

Abstract
Enterohemorrhagic E.coli (EHEC) is an important foodborne pathogen in the developed world and can cause life-threatening disease particularly in children. EHEC persists in the human gut by adhering intimately to colonic epithelium and forming characteristic attaching/effacing lesions. In this study, we investigated the innate immune response to EHEC infection with particular focus on antimicrobial peptide and protein expression by colonic epithelium. Using a novel human colonic biopsy model and polarized T84 colon carcinoma cells, we found that EHEC infection induced expression of human β-defensin 2 (hBD2), whereas hBD1, hBD3, LL-37, and lysozyme remained unchanged. Infection with specific EHEC deletion mutants demonstrated that this was dependent on flagellin, and apical exposure to purified flagellin was sufficient to stimulate hBD2 and also interleukin (IL)-8 expression ex vivo and in vitro. Flagellin-mediated hBD2 induction was significantly reduced by inhibitors of NF-κB, MAP kinase p38 and JNK but not ERK1/2. Interestingly, IL-8 secretion by polarized T84 cells was vectorial depending on the side of stimulation, and apical exposure to EHEC or flagellin resulted in apical IL-8 release. Our results demonstrate that EHEC only induces a modest immune response in human colonic epithelium characterized by flagellin-dependent induction of hBD2 and low levels of IL-8.
Keywords: EHEC, flagellin, colon, β-defensin, interleukin-8, inflammation
Introduction
Enterohemorrhagic E. coli (EHEC) is a foodborne pathogen of worldwide importance (Croxen et al., 2013). Although infections with EHEC are rare compared with Campylobacter and Salmonella, they can lead to severe systemic hemolytic uremic syndrome (HUS) resulting in kidney failure and death (Tarr et al., 2005). EHEC mainly affects young children and the elderly, and the predominant serotype in most parts of the world is O157:H7 (Croxen and Finlay, 2010).
EHEC causes diarrhea by adhering to human intestinal epithelium, particularly the colon, and forming attaching and effacing (A/E) lesions (Golan et al., 2011; Lewis et al., 2015). These are characterized by intimate bacterial attachment to the host cell membrane and effacement of underlying microvilli. A/E lesion formation is dependent on a bacterial type III secretion system (T3SS) which enables EHEC to inject effector proteins into the host cell (Jarvis and Kaper, 1996). Around 50 EHEC effector proteins have been identified so far (Tobe et al., 2006), affecting many different host cell functions such as maintenance of epithelial barrier integrity, water and ion transport, and immune response to infection (Viswanathan et al., 2009). In addition to the T3SS, EHEC also produces Shiga toxins (Stxs) which are linked to HUS and strongly cytotoxic to kidney cells (Obrig and Karpman, 2012). After release into the gut lumen, Stxs transverse the intestinal epithelium and are transported to the kidneys via the bloodstream (Schüller, 2011).
EHEC infection in the human gut is accompanied by neutrophil recruitment to the intestinal mucosa (Griffin et al., 1990; Kelly et al., 1990), and high levels of the neutrophil chemoattractant interleukin (IL)-8 have been detected in HUS patients (Fitzpatrick et al., 1992; Murata et al., 1998) indicating a key role of this cytokine in the innate immune response to EHEC infection. In addition to cytokine production which recruits phagocytes to the site of infection, secretion of antimicrobial peptides and proteins (AMPs) such as defensins, cathelicidins, and lysozyme by intestinal epithelium constitutes an important part in the innate immune defense against intestinal pathogens. Pore-forming AMPs directly kill bacteria and also promote an inflammatory immune response by acting as chemoattractants (Wassing et al., 2014). Human colonic epithelium, the primary target site of EHEC, produces lysozyme and LL-37, which is the only cathelicidin in humans and expressed by upper crypt epithelial cells. In addition, four types of defensins are secreted by human colonic epithelium: While human β-defensin (hBD)1 is constitutively expressed, hBD2-4 are induced by infection and inflammation (Eckmann and Kagnoff, 2005; Muniz et al., 2012).
Previous studies have demonstrated that infection with intestinal pathogens can modulate expression of AMPs and cytokines by the host epithelium, thereby resulting in an induced or suppressed immune response (Islam et al., 2001; Zilbauer et al., 2005; Sharma et al., 2006; Sperandio et al., 2008). In this study, we have investigated the influence of EHEC infection on AMP and IL-8 expression in physiologically relevant colonic biopsy and polarized cell culture models.
Materials and methods
Bacterial strains and flagellin
Bacterial strains used in this study are listed in Table 1. Bacteria were grown standing in LB broth overnight at 37°C. Deletion mutants were selected with kanamycin (50 μg/ml) or chloramphenicol (30 μg/ml). Bacteria were spun down before infection and suspended in serum-free culture medium. LPS-free purified H7 monomeric flagellin was kindly provided by David Gally, Roslin Institute, Edinburgh.
Table 1.
