

Abstract
The use of chimeric antigen receptor (CAR)-T cell therapy for the treatment of hematologic malignancies has generated significant excitement over the last several years. From a transfusion medicine perspective, the implementation of CAR-T therapy as a potential mainstay treatment for not only hematologic but also solid-organ malignancies represents a significant opportunity for growth and expansion. In this review, we will describe the rationale for the development of genetically re-directed T-cells as a cancer therapeutic, the different elements that are required to engineer these cells, as well as an overview of the process by which patient cells are harvested and processed to create and subsequently validate CAR-T cells. Finally, we will briefly describe some of the toxicities and clinical efficacy of CAR-T cells in the setting of patients with advanced malignancy.
Keywords: T cells, adoptive immunotherapy, gene transfer, leukapheresis, cell processing
History of immunotherapy for cancer
The concept of cancer immunotherapy, or the harnessing of the immune system to treat malignancy, grew out of the realization that the immune system functions not only to protect the body from external disease-causing pathogens but also to play a central and active role in preventing the formation of primary malignancies[1]. Early observations that organ transplant recipients on long-term immunosuppression were highly susceptible to developing cancers[2] and that tumor-infiltrating lymphocytes in certain types of tumors were associated with variable prognoses[3] intimated the immune system in controlling cancers. However, the result that allogeneic (relative to autologous) bone marrow transplants was associated with lower occurrence of relapsed disease was integral in functionally proving that immune cells can have potent anti-cancer activity[4]. It further suggested the existence of specific tumor epitopes that could be recognized and targeted by allogeneic immune cells where syngeneic immune cells may not be as able to readily discriminate tumor from self.
These initial discoveries spurred the development of multiple methods by which to modulate the immune system to better target tumors. The identification, characterization, and subsequent utilization of different cytokines (proteins that can drive immune cell proliferation, differentiation, and activation) such as interleukin-2 and interferon-alpha, lead to the first efficacious cancer immunotherapy medications for the treatment of metastatic melanoma and renal cell carcinomas[5]. Additionally, it became possible to isolate and produce large amounts of monoclonal antibody targeting tumor-specific epitopes[6], allowing for directed killing of cancer cells primarily through either antibody-dependent cell-mediated cytotoxicity via NK-cells or complement-dependent cytotoxicity. These advances led to the development of numerous successful drugs targeting a variety of malignancies based on tumor surface marker expression, the first of which was Rituximab directed towards the B-cell surface marker CD20, approved first for the treatment of non-Hodgkin lymphoma and subsequently for chronic lymphocytic leukemia[7].
Adoptive immunotherapy
Classical adoptive immunotherapy consists of passive transfer of either autologous patient-derived tumor-infiltrating lymphocytes or infusion of allogeneic donor lymphocytes in the context of refractory malignant disease following bone marrow transplantation[8]. Early studies showed that ex vivo activation and expansion of tumor-specific T-cells using cytokines and growth factors as well as host immune-depletion can be combined to achieve significant immune cell-mediated cancer regression in a limited proportion of patients[9]. However, T-cell repertoire deficiencies along with the inherent difficulty of isolating and expanding tumor-reactive T-cells from patients represented significant obstacles against this approach. Immune repertoire deficiencies were first addressed through direct conferral pre-selected T-cell receptors on autologous T-cells[10]. However, TCR reactivity is constrained by the human leukocyte antigens (HLA) type of the major histocompatibility complex (MHC) expressed by a given tumor, limiting the generalizable utility of any given TCR. The development of single-chain variable fragments[11], usually derived from a mouse monoclonal antibody fused to TCR domains, redirect T cells with antibody-like specificity to enable T-cell activation and cytotoxic killing without MHC-restriction[11]. Promisingly, early proof-of-concept studies with CAR-T cells targeting CD4+ cells in HIV patients showed active tissue and cell targeting with long-term, safe persistence of re-directed T-cells[12, 13].
Chimeric antigen receptors can be conceptualized as combination of customizable antigen-recognition and signal transduction domains. Most CAR specificity has been conferred through the use of antibody-derived single chain proteins which, to date, have targeted mostly hematologic markers such as CD19 and CD20 although new antigens and specificities are of intense interest and continue to be developed[14]. First generation CARs, analogous to a traditional TCR, utilized a single CD3 signaling domain for signal transduction. However limited CAR-T cell persistence was observed in patients, leading to continued receptor re-design and modification. In order to further T-cell activation, proliferation, and persistence in vivo, extra cytoplasmic domains have been placed in various configurations, with second generation receptors containing a single additional cytoplasmic domain, such as 4-1BB or CD28 and third generation CAR T-cell receptors containing combinations of two or more of 4-1BB, CD28, CD27, ICOS, or OX40 of the co-stimulating domains (Figure 1). Together, the advances in receptor design along with a growing understanding of T-cell biology laid the groundwork for building a toolbox that has driven large-scale ex vivo manipulation and purposeful re-direction of immune cells for the purposes of targeted cancer therapy.
