 μM efavirenz, followed by treatment with 50 μM of the MAO-A substrate kynuramine for 60 min at 37°C. The reaction was terminated at by the addition of 6 N NaOH and the fluorescent 4-hydroxyquinoline cleavage product (Ex325/Em465) served as a measure of MAO-A activity.
2.8. Protein Assay
Membrane protein concentrations were measured by bicinchoninic acid assay (Pierce, IL) according to the manufacturer’s instructions. Protein standard curves were constructed using purified bovine serum albumin.
2.9. Measurement of 5-HT3A and GABAA receptor currents using electrophysiology
Currents were obtained in the whole cell configuration from HEK293 cells stably expressing the rat or human GABAA receptors (α1β2γ2 (short isoform of the γ2 subunit) [26], or the mouse 5HT3A receptors transiently transfected into HEK293 cells using a modified polyethylenimine method described previously [27]. Cell were voltage-clamped at −60 mV and the recordings were made at room temperature in the whole cell configuration using borosilicate pipettes with a tip resistance of 1–2.5 MΩ. The pipette solution contained 140 mM CsCl, 10 mM EGTA, 10 mM HEPES, 4 mM Mg-ATP, pH = 7.2. Coverslips containing cultured cells were placed in a 1.5 mL chamber on the stage of an inverted light microscope and superfused continuously (5–8 mL/min) with an extracellular solution containing 125 mM NaCl, 5.5 mM KCl, 0.8 mM MgCl2, CaCl2 (1.2 mM in 5-HT3A studies and 3 mM in GABAA studies), 10 mM HEPES, 10 mM D-glucose pH = 7.3. GABA or 5-HT-induced currents were low-pass filtered at 5 kHz, monitored on an oscilloscope and a chart recorder, and stored for subsequent analysis. A 60–80% series resistance compensation was applied at the amplifier. Any change in access resistance observed during the recording period resulted in the patch being aborted and that data was excluded from the analysis.
5-HT or GABA, with or without antiretroviral drugs, was dissolved in the extracellular solution and then applied to cells for 10–20 sec from independent reservoirs by gravity flow using a Y-shaped tube positioned within 100 μm of the cells. Using this system, the 10–90% rise time of the junction potential at the open tip is 12–51 milliseconds [28]. Receptors were activated with approximately an EC30 concentration of the appropriate neurotransmitter (0.6 μM for 5-HT3A receptors, 5 μM for rat α1β2γ2 and 10 μM for human α1β2γ2 GABAA receptors), because this level of occupancy elicits minimal desensitization. Once a positive (baseline) control GABA or 5-HT response was established, the effect of ARV drug was examined by co-application with GABA or 5-HT. 5-HT or GABA responses were again monitored after each ARV drug application to ensure adequate washout and to ensure no loss of signal and integrity of the patch. Using this strategy, multiple concentrations of ARV drug could generally be tested for each cell studied. ARV drug stocks were dissolved in DMSO and diluted in saline so that the final DMSO concentration (v/v) was ≤ 0.2%. The concentration-response curve for efavirenz’s modulatory effect on 5-HT3A receptors was fitted to the following equation: I/Imax= 1/(1+(IC50/[efavirenz])n), where I is the peak 5-HT current normalized to control at a given efavirenz concentration and Imax represents the normalized 5-HT-induced current, IC50 is the half-maximal blocking concentration, and n is the Hill coefficient. All data are presented as mean values ± SEM of a minimum of six individual experiments. A student’s paired t-test was used to determine statistical significance (p< 0.05) for experiments comparing responses in the presence or absence of a single concentration ARV drug.
3. Results
In a previous report [9], CNS targets for efavirenz were identified that may contribute to its attractiveness as a drug of abuse. In this report, the molecular mechanisms of action for efavirenz were examined across a broader range of serotonergic targets in an effort to enhance our understanding of some of the observed NPAEs in patients taking it as prescribed. This included a comparison of efavirenz’s mode of action with those of the other HIV-1 medications FTC, NVP and ZDV.
3.1. Efavirenz interacts with a number of receptors in the 5-HT family
Potential interactions of efavirenz with serotonin receptor subtypes from each of the seven subfamilies, 5-HT1 through 5-HT7, were probed initially by measuring the ability of a fixed concentration of efavirenz (10 μM) to displace radioligands specifically bound to individual receptor subtypes (Figure 1). The concentration selected was based upon our investigation of numerous receptor systems, including the 5-HT2A and 5HT2C receptors, and the estimated levels of efavirenz’s brain exposure (> 40 μM) in those at risk for experiencing NPAEs following a standard oral dose (see Introduction). Using this assay format, we showed in our previous report that efavirenz interacted with the 5-HT2A and 5-HT2C receptors, and in this report we show that efavirenz additionally had interactions with the 5-HT2B, and 5-HT6 receptors, which reached statistical significance with a stringent cutoff (P < 0.001, ANOVA followed by a Bonferroni post-hoc) (Figure 1). Of these interactions, the most robust were with the 5-HT2A, 5-HT2C, and 5-HT6 receptors as efavirenz displaced significantly more radioligand from these three 5-HT receptor subtypes than from the 5-HT2B receptor (P < 0.05, ANOVA followed by a Bonferroni post-hoc).
Figure 1. Efavirenz has significant interactions with the cloned metabotropic 5-HT2A, 5-HT2B, 5-HT2C and 5-HT6 receptors.
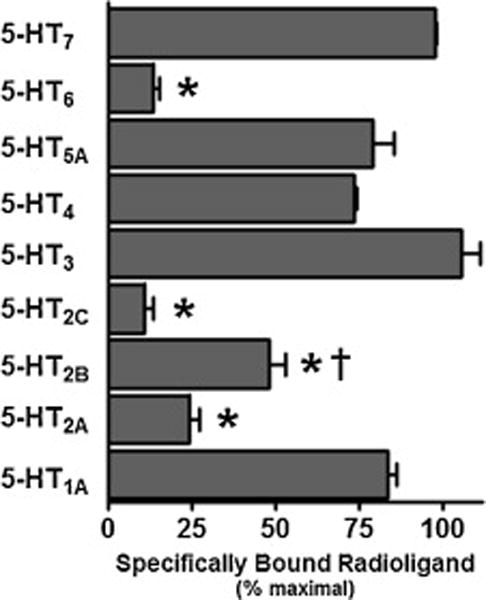
At a concentration of 10 μM, efavirenz displaced greater than 50% of specifically bound radioligand from the 5-HT6 receptor and 5-HT2 subfamily of receptors, while having a less pronounced or no detectable interaction with the other serotonin receptor subtypes: 5-HT1A, 5-HT3A, 5-HT4, 5-HT5, 5-HT7. A one-way ANOVA followed by a Bonferroni post-hoc analysis set to high stringency (P < 0.001) indicated significant displacement for those groups having an asterisk. When considered as a group, efavirenz has similar robust interactions with 5-HT2A, 5-HT2C and 5-HT6 receptors, but significantly weaker interactions with the 5-HT2B receptor as determined by a one-way ANOVA followed by a Bonferroni post-hoc analysis (P < 0.05) and indicated by the dagger. All data are presented as averaged values from multiple experiments ± SEM. Displacement data for the 5-HT1A, 5-HT2A and 5HT2C receptors were adapted from Gatch et al., 2013 [9] and shown here for completeness.
Those receptors experiencing ≥ 75% displacement of radioligand in the presence of 10 μM efavirenz (i.e., 5-HT2A, 5-HT2C and 5-HT6 receptors) were examined further in competition-type concentration-response curves to measure their affinity for efavirenz (Figure 2). The calculated affinities (Ki) for efavirenz were similar in magnitude ranging from 1.7 to 4.4 μM. The highest affinity was for the 5-HT2C receptor and the lowest for the 5-HT2A, which had significantly lower affinity than for the other two receptors (P < 0.05 ANOVA with a Bonferroni post-hoc). While a variable slope model was not the statistically preferred model over a model with the pseudo Hill slope fixed to unity (e.g. −1.0), in order to evaluate the pseudo Hill slope parameter all the slopes were treated as a variable (instead of a fixed value) for the purpose of curve fitting. Using this approach, in all cases the pseudo Hill slopes were moderately greater than unity (−1.4 to −1.2) but the variance was such that none of them deviated significantly from unity when strict criteria were applied (P < 0.001, t-test), and further none of them differed significantly from one another (P < 0.05 ANOVA with a Bonferroni post-hoc). Slopes not different from unity are consistent with efavirenz binding to a single site on each of the 5-HT2A, 5-HT2C, and 5-HT6 receptors. As a reference, the affinities for serotonin at the 5-HT2A, 5-HT2C and 5-HT6 receptors were measured as well, yielding Ki ± SEM values equal to 360 ± 29 nM, 16 ± 3.5 nM and 105 ± 8 nM, respectively, which are consistent with previous reports [29–31].
Figure 2. Efavirenz has low micromolar affinity for cloned metabotropic 5-HT2A, 5-HT2C and 5-HT6 receptors.
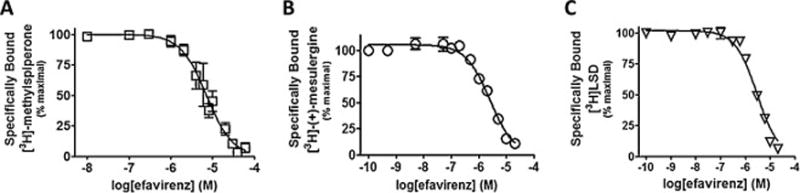
The affinity of efavirenz for each receptor was determined at equilibrium by concentration-dependent competition binding of radiolabeled receptors using rapid filtration. All data are presented as averaged values from multiple experiments ± SEM. A, The 5-HT2A receptor binds to efavirenz with an affinity (Ki) = 4.4 ± 0.43 μM and a pseudo Hill slope = −1.39 ± −0.18. B, The 5-HT2C receptor binds to efavirenz with an affinity (Ki) = 1.7 ± 0.28 μM and a pseudo Hill slope = −1.22 ± −0.18. C, The 5-HT6 receptor binds to efavirenz with an affinity (Ki) = 2.2 ± 0.27 μM and a pseudo Hill slope = −1.36 ± −0.14.
3.2. Efavirenz functions as a 5-HT2A and 5-HT2C receptor antagonist, a 5-HT6 receptor inverse agonist, a 5-HT3 receptor pore blocker, and an MAO-A inhibitor
The individual functional properties of the stably expressed cloned Gq-coupled 5-HT2A and 5-HT2C, and the Gs-coupled 5-HT6 receptors were assessed starting with their ability to be stimulated by the endogenous full agonist serotonin (Figure 3). Serotonin activated the 5-HT2A receptor with a potency that was unaffected by the serum condition in the culture media: 13.3 ± 2.5 nM in absence of serum, 13.9 ± 4.1 nM in presence of dialyzed FBS, and 17.2 ± 7.5 nM in presence of FBS. In stark contrast, the potency of 5-HT at the 5-HT2C receptor spanned from the sub-nanomolar to the low nanomolar range: 0.039 ± 0.0039 nM in absence of serum, 1.5 ± 0.18 nM in presence of dialyzed FBS, and undetectable in the presence of FBS. Most notable was that no 5-HT response above baseline was observed when the 5-HT2C receptor was maintained in FBS prior to measuring the Ca2+ response. Serotonin had potency in the lower nanomolar range, 12.7 nM, for the 5-HT6 receptor. The 5-HT2C receptor had a significantly higher potency for serotonin under serum free conditions compared with the 5-HT2A receptor (P < 0.05 Student’s t-test) or the 5-HT6 receptor (P < 0.05 Student’s t-test). Large differences in pseudo Hill slope values were measured as well ranging from 0.55 to 1.5, with the 5HT2A receptor having the shallowest slope and the 5HT2C receptor having the steepest slope. However, due to the variance in the data, these pseudo Hill slopes were not significantly different between receptors (P > 0.05 ANOVA with a Bonferroni post-hoc) and did not differ significantly from unity (P > 0.05, Student’s t-test). More importantly, these values set a baseline for a full agonist response under our assays conditions and were useful for comparing and contrasting the ARV drug responses mediated by these receptors. Also, the mean EC50 values reported here for the 5-HT2A, 5-HT2C (for each of the serum conditions tested) and the 5-HT6 receptors are in agreement with previously published values [32–34].