E. coli strains used in this study.
| Name | Description | Source or references |
|---|---|---|
| EDL933 | Wild-type EHEC O157:H7 | Riley et al., 1983 |
| EDL933 ΔescN | EDL933 escN deletion mutant | Jarvis and Kaper, 1996 |
| EDL933 ΔfliC | EDL933 fliC deletion mutant | Gobert et al., 2008 |
| EDL933 Δstx | EDL933 stx1 stx2 deletion mutant | Gobert et al., 2007 |
| 85–170 | Stx-negative EHEC O157:H7 | Tzipori et al., 1987 |
| 85–170 ΔhcpA | 85–170 hcpA deletion mutant | Xicohtencatl-Cortes et al., 2007 |
| 85–170 ΔlpfA1 | 85–170 lpfA1 deletion mutant | Fitzhenry et al., 2006 |
| Walla-1 | EHEC O157:H7 | Ostroff et al., 1990 |
| H0-7184-0336 | EHEC O157:H7 | G. Smith, Public Health England |
Cell culture and infection
Human colon carcinoma T84 cells (ATCC CCL-248) were cultured in DMEM/F-12 mixture supplemented with 10% fetal bovine serum (Sigma) and used between passage 45 and 65. To obtain polarized monolayers, cells were seeded out on collagen-coated Transwell filter inserts (12 mm diameter, 0.4 μm pore; Corning Costar) at a density of 5 × 105 cells/insert. Transepithelial electrical resistance (TER) was monitored using an EVOM2 resistance meter with an STX2 electrode (World Precision Instruments), and values above 1500 Ω × cm2 after 7–10 d of differentiation indicated establishment of epithelial barrier function. Polarized T84 cells were infected with approximately 6 × 107 bacteria in plain DMEM/F-12 medium and incubated for 3 h. After that, medium was exchanged every hour to prevent bacterial overgrowth and acidification of the medium. For 24 h infections, gentamicin (200 μg/ml, Sigma) was added 1 h after infection to slow down bacterial growth, and incubations were continued without medium exchange for up to 24 h. For incubations with flagellin and IL-1β (Sigma), no medium exchange was performed. For signal transduction studies, cells were incubated with the chemical inhibitors quinazoline (NF-κB) (28 μM, Sigma), SB203580 (p38), SP600125 (JNK), or PD98059 (ERK 1/2) (25 μM, Merck Millipore) for 1 h before flagellin (0.1 μg/ml) was added for 6 h. Cells were incubated at 37°C in a 5% CO2 atmosphere. At the end of the experiment, cells were washed twice in PBS to remove non-adherent bacteria and processed according to further analysis.
Polarized In vitro organ culture (pIVOC)
This study was performed with approval from the University of East Anglia Faculty of Medicine and Health Ethics Committee (ref 2010/11-030). All samples were provided through the Norwich Biorepository, which has NRES approval (ref 08/h0304/85+5). Up to four biopsy samples from the transverse colon were obtained with informed consent during colonoscopy of 23 adult patients (24–77 years old) during routine investigation of potential intestinal disorders. Samples were taken from macroscopically normal areas, transported to the laboratory in IVOC medium and processed within the next hour. Polarized IVOC was performed as described previously (Schüller et al., 2009). Briefly, biopsies were mounted mucosal side upwards on a circular cellulose nitrate filter and sandwiched between two Perspex disks with a 2 mm central aperture (manufactured by the School of Environmental Sciences workshop, University of East Anglia). To prevent bacterial leakage, the apical disk was sealed to the mucosal side of the biopsy with Histoacryl tissue glue (Braun Medical). The sandwich holding the biopsy was then mounted in a Snapwell support (Corning Costar) and inserted in a six-well culture plate. Apical and basal compartments were filled with IVOC medium, and 20 μl of bacterial overnight culture (~107 bacteria), purified monomeric H7 flagellin or IL-1β were added apically. A specimen inoculated with medium only was included with each experiment to exclude in vivo bacterial colonization. Samples were incubated in a 5% CO2 atmosphere at 37°C on a rocking platform for 7 h. Bacterial leakage into the basolateral compartment was assessed by bacterial growth and turbidity of the medium, and respective samples were excluded from analysis. At the end of the experiment, biopsies were removed from the Snapwell support, washed in PBS to remove mucus and non-adherent bacteria and processed for further analysis.
RNA isolation and quantitative real-time PCR (qPCR) analysis
Total RNA from cells and biopsies was isolated using the RNeasy Mini kit with on-column DNase digestion (Qiagen) according to the manufacturer's instructions. Biopsy tissue was homogenized with a pestle (Kimble Chase) before extraction. RNA quality was assessed by gel electrophoresis and OD260∕280 determination. RNA was quantified using a Nanodrop ND-1000 spectrophotometer, and 1 μg RNA was converted to cDNA using the qScript cDNA supermix (Quanta Biosciences) in a 20 μl reaction. Quantitative real-time PCR was performed using an ABI 7500 PCR system (Applied Biosystems). Primers were purchased from Sigma-Genosys. Gene-specific sequences (Table 2) were obtained from published studies or designed using PrimerBLAST software (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Up to 4 μl cDNA were amplified in a 10 μl reaction containing 0.5 μM of each primer and 5 μl of 2 × SYBR Green JumpStart Taq ReadyMix (Sigma). Cycling parameters were as follows: 2 min at 95°C (initial denaturation); 30 s at 95°C, 30 s at 60°C, 35 s at 72°C (40 cycles); 5 min at 72°C (final elongation); 15 s at 95°C, 60 s at 60°C, 15 s at 95°C, 15 s at 60°C (dissociation for melt curve analysis). PCR product specificity was confirmed by melt curve analysis and agarose gel electrophoresis. Relative quantification of gene expression was performed using the comparative Ct method. Ct values for genes of interest were normalized using the geometric mean Ct of two housekeeper genes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and RNA polymerase II polypeptide A (POLR2A). Fold expression levels in treated samples were calculated relative to matched non-treated controls using the formula 2−ΔΔCt.
Table 2.
Primer sequences used in this study.
| Target | Primer sequence | References |
|---|---|---|
| hBD1 | F 5′-CTGCTGTTTACTCTCTGCTTACTTTT-3′ | Fahlgren et al., 2003 |
| R 5′-CCTCCACTGCTGACGCA-3′ | ||
| hBD2 | F 5′-CTCGTTCCTCTTCATATTCCTGA-3′ | Fahlgren et al., 2003 |
| R 5′-CTAGGGCAAAAGACTGGATGAC-3′ | ||
| hBD3 | F 5′-TGAAGCCTAGCAGCTATGAGGATC-3′ | Fahlgren et al., 2004 |
| R 5′-CCGCCTCTGACTCTGCAATAA-3′ | ||
| hBD4 | F 5′-CCCAGCATTATGCAGAGACTT-3′ | Fahlgren et al., 2004 |
| R 5′-ACCACATATTCTGTCCAATTCAAAT-3′ | ||
| IL-8 | F 5′-TTGAGAGTGGACCACACTGC-3′ | Ou et al., 2009 |
| R 5′-TGCACCCAGTTTTCCTTGG-3′ | ||
| GAPDH | F 5′-AGGTCGGAGTCAACGGATTT-3′ | Schüller et al., 2009 |
| R 5′-TGGAAGATGGTGATGGGATTT-3′ | ||
| LL-37 | F 5′-GTGCCCCAGGACGACACAGC-3′ | This study |
| R 5′-CCCCTGGCCTGGTTGAGGGT-3′ | ||
| Lysozyme | F 5′-AAAACCCCAGGAGCAGTTAAT-3′ | Fahlgren et al., 2003 |
| R 5′-CAACCCTCTTTGCACAAGCT-3′ | ||
| POLR2A | F 5′-GATGGGCAAAAGAGTGGACTT-3′ | Schüller et al., 2009 |
| R 5′-GGGTACTGACTGTTCCCCCT-3′ |
Forward (F) and reverse (R) primer sequences.