Figure 1.

Design of chimeric antigen receptors.
Apheresis collection for CAR T cell therapy
Apheresis collection of the mononuclear cell (MNC) layer has been shown to be a safe and efficient method of collecting the large number of T lymphocytes necessary to initiate CART cell culture. Apheresis involves application of centrifugal force to a continuous or semi-continuous flow of anti-coagulated whole blood. As cell layers separate by density, individual layers may be selectively and efficiently removed or replaced. The mononuclear cell layer is located between the dense polymorphonuclear cell / red blood cell layers and the less dense platelet layer (Figure 2). Circulating mature lymphocytes can be found within the MNC layer; therefore, isolation of this layer provides the cells to begin CAR-T cell manufacture.
Figure 2.
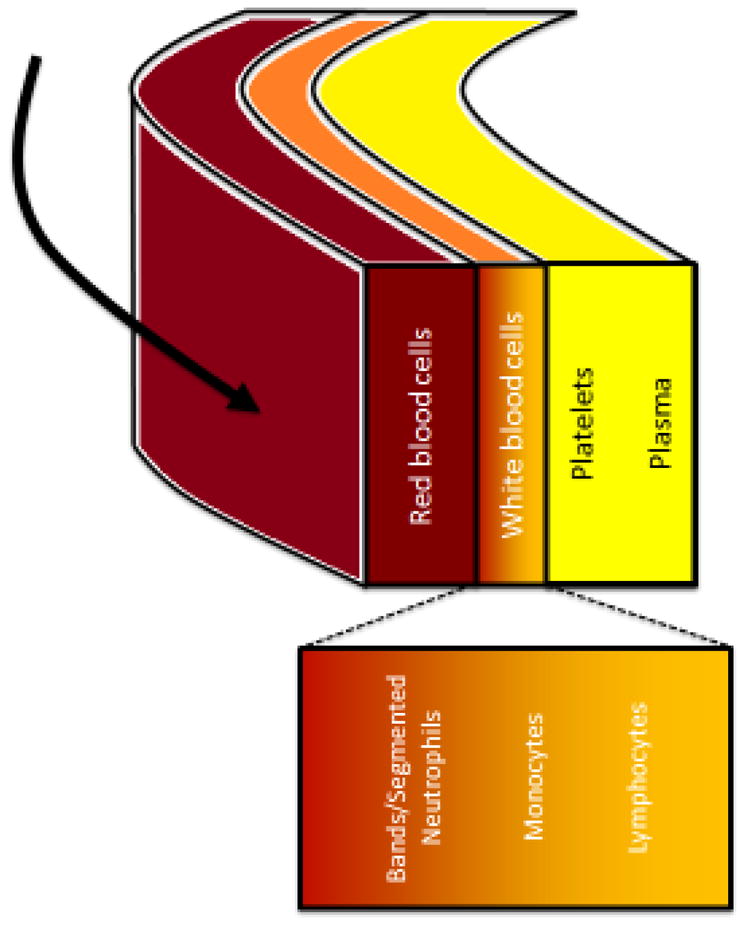
Peripheral blood separation via leukapheresis.
Several FDA-cleared systems are available to perform apheresis MNC collection, including the COBE Spectra and Spectra Optia Apheresis systems from TerumoBCT Inc. and the Amicus Cell Separator from Fenwal Inc./Fresenius Kabi AG. While the available systems are similar, product characteristics may differ slightly depending on the approach[16]. When selecting a particular collection method for CAR-T cell production many factors must be considered including the availability of instruments, kits, reagents, and trained staff. Furthermore, downstream processing may influence the choice of collection and collection parameters. For example, protocols that include efficient downstream enrichment of lymphocytes should prioritize yield over purity, whereas protocols with robust expansion may target purity over yield. Importantly, because different apheresis centers may have access to only one type of instrument, multi-site trials must demonstrate consistent collection of comparable products across all sites to ensure reliable cell manufacturing.
Optimal MNC collection parameters for CAR-T cell manufacture have yet to be determined. Apheresis protocol development has largely focused on optimal collection of circulating hematopoietic progenitor cells (HPCs) in the transplant setting. Targeting large, immature HPCs, whether benign or malignant has long been a focus of therapeutic apheresis. In fact, the first automated leukapheresis instruments were developed to selectively remove circulating large, immature leukemic cells[17]. Symptomatic leukostasis continues to be a leading indication for therapeutic leukapheresis[18, 19]. Collection of circulating CD34+ HPCs is now the most common source of HPCs for transplantation[20]. With decades of experience, the optimal apheresis parameters in these settings have been determined.
The optimal parameters for HPC collection may not be applicable to collection of mature T cells for CAR-T manufacture for several reasons. First, non-mobilized CAR-T cell patients often have low total white blood cell counts making identification and continued isolation of the RBC-plasma interface challenging. Second, mature lymphocytes are smaller and denser than immature HPCs, which makes red blood cell removal more challenging.