Figure 3. The cloned metabotropic 5-HT2A, 5-HT2C and 5-HT6 receptors are potently activated by serotonin.
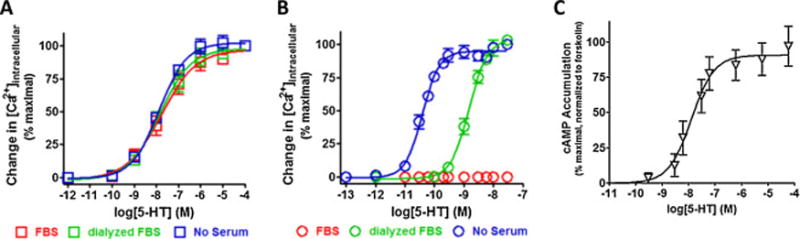
Functional dose-responses to serotonin for the Gq-coupled 5-HT2 receptors were measured as an increase in intracellular calcium using the calcium sensitive fluorescent dye Calcium-6 QF (Ex485/Em525), while the functional dose-response for the Gs-coupled 5-HT6 receptor was measured as an increase in cAMP detected using the bioluminescent cAMPGlo assay. All data are presented as averaged values from multiple experiments ± SEM. A, Serotonin activates the 5-HT2A receptor with an average potency (EC50) range from 13–17 nM and a pseudo Hill slope range from 0.55–0.70 that was insensitive the serum condition used for culturing (P > 0.05 ANOVA with with a Bonferroni post-hoc). B, Serotonin activates the 5-HT2C receptor with a potency (EC50) = 1.5 ± 0.18 nM and a pseudo Hill slope = 1.4 ± 0.19 when cells were cultured in dialyzed FBS and a potency (EC50) = 0.039 ± 0.0039 nM and a pseudo Hill slope = 1.5 ± 0.17 when cells were cultured in absence of serum. No response was observed when cells were culture in FBS. C, Serotonin activates the 5-HT6 receptor with a potency (EC50) = 12.7 ± 6.2 nM and a pseudo Hill slope = 1.02 ± 0.35.
In a previous collaborative report, efavirenz was determined to have LSD-like properties mediated by the 5-HT2A receptor based primarily upon its behavioral properties in rodents [9]. In this report, we employed detection of intracellular calcium or IP1 accumulation as robust measures of 5-HT2A–mediated activation of the Gq-PLC-IP3-[Ca2+]i signaling pathway. When a change in intracellular Ca2+ was employed as the measure of Gq-signaling, efavirenz did not stimulate the 5-HT2A receptor at the maximum concentration (10 μM efavirenz) that could be tested as an agonist under the assay conditions (Figure 4A). To completely rule out the possibility that efavirenz might be a weak partial agonist, a higher concentration needed to be tested; due to efavirenz’s solubility restriction in the Ca2+ assay, the IP-One assay was employed. Using the maximum concentration that could be tested, 100 μM efavirenz did not activate the Gq-pathway when IP-One was used as the readout of Gq activity (Figure 4B). This prompted us to test whether efavirenz might have other actions given that the radioligand binding data indicated that it interacts with the 5-HT2A receptor. Accordingly, efavirenz was tested for its ability to modify functional responses of different classes of the 5-HT2A receptor agonists: 5-HT, TCB-2 and α-methyl-5-hydroxytryptamine (α-Me-5HT). Like the selective 5-HT2A receptor antagonist MDL100,907, efavirenz was able to antagonize the effects of all three agonists (Figure 4A). Given these results, efavirenz was also tested for its ability to modify the in vitro functional response of the well-known hallucinogens (±)-DOI and LSD by measuring changes in intracellular calcium. Both (±)-DOI and LSD behaved as partial agonists (Figure 5B & 5E), which is consistent with previously published reports [35–37]. In the presence of increasing concentrations of efavirenz, both (±)-DOI and LSD functional responses were progressively shifted to the right without a significant decrease in the maximal response (Figure 5A, P > 0.05 ANOVA followed by Bonferroni’s post-hoc for maximal responses) suggestive of either a negative heterotropic allosteric modulation with high cooperativity or a purely competitive interaction at a single site. Schild plot transformation of the dose-ratio shift data yielded slopes equal to 1.06 ± 0.054 and 1.20 ± 0.16 for (±)-DOI and LSD, respectively, that were not significantly different from unity (P > 0.05 t-test) suggesting that efavirenz competes with (±)-DOI and LSD for binding to a single site on the 5-HT2A receptor (Figure 5C, 5E). From the Schild plots, the pA2 value (also known as the antagonist potency (KB)) for efavirenz was calculated to be 1.3 ± 0.2 μM and 0.31 ± 0.18 μM using either (±)-DOI or LSD, respectively, as the agonist in competition with efavirenz. However, these efavirenz antagonist potency values were not statistically different from one another or from the measured affinity (Ki = 4.37 ± 0.43 μM, Figure 2A) of efavirenz for the 5-HT2A receptor (P > 0.01 ANOVA with a Bonferroni post-hoc). Collectively, the results suggest that efavirenz competitively interacts with a single site on the 5-HT2A receptor that is indistinguishable from the site where LSD and (±)-DOI bind.
Figure 4. Efavirenz is a 5-HT2A receptor antagonist of Gq signaling.
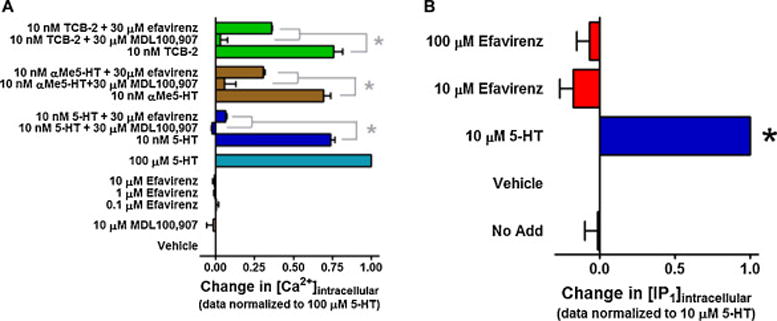
All data are presented as averaged values from multiple experiments ± SEM. A, Efavirenz behaves as an antagonist at the cloned 5-HT2A receptor when a change in intracellular calcium is used as a measure of Gq-signaling and the 5-HT2A receptor is stimulated with different classes of agonists: 5-HT, TCB-2 and α-Me-5HT. MDL100,907, a potent and selective 5-HT2A receptor antagonist was used as a positive control for antagonist activity. A one-way ANOVA followed by a Bonferroni post-hoc analysis set to high stringency (*P < 0.001) indicated significant inhibition of agonist responses by both 30 μM efavirenz and 30 μM MDL100,907. B, To rule out the possibility that weak partial agonist properties of efavirenz might only be observed at a very high concentration, a higher maximal concentration of efavirenz was tested using IP-One assay conditions than was possible under the Ca2+ assay conditions. Nevertheless, at the highest concentration that could be tested (100 μM) in the IP-One assay, efavirenz did not display any agonist activity. A one-way ANOVA followed by Dunnett’s multiple comparison test set to high stringency (P < 0.001) indicated by an asterisk revealed only the response by 5-HT to be significantly different from vehicle.
Figure 5. Schild shift analysis of efavirenz at the cloned 5-HT2A receptor suggests that it competes for the same binding site as (±)-DOI and LSD.
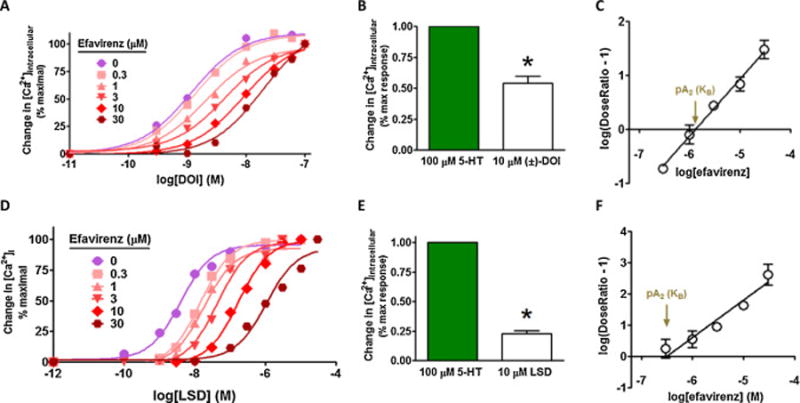
A & D, Gq-signaling was measured as a change in intracellular calcium. Data are presented as averaged values from multiple experiments ± SEM. For clarity error bars are not shown and the Schild data are normalized to 100% response, even though both (±)-DOI and LSD are partial agonists of the 5-HT2A receptor, P < 0.01, two-tailed t-test as shown in B and E. (±)-DOI and LSD concentration-response curves of the Gq-mediated functional responses in the presence of increasing fixed concentrations of efavirenz induce a progressive shift to the right with no reduction in efficacy. C, Schild plot transformation of the data shown in panel A suggests that efavirenz’s interaction with (±)-DOI is purely competitive given a slope equal to 1.06 ± 0.054. The pA2 or antagonist potency (KB) value for efavirenz competition of (±)-DOI is 1.3 ± 0.20 μM. Panel F suggests that efavirenz’s interaction with LSD is also purely competitive with a slope of 1.2 ± 0.16. The pA2 value for efavirenz competition of LSD is 0.31 ± 0.18 μM. In both panels C and F, the slopes of the Schild plot lines are not significantly different from unity (P > 0.05) and a two-site model is not significantly better than a one site model (P > 0.05).