Scanning electron microscopy
Samples were fixed with 2.5% glutaraldehyde in PBS and dehydrated through graded acetone series. Specimens were dried using tetramethylsilane (Sigma), mounted on aluminum stubs, sputter-coated with gold (Polaron SC7640 sputter coater, Quorum Technologies), and viewed with a JEOL JSM 4900 LV scanning electron microscope.
Immunofluorescence staining
Biopsy samples were fixed in 3.7% formaldehyde in PBS for 30 min, cryoprotected in 15 and 30% sucrose in PBS for 10 min, embedded in OCT compound (Sakura), snap-frozen in a dry ice/ethanol bath and stored at −70°C until use. Serial sections of 7 μm were cut with a Microm HM550 cryostat (Thermo Scientific), picked up on poly L-lysine-coated slides and air-dried. Tissue sections were blocked with 0.5% BSA in PBS for 20 min. Cryosections were subsequently incubated in rabbit anti-hBD2 (abcam) overnight at 4°C, washed and incubated in Alexa Fluor 488-conjugated secondary antibody (Life Technologies) for 30 min. Cell nuclei were counterstained with DAPI (Roche). Samples were mounted in Vectashield (Vector Laboratories) and analyzed using a fluorescence light microscope (Axiovert 200 M, Zeiss).
IL-8 ELISA
Polarized T84 cells were lysed on ice in 1% Triton X-100 and 1μl/200 μl protease inhibitor cocktail (Sigma) in PBS, and Triton-insoluble proteins were removed by centrifugation. IL-8 concentrations in lysates and supernatants were determined using a human IL-8 ELISA kit (PeproTech) according to the manufacturer's instructions.
Statistics
Statistical analysis was performed using GraphPad Prism software (version 5). qPCR data were log transformed before analysis. For parametric T84 cell data, one-way ANOVA with Tukey's multiple comparisons test was used to determine differences between multiple groups. For non-parametric biopsy data, Wilcoxon's signed-rank test or Kruskall–Wallis with Dunn's multiple comparisons test was used to determine differences between two or multiple groups, respectively. A P < 0.05 was considered significant.
Results
Apical EHEC infection of polarized T84 cells induces hBD2 and IL-8 expression
To determine the influence of EHEC infection on AMP and IL-8 expression by human colonic epithelium, polarized T84 human colon carcinoma cells were infected with EHEC O157:H7 strains EDL933, Walla-1, and H0-7184-0336 (H07184) on the apical side. Bacterial overgrowth and loss of epithelial integrity was prevented by removal of bacteria after 3 h and subsequent medium exchange at hourly intervals. As shown in Figure 1A, this protocol resulted in maintenance of epithelial barrier function for up to 9 h as determined by TER. Expression of hBD1-4, LL-37, lysozyme, and IL-8 was determined by qPCR analysis. Results in Figure 1B show that apical EHEC infection resulted in a significant induction of hBD2 (21.5 ± 9.1 fold for EDL933, 70.4 ± 21.8 fold for Walla-1, and 31.5 ± 2.0 fold for H07184) and IL-8 (6.0 ± 1.6 fold for EDL933, 8.3 ± 2.4 fold for Walla-1, and 8.2 ± 3.9 fold for H07184) mRNA expression compared with non-infected (NI) controls whereas no significant change was observed for hBD1, hBD3, LL-37, and lysozyme. No specific amplification product was detected for hBD4. Subsequent kinetic analysis of hBD2 and IL-8 mRNA expression indicated that levels of both transcripts were first significantly induced at 6 h post-infection and showed a continuous increase during a 12 h period of infection (Supplementary Figure 1). For practical reasons, infections of polarized T84 cells were performed for 9 h in subsequent experiments.
Figure 1.
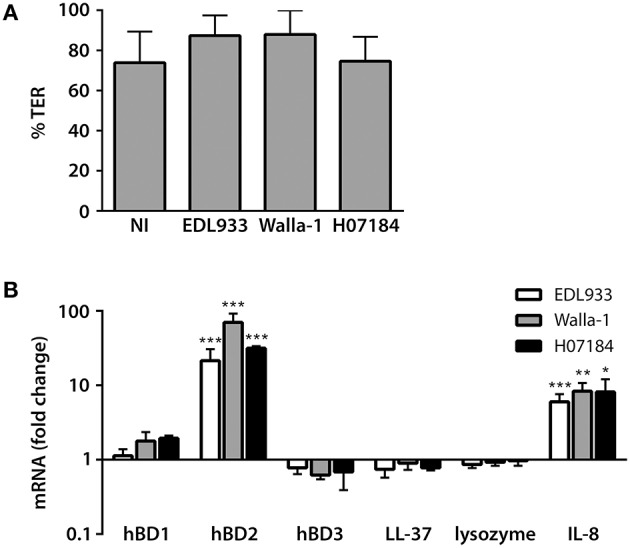
EHEC infection induces hBD2 and IL-8 expression in polarized T84 cells. Polarized T84 cells were apically infected with EHEC strains EDL933, Walla-1, H07184, or left non-infected (NI) for 9 h. (A) EHEC infection did not compromise epithelial barrier function. TER after infection is expressed as percentage of TER before infection. (B) AMP and IL-8 mRNA levels were quantified by qPCR and expressed as fold changes relative to NI controls. Data are shown as means ± standard errors of the means (SEM) of four independent experiments performed in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NI control.