While HPC collection and collection for CAR-T cell therapy differ significantly, MNC collection for donor lymphocyte infusion (DLI) share some features. DLI is used in the setting of relapse post-allogeneic hematopoietic stem cell transplant to harness the graft-versus-tumor effect. For both DLI and CAR-T cell manufacturing, mature lymphocytes are the target cells for collection; however, the status of the donor may differ: whereas DLI donor are healthy allogeneic donors, most CAR-T cell donors have an underlying hematologic malignancy (or other cancer), which affects their lymphocytes either due to their disease or their prior treatment. Therefore, DLI parameters are a good starting point to investigate CAR-T cell collection, but certain parameters, such as collection efficacy, target volumes and optimal targeting within the mononuclear cell layer, will likely require further optimization to ensure high quality collection.
Technologic advances in the field of apheresis will allow for improved collection for CAR-T cell therapy. The Spectra Optia, successor to the COBE Spectra, is currently FDA-cleared for MNC collection. This instrument offers the ability to isolate particular portions of the MNC layer. Some have reported superior purity and yield with the Spectra Optia compared to the COBE Spectra in collection of granulocytes[21] and allogeneic HPCs[22]. Even more encouraging, the Spectra Optia has been shown to be capable of collecting mature lymphocytes for DLI with high efficiency and low contaminating red cells, granulocytes and platelets[23, 24], which is likely to apply well to collections for CAR-T cell products.
Cell processing in CAR-T cell manufacturing
Collection of the MNC layer yields not only lymphocytes, but also a significant amount of plasma, PMNs, monocytes, RBCs and platelets. This is particularly the case when the lymphocyte count is low and the MNC layer is narrow, as is often the case in patients with hematologic malignancies. Additionally, because monocytes are only minimally denser than lymphocytes, even with a readily identifiable MNC layer, apheresis will not distinguish monocytes from lymphocytes. Ultimately, to obtain sufficient yield, heterogeneous products must be accepted.
Heterogeneous mononuclear cell products can prove challenging for CAR-T cell production. Red cells and platelets can make accurate lymphocyte enumeration difficult and/or confound flow cytometry[25]. Plasma components, platelets, monocytes and granulocytes may induce clumping in culture, requiring filtration and/or the addition of DNase[26]. Red cells and monocytes have also been shown to interfere with the clinical efficacy of some types of therapeutic cells[27–30]. For these reasons, pre-culture processing of MNC products is necessary to provide a relatively pure lymphocyte population.
Removal of troublesome plasma and cellular components can be achieved through washing and selection methods. To limit open processes, closed-system, automated cell washers are preferred in the GMP laboratory. Commercially available equipment specifically designed for optimal recovery of lymphocytes is not readily available. Current practice largely involves adaptation of legacy instruments. Among others, the Cell Saver 5+ (Haemonetics Corp.) and the COBE 2991 Cell Processor (TerumoBCT Inc.) have been employed in GMP laboratories to wash and concentrate cells for CAR-T cell culture. Next generation equipment, such as the Lovo Cell Processing System (Fresenius Kabi) offer potential advantages in more closed, automated washing with greater cell recovery[31].
Selection of specific cell constituents with or without a wash step allows for purification of cell populations prior to seeding CAR-T cell culture. Cell selection is achieved primarily by cell density, cell size or immunophenotypic features. Sedimentation agents, such as Ficoll-Hypaque, use density to purify lymphocytes from MNC products. The Ficoll-Hypaque gradient technique involves a high-molecular weight sucrose polymer (Ficoll) and a dense, organic compound (Hypaque)[32–34]. Because Ficoll-Hypaque is denser than lymphocytes, monocytes and platelets, but less dense than granulocytes and RBCs, this technique allows granulocyte and RBC depletion of MNC products. Ficoll-Hypaque has been in use for decades and is highly efficient[32, 34], however it cannot be used to separate lymphocytes from monocytes or platelets. In addition, until recently, large-scale, Ficoll-based separation required an open system with extensive manipulation. New devices and adapted legacy instruments have been developed to conduct GMP-compliant, closed system Ficoll separation[35, 36].
Density gradients alone cannot distinguish monocytes and lymphocytes. Because monocytes can inhibit T cell activation and expansion it is necessary to isolate lymphocytes from monocyte contaminated products prior to culture[28–30]. Two approaches have been used to isolate lymphocytes from monocytes in apheresis products: elutriation and antibody-bead conjugate selection (Figure 3). Elutriation is the process by which centrifugal force and counter-flow fluid separate cell components based on both size and density. Importantly, because elutriation takes cell size into account, this method is capable of distinguishing between monocytes and lymphocytes in addition to RBC reduction. Elutriation has already been used extensively in dendritic cell vaccine production[16, 37, 38] and more recently adapted to isolate lymphocytes for T cell immunotherapy[39, 40]. Scalable, GMP-compliant, closed system elutriation instruments are commercially available (Elutra Cell Separation System, TerumoBCT Inc.). To date, elutriation has been widely used in CAR-T cell clinical trials with little apparent negative influence on clinical efficacy[41, 42]. Alternatively, monoclonal antibodies fused to magnetic beads can be used to perform either positive or negative selection of target cells. GMP-grade, antibody-conjugated beads are readily available and target a wide array of cell surface markers (Miltenyi Biotec, Bergisch Gladbach, Germany). In addition to selection, antibody-bead conjugates have been shown to efficiently provide primary and co-stimulatory signal to expand T cells in culture[43]. However, this costly technique may mask important cellular epitopes, alter therapeutic cell function[44] and require additional processing to remove the beads. Newly developed biodegradable beads may abrogate the need for such de-beading (MicroBeads, Miltenyi Biotec).