While efavirenz produces NPAEs in the majority of patients, the initial clinical finding was that in many of those affected, the NPAEs appear to lessen or disappear after several weeks [7]. Thus, we attempted to model a chronic exposure scenario in vitro and evaluate the possibility that efavirenz might induce a pharmacodynamic tachyphylaxis type response, meaning that its ability to antagonize a 5-HT2A receptor response would diminish upon repeated exposure. HEK293 cells stably expressing the cloned human 5-HT2A receptor were exposed to vehicle, 30 μM 5-HT, or 30 μM efavirenz for 3 days. At the end of the 3 day treatment, cells were thoroughly washed and suppression of 5-HT-induced Ca2+ response by efavirenz was assessed. In all the treatment groups, 10 μM efavirenz treatment resulted in a 48–49% suppression of the calcium response induced by a submaximal concentration of 5-HT (100 nM). This suppression was statistically significant in the two vehicle and the serotonin pre-treatment groups (Figure 6A, P < 0.05 ANOVA followed by Bonferroni post-hoc), but was not significant in the 3 day 30 μM efavirenz pre-treatment group (Figure 6A, P > 0.05 ANOVA followed by Bonferroni post-hoc) even though a 49% suppression is evident. Unexpectedly, the overwhelming effect of chronic treatment with efavirenz was a large (81%) and significant reduction in the 5-HT2A receptor’s responsiveness to 5-HT (Figure 6A, P < 0.05 ANOVA followed by Bonferroni post-hoc). This drastic reduction was not caused by the inability of efavirenz to dissociate from the receptor, because the responses in cells treated with 30 μM efavirenz for 15 min after 3 day treatment with DMSO were not significantly different from the responses in cells that underwent the 3 day treatment with DMSO only. To assess if the reduction in the 5-HT response in the 3 day efavirenz treatment group was due to the reduction in receptor density, Bmax was estimated following the same series of 3 day treatments. Three day exposure to efavirenz resulted in a statistically significant 52% reduction in Bmax (Figure 6B, P < 0.05 ANOVA followed by Bonferroni post-hoc), which is consistent with the observed reduction in the functional response to agonist following 3 days chronic treatment with efavirenz.
Figure 6. Chronic exposure to efavirenz reduces the 5-HT2A receptor density and 5-HT mediated Gq activation at the 5-HT2A receptor.
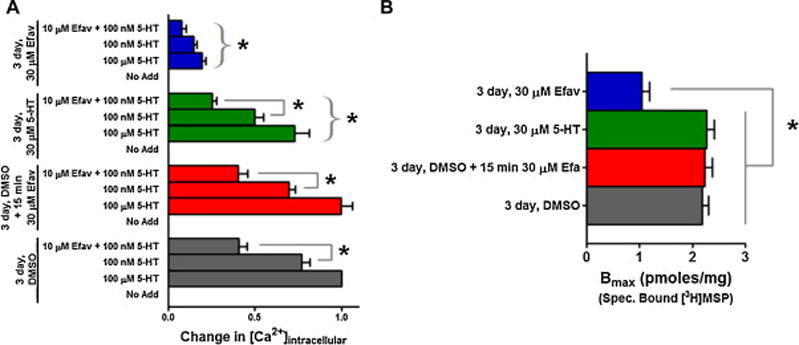
Cells were exposed (pretreated) for 3 days to DMSO vehicle [gray bars], serotonin (30 μM) [green bars] or efavirenz (30 μM) [blue bars], or to DMSO vehicle for 3 days and efavirenz for only 15 min on the third day [red bars]. The latter category was to ensure that our wash procedure was able to effectively remove all traces of efavirenz. After extensive washes, the cells from all groups were tested for their ability to mount functional responses to serotonin in the absence and presence of efavirenz (10 μM). All data were normalized to a maximal response generated by 100 μM 5-HT following the 3-day DMSO pretreatment and are presented as averaged values from multiple experiments ± SEM. A, Cells exposed to DMSO vehicle for 3 days elicited a robust calcium response when stimulated with a submaximal concentration of 5-HT (100 nM), and 10 μM efavirenz was able to antagonize this response by 48%. The same result was obtained when cells were exposed to efavirenz for only 15 min following 3 day exposure to DMSO, demonstrating that efavirenz could be easily removed by washing. Compared to the 3 day vehicle treated group, three day exposure to 30 μM 5-HT reduced the 100 μM maximal 5-HT response by 27%, and 10 μM efavirenz antagonized the 100 nM 5-HT submaximal response by 49%. In contrast to the other groups, 3 day exposure to 30 μM efavirenz drastically reduced the maximal 5-HT response by 81%, but the magnitude of efavirenz’s antagonist effect on the 100 nM 5-HT submaximal response was similar to all the other groups (i.e, a 49% reduction). The comparisons being made between specified groups within a category are indicated by interconnecting gray lines and show a significant inhibition by efavirenz. A bracket with asterisk indicates a significant difference between the 3 day DMSO category and the remaining categories for comparisons between matched groups. A two-way ANOVA followed by Bonferroni multiple comparisons test was performed to identify significant differences within and between the various pretreatment categories at P < 0.05. B, Three day exposure to 30 μM efavirenz resulted in a significant 52% decrease in receptor density (Bmax). However, none of the other exposure conditions had a significant effect on Bmax. A one way ANOVA followed by Bonferroni’s multiple comparisons test was performed to identify significant differences between various pretreatment categories. * P < 0.05 between the 3 day efavirenz pretreatment category and all the other pretreatment categories.
Like at the 5-HT2A receptor, efavirenz functioned as an antagonist at the 5-HT2C receptor. Given that serum had a drastic effect on 5-HT’s potency at the 5-HT2C receptor (Figure 3B), we tested efavirenz’s ability to modify the in vitro functional response to 5-HT in the absence of serum. Using an EC50 concentration of 5-HT (0.03 nM) to stimulate the 5-HT2C receptor, up to 3 μM efavirenz had no significant effect on 5-HT responses; however, at concentrations ≥ 10 μM efavirenz inhibited the 5-HT agonist effect, and at a concentration ≥ 60 μM efavirenz completely suppressed it (Figure 7A, P < 0.001 ANOVA followed by Dunnett’s multiple comparison). To verify the specificity of the results, we tested efavirenz’s ability to modulate the activity of an endogenous Gq-coupled M3 muscarinic receptors known to be expressed in HEK293 cells [22–24]. While efavirenz alone had no effect on intracellular Ca2+ levels in untransfected HEK293 cells (Figure 8A), at concentrations ≥ 10 μM it was able to partially suppress the Gq response mediated by an EC50 concentration of acetylcholine (ACh) (Figure 8B, P < 0.05 ANOVA followed by Dunnett’s multiple comparison). Since it was previously reported based upon binding studies that efavirenz does not interact with muscarinic receptors [9], and our data indicated that it inhibits the agonist responses at the human M3 receptor, we investigated if efavirenz interacts with a different Gq-coupled muscarinic receptor. The M1 receptor was selected because it is the predominant muscarinic receptor in the brain, and because it plays an important role in cognition and learning and memory [38–41]. CHO cells stably expressing the human M1 receptor were used, to test for possible interactions of efavirenz with the M1 receptor. While efavirez alone failed to stimulate the M1 receptor, at concentrations ≥ 10 μM it suppressed the Gq response mediated by an EC50 concentration of carbachol, and concentrations ≥ 30 μM resulted in complete inhibition (Figure 8C–D, P < 0.05 ANOVA followed by Dunnett’s multiple comparison). No Gq-mediated response was elicited by efavirenz, acetycholine or carbachol in untransfected CHO cells (data not shown).
Figure 7. Efavirenz is an antagonist of the cloned 5-HT2C receptor.
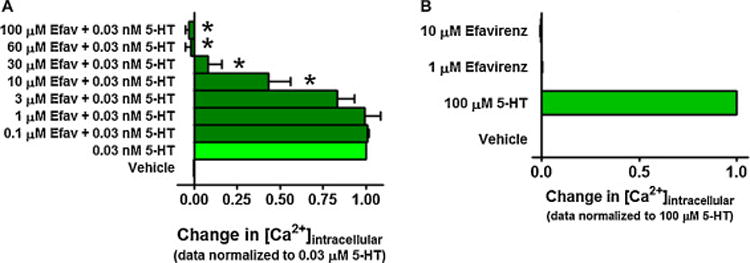
Gq-signaling was measured as a change in intracellular calcium. All data are presented as averaged values from multiple experiments ± SEM. A, Efavirenz dose-dependently antagonizes agonist responses produced by an EC50 concentration of 5-HT (EC50 data normalized to 100%). For these experiments cells were maintained in serum free media for at least 12 hours, pretreated with efavirenz, then stimulated with 0.03 nM serotonin, which is the EC50 for 5-HT for this receptor under serum free conditions (see Figure 3B). Values significantly different from 0.03 nM 5-HT, (Dunnett’s multiple comparison test set to high stringency, P < 0.001) are denoted by asterisks. B, Efavirenz did not have any agonist activity at the 5-HT2C receptor under similar conditions.
Figure 8. Efavirenz partially inhibits the activation of the M3 muscarinic receptor, and completely inhibits the activation of the M1 muscarinic receptor.
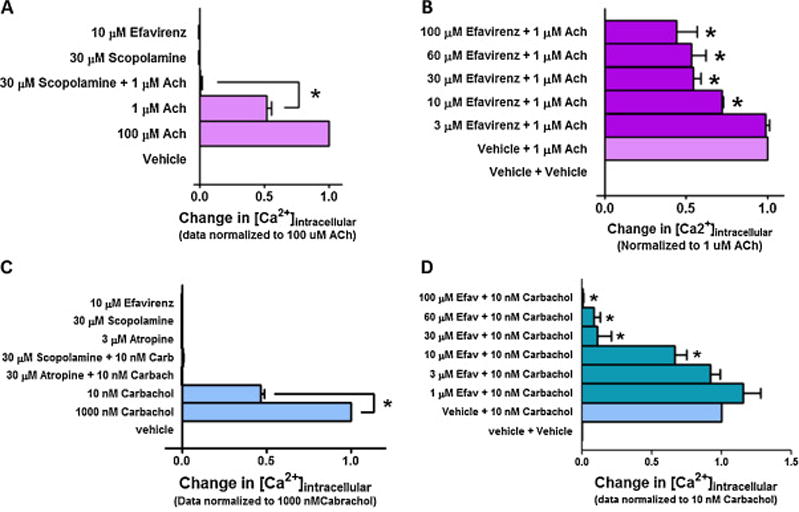
Gq-signaling was measured as a change in intracellular calcium. All data are presented as averaged values from multiple experiments ± SEM. A, In untransfected HEK293 cells, efavirenz does not stimulate Gq-coupling as measured by changes in intracellular Ca2+. However, these cells express an endogenous Gq-coupled M3 muscarinic receptor [22–24] that was activated by acetylcholine, and this response was blocked by the muscarinic receptor antagonist scopolamine. B, Efavirenz partially suppresses ACh mediated M3 receptor activation at concentrations ≥ 10 μM but was unable to produce a complete block at the highest concentration that could be tested under these assay conditions. C, Efavirenz fails to stimulate Gq-coupling in CHO cells stably expressing the human M1 receptor, but this receptor can be stimulated with the muscarinic receptor agonist carbachol and the muscarinic receptor antagonists scopolamine and atropine completely block carbachol’s effect. D, Efavirenz antagonizes carbachol-mediated M1 receptor activation in a concentration dependent manner. At 10 μM there is a significant inhibition of the response produced by an EC50 concentration of carbachol (normalized to 100%) and a complete inhibition at concentrations ≥ 30 μM. A one-way ANOVA followed by Dunnett’s multiple comparison test was performed to identify significant differences between the agonist response in the absence of efavirenz compared to the agonist response in the presence of different concentrations of efavirenz. * P < 0.05 for those responses in the presence of efavirenz that are significantly different from the agonist response in the absence of efavirenz.