Apical EHEC infection results in hBD2 and IL-8 induction in a polarized human colonic biopsy model
Our recent studies have demonstrated EHEC adherence to human colonic biopsy epithelium by forming typical A/E lesions (Lewis et al., 2015). To investigate the effect of EHEC infection on AMP and IL-8 expression ex vivo, a pIVOC model, which restricts bacterial access to the mucosal side of the biopsy, was developed. This was based on the pIVOC system established earlier to investigate the inflammatory response in duodenal biopsies (Schüller et al., 2009). Colonic biopsy samples were mounted in Snapwell supports, infected with EDL933, Walla-1, or H07184 on the mucosal side and incubated for up to 7 h. Tissue preservation and EHEC adherence were evaluated by scanning electron microscopy. As shown in Figure 2A, good tissue morphology was observed within the area of the central aperture (white circle) with intact surface epithelium visible at higher magnifications (Figure 2B). In addition, EHEC adhered intimately to colonic epithelium as described previously (Figure 2C; Lewis et al., 2015). Analysis of AMP and IL-8 expression by qPCR demonstrated a significant increase in hBD2 (median fold increase = 8.1 for EDL933 and 5.7 for H07184) and IL-8 expression (median fold increase = 6.6 for EDL933, 5.2 for Walla-1, and 4.8 for H07184) in EHEC-infected samples compared with non-infected controls. In contrast, transcript levels of hBD1, LL-37, and lysozyme were not significantly affected by EHEC infection (Figure 2D). Amplification levels for hBD3 and hBD4 were generally below detection threshold levels. However, amplification products of corresponding sizes were detected in some experiments, thereby confirming primer specificity and functionality of the qPCR assay (data not shown).
Figure 2.

EHEC induction of hBD2 and IL-8 in a polarized human colonic biopsy model. Human colonic biopsy samples were sandwiched between two Perspex disks with a central aperture and mounted in Snapwell supports as described in Experimental Procedures. Mucosal sides of matched biopsies were infected with EHEC strains EDL933, Walla-1, H07184, or left NI for 7 h. (A) Scanning electron micrograph of colonic biopsy after 7 h of incubation. The white circle indicates the central aperture of the disk. Bar = 200 μm. (B) Intact colonic surface epithelium at higher magnification. Bar = 50 μm. (C) EDL933 intimately adhering to colonic biopsy epithelium. Bar = 2 μm. Images are representative of three experiments performed in duplicate. (D) AMP and IL-8 mRNA levels were quantified by qPCR and expressed as fold changes relative to matched NI controls. Data points represent individual biopsies from a total of six patients. Medians are indicated by a line. *P < 0.05, **P < 0.01 vs. NI control.
EHEC induction of colonic epithelial hBD2 and IL-8 expression is dependent on flagellin
We next determined which EHEC proteins were involved in hBD2 and IL-8 induction in colonic epithelium. Polarized T84 cells were apically infected with wild-type (wt) EDL933 or isogenic deletion mutants in fliC (flagellin, main component of EHEC flagellum), escN (cytoplasmic ATPase of T3SS), or stx (Stxs) for 9 h. In addition, the contribution of long polar fimbriae (Lpf) and hemorrhagic coli pili (HCP) was examined by using isogenic deletion mutants in lpfA1 and hcp of EHEC strain 85–170. None of the strains significantly compromised epithelial barrier function as determined by TER (Supplementary Figure 2A). Expression levels of hBD2 and IL-8 were determined by qPCR. Whereas, infection with EDL933 wt, ΔescN, and Δstx showed a significant induction of hBD2 (48.6 ± 12.3 fold for wt, 60.5 ± 15.6 fold for ΔescN, and 41.5 ± 8.3 fold for Δstx) and IL-8 (5.6 ± 1.3 fold for wt, 7.5 ± 1.2 fold for ΔescN, and 4.9 ± 0.3 fold for Δstx) expression vs. NI controls, no significant effect was observed in EDL933 ΔfliC-infected cells (Figure 3A). For infections with 85–170, all strains significantly increased expression of hBD2 (195.9 ± 50.1 fold for wt, 168.6 ± 40.4 fold for Δhcp, and 163.4 ± 26.8 fold for ΔlpfA1) and IL-8 (84.3 ± 21.8 fold for wt, 157.5 ± 45.7 fold for Δhcp, and 88.4 ± 28.0 fold for ΔlpfA1) vs. NI controls (Figure 3B). Notably, induction levels of hBD2 and IL-8 expression by EHEC strain 85–170 were considerably higher compared with EDL933 (Figure 3).
Figure 3.

EHEC induction of hBD2 and IL-8 expression in polarized T84 cells is dependent on flagellin. Polarized T84 cells were apically infected with EHEC EDL933 wild-type (WT) and isogenic deletion mutants in fliC, escN, or stx (A) or EHEC 85–170 wild-type (WT) and isogenic deletion mutants in hcp or lpfA1 (B) for 9 h. IL-8 and hBD2 mRNA levels were quantified by qPCR and expressed as fold changes relative to NI controls. Data are shown as means ± SEM of three independent experiments performed in duplicate. ***P < 0.001 vs. NI control.
We also determined the effect of flagellin, T3S and Stxs on hBD2 and IL-8 expression in colonic pIVOC. As shown in Figure 4, EDL933 wt, ΔescN, and Δstx significantly enhanced hBD2 (median fold increase = 15.9 for wt, 14.3 for ΔescN, and 17.8 for Δstx) and IL-8 (median fold increase = 3.6 for wt, 3.1 for ΔescN, and 4.9 for Δstx) expression in colonic biopsies whereas no significant effect was observed for the ΔfliC mutant.
Figure 4.