Figure 3.

Cell separation by elutriation and antibody-bead conjugate selection.
a. Elutriation begins with the introduction of a heterogeneous cell population into the elutriation chamber. i. Centrifugal force, applied to the cells by spinning the chamber, begins to separate cell populations based on density. ii. Buffer is introduced counter to the direction of centrifugal force. Counter flow buffer separates cells by size and density. iii. The separated cell populations can be sequentially removed in fractions to obtain enriched final cell populations. While certain cell types are typically found in specific fractions, a variety of factors influence which fractions contain target cell populations. b. i. Antibody-bead conjugate selection also begins with a heterogeneous cell population. ii. A monoclonal antibody-magnetic bead conjugate is applied to the cells, coating the target cell population. iii. The cells are then placed in a magnetic field as they flow through a semi-porous column. iv. Non-coated cells are separated as waste. v. The column can then be removed from the magnetic field and flushed to obtain an enriched cell population. This approach can be used to depletion undesirable cell populations as well, in which case the flow through cells would be retained.
Given the significant time and manipulation that early cell processing requires, automated, scalable and fully closed systems are extremely attractive for CAR-T cell manufacturing. Some have taken the “all-in-one” approach, such as with Miltenyi’s CliniMACS Prodigy, a GMP-compliant, cell manufacturing system that allows cell processing, gene modification and expansion in an automated, closed system. This likely represents the future of small-batch therapeutic T cell manufacturing and is directly applicable to CAR-T production. Alternatively, advanced modular components such as the gas-permeable rapid expansion cultureware (G-Rex, Wilson Wolf) allow for highly-scalable and custom fit manufacturing[45]. These new technologies will enable even more rapid and agile development of CART cell therapy.
Validation of CAR-T cell processes
Proper validation is essential to successful production of CAR-T cells. Furthermore, documented validation is a regulatory requirement of GMP production per the Title 21 Code of Federal Regulations. Validation is necessary to ensure that the manufacturing facility is capable of consistently providing a quality product. Recently, the FDA offered updated guidance for conducting proper validation[46]. In this guidance, the FDA recommended that validation occur in three stages: (1) process design, (2) process qualification and (3) continued process verification. Both qualification and verification may force re-design of a given process. Through careful design and proper evaluation, a process validation should produce scientific evidence that demonstrates consistent production of a quality product.
CAR-T cell manufacturing facilities must validate any process, policy or procedure that has potential to impact the quality of the product. In practice, CAR-T cell process validation can present unique challenges in each stage of validation. During process design, understanding and control of the process must be established. For most CAR-T cell applications, this involves translation of bench-developed protocols to clinical grade manufacturing. Even at this early stage, GMP requirements may limit or even entirely prevent translation of a given CAR-T cell protocol, as some research grade reagents do not have functional GMP-grade alternatives. In addition, autologous cell products often display a high degree of variation. This presents challenges in choosing appropriate controls. Process control via operational limits becomes crucial in this setting.
The second stage of validation, process qualification, requires design and qualification of infrastructure and equipment. Installation and operation qualification of utilities and equipment must occur prior to obtaining data for validation studies. Manufacture of CAR-T cells predominantly relies on adapted legacy instruments. Cell therapy specific instrumentation is a small but growing market and is currently not widely available. Therefore, when selecting equipment to implement new cell therapy applications, careful consideration must be given to the proper approach to qualification. Acceptable performance characteristics may be determined through a combination of pre-clinical studies, engineering runs, production of similar but non-CAR-T cell products and published data from other groups. Ultimately, the equipment and global process must be qualified to manufacture a product to expected specifications.
The final stage of validation, continued process verification, also must be implemented when validating CAR-T cell manufacture. As indicated, this is a continual monitoring to ensure that the process continues to perform as initially validated. Obviously, many factors may drive production out of specification. As such, a system must be implemented to collect and analyze process data over time. As noted above, significant variation between individual products makes this stage challenging for CAR-T cell manufacture. Rigorous and regular statistical analysis can aid in identifying variation beyond that which is expected.