In HeLa cells stably expressing the Gs-coupled rat 5-HT6 receptor, efavirenz significantly reduced the basal level of cAMP activity (Figure 9A). At a lower concentration (3 μM), efavirenz first diminished 5-HT’s agonist effect, and at a higher concentration (30 μM), not only completely overcame the agonist effect of 5-HT but also reduced activity to below basal levels (Figure 9A). Clozapine is an atypical antipsychotic drug that has been reported to interact with the 5-HT6 receptor [42] acting as an inverse agonist [34]. Like efavirenz, a high concentration of clozapine (30 μM) significantly reduced basal levels of cAMP, and not only completely overcame the agonist effects of 5-HT but also reduced cAMP levels to below basal (Figure 9A). However, in untransfected HeLa cells, which lack the 5-HT6 receptor, neither efavirenz (30 μM), clozapine (30 μM), nor 5-HT (1 μM) produced significant functional effects compared with the vehicle control (Figure 9B). This demonstrates that the functional responses we observed for efavirenz, clozapine, and serotonin were mediated via the 5-HT6 receptor. HeLa cells have been reported to express an endogenous Gs-coupled β-adrenergic receptor [43]. Thus, as a further control, we invoked a cAMP functional response in untransfected HeLa cells by stimulating an endogenous Gs-coupled β-adrenergic receptor with (−)-epinephrine, and this effect was blocked by the β-adrenergic receptor antagonist timolol (Figure 9B). In this case though, efavirenz failed to significantly modulate (−)-epinephrine’s stimulation of the β-adrenergic receptor response (Figure 9B). Thus, efavirenz exerts no cAMP-mediated functional effect in untransfected HeLa cells and it additionally failed to modulate functional responses produced by stimulation of the endogenous β-adrenergic receptors.
Figure 9. Efavirenz behaves as an inverse agonist of the cloned 5-HT6 receptor.
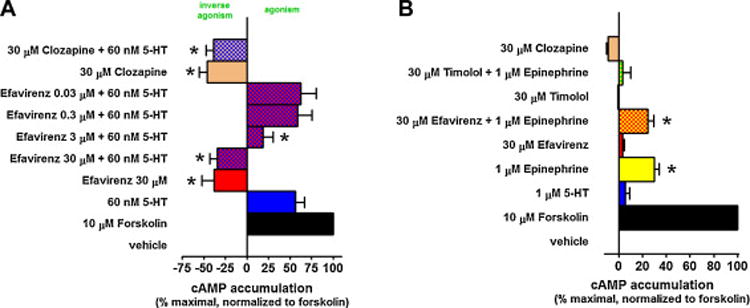
Gs-signaling was measured as a change in intracellular cyclic AMP (cAMP) levels. All data are presented as averaged values from multiple experiments ± SEM. A, Efavirenz (30 μM) acts as an inverse agonist of the cloned 5-HT6 receptor stably expressed in HeLa cells as it lowers the basal levels of cAMP. Only at the higher concentrations (3–30 μM) can efavirenz overcome the stimulatory effect of 60 nM serotonin. The atypical antipsychotic clozapine (30 μM), a known inverse agonist, was used as a positive control for the 5-HT6 inverse agonist response. One-way ANOVA followed by a Dunnett’s post-hoc analysis comparing the 60 nM 5-HT group to groups containing increasing concentrations of efavirenz indicated that cAMP levels were significantly lower in experimental groups containing ≥ 3 μM efavirenz (P < 0.05). B, There is no stimulatory effect of 5-HT or inverse agonism by efavirenz or clozapine in untransfected HeLa cells. HeLa cells express endogenous β-adrenergic receptors whose stimulation by (−)-epinephrine can be blocked by the β-adrenergic receptor antagonist timolol but not by efavirenz.
Although efavirenz did not significantly displace the binding of the 5-HT3 receptor antagonist [3H]-granisetron from the 5-HT3A receptor (Figure 1), we reasoned that since this receptor subtype is a ligand-gated ion channel efavirenz might act as a pore blocker or a direct channel activator, and thus a follow-up functional assay was warranted. Consistent with the idea that it might act as a pore blocker, efavirenz (10 μM) alone had no effect (n = 5, data not shown), but when co-applied with an EC30 concentration of 5-HT, which will open the channel pore, efavirenz inhibited the 5-HT3A receptor currents in a concentration-dependent manner (0.1–30 μM, Figure 10B). At higher concentrations, efavirenz had a greater blocking effect on the steady state current than peak current (Figure 10A), with 30 μM producing a complete inhibition of the steady-state currents. The efavirenz-induced inhibition of 5-HT activated current was completely reversible when treated with concentrations of efavirenz up to 10 μM and partially recovered at 30 μM after 2–3 min washout (data not shown).
Figure 10. Efavirenz inhibits cloned 5-HT3A receptor currents.
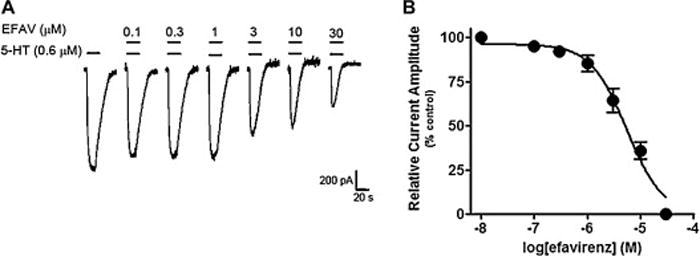
All recording were performed on HEK293 cells transiently expressing mouse 5-HT3A receptors. A, Representative traces showing whole-cell currents activated by an EC30 concentration 0.6 μM 5-HT in the absence and presence of co-applied (20 sec) increasing concentrations (0.1–30 μM) of efavirenz. Note that efavirenz produces a greater inhibition on the steady-state current amplitude as the concentration increases. B, Summary data for effect of efavirenz on 5-HT-induced currents. The currents at the end of ligand application were normalized to the current amplitude at the same time point as in the control (5-HT alone). Efavirenz’s inhibitory concentration (IC50) is 4.9 ± 0.7 μM with a Hill coefficient of 1.2 ± 0.19 (n = 10). All data are presented as means ± SEM.
We previously demonstrated [9] that efavirenz (10 μM) blocks reuptake of serotonin via the serotonin transporter (SERT). Here we demonstrate that the same concentration of efavirenz also significantly, though modestly, inhibits (36%) monoamine oxidase A, an enzyme responsible for metabolizing 5-HT (Figure 11).
Figure 11. Efavirenz modestly inhibits monoamine oxidase-A.
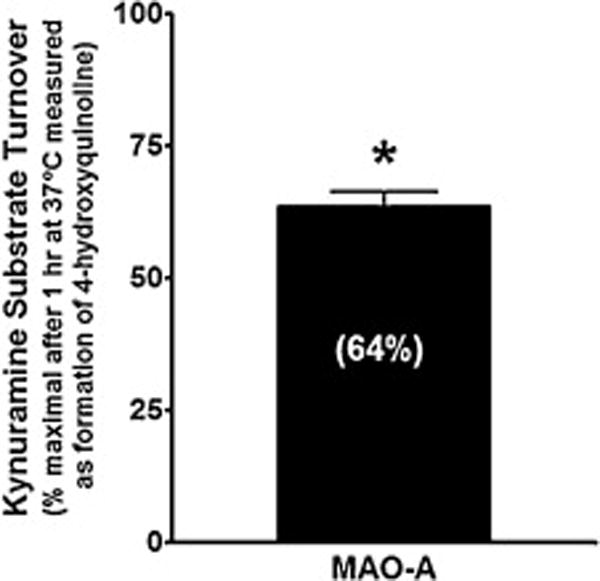
Efavirenz (10 μM) decreases the activity of MAO-A by 36 ± 1.7%. The mean value from multiple experiments ± SEM is plotted. Student’s t-test was used to determine statistical significance (P < 0.05).
3.3. Other ARVs (FTC, NVP, ZDV) have a different receptor profile than efavirenz
In light of the findings that efavirenz has the most pronounced binding interactions with the 5-HT2A, 5-HT2C, and 5-HT6 receptors, we sampled other ARV drugs to see if they also had notable interactions with these receptor subtypes. Although no significant interactions for ZDV, FTC or NVP were observed for any of these receptors at the original profiling concentration of 10 μM (data not shown), we tested these other ARVs to their limit of solubility for the particular assay (60 μM). At these highest concentrations, only NVP displayed a significant, albeit comparatively weak interaction, with the 5-HT6 receptor (Figure 12).
Figure 12. Nevirapine, but not emtricitabine or zidovudine, interacts with the cloned 5-HT6 receptor.
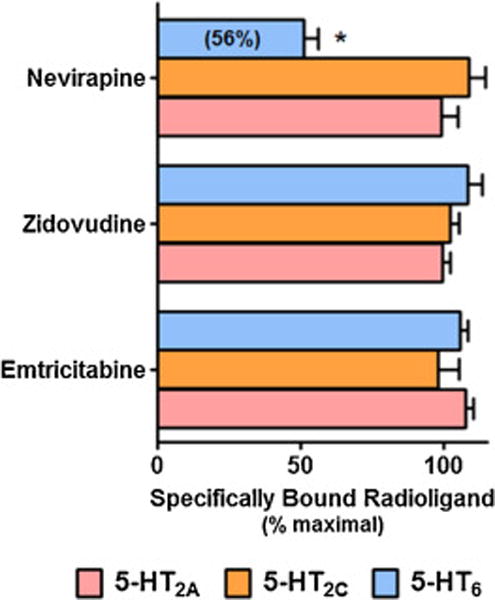
All of the ARV drugs were tested at the highest concentration (60 μM) that would stay in solution under the assay conditions. All data are presented as averaged values from multiple experiments ± SEM. A two-way ANOVA followed by a Bonferroni post-hoc indicated that interaction of nevirapine with the 5HT6 receptor is significantly less than its vehicle control (P < 0.001).
Given that efavirenz was shown previously to potentiate GABAA receptor currents [9], we also investigated the effects of these same three ARV drugs on the GABAA receptor. Both ZDV and FTC inhibited the GABA-activated currents in α1β2γ2GABAA receptors with reductions of 36–50% of control at 30 μM (Figure 13). The inhibitory effect had a very rapid onset (msec), and was reversible upon removal of the drugs (Figure 13A). In contrast, co-application of NVP (30 μM) with GABA, caused only a small, though significant, 8% potentiation of GABA-activated currents that was also rapid and reversible (Figure 13B).
Figure 13. The ARV drugs zidovudine (ZDV), emtricitabine (FTC), but not nevirapine (NVP), inhibit cloned GABAA receptor currents.
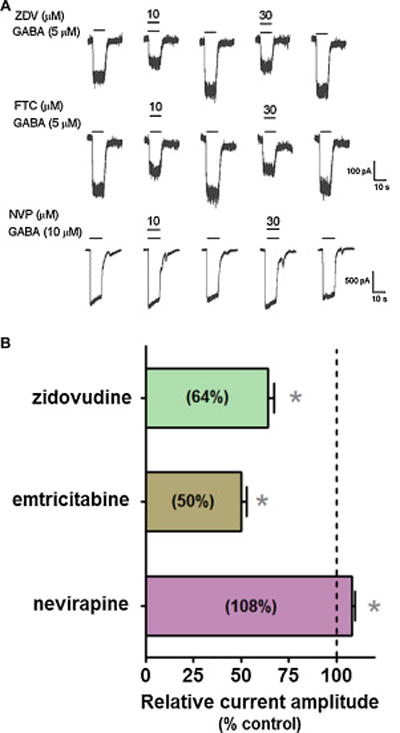
A, Representative traces showing whole cell currents activated by an EC30 concentration of GABA (5 μM) in HEK293 cells stably expressing α1β2γ2GABAA receptors. Efavirenz was co-applied with GABA for 10 sec. Note that ARV drug-induced action was rapid and reversible. B, Summary data of the modulatory effect induced by ZDV, FTC and NVP (30 μM). All currents were normalized to the GABA control response (GABA alone). *P < 0.05, paired t-test, compared to the control, n=6–8. Data are presented as means ± SEM.