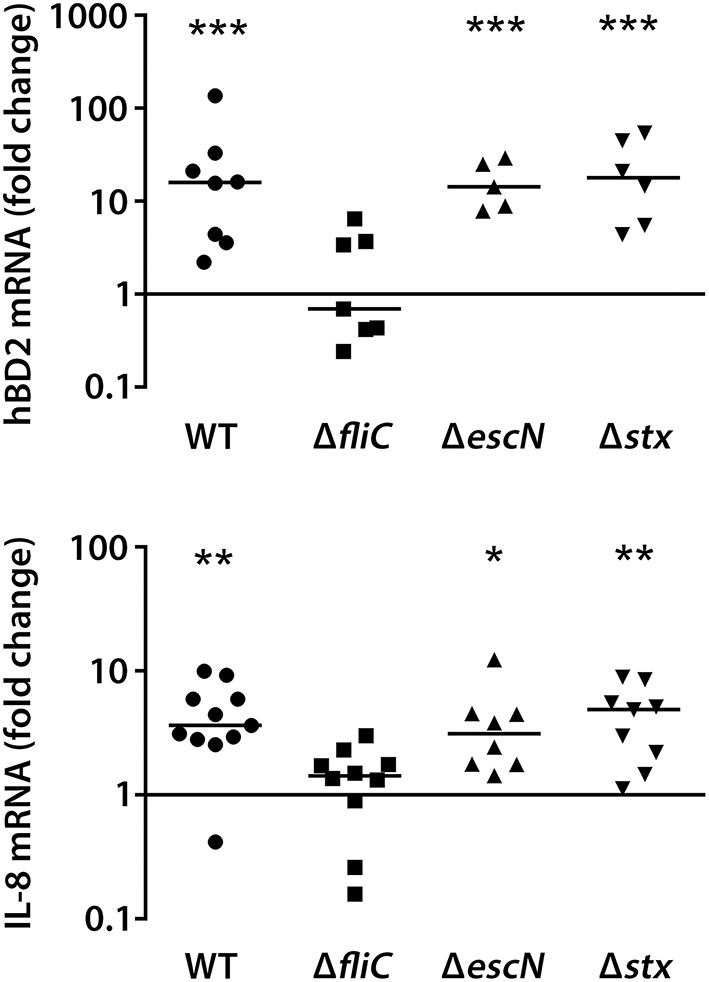
EHEC stimulation of IL-8 and hBD2 expression in colonic biopsies requires flagellin. Colonic biopsies were infected with EHEC EDL933 wild-type (WT), isogenic deletion mutants in fliC, escN, or stx or left NI on the mucosal side, and incubations were performed for 7 h. IL-8 and hBD2 mRNA levels were quantified by qPCR and expressed as fold changes relative to matched NI controls. Data points represent individual biopsies from a total of four patients. Medians are indicated by a line. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NI control.
Purified flagellin induces hBD2 and IL-8 expression in colonic epithelium
To examine whether EHEC flagellin alone was sufficient to increase hBD2 and IL-8 expression, polarized T84 cells were infected with EDL933 or incubated with different concentrations of purified LPS-free monomeric H7 flagellin on the apical side for 9 h. No significant effect on TER was observed under any of the conditions tested (Supplementary Figure 2A). As demonstrated in Figure 5A, a dose-dependent increase in hBD2 mRNA expression was observed after 9 h of incubation with similar induction levels for 1 μg/ml flagellin and infection with EDL933 (101.5 ± 24.0 fold for flagellin and 99.0 ± 23.0 fold for EDL933). In contrast, only very low induction levels were noted for IL-8 (Figure 5B, data shown for 0.1 μg/ml flagellin). Therefore, kinetic analyses were performed, and cells were apically exposed to flagellin for 1–9 h. Whereas, highest induction levels for hBD2 were observed after 6–9 h of flagellin exposure, IL-8 mRNA expression was induced much earlier and peaked at 1–3 h of exposure (Figure 5B). IL-8 induction was subsequently examined after 2 h of flagellin exposure, and a dose-dependent response was noted with induction levels for 1 μg/ml flagellin approximating those observed during infection with EDL933 (62.6 ± 8.9 fold for flagellin and 80.8 ± 10.5 fold for EDL933; Figure 5C).
Figure 5.
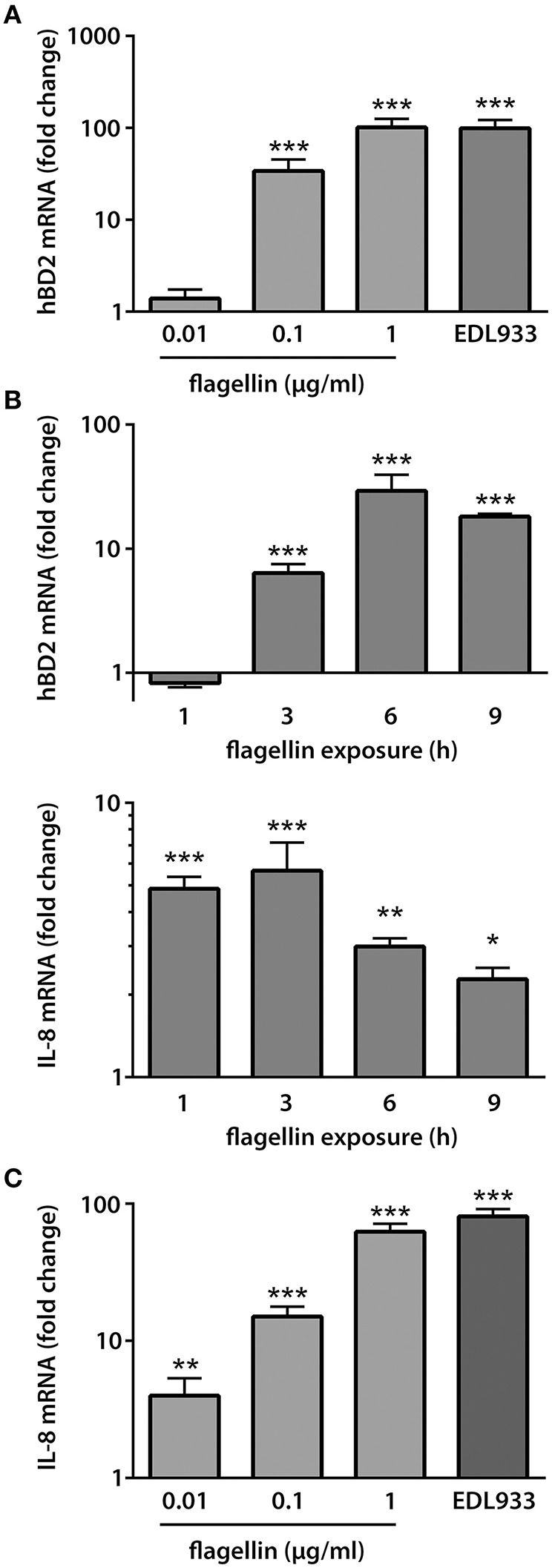
EHEC flagellin is sufficient to induce hBD2 and IL-8 expression in polarized T84 cells. Polarized T84 cells were incubated with EDL933, purified flagellin or left NI on the apical side. IL-8 and hBD2 mRNA levels were quantified by qPCR and expressed as fold changes relative to NI controls. (A) Purified flagellin induced a dose-dependent increase in hBD2 mRNA expression after 9 h. Data are shown as means ± SEM of three independent experiments performed in duplicate. (B) Kinetics of IL-8 and hBD2 induction by purified flagellin (0.1 μg/ml). Data are shown as means ± SEM of two independent experiments performed in duplicate. (C) Purified flagellin induced a dose-dependent increase in IL-8 mRNA expression after 2 h. Data are shown as means ± SEM of two independent experiments performed in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NI control.