Despite the unique challenges posed by CAR-T cell production, well controlled, properly validated manufacturing protocols can and have been implemented[41, 47, 48]. As we accumulate more clinical data, new or modified metrics may be incorporated into validation studies. For instance, to date there is no standard determination of CAR-T cell potency. FDA draft guidance acknowledges that cell therapy complexity may make it impossible to meet the requirements for demonstration of potency of licensed biological products[49]. Nonetheless, the
FDA recommends submission of available data to demonstrate potency[49]. This is of particular concern given that Kunkele et al. demonstrated discordance between in vitro and in vivo antitumor effect in both chromium release and bioluminescence cytotoxicity assays[50]. These data suggest the need for a more predictive assay of effector function. Some have suggested that molecular assays may provide accurate cell product functional data[51]. If developed, such an assay could become an accepted standard assay in process performance validation of CAR-T cells.
CAR-T Transduction
The effectiveness of CAR-T cells for cancer immunotherapy is dependent on the safe and efficient delivery of novel antigen-specificity into substantial population of a patient’s immune effector cells. Following apheresis collection of autologous T lymphocytes, it is necessary to functionalize these cells through the introduction of exogenous genetic elements. This can be accomplished through a variety of approaches, each with its own strengths and limitations that should be taken into consideration when choosing a particular gene delivery mechanism: ease of production, efficiency of gene delivery to target cells, danger of host genome mutagenesis and subsequent transformation, vector immunogenicity, and duration of transgene expression.
Most gammaretroviral, lentiviral, and adenoviral vectors that have been engineered for gene delivery possess an ability to transduce many different types of cells with wide tissue trophism with high efficiency. Stable transgene expression and persistence of re-directed T cells in vivo has been observed to be associated with durable responses[52, 53] which in general requires integration of the genetic element into the host genome. As such, gammaretroviral and lentiviral gene delivery vectors have become the current gold-standard for modifying T-cells for immunotherapy. However, genomic integration represents a potential for malignant transformation of transduced cells that has been clinically observed in the case of gammaretroviruses[54], remains a possibility with lentiviral vectors, and to a much lesser degree, DNA plasmid transfections. In contrast, adenoviral vectors and mRNA transfection are both highly efficient non-integrating means of gene delivery but are limited by transient gene expression that would require repeat dosing in many applications. Viral vector-related immunogenicity can present as pre-existing neutralizing antibodies that may sequester viral particles and hinder viral infection of target cells as well as can predispose a patient to severe allergic-type reactions that precludes re-exposure to those means of delivery.
From a manufacturing perspective, in order for gene delivery to become a common procedure, it will be necessary to develop highly reliable, safe, and reproducible means of generating large amounts of deliverable gene product. Moreover, the efficiency with which a given delivery method is capable of entering the relevant cell population is of utmost importance as this can be the key step when determining the rapidity or even feasibility of downstream cell production. Optimal processing should maximize both the therapeutic dose and in vivo persistence [55]. While a variety of strategies have been tried, head-to-head comparison of manufacturing techniques with in vivo efficacy remain to be determined. To this end, there are numerous academic and biotechnology companies that are dedicated to the development and manufacture of both GMP-compliant vectors for gene transfer and modified T-lymphocytes for transfusion.
Patient Outcomes
Early clinical trials utilizing CD19-targeting CAR-T cells in first chronic lymphoid leukemia and then acute lymphoid leukemia showed remarkably promising results in a small number of patients with advanced, refractory disease[15, 48] and spurred further clinical trials and the continued development of the technology. A recent meta-analysis of clinical trials from 1991 through 2014 using CD19-targeting CAR-T cells in refractory B-cell malignancies encompassed fourteen Phase I clinical trials, 119 patients, including a variety of different gene delivery mechanisms, cellular infusion doses, and treatment protocols[56]. They reported an overall pooled response rate of 73% and observed that lymphodepletion, higher T-cell infusion dose, sustained modified T-cell persistence in vivo greater than 2 months, and lack of concurrent IL-2 treatment were all associated with better prognosis in these early trials. These findings underscored the importance of developing robust protocols for generating sufficient numbers of modified T-cells and the need for certain adjuvant procedures during the infusion process that may help the cells remain active and engraft better.
The most serious and frequently observed toxicity of CAR-T cells directed to CD19 is cytokine release syndrome (CRS), an inflammatory process characterized by marked elevation in cytokine levels with presentation ranging from flu-like symptoms to systemic shock and multi-organ failure, is the most common and potentially serious toxicity[57]. More mild symptoms typically present within 4 days of infusions with severe reactions presenting significantly earlier, usually within one day of infusion. Severity has also been correlated with disease-burden and, to a lesser degree, efficacy of treatment and is usually associated with marked elevation of the cytokine interleukin-6 (IL-6). Management is primarily supportive although IL-6 blockade with tocilizumab has been associated with rapid defervescence and stabilization of blood pressure within 1–3 days[58]. Additionally, neurological toxicities have been reported with T-cell targeting therapies and have been observed with CAR-T applications. Global encephalopathy is most frequently observed in addition to delirium, most frequently in the context of high fever. The effects have so far been transient and self-resolving over several days without long-term effects. Finally, in the context of using CAR-T cells targeting CD19, a universal B-cell marker, there is significant on-target, off-tumor toxicity that presents as immunodeficiency secondary to B-cell aplasia and was observed in 100% of successfully treated patients. This effect is persistent for as long as CAR-modified T cells remain present and requires immunoglobulin replacement therapy for prevention of infectious complications[58].