4. Discussion
In a previous study focusing on its recreational use [9], efavirenz was reported to interact with a number of known molecular targets for drugs of abuse (e.g., GABAA and 5-HT2A receptors, and DAT, SERT and VMAT2 transporters), and to have a prevailing behavioral effect in rodents most similar to an LSD-like activity mediated by the 5-HT2A receptor. Here our attention was primarily on discovering molecular mechanisms potentially relevant to the reported CNS toxicity of efavirenz in those taking a prescribed dose for the treatment of HIV-1. In this context, we additionally refined our focus on serotonergic systems as it was suspected that other components of this signaling family were likely involved. Indeed, efavirenz was found to interact with the entire 5-HT2 subfamily of receptors, the ionotropic 5-HT3A receptor, the 5-HT6 receptor and MAO-A. The most prominent of the newly discovered interactions were followed up here with detailed mechanistic studies.
While efavirenz was reported to interact with the 5-HT2C receptor [9], its activity at that receptor was never elucidated. We report here for the first time that efavirenz acts as an antagonist of the 5-HT2C receptor as it has no agonist activity alone but it concentration-dependently blocked the agonist response by serotonin. Given that ligands which interact with both the 5-HT2C and 5-HT2A receptors typically display similar functional properties, we re-evaluated the activity of efavirenz on the 5-HT2A receptors. Using cloned 5-HT2A receptors and either changes in intracellular calcium or IP1 accumulation as measures of activation of the Gq-PLC-IP3-[Ca2+]i signaling pathway, efavirenz had no detectable agonist activity, but instead blocked multiple different agonists (i.e., 5-HT, α-methyl-5-HT, TCB-2, (±)-DOI, and LSD) from activating the 5-HT2A receptor. Though it is unclear what the exact reason might be for the discrepancy in efavirenz’s activity reported here and a previous collaborative report employing different methodology and conditions [9], it cannot be due to a greater sensitivity of activation as measured by radiolabeled inositol phosphates because measurements of IP3-mediated changes in intracellular calcium are much more sensitive than measures of changes in inositol phosphates (e.g., IP3) [44]. Since radioligand binding assays clearly demonstrate efavirenz interacts with the 5-HT2A receptor and in the present report efavirenz acts as an antagonist of Gq-mediated agonist responses, Schild analysis was performed to determine its antagonist potency (KB). No significant difference in efavirenz’s antagonist potency (KB) was observed using either (±)-DOI or LSD as the agonist, indicating an absence of allosteric probe dependence [45, 46]. Regardless of whether the agonist employed was (±)-DOI or LSD, the slopes of the Schild plots for efavirenz were not different from unity. The implication is that efavirenz competes for the same binding site on the 5-HT2A receptor as either of these known hallucinogens. Our finding that efavirenz behaves as an antagonist of the 5-HT2A receptor-mediated Gq signaling is consistent with efavirenz affecting human sleep patterns in a manner similar to the 5-HT2A receptor antagonist MK-0454 [47].
In large clinical studies (1008 patients), hallucinations have been reported as one of the NPAEs associated with efavirenz-containing treatment regimes, but the incidents were relatively low (1.2%) compared to other NPAEs [7]. Further, the frequency and severity of all NPAEs are highest during the first couple weeks of treatment and tend to disappear with continued treatment in some, but not all patients [5, 48]. An even higher frequently (6–31%) of hallucinations associated with efavirenz treatment have been reported in three other clinical studies, each involving between 114–197 patients [49–51]. An HIV-negative individual with a history of psychedelic drug experience reported, upon first time experimental ingestion of efavirenz (initially 300 mg stepped up to a full 600 mg dose p.o.), that it has a classical psychedelic quality with acute effects somewhat comparable to the hallucinogen mescaline [52]. Thus, it seems clear that efavirenz, at the prescribed dose is capable of inducing hallucinations in humans in some instances. Given our finding that efavirenz competitively antagonizes the 5-HT2A receptor-mediated Gq signaling responses produced by the hallucinogenic 5-HT2A receptor partial agonists (±)-DOI and LSD, the question becomes whether efavirenz mediates hallucinogenic responses via the 5-HT2A receptor, as is the case for classical hallucinogenic drugs like LSD and DOI, or via some other receptor mechanism.
The 5-HT2A receptor is an established site for hallucinogenic drug action. The hallucinogenic indoleamine psilocybin increases cerebral metabolic rate in the frontal cortex and anterior cingulate cortex [53], and pretreatment with the 5-HT2A antagonist ketanserin dose-dependently prevents hallucinations caused by psilocybin in humans [54]. The frontal cortex-mediated headtwitch response [55, 56], a hallucinogenic behavioral proxy in rodents, is absent in 5-HT2A knockout mice [57]. In wild type mice the head-twitch response is blocked by the 5-HT2A antagonists, MDL100,907 and ketanserin, as is the increase in synaptic activity in the frontal cortex neurons induced by application of the hallucinogenic 5-HT2A agonists DOI and α-methyl-5-HT [37, 58]. To our knowledge, all known hallucinogens which signal via the 5-HT2A receptor happen to also act as Gq signaling agonists; however, we show here that efavirenz functions as an antagonist of Gq signaling. In Gq homozygous knockout mice, DOI is still capable of producing a significant, albeit blunted, head-twitch response, which suggests that the Gq signaling pathway is not the sole mediator of the 5-HT2A receptor dependent behavioral effects of hallucinogens [59]. In a previous report, efavirenz was shown to invoke a head-twitch response of remarkably reduced duration and intensity compared to other hallucinogens like LSD and DOI in wild type mice, but failed to induce this response in 5-HT2A knockout mice [9]. One possibility is that the comparatively diminished behavioral response for efavirenz, relative to other hallucinogens, might be related to efavirenz’s lack of Gq-mediated agonist signaling. Efavirenz also has other behavioral properties similar to LSD, including in tests of discrimination where efavirenz partially substituted for LSD and vice versa [9]. Taken together, it seems clear that efavirenz interacts with the 5-HT2A receptor and has psychoactive properties, though it also has properties distinct from classical hallucinogens including that it fails to stimulate Gq signaling via the 5-HT2A receptor.
An alternative explanation is that the hallucinogenic properties of efavirenz are not exerted via its interactions with the 5-HT2A receptor, but instead might be attributed to its interactions with a different receptor system. A previous study using individual cloned receptors capable of mediating hallucinations revealed that efavirenz (10 μM) did not have significant interactions with dopamine receptors or NMDA receptors [9]. That same study also reported that efavirenz (10 μM) did not have high affinity interactions with mixed populations of muscarinic receptors present in brain tissue [9]; however, this mixed receptor population approach utilizing a radiolabeled antagonist is not well suited to reveal moderate interactions with a single subtype nor would it reveal allosteric interactions with agonists. Though efavirenz alone had no effect on intracellular calcium levels in untransfected HEK293 cells, during the course of evaluating the possible modulation of endogenous Gq-coupled receptor-mediated responses in untransfected HEK293 cells, we discovered that efavirenz is able to partially antagonize agonist responses produced by endogenous M3 muscarinic receptors. The plateauing of the inhibitory response for efavirenz at the muscarinic M3 receptor and the lack of any 5-HT response in untransfected cells, clearly demonstrates efavirenz has a different mechanistic profile at the endogenous M3 receptor than at the exogenously expressed 5-HT2A and 5-HT2C receptors. Given that efavirenz exerted a modest and partial attenuation of agonist actions on the M3 receptor, we investigated whether efavirenz might also affect the cloned human M1 receptor. In contrast to its effect on the M3 receptor, efavirenz was able to completely block agonist activation of the M1 receptor, like the muscarinic receptor antagonists scopolamine and atropine. In this context it is worth noting that scopolamine [60] has psychoactive properties that include hallucinations, vivid dreams, and memory impairment [61–65]. Similarly, atropine, another muscarinic receptor antagonist [66], is also known to cause hallucinations [67–69]. Hence, partial and complete blockade of the M3 and M1 receptors, respectively, by efavirenz might account for or contribute to its hallucinogenic properties. The M1 receptor is the most abundant muscarinic subtype in the brain and is known to play a role in cognition, learning and memory, and vigilance. M1 knockout mice exhibit cognitive deficits [38, 39], hence blockade of the M1 receptor by efavirenz may contribute to the cognitive impairment observed in patients taking efavirenz [7, 70].
Although efavirenz reduces the 5-HT3A receptor currents, like a number of 5-HT3 receptor antagonists, it did not displace [3H]granisetron (also known as [3H]BRL-43694) binding and thus is not a competitive antagonist for the 5-HT ligand binding site like granisetron is [71]. However, efavirenz does block 5-HT3 receptor mediated currents in the presence of 5-HT, but in contrast to ondansetron, a competitive antagonist of the 5-HT site that reduces peak currents more strongly than steady-state currents [72], efavirenz reduces steady-state currents (as measured by end-application of current) more strongly than peak currents. This latter inhibition profile by efavirenz suggests it might act as an open-channel blocker of the 5-HT3A receptor similar to picrotoxin [73], though the precise mechanism remains to be elucidated and we cannot rule out that efavirenz behaves as some sort of complex allosteric modulator.
In contrast to its actions on the 5-HT2A, 5-HT2C, and 5-HT3A receptors, at the 5-HT6 receptor efavirenz functions as an inverse agonist as evidenced by its ability to significantly reduce basal levels of cAMP in the absence of 5-HT. Also consistent with an inverse agonist, the 5-HT responses were first diminished then completely overcome by increasing concentrations of efavirenz even to the point where basal levels of activity were reduced. Clozapine, an accepted 5-HT6 receptor inverse agonist [34], produced the same functional effect as efavirenz and served as our positive control for inverse agonism. These effects were observed using a rat receptor stably expressed in HeLa cells and did not require special measures to observe the inverse agonist activity as has been reported when the rat 5HT6 receptor is expressed in other cell lines [74]. The functional responses we observed for efavirenz, clozapine, and serotonin were all mediated by the cloned 5-HT6 receptor, since these compounds produced no significant changes in cAMP levels in untransfected HeLa cells which lack any detectable level of 5-HT6 expression. Further, the agonist-induced increase in cAMP that is mediated by endogenous β-adrenergic receptors was unaffected by efavirenz and suggests that efavirenz is not exerting a non-specific effect in these cells.
4.1 CNS targets of other ARVs
Given that efavirenz has multiple off-target activities and some of these appear to correlate with certain NPAEs observed in humans, we investigated other ARV drugs for possible interactions with some of these off-target receptors. ZDV and NVP have been occasionally associated with reports of CNS toxicity, while FTC has not. For example, ZDV use has been associated with malaise, fatigue and insomnia [75, 76]. Further, a number of case reports have been published describing the onset of dose-related NPAEs associated with ZDV for the treatment of HIV-1, including depressive and manic syndromes primarily accompanied by auditory hallucinations and paranoid delusions [77, 78]. Case reports of NPAEs for NVP include hallucinations, vivid dreaming or psychosis in conjunction with depression and concomitant delusions soon after initiating NVP in patients with no previous history of psychiatric illness or substance use [79, 80]. These NPAEs subsided after withdrawal of NVP. We report here that, in general, ZDV, NVP and FTC did not share the same complex profile as efavirenz with respect to interactions with 5-HT2A, 5-HT2C and 5-HT6 receptors. In fact, only in the case of the 5-HT6 receptor and NVP, and only at a much higher concentration, was an interaction even detectable (estimated affinity would correspond to ~65 μM or approximately 30-fold lower than for efavirenz). Since we had found previously that efavirenz acted as an allosteric potentiator of GABAA receptors [9], the actions of ZDV, NVP and FTC on this receptor were examined as well. The most pronounced modulatory effect was observed for ZDV and FTC, and, in contrast to efavirenz, these ARV drugs inhibited, rather than potentiated the GABA response. While the investigation of other ARV drugs was limited in scope, it seems clear that the complex multiple off-target profile for efavirenz is unique amongst the ARV drugs examined. Though no rodent behavior clearly related to the GABAA receptor potentiating effects of efavirenz has been elucidated, the finding that efavirenz potentiates and ZDV and FTC inhibit GABA currents might still have relevance for HAART therapy given that the standard of HIV-1 care is combined drug treatments.