Similar to findings in polarized T84 cells, purified flagellin was also sufficient to induce hBD2 (median fold increase = 5.3) and IL-8 (median fold increase = 2.2) expression in colonic pIVOC (Figure 6).
Figure 6.
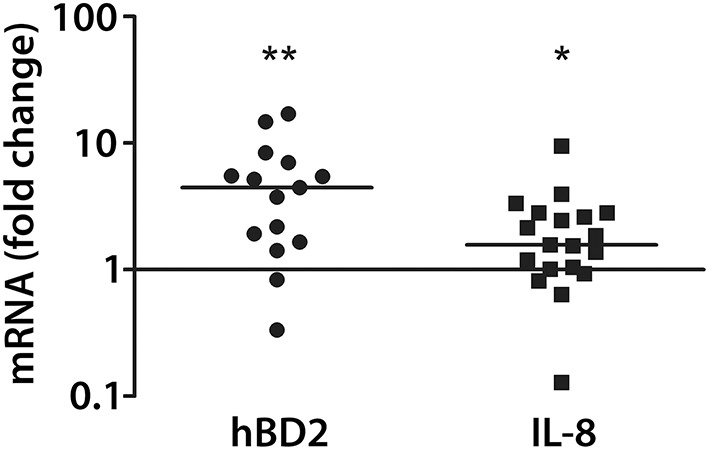
EHEC flagellin induces hBD2 and IL-8 expression in colonic biopsies. Colonic biopsies were incubated with purified flagellin (1 μg/ml) or left NI on the mucosal side for 7 h. IL-8 and hBD2 mRNA levels were quantified by qPCR and expressed as fold changes relative to matched NI controls. Data points represent individual biopsies from a total of four patients. Medians are indicated by a line. *P < 0.05, **P < 0.01 vs. NI control.
Flagellin-induced hBD2 expression is mediated by NF-κB, map kinases p38 and JNK
We further characterized the signaling pathways involved in flagellin-induced hBD2 and IL-8 expression. To this aim, polarized T84 cells were treated with specific chemical inhibitors before flagellin (0.1 μg/ml) was added. Gene expression was quantified by qPCR. None of the chemical inhibitors compromised epithelial barrier function (Figure 7A). While inhibition of NF-κB or the MAPKs p38 and JNK significantly reduced hBD2 gene expression, treatment with the ERK1/2 inhibitor did not have any significant effect (Figure 7B). For IL-8, all inhibitors reduced gene expression, but this did not reach significance (Figure 7B).
Figure 7.
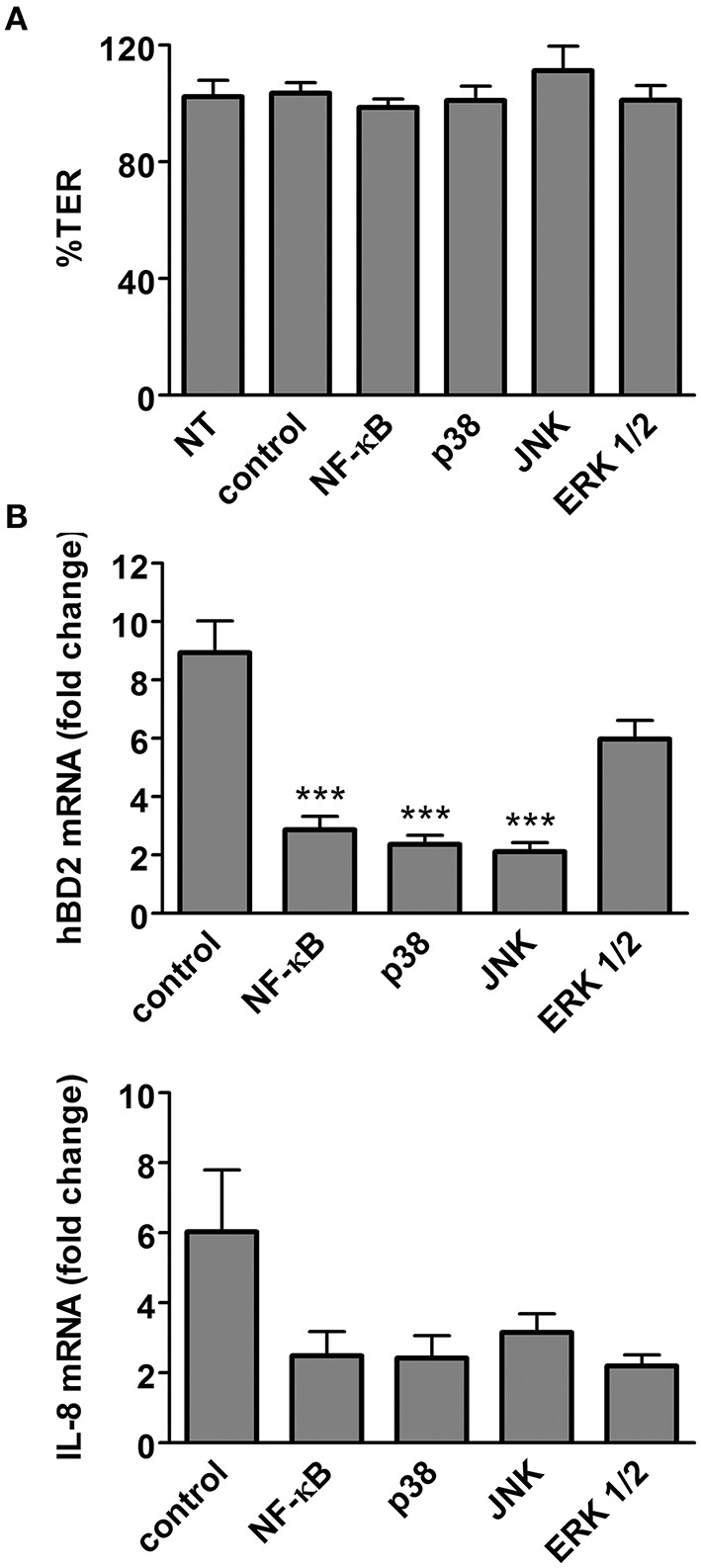
Flagellin-mediated hBD2 induction is dependent on NF-κB, MAPK p38, and JNK. Polarized T84 cells were incubated with specific inhibitors of NF-κB, p38, JNK, ERK1/2, or solvent (control) for 1 h before purified flagellin (0.1 μg/ml) was added for 6 h. Alternatively, T84 cells were left untreated (NT) for 7 h. (A) Influence of chemical inhibitors on epithelial barrier function. TER after treatment is expressed as percentage of TER before treatment. (B) hBD2 and IL-8 mRNA levels were quantified by qPCR and expressed as fold changes relative to NT samples. ***P < 0.001 vs. control. Data are shown as means ± SEM of three independent experiments performed in duplicate.
EHEC infection induces epithelial hBD2 peptide expression in colonic biopsies
Having shown that EHEC flagellin induced hBD2 mRNA expression, we investigated whether this also affected hBD2 peptide levels. Colonic biopsies were infected with EDL933 or incubated with purified flagellin (1 μg/ml) on the mucosal side, and hBD-2 peptide expression was assessed by immunofluorescence staining. As shown in Figure 8, elevated hBD2 peptide expression was observed in epithelial cells of colonic biopsies incubated with EDL933 or flagellin vs. NI controls.
Figure 8.
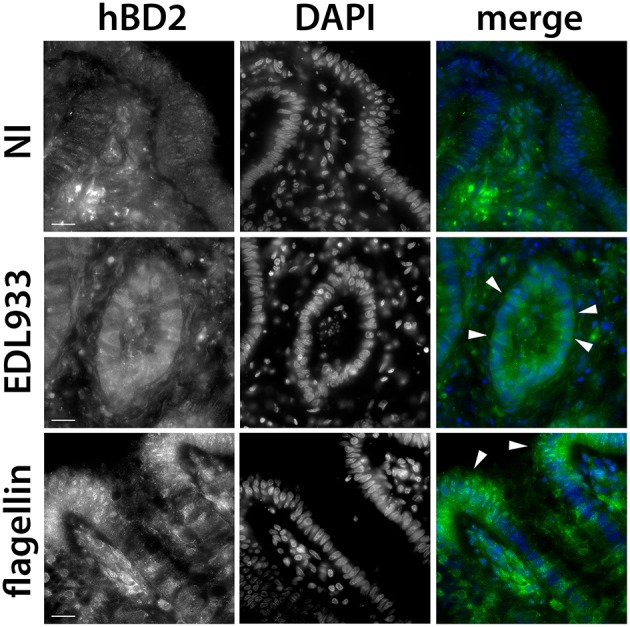