Relapsed disease following CAR-T cell therapy can be broadly divided into antigen-positive and antigen-negative disease. Antigen-positive disease is usually seen in the context of rapid loss or engraftment failure of re-directed T-cells. Alternatively, the modified T-cells could have decreased functional activity secondary to immunosuppression by the tumor microenvironment. Efforts are already underway to develop next generation CAR-T cells through further receptor domain engineering[59, 60] and continued optimization of the manufacturing process[61] to facilitate prolonged in vivo persistence of the infused cells. Conversely, antigen-negative disease occurs when the tumor has been able to down-regulate or eliminate expression of the target marker, thus evading immune-surveillance. This necessitates the validation of additional tumor markers with concurrent development of high-affinity receptors for targeting those epitopes of interest, such as the development of CAR-T cells targeting CD22, another B-cell antigen[62]. These could be used in combination with existing CAR-T therapies to prevent the development of relapsed disease or following relapse to target escaped tumor cells. Ultimately, the development of new chimeric antigen receptors targeting additional antigens will be critical not only for treatment of relapsed disease, but also for expanding the number of indications for which CAR-T therapy is an option.
While it is known that CAR-T cells need to persist for at least several weeks for efficacy, the optimal duration of persistence is not yet known. It may be desirable to eliminate CAR-T cells under various different circumstances, such as unacceptable on-target toxicity or off-target toxicity. One example of an on-target toxicity is when carbonic anhydrase IX targeting (CAIX) CAR-T cells were used in patients with renal cell carcinoma, but the patients also developed significant liver toxicity due to low level expression of CAIX[63]; fortunately, the CAR T cells in this instance did not persist for very long and the patients recovered. Rapid off-target toxicity may also occur, as was observed with TCR-redirected cells targeting MAGE-A3[64] but cross-reacting with cardiac muscle. Efforts to develop CAR-T cells that can either be “turned off” with activation of a “suicide gene”[65] or “turned on” through conditional dimerization[66, 67] are underway. Although these systems have obvious use in the setting of unacceptable toxicity, it remains to be determined when the “trigger” should be used in the setting of more acceptable but longer-lasting effects, such as B cell aplasia.
Future Outlook
CAR-T cell therapy is poised to become widely available and utilized, but is still in its early stages of dissemination and transfer to industry-scale manufacturing. Being able to efficiently expand the population of targeted cells to a therapeutic dose that is ready to be released to the patient in a timely manner remains a significant challenge. Large-scale manufacturing, distribution, and delivery are will require a robust and scalable infrastructure, both within industry and at health care delivery sites.
Highlights.
Design and pre-clinical testing of CAR-T cells.
Manufacturing processes and requirements for CAR T cells.
Clinical outcomes of CAR-T cells in patients with cancer.
Acknowledgments
MVM was supported by NCI K08 CA166039.
ADF is supported by National Institutes of Health grant 5T32HL007775.
Footnotes
Conflict of interest
MVM and DLS are inventors on issued patents and/or patent applications related to the design, manufacture, and use of CAR T cells; patents are held by the University of Pennsylvania and/or Novartis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Thomas L. On immunosurveillance in human cancer. Yale J Biol Med. 1982;55:329–333. [PMC free article] [PubMed] [Google Scholar]
- 3.Oble DA, Loewe R, Yu P, Mihm MC., Jr Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun. 2009;9:3. [PMC free article] [PubMed] [Google Scholar]
- 4.Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, Storb R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300:1068–1073. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 5.Dutcher J. High-dose interleukin-2 in metastatic disease: renal cell carcinoma and melanoma. Oncology (Williston Park) 2002;16:3. [PubMed] [Google Scholar]
- 6.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 7.Grillo-Lopez AJ, White CA, Varns C, Shen D, Wei A, McClure A, Dallaire BK. Overview of the clinical development of rituximab: first monoclonal antibody approved for the treatment of lymphoma. Semin Oncol. 1999;26:66–73. [PubMed] [Google Scholar]
- 8.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–513. [PubMed] [Google Scholar]