4.2 Speculation concerning efavirenz’s CNS targets possibly contributing to NPAEs
Given the molecular mechanistic insights gained from our studies, we became curious about which one(s) of the prominent NPAEs might be most likely to be mediated by 5-HT3A and 5-HT6 receptors. The following is educated speculation based upon inference drawn from the in vitro receptor pharmacology. Dizziness and headache are amongst the most frequently reported adverse events for 5-HT3 receptor antagonists [81–83]. Since efavirenz blocks 5-HT3 receptor currents, it seems reasonable that 5-HT3 receptor antagonism may contribute in part to efavirenz-induced dizziness and headache [4, 5, 7, 84]. HIV-1 patients are advised to take efavirenz before going to bed and sleep disturbances are commonly reported NPAEs including both insomnia and somnolence [4, 7, 85–87]. Several serotoninergic targets for efavirenz have been identified in this and a previous report (5-HT2A, 5-HT2C, 5-HT3, 5-HT6, 5-HT2B, MAO-A, and SERT), and given the complex role that serotonin plays in the regulation of sleep and wakefulness and the known involvement of each of these serotonergic targets [88–93], it seems reasonable to infer that one or more of these targets would be implicated in efavirenz’s sleep-related NPAEs. Consistent with this idea, and as noted above, efavirenz alters certain aspects of human sleep patterns in a manner similar to that of a 5-HT2A receptor antagonist [47].
Other reported NPAEs for efavirenz include depression, anxiety and feelings of stress [4, 6, 7, 85, 94]. Homozygous 5-HT2A knockout mice exhibit an anxiodepressive-like phenotype compared to wild type mice, possibly due to diminished 5-HT2A receptor mediated transmission [95]. Though we demonstrated that efavirenz acts as an antagonist of 5-HT2A/2C–mediated Gq signaling, we also discovered that chronic application of efavirenz reduces 5-HT2A receptor density and the responsiveness of this receptor to serotonin. The inference here would be that efavirenz could also be reducing 5-HT2A mediated neurotransmission thus precipitating an anxiodepressive-like phenotype.
5. Conclusion
In this report, we detail the molecular mechanisms of action of efavirenz across a family of serotonergic targets. Our radioligand binding studies revealed efavirenz’s interactions with the 5-HT2A, 5-HT2B, 5-HT2C and 5-HT6 receptors. Functional studies showed that efavirenz acts as an antagonist of Gq-coupling at the 5-HT2A and 5-HT2C receptors, an inverse agonist at the Gs-coupled 5-HT6 receptor, a 5-HT3A receptor pore blocker, and a modest inhibitor of MAO-A. Within a similar concentration range, efavirenz also completely or partially blocks agonist responses at the M1 and M3 muscarinic receptors, respectively. Schild shift analysis suggests that efavirenz competes with LSD and (±)-DOI for binding to the same site on the 5-HT2A receptor. 5-HT2A receptor density and responsiveness to 5-HT are reduced upon prolonged treatment with efavirenz. Other ARVs like NVP, FTC, and ZDV did not share the same complex pharmacology as efavirenz; only NVP interacted with the 5-HT6 receptor and potentiated GABAA receptors currents, like efavirenz, albeit with much smaller effect sizes. FTC and ZDV inhibited, instead of potentiating, GABAA receptor currents. Overall, the data suggests that efavirenz has complex receptor pharmacology with varied functional responses that is unique among ARV drugs tested in this report.
Acknowledgments
We thank the authors’ colleagues for providing the following as generous gifts: Dr. David Julius (USCF, San Francisco, CA) for cDNA for the mouse 5-HT3A receptor (GenBank accession no. S41757), Dr. David R. Sibley (NIH, Bethesda, MD) for the stable line expressing cloned rat 5-HT6 receptor, and Dr. Juergen Wess (NIH, Bethesda, MD) for the stable line expressing the cloned human M1 receptor. We also thank Dr. Glenn Dillon and Cathy Bell-Horner for contributions to some of the preliminary aspects of this work. This work was supported in part by the National Institutes of Health [Grant R01-MH063162 (JAS); Grant R41AG043243 (JAS)], ADDF grant 20140803 (JAS), UNTHSC Intramural Grant RI-6015 (JAS) and institutional funds 67673 (JAS).
Abbreviations
- NPAEs
Neuropsychiatric adverse events
- ARV
Antiretroviral
- HAART
Highly active antiretroviral therapy
- ZDV
Zidovudine
- FTC
Emtricitabine
- NVP
Nevirapine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds: Serotonin; 5-Hydroxytryptamine (PubChem CID, 5202); Efavirenz (PubChem CID, 64139); DOI (PubChem CID, 170617); LSD Tartrate (PubChem CID, 159828); Nevirapine (PubChem CID, 4463); Emtricitabine (PubChem CID, 60877); Zidovudine (PubChem CID, 35370); TCB-2 (PubChem CID, 71433791); α-Methyl-5HT (PubChem CID, 2107); Atropine (PubChem CID, 174174); Scopolamine (PubChem CID, 3000322)
Author Contribution:
Dhwanil A. Dalwadi – designed, performed experiments, and helped with data interpretation and writing of the manuscript
Seongcheol Kim – performed experiments for early phases of this work
Shahnawaz M. Amdani – performed experiments for early phases of this work
Zhenglan Chen – performed experiments
Ren-Qi Huang – designed experiments, interpreted data, and helped write the manuscript
John A. Schetz – conceived of the project, designed experiments, interpreted data, and wrote the manuscript
Conflict of Interest:
Authors declare no competing financial interest.
References
- 1.Young SD, Britcher SF, Tran LO, Payne LS, Lumma WC, Lyle TA, et al. L-743, 726 (DMP-266): a novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1995;39:2602–5. doi: 10.1128/aac.39.12.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. Consolidated guidelines on the use of antiretroviral drug for the treatment and preventing HIV infection. Recommendations for a public health approach. 2013;2015:272. [PubMed] [Google Scholar]
- 3.Gutierrez F, Navarro A, Padilla S, Anton R, Masia M, Borras J, et al. Prediction of neuropsychiatric adverse events associated with long-term efavirenz therapy, using plasma drug level monitoring. Clin Infect Dis. 2005;41:1648–53. doi: 10.1086/497835. [DOI] [PubMed] [Google Scholar]
- 4.Cohen C, Wohl D, Arribas JR, Henry K, Van Lunzen J, Bloch M, et al. Week 48 results from a randomized clinical trial of rilpivirine/emtricitabine/tenofovir disoproxil fumarate vs. efavirenz/emtricitabine/tenofovir disoproxil fumarate in treatment-naive HIV-1-infected adults. AIDS. 2014;28:989–97. doi: 10.1097/QAD.0000000000000169. [DOI] [PubMed] [Google Scholar]
- 5.Mills AM, Antinori A, Clotet B, Fourie J, Herrera G, Hicks C, et al. Neurological and psychiatric tolerability of rilpivirine (TMC278) vs. efavirenz in treatment-naive, HIV-1-infected patients at 48 weeks. HIV Med. 2013;14:391–400. doi: 10.1111/hiv.12012. [DOI] [PubMed] [Google Scholar]
- 6.Munoz-Moreno JA, Fumaz CR, Ferrer MJ, Gonzalez-Garcia M, Molto J, Negredo E, et al. Neuropsychiatric symptoms associated with efavirenz: prevalence, correlates, and management. A neurobehavioral review. AIDS Rev. 2009;11:103–9. [PubMed] [Google Scholar]
- 7.Sustiva. Sustiva product insert: Highlights of prescribing information. 1998;2015:21. [Google Scholar]
- 8.Shubber Z, Calmy A, Andrieux-Meyer I, Vitoria M, Renaud-Thery F, Shaffer N, et al. Adverse events associated with nevirapine and efavirenz-based first-line antiretroviral therapy: a systematic review and meta-analysis. AIDS. 2013;27:1403–12. doi: 10.1097/QAD.0b013e32835f1db0. [DOI] [PubMed] [Google Scholar]
- 9.Gatch MB, Kozlenkov A, Huang RQ, Yang W, Nguyen JD, Gonzalez-Maeso J, et al. The HIV antiretroviral drug efavirenz has LSD-like properties. Neuropsychopharmacology. 2013;38:2373–84. doi: 10.1038/npp.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tozzi V, Balestra P, Salvatori MF, Vlassi C, Liuzzi G, Giancola ML, et al. Changes in cognition during antiretroviral therapy: comparison of 2 different ranking systems to measure antiretroviral drug efficacy on HIV-associated neurocognitive disorders. J Acquir Immune Defic Syndr. 2009;52:56–63. doi: 10.1097/qai.0b013e3181af83d6. [DOI] [PubMed] [Google Scholar]
- 11.Yilmaz A, Watson V, Dickinson L, Back D. Efavirenz pharmacokinetics in cerebrospinal fluid and plasma over a 24-hour dosing interval. Antimicrob Agents Chemother. 2012;56:4583–5. doi: 10.1128/AAC.06311-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tashima KT, Caliendo AM, Ahmad M, Gormley JM, Fiske WD, Brennan JM, et al. Cerebrospinal fluid human immunodeficiency virus type 1 (HIV-1) suppression and efavirenz drug concentrations in HIV-1-infected patients receiving combination therapy. J Infect Dis. 1999;180:862–4. doi: 10.1086/314945. [DOI] [PubMed] [Google Scholar]
- 13.Best BM, Koopmans PP, Letendre SL, Capparelli EV, Rossi SS, Clifford DB, et al. Efavirenz concentrations in CSF exceed IC50 for wild-type HIV. J Antimicrob Chemother. 2011;66:354–7. doi: 10.1093/jac/dkq434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avery LB, Sacktor N, McArthur JC, Hendrix CW. Protein-free efavirenz concentrations in cerebrospinal fluid and blood plasma are equivalent: applying the law of mass action to predict protein-free drug concentration. Antimicrob Agents Chemother. 2013;57:1409–14. doi: 10.1128/AAC.02329-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka R, Hanabusa H, Kinai E, Hasegawa N, Negishi M, Kato S. Intracellular efavirenz levels in peripheral blood mononuclear cells from human immunodeficiency virus-infected individuals. Antimicrob Agents Chemother. 2008;52:782–5. doi: 10.1128/AAC.01613-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dirson G, Fernandez C, Hindlet P, Roux F, German-Fattal M, Gimenez F, et al. Efavirenz does not interact with the ABCB1 transporter at the blood-brain barrier. Pharm Res. 2006;23:1525–32. doi: 10.1007/s11095-006-0279-5. [DOI] [PubMed] [Google Scholar]