EHEC stimulates hBD2 peptide expression in colonic biopsy epithelium. Colonic biopsies were incubated with EDL933, purified flagellin (1 μg/ml) or left NI for 8 h. Cryosections from snap-frozen tissue were stained with anti-hBD2 (green), and cell nuclei were counterstained with DAPI (blue). Arrowheads show epithelial cells with induced hBD2 expression. Images are displayed as separate monochrome images of each fluorescence channel (hBD2, DAPI) and merged color images from both channels (merge). Shown are representative images from two independent experiments performed in duplicate. Bars = 10 μm.
EHEC-induced IL-8 protein secretion in polarized T84 cells is directional and depends on the side of exposure
We further investigated EHEC-induced IL-8 expression at the protein level. Polarized T84 cells were apically infected with EDL933 or incubated with the potent IL-8 inducer IL-1β (10 ng/ml) for 24 h. This time point was chosen according to previous IL-8 secretion studies on EHEC and enteropathogenic E. coli (EPEC) (Hauf and Chakraborty, 2003; Baruch et al., 2011; Sham et al., 2011). To prevent cell damage and breach of barrier function due to bacterial overgrowth and acidification, EHEC-infected monolayers were treated with gentamicin (200 μg/ml) after 1 h. This treatment did not result in complete killing, but reduced bacterial growth so that infections could be continued for up to 24 h. TER was not significantly affected by any of the treatments (Supplementary Figure 2B) indicating maintenance of epithelial barrier function. At the end of the incubation, IL-8 protein levels were determined in cell lysates and apical and basolateral supernatants. As shown in Figure 9A, apical EHEC infection significantly increased IL-8 protein levels in apical supernatants (240.9 ± 27.5 vs. 32.0 ± 6.2 pg/well in NI controls) and to a lesser extent in cell lysates (88.9 ± 21.5 vs. 16.6 ± 1.4 pg/well in NI controls) but not in basolateral media. Similarly, apical IL-1β exposure resulted in significantly enhanced amounts of IL-8 in apical supernatants (102.3 ± 14.9 vs. 32.0 ± 6.2 pg/well in NI controls) but not in cell lysates or basolateral media. We further extended these studies and investigated the influence of the side of exposure on the direction of IL-8 release into the media. To this aim, polarized T84 cells were incubated with EDL933 or purified flagellin (1 μg/ml) on the apical or basolateral side for 24 h, and IL-8 levels were evaluated in apical and basolateral supernatants. None of the treatments significantly affected the TER (Supplementary Figure 2B). As demonstrated in Figure 9B, apical exposure to EDL933 or flagellin led to a significant increase of IL-8 secretion into apical supernatants (569.5 ± 46.2 or 761.2 ± 8.5 vs. 108.2 ± 16.9 pg/well in NI controls, respectively) whereas IL-8 levels in basolateral media were not significantly affected. On the other hand, basolateral exposure of polarized T84 cells to EDL933 or flagellin significantly stimulated IL-8 protein release into basolateral compartments (506.6 ± 103.2 or 1119.0 ± 181.4 vs. 128.6 ± 8.1 pg/well in NI controls, respectively) whereas IL-8 levels in apical media were not significantly affected (Figure 9B).