- 11.Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A. 1988;85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, Mitsuyasu RT, Bernstein WB, Aronson NE, Levine BL, Bushman FD, June CH. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra153. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, Lin AA, Pennathur-Das R, Hege KM. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 14.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter D, Rheingold S, Teachey D, Chew A, Hauck B, Wright J, Milone M, Levine B, June C. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strasser EF, Eckstein R. Optimization of leukocyte collection and monocyte isolation for dendritic cell culture. Transfus Med Rev. 2010;24:130–139. doi: 10.1016/j.tmrv.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Jones AL. The IBM Blood Cell Separator and Blood Cell Processor: a personal perspective. J Clin Apher. 1988;4:171–182. doi: 10.1002/jca.2920040408. [DOI] [PubMed] [Google Scholar]
- 18.Bug G, Anargyrou K, Tonn T, Bialleck H, Seifried E, Hoelzer D, Ottmann OG. Impact of leukapheresis on early death rate in adult acute myeloid leukemia presenting with hyperleukocytosis. Transfusion. 2007;47:1843–1850. doi: 10.1111/j.1537-2995.2007.01406.x. [DOI] [PubMed] [Google Scholar]
- 19.Kasner MT, Laury A, Kasner SE, Carroll M, Luger SM. Increased cerebral blood flow after leukapheresis for acute myelogenous leukemia. Am J Hematol. 2007;82:1110–1112. doi: 10.1002/ajh.21006. [DOI] [PubMed] [Google Scholar]
- 20.Pasquini MC, Zhu X. Current uses and outcomes of hematopoietic stem cell transplantation: 2014 CIBMTR Summary Slides [Google Scholar]
- 21.Cancelas JA, Padmanabhan A, Le T, Ambruso DR, Rugg N, Worsham DN, Pinkard SL, Graminske S, Buck J, Goldberg J, Bill J. Spectra Optia granulocyte apheresis collections result in higher collection efficiency of viable, functional neutrophils in a randomized, crossover, multicenter trial. Transfusion. 2014 doi: 10.1111/trf.12907. [DOI] [PubMed] [Google Scholar]
- 22.Brauninger S, Bialleck H, Thorausch K, Felt T, Seifried E, Bonig H. Allogeneic donor peripheral blood “stem cell” apheresis: prospective comparison of two apheresis systems. Transfusion. 2012;52:1137–1145. doi: 10.1111/j.1537-2995.2011.03414.x. [DOI] [PubMed] [Google Scholar]
- 23.Loaiza S, Haynes R, Bray E, Finn SA, Rezvani K, Apperley J, Davis J. Donor lymphocyte collections using the spectra Optia MNC version 5. Transfus Apher Sci. 2013;48:171. doi: 10.1016/j.transci.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Schulz M, Bialleck H, Thorausch K, Bug G, Dunzinger U, Seifried E, Bonig H. Unstimulated leukapheresis in patients and donors: comparison of two apheresis systems. Transfusion. 2014;54:1622–1629. doi: 10.1111/trf.12506. [DOI] [PubMed] [Google Scholar]
- 25.McFarland DC, Zhang C, Thomas HC, LRT Confounding effects of platelets on flow cytometric analysis and cell-sorting experiments using blood-derived cells. Cytometry A. 2006;69:86–94. doi: 10.1002/cyto.a.20207. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Pineres AJ, Hildesheim A, Williams M, Trivett M, Strobl S, Pinto LA. DNAse treatment following thawing of cryopreserved PBMC is a procedure suitable for lymphocyte functional studies. J Immunol Methods. 2006;313:209–213. doi: 10.1016/j.jim.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 27.Assmus B, Tonn T, Seeger FH, Yoon CH, Leistner D, Klotsche J, Schachinger V, Seifried E, Zeiher AM, Dimmeler S. Red blood cell contamination of the final cell product impairs the efficacy of autologous bone marrow mononuclear cell therapy. J Am Coll Cardiol. 2010;55:1385–1394. doi: 10.1016/j.jacc.2009.10.059. [DOI] [PubMed] [Google Scholar]
- 28.Bryn T, Yaqub S, Mahic M, Henjum K, Aandahl EM, Tasken K. LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+ T cells through a COX-2-PGE2-dependent mechanism. Int Immunol. 2008;20:235–245. doi: 10.1093/intimm/dxm134. [DOI] [PubMed] [Google Scholar]
- 29.Ino K, Ageitos AG, Singh RK, Talmadge JE. Activation-induced T cell apoptosis by monocytes from stem cell products. Int Immunopharmacol. 2001;1:1307–1319. doi: 10.1016/s1567-5769(01)00062-5. [DOI] [PubMed] [Google Scholar]
- 30.Ino K, Singh RK, Talmadge JE. Monocytes from mobilized stem cells inhibit T cell function. J Leukoc Biol. 1997;61:583–591. doi: 10.1002/jlb.61.5.583. [DOI] [PubMed] [Google Scholar]
- 31.Wegener C, Heber C, Min K. Novel cell washing device using spinning membrane filtration. Cytotherapy. 2013;15:S27–S27. [Google Scholar]
- 32.Ferrante A, Thong YH. Optimal conditions for simultaneous purification of mononuclear and polymorphonuclear leucocytes from human blood by the Hypaque-Ficoll method. J Immunol Methods. 1980;36:109–117. doi: 10.1016/0022-1759(80)90036-8. [DOI] [PubMed] [Google Scholar]
- 33.Fuss IJ, Kanof ME, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol. 2009;Chapter 7(Unit7.1) doi: 10.1002/0471142735.im0701s85. [DOI] [PubMed] [Google Scholar]