- 17.Romao PR, Lemos JC, Moreira J, de Chaves G, Moretti M, Castro AA, et al. Anti-HIV drugs nevirapine and efavirenz affect anxiety-related behavior and cognitive performance in mice. Neurotox Res. 2011;19:73–80. doi: 10.1007/s12640-009-9141-y. [DOI] [PubMed] [Google Scholar]
- 18.Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS. 2001;15:71–5. doi: 10.1097/00002030-200101050-00011. [DOI] [PubMed] [Google Scholar]
- 19.Corne SJ, Pickering RW, Warner BT. A method for assessing the effects of drugs on the central actions of 5-hydroxytryptamine. Br J Pharmacol Chemother. 1963;20:106–20. doi: 10.1111/j.1476-5381.1963.tb01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colpaert FC, Meert TF, Niemegeers CJ, Janssen PA. Behavioral and 5-HT antagonist effects of ritanserin: a pure and selective antagonist of LSD discrimination in rat. Psychopharmacology (Berl) 1985;86:45–54. doi: 10.1007/BF00431683. [DOI] [PubMed] [Google Scholar]
- 21.Singleton C, Marsden CA. Circadian variation in the head twitch response produced by 5-methoxy-N1,N1-dimethyltryptamine and p-chloroamphetamine in the mouse. Psychopharmacology (Berl) 1981;74:173–6. doi: 10.1007/BF00432688. [DOI] [PubMed] [Google Scholar]
- 22.Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. 2164-12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ancellin N, Preisser L, Le Maout S, Barbado M, Creminon C, Corman B, et al. Homologous and heterologous phosphorylation of the vasopressin V1a receptor. Cell Signal. 1999;11:743–51. doi: 10.1016/s0898-6568(99)00035-2. [DOI] [PubMed] [Google Scholar]
- 24.Luo J, Busillo JM, Benovic JL. M3 muscarinic acetylcholine receptor-mediated signaling is regulated by distinct mechanisms. Mol Pharmacol. 2008;74:338–47. doi: 10.1124/mol.107.044750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cummings DF, Canseco DC, Sheth P, Johnson JE, Schetz JA. Synthesis and structure-affinity relationships of novel small molecule natural product derivatives capable of discriminating between serotonin 5-HT1A, 5-HT2A, 5-HT2C receptor subtypes. Bioorg Med Chem. 2010;18:4783–92. doi: 10.1016/j.bmc.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawkinson JE, Drewe JA, Kimbrough CL, Chen JS, Hogenkamp DJ, Lan NC, et al. 3 alpha-Hydroxy-3 beta-trifluoromethyl-5 alpha-pregnan-20-one (Co 2-1970): a partial agonist at the neuroactive steroid site of the gamma-aminobutyric acidA receptor. Mol Pharmacol. 1996;49:897–906. [PubMed] [Google Scholar]
- 27.Hsu CY, Uludag H. A simple and rapid nonviral approach to efficiently transfect primary tissue-derived cells using polyethylenimine. Nat Protoc. 2012;7:935–45. doi: 10.1038/nprot.2012.038. [DOI] [PubMed] [Google Scholar]
- 28.Huang RQ, Dillon GH. Effect of extracellular pH on GABA-activated current in rat recombinant receptors and thin hypothalamic slices. J Neurophysiol. 1999;82:1233–43. doi: 10.1152/jn.1999.82.3.1233. [DOI] [PubMed] [Google Scholar]
- 29.Cussac D, Newman-Tancredi A, Quentric Y, Carpentier N, Poissonnet G, Parmentier JG, et al. Characterization of phospholipase C activity at h5-HT2C compared with h5-HT2B receptors: influence of novel ligands upon membrane-bound levels of [3H]phosphatidylinositols. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:242–52. doi: 10.1007/s00210-001-0505-y. [DOI] [PubMed] [Google Scholar]
- 30.Hirst WD, Abrahamsen B, Blaney FE, Calver AR, Aloj L, Price GW, et al. Differences in the central nervous system distribution and pharmacology of the mouse 5-hydroxytryptamine-6 receptor compared with rat and human receptors investigated by radioligand binding, site-directed mutagenesis, and molecular modeling. Mol Pharmacol. 2003;64:1295–308. doi: 10.1124/mol.64.6.1295. [DOI] [PubMed] [Google Scholar]
- 31.Johnson MP, Loncharich RJ, Baez M, Nelson DL. Species variations in transmembrane region V of the 5-hydroxytryptamine type 2A receptor alter the structure-activity relationship of certain ergolines and tryptamines. Mol Pharmacol. 1994;45:277–86. [PubMed] [Google Scholar]
- 32.Chen G, Cho SJ, Huang XP, Jensen NH, Svennebring A, Sassano MF, et al. Rational Drug Design Leading to the Identification of a Potent 5-HT(2C) Agonist Lacking 5-HT(2B) Activity. ACS Med Chem Lett. 2011;2:929–32. doi: 10.1021/ml200206z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porter RH, Benwell KR, Lamb H, Malcolm CS, Allen NH, Revell DF, et al. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Br J Pharmacol. 1999;128:13–20. doi: 10.1038/sj.bjp.0702751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purohit A, Herrick-Davis K, Teitler M. Creation, expression, and characterization of a constitutively active mutant of the human serotonin 5-HT6 receptor. Synapse. 2003;47:218–24. doi: 10.1002/syn.10157. [DOI] [PubMed] [Google Scholar]
- 35.Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- 36.Egan CT, Herrick-Davis K, Miller K, Glennon RA, Teitler M. Agonist activity of LSD and lisuride at cloned 5HT2A and 5HT2C receptors. Psychopharmacology (Berl) 1998;136:409–14. doi: 10.1007/s002130050585. [DOI] [PubMed] [Google Scholar]
- 37.Marek GJ, Aghajanian GK. LSD and the phenethylamine hallucinogen DOI are potent partial agonists at 5-HT2A receptors on interneurons in rat piriform cortex. J Pharmacol Exp Ther. 1996;278:1373–82. [PubMed] [Google Scholar]
- 38.Gould RW, Dencker D, Grannan M, Bubser M, Zhan X, Wess J, et al. Role for the M1 Muscarinic Acetylcholine Receptor in Top-Down Cognitive Processing Using a Touchscreen Visual Discrimination Task in Mice. ACS Chem Neurosci. 2015;6:1683–95. doi: 10.1021/acschemneuro.5b00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang S, Li Y, Zhang C, Zhao Y, Bu G, Xu H, et al. M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neurosci Bull. 2014;30:295–307. doi: 10.1007/s12264-013-1406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange HS, Cannon CE, Drott JT, Kuduk SD, Uslaner JM. The M1 Muscarinic Positive Allosteric Modulator PQCA Improves Performance on Translatable Tests of Memory and Attention in Rhesus Monkeys. J Pharmacol Exp Ther. 2015;355:442–50. doi: 10.1124/jpet.115.226712. [DOI] [PubMed] [Google Scholar]
- 41.Puri V, Wang X, Vardigan JD, Kuduk SD, Uslaner JM. The selective positive allosteric M1 muscarinic receptor modulator PQCA attenuates learning and memory deficits in the Tg2576 Alzheimer’s disease mouse model. Behav Brain Res. 2015;287:96–9. doi: 10.1016/j.bbr.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 42.Monsma FJ, Jr, Shen Y, Ward RP, Hamblin MW, Sibley DR. Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol Pharmacol. 1993;43:320–7. [PubMed] [Google Scholar]
- 43.Tallman JF, Smith CC, Henneberry RC. Induction of functional beta-adrenergic receptors in HeLa cells. Proc Natl Acad Sci U S A. 1977;74:873–7. doi: 10.1073/pnas.74.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dickson EJ, Falkenburger BH, Hille B. Quantitative properties and receptor reserve of the IP(3) and calcium branch of G(q)-coupled receptor signaling. J Gen Physiol. 2013;141:521–35. doi: 10.1085/jgp.201210886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armour SL, Foord S, Kenakin T, Chen WJ. Pharmacological characterization of receptor-activity-modifying proteins (RAMPs) and the human calcitonin receptor. J Pharmacol Toxicol Methods. 1999;42:217–24. doi: 10.1016/s1056-8719(00)00074-5. [DOI] [PubMed] [Google Scholar]
- 46.Kenakin T. What is pharmacological ‘affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol Sci. 2014;35:434–41. doi: 10.1016/j.tips.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Simen AA, Ma J, Svetnik V, Mayleben D, Maynard J, Roth A, et al. Efavirenz modulation of sleep spindles and sleep spectral profile. J Sleep Res. 2015;24:66–73. doi: 10.1111/jsr.12196. [DOI] [PubMed] [Google Scholar]
- 48.Nelson MR, Elion RA, Cohen CJ, Mills A, Hodder SL, Segal-Maurer S, et al. Rilpivirine versus efavirenz in HIV-1-infected subjects receiving emtricitabine/tenofovir DF: pooled 96-week data from ECHO and THRIVE Studies. HIV Clin Trials. 2013;14:81–91. doi: 10.1310/hct1403-81. [DOI] [PubMed] [Google Scholar]
- 49.Gutierrez-Valencia A, Viciana P, Palacios R, Ruiz-Valderas R, Lozano F, Terron A, et al. Stepped-dose versus full-dose efavirenz for HIV infection and neuropsychiatric adverse events: a randomized trial. Ann Intern Med. 2009;151:149–56. doi: 10.7326/0003-4819-151-3-200908040-00127. [DOI] [PubMed] [Google Scholar]
- 50.Lochet P, Peyriere H, Lotthe A, Mauboussin JM, Delmas B, Reynes J. Long-term assessment of neuropsychiatric adverse reactions associated with efavirenz. HIV Med. 2003;4:62–6. doi: 10.1046/j.1468-1293.2003.00136.x. [DOI] [PubMed] [Google Scholar]
- 51.Mukonzo JK, Okwera A, Nakasujja N, Luzze H, Sebuwufu D, Ogwal-Okeng J, et al. Influence of efavirenz pharmacokinetics and pharmacogenetics on neuropsychological disorders in Ugandan HIV-positive patients with or without tuberculosis: a prospective cohort study. BMC Infect Dis. 2013;13:261. doi: 10.1186/1471-2334-13-261. 2334-13–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris H. Getting high on HIV medication (Full documentary) 2014;2015 [Google Scholar]
- 53.Vollenweider FX, Leenders KL, Scharfetter C, Maguire P, Stadelmann O, Angst J. Positron emission tomography and fluorodeoxyglucose studies of metabolic hyperfrontality and psychopathology in the psilocybin model of psychosis. Neuropsychopharmacology. 1997;16:357–72. doi: 10.1016/S0893-133X(96)00246-1. [DOI] [PubMed] [Google Scholar]
- 54.Vollenweider FX, Vollenweider-Scherpenhuyzen MF, Babler A, Vogel H, Hell D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport. 1998;9:3897–902. doi: 10.1097/00001756-199812010-00024. [DOI] [PubMed] [Google Scholar]
- 55.Vickers SP, Easton N, Malcolm CS, Allen NH, Porter RH, Bickerdike MJ, et al. Modulation of 5-HT(2A) receptor-mediated head-twitch behaviour in the rat by 5-HT(2C) receptor agonists. Pharmacol Biochem Behav. 2001;69:643–52. doi: 10.1016/s0091-3057(01)00552-4. [DOI] [PubMed] [Google Scholar]
- 56.Willins DL, Meltzer HY. Direct injection of 5-HT2A receptor agonists into the medial prefrontal cortex produces a head-twitch response in rats. J Pharmacol Exp Ther. 1997;282:699–706. [PubMed] [Google Scholar]
- 57.Gonzalez-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–52. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Beique JC, Imad M, Mladenovic L, Gingrich JA, Andrade R. Mechanism of the 5-hydroxytryptamine 2A receptor-mediated facilitation of synaptic activity in prefrontal cortex. Proc Natl Acad Sci U S A. 2007;104:9870–5. doi: 10.1073/pnas.0700436104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garcia EE, Smith RL, Sanders-Bush E. Role of G(q) protein in behavioral effects of the hallucinogenic drug 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane. Neuropharmacology. 2007;52:1671–7. doi: 10.1016/j.neuropharm.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Golding JF, Stott JR. Comparison of the effects of a selective muscarinic receptor antagonist and hyoscine (scopolamine) on motion sickness, skin conductance and heart rate. Br J Clin Pharmacol. 1997;43:633–7. doi: 10.1046/j.1365-2125.1997.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cotroneo C. Devil’s Breath: Scopolamine, AKA Burundanga, Hailed As ‘World’s Scariest Drug’ 2013. 2015 [Google Scholar]
- 62.Lin YG, Chen PH, Chang FY, Wu LT, Liao KY, Wu TC. Delirium due to scopolamine patch in a 4-year-old boy. J Formos Med Assoc. 2011;110:208–11. doi: 10.1016/S0929-6646(11)60031-4. [DOI] [PubMed] [Google Scholar]
- 63.Jalali F, Afshari R, Babaei A. Smoking crushed hyoscine/scopolamine tablets as drug abuse. Subst Use Misuse. 2014;49:793–7. doi: 10.3109/10826084.2014.880178. [DOI] [PubMed] [Google Scholar]
- 64.Seo SW, Suh MK, Chin J, Na DL. Mental confusion associated with scopolamine patch in elderly with mild cognitive impairment (MCI) Arch Gerontol Geriatr. 2009;49:204–7. doi: 10.1016/j.archger.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 65.Vallersnes OM, Lund C, Duns AK, Netland H, Rasmussen IA. Epidemic of poisoning caused by scopolamine disguised as Rohypnol tablets. Clin Toxicol (Phila) 2009;47:889–93. doi: 10.3109/15563650903333804. [DOI] [PubMed] [Google Scholar]
- 66.Ma K, Yang ZH, Yang LM, Chen HZ, Lu Y. Activation of M1 mAChRs by lesatropane rescues glutamate neurotoxicity in PC12 cells via PKC-mediated phosphorylation of ERK1/2. Bosn J Basic Med Sci. 2013;13:146–52. doi: 10.17305/bjbms.2013.2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arthurs GJ, Davies R. Atropine–a safe drug. Anaesthesia. 1980;35:1077–9. doi: 10.1111/j.1365-2044.1980.tb05046.x. [DOI] [PubMed] [Google Scholar]
- 68.Baker JH, Silver JR. Atropine toxicity in acute cervical spinal injury. Paraplegia. 1984;22:379–82. doi: 10.1038/sc.1984.61. [DOI] [PubMed] [Google Scholar]
- 69.Fisher CM. Visual hallucinations on eye closure associated with atropine toxicity. A neurological analysis and comparison with other visual hallucinations. Can J Neurol Sci. 1991;18:18–27. doi: 10.1017/s0317167100031255. [DOI] [PubMed] [Google Scholar]
- 70.Ciccarelli N, Fabbiani M, Di Giambenedetto S, Fanti I, Baldonero E, Bracciale L, et al. Efavirenz associated with cognitive disorders in otherwise asymptomatic HIV-infected patients. Neurology. 2011;76:1403–9. doi: 10.1212/WNL.0b013e31821670fb. [DOI] [PubMed] [Google Scholar]
- 71.Rojas C, Stathis M, Thomas AG, Massuda EB, Alt J, Zhang J, et al. Palonosetron exhibits unique molecular interactions with the 5-HT3 receptor. Anesth Analg. 2008;107:469–78. doi: 10.1213/ane.0b013e318172fa74. [DOI] [PubMed] [Google Scholar]
- 72.Rammes G, Eisensamer B, Ferrari U, Shapa M, Gimpl G, Gilling K, et al. Antipsychotic drugs antagonize human serotonin type 3 receptor currents in a noncompetitive manner. Mol Psychiatry. 2004;9:846. doi: 10.1038/sj.mp.4001490. 58, 818. [DOI] [PubMed] [Google Scholar]
- 73.Das P, Dillon GH. Molecular determinants of picrotoxin inhibition of 5-hydroxytryptamine type 3 receptors. J Pharmacol Exp Ther. 2005;314:320–8. doi: 10.1124/jpet.104.080325. [DOI] [PubMed] [Google Scholar]
- 74.Romero G, Sanchez E, Pujol M, Perez P, Codony X, Holenz J, et al. Efficacy of selective 5-HT6 receptor ligands determined by monitoring 5-HT6 receptor-mediated cAMP signaling pathways. Br J Pharmacol. 2006;148:1133–43. doi: 10.1038/sj.bjp.0706827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fischl MA, Richman DD, Hansen N, Collier AC, Carey JT, Para MF, et al. The safety and efficacy of zidovudine (AZT) in the treatment of subjects with mildly symptomatic human immunodeficiency virus type 1 (HIV) infection. A double-blind, placebo-controlled trial. The AIDS Clinical Trials Group. Ann Intern Med. 1990;112:727–37. doi: 10.7326/0003-4819-112-10-727. [DOI] [PubMed] [Google Scholar]
- 76.McLeod GX, Hammer SM. Zidovudine: five years later. Ann Intern Med. 1992;117:487–501. doi: 10.7326/0003-4819-117-6-487. [DOI] [PubMed] [Google Scholar]
- 77.Maxwell S, Scheftner WA, Kessler HA, Busch K. Manic syndrome associated with zidovudine treatment. JAMA. 1988;259:3406–7. [PubMed] [Google Scholar]
- 78.Wright JM, Sachdev PS, Perkins RJ, Rodriguez P. Zidovudine-related mania. Med J Aust. 1989;150:339–41. doi: 10.5694/j.1326-5377.1989.tb136499.x. [DOI] [PubMed] [Google Scholar]
- 79.Morlese JF, Qazi NA, Gazzard BG, Nelson MR. Nevirapine-induced neuropsychiatric complications, a class effect of non-nucleoside reverse transcriptase inhibitors? AIDS. 2002;16:1840–1. doi: 10.1097/00002030-200209060-00023. [DOI] [PubMed] [Google Scholar]
- 80.Wise ME, Mistry K, Reid S. Drug points: Neuropsychiatric complications of nevirapine treatment. BMJ. 2002;324:879. doi: 10.1136/bmj.324.7342.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haus U, Spath M, Farber L. Spectrum of use and tolerability of 5-HT3 receptor antagonists. Scand J Rheumatol Suppl. 2004;119:12–8. [PubMed] [Google Scholar]
- 82.Tralongo P, Dimari A, Conti G, Aiello R, Mauceri G. Central nervous system side-effects of 5-HT3-receptor antagonists in elderly cancer patients treated with chemotherapy. Ann Oncol. 2004;15:987–8. doi: 10.1093/annonc/mdh223. [DOI] [PubMed] [Google Scholar]
- 83.Goodin S, Cunningham R. 5-HT(3)-receptor antagonists for the treatment of nausea and vomiting: a reappraisal of their side-effect profile. Oncologist. 2002;7:424–36. doi: 10.1634/theoncologist.7-5-424. [DOI] [PubMed] [Google Scholar]
- 84.Smith SR, Prosser WA, Donahue DJ, Morgan ME, Anderson CM, Shanahan WR, et al. Lorcaserin (APD356), a selective 5-HT(2C) agonist, reduces body weight in obese men and women. Obesity (Silver Spring) 2009;17:494–503. doi: 10.1038/oby.2008.537. [DOI] [PubMed] [Google Scholar]
- 85.Waters L, Fisher M, Winston A, Higgs C, Hadley W, Garvey L, et al. A phase IV, double-blind, multicentre, randomized, placebo-controlled, pilot study to assess the feasibility of switching individuals receiving efavirenz with continuing central nervous system adverse events to etravirine. AIDS. 2011;25:65–71. doi: 10.1097/QAD.0b013e328341685b. [DOI] [PubMed] [Google Scholar]
- 86.Clifford DB, Evans S, Yang Y, Acosta EP, Goodkin K, Tashima K, et al. Impact of efavirenz on neuropsychological performance and symptoms in HIV-infected individuals. Ann Intern Med. 2005;143:714–21. doi: 10.7326/0003-4819-143-10-200511150-00008. [DOI] [PubMed] [Google Scholar]
- 87.Nunez M, Gonzalez de Requena D, Gallego L, Jimenez-Nacher I, Gonzalez-Lahoz J, Soriano V. Higher efavirenz plasma levels correlate with development of insomnia. J Acquir Immune Defic Syndr. 2001;28:399–400. doi: 10.1097/00126334-200112010-00015. [DOI] [PubMed] [Google Scholar]
- 88.Monti JM, Jantos H. Effects of the 5-HT(6) receptor antagonists SB-399885 and RO-4368554 and of the 5-HT(2A) receptor antagonist EMD 281014 on sleep and wakefulness in the rat during both phases of the light-dark cycle. Behav Brain Res. 2011;216:381–8. doi: 10.1016/j.bbr.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 89.Monti JM. Serotonin control of sleep-wake behavior. Sleep Med Rev. 2011;15:269–81. doi: 10.1016/j.smrv.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 90.Real C, Popa D, Seif I, Callebert J, Launay JM, Adrien J, et al. Sleep apneas are increased in mice lacking monoamine oxidase A. Sleep. 2007;30:1295–302. doi: 10.1093/sleep/30.10.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wisor JP, Wurts SW, Hall FS, Lesch KP, Murphy DL, Uhl GR, et al. Altered rapid eye movement sleep timing in serotonin transporter knockout mice. Neuroreport. 2003;14:233–8. doi: 10.1097/00001756-200302100-00015. [DOI] [PubMed] [Google Scholar]
- 92.Morairty SR, Hedley L, Flores J, Martin R, Kilduff TS. Selective 5HT2A and 5HT6 receptor antagonists promote sleep in rats. Sleep. 2008;31:34–44. doi: 10.1093/sleep/31.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ponzoni A, Monti JM, Jantos H. The effects of selective activation of the 5-HT3 receptor with m-chlorophenylbiguanide on sleep and wakefulness in the rat. Eur J Pharmacol. 1993;249:259–64. doi: 10.1016/0014-2999(93)90520-r. [DOI] [PubMed] [Google Scholar]
- 94.Rihs TA, Begley K, Smith DE, Sarangapany J, Callaghan A, Kelly M, et al. Efavirenz and chronic neuropsychiatric symptoms: a cross-sectional case control study. HIV Med. 2006;7:544–8. doi: 10.1111/j.1468-1293.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 95.Petit AC, Quesseveur G, Gressier F, Colle R, David DJ, Gardier AM, et al. Converging translational evidence for the involvement of the serotonin 2A receptor gene in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:76–82. doi: 10.1016/j.pnpbp.2014.04.013. [DOI] [PubMed] [Google Scholar]