- 34.Thong YH, Currell JM, Rodwell RL. The rapid one-step gradient centrifugation procedure for simultaneous isolation of granulocytic and mononuclear leukocytes from human blood: biological, physical and chemical bases. Med Hypotheses. 1983;12:103–111. doi: 10.1016/0306-9877(83)90072-5. [DOI] [PubMed] [Google Scholar]
- 35.Aktas M, Radke TF, Strauer BE, Wernet P, Kogler G. Separation of adult bone marrow mononuclear cells using the automated closed separation system Sepax. Cytotherapy. 2008;10:203–211. doi: 10.1080/14653240701851324. [DOI] [PubMed] [Google Scholar]
- 36.Janssen WE, Ribickas A, Meyer LV, Smilee RC. Large-scale Ficoll gradient separations using a commercially available, effectively closed, system. Cytotherapy. 2010;12:418–424. doi: 10.3109/14653240903479663. [DOI] [PubMed] [Google Scholar]
- 37.Berger TG, Strasser E, Smith R, Carste C, Schuler-Thurner B, Kaempgen E, Schuler G. Efficient elutriation of monocytes within a closed system (Elutra) for clinical-scale generation of dendritic cells. J Immunol Methods. 2005;298:61–72. doi: 10.1016/j.jim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Kim S, Kim HO, Baek EJ, Choi Y, Kim HS, Lee MG. Monocyte enrichment from leukapheresis products by using the Elutra cell separator. Transfusion. 2007;47:2290–2296. doi: 10.1111/j.1537-2995.2007.01470.x. [DOI] [PubMed] [Google Scholar]
- 39.Powell DJ, Jr, Brennan AL, Zheng Z, Huynh H, Cotte J, Levine BL. Efficient clinical-scale enrichment of lymphocytes for use in adoptive immunotherapy using a modified counterflow centrifugal elutriation program. Cytotherapy. 2009;11:923–935. doi: 10.3109/14653240903188921. [DOI] [PubMed] [Google Scholar]
- 40.Stroncek DF, Fellowes V, Pham C, Khuu H, Fowler DH, Wood LV, Sabatino M. Counter-flow elutriation of clinical peripheral blood mononuclear cell concentrates for the production of dendritic and T cell therapies. J Transl Med. 2014;12:241. doi: 10.1186/s12967-014-0241-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, June CH. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–5930. [PubMed] [Google Scholar]
- 44.Elkord E, Williams PE, Kynaston H, Rowbottom AW. Human monocyte isolation methods influence cytokine production from in vitro generated dendritic cells. Immunology. 2005;114:204–212. doi: 10.1111/j.1365-2567.2004.02076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vera JF, Brenner LJ, Gerdemann U, Ngo MC, Sili U, Liu H, Wilson J, Dotti G, Heslop HE, Leen AM, Rooney CM. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex) J Immunother. 2010;33:305–315. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.F.a.D.A.C.f.B.E.a. Research. Guidance for Industry: Process Validation: General Principles and Practices. 2011. [Google Scholar]
- 47.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.F.a.D.A.C.f.B.E.a. Research. Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. 2008. [Google Scholar]
- 50.Kunkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, Jensen MC. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res. 2015;3:368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 51.Stroncek DF, Jin P, Wang E, Jett B. Potency analysis of cellular therapies: the emerging role of molecular assays. J Transl Med. 2007;5:24. doi: 10.1186/1479-5876-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, Brentjens R. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 55.Levine BL. T lymphocyte engineering ex vivo for cancer and infectious disease. Expert Opin Biol Ther. 2008;8:475–489. doi: 10.1517/14712598.8.4.475. [DOI] [PubMed] [Google Scholar]
- 56.Zhu Y, Tan Y, Ou R, Zhong Q, Zheng L, Du Y, Zhang Q, Huang J. Anti-CD19 chimeric antigen receptor-modified T cells for B cell malignancies: A systematic review of efficacy and safety in clinical trials. Eur J Haematol. 2016;96:389–396. doi: 10.1111/ejh.12602. [DOI] [PubMed] [Google Scholar]
- 57.Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campana D, Schwarz H, Imai C. 4-1BB chimeric antigen receptors. Cancer J. 2014;20:134–140. doi: 10.1097/PPO.0000000000000028. [DOI] [PubMed] [Google Scholar]
- 60.Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J. 2014;20:127–133. doi: 10.1097/PPO.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barrett DM, Singh N, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy. 2014;16:619–630. doi: 10.1016/j.jcyt.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, Wayne AS, Mackall CL, Orentas RJ. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R, Gratama JW. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH. Cardiovascular toxicity and titin cross-reactivity of affinity enhanced T cells in myeloma and melanoma. Blood. 2013 doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, Duchateau P. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep. 2016;6:18950. doi: 10.1038/srep18950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487:64–69. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]