Figure 9.
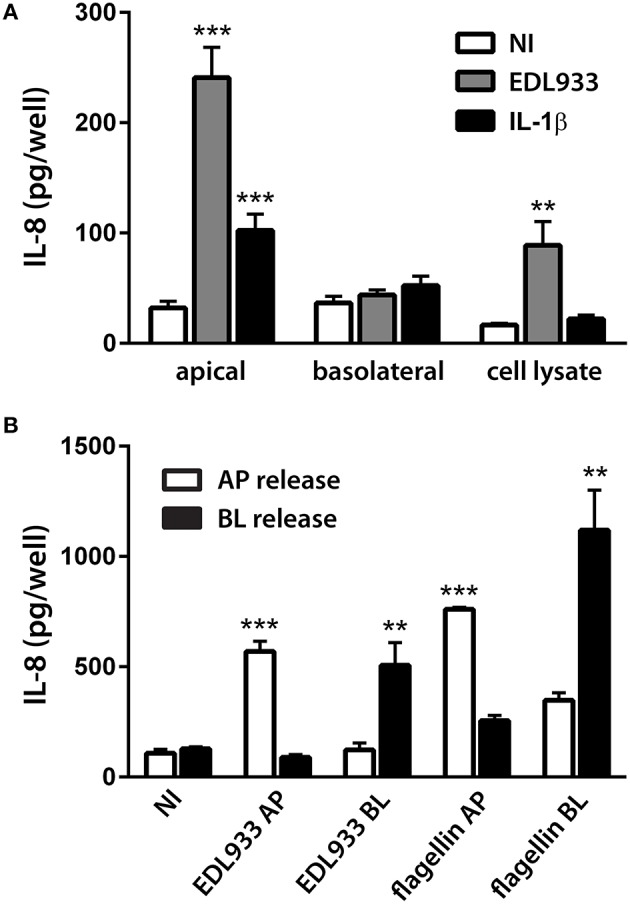
EHEC-induced IL-8 release from polarized T84 cells is directional and depends on the side of exposure. Polarized T84 cells were incubated with EDL933, IL-1β (10 ng/ml), flagellin (1 μg/ml), or left NI on the apical (A,B) or basolateral side (B) for 24 h. IL-8 protein levels in cell lysates, apical and basolateral supernatants were determined by ELISA. (A) Apical exposure to EDL933 or IL-1β stimulated apical IL-8 release. Data are shown as means ± SEM of three independent experiments performed in duplicate. (B) Apical (AP) exposure to EDL933 or flagellin induced apical IL-8 release (white columns), whereas basolateral (BL) exposure to EDL933 or flagellin induced basolateral IL-8 release (black columns). Data are shown as means ± SEM of two independent experiments performed in triplicate. **P < 0.01, ***P < 0.001 vs. NI control.
Discussion
The intestinal epithelium is the first line of defense against enteric pathogens, and the expression and release of AMPs represents an important part of the innate immune response during intestinal infections. In the colon, the main target site of EHEC, epithelial cells express hBD1-4, LL-37, and lysozyme. In this study, we have investigated the effect of EHEC infection on AMP expression in two physiologically relevant infection models: human colonic biopsies and polarized T84 cells. As demonstrated by our recent studies, IVOC of human colonic biopsies supports EHEC A/E lesion formation (Lewis et al., 2015). However, standard IVOC is not suitable to study the host immune response as it allows bacterial access and stimulation of the submucosal tissue surface. Therefore, we adopted a polarized IVOC method developed previously for small intestinal biopsy samples (Schüller et al., 2009). In addition to IVOC, we used T84 human colon carcinoma cells which lack expression of the Stx receptor Gb3 similar to human colonic epithelium (Schüller et al., 2004) and form highly polarized epithelia with high TER when grown on membrane supports (Madara et al., 1987).
Our results show that apical EHEC infection induced hBD2 expression in both colonic biopsies and polarized T84 cells whereas all other AMPs remained unaffected. This is of relevance as hBD2 has been demonstrated to directly kill E. coli and also promote the release of immuno-modulating adenosine (Estrela et al., 2013). In contrast to hBD1 which is constitutively expressed in human colonic epithelium and thus confers a baseline protection against bacterial pathogens, hBD2 is undetectable or only expressed at very low levels in healthy children and adults, but elevated in patients with inflammatory bowel disease (Fahlgren et al., 2003; Zilbauer et al., 2010). This might be due to enhanced contact of the gut microbiota with intestinal epithelium, and previous studies have demonstrated induction of hBD2 by both commensal and pathogenic bacteria. Most of these studies were performed using intestinal epithelial cell lines (e.g., Caco-2 and HT-29) and have demonstrated stimulation of hBD2 expression by enteroinvasive pathogens such as Salmonella spp., Shigella flexneri and dysenteriae, enteroinvasive E. coli, and Campylobacter jejuni (O'Neil et al., 1999; Ogushi et al., 2001; Hase et al., 2002; Zilbauer et al., 2005). In addition, non-invasive EPEC and probiotic E. coli Nissle 1917 have also been shown to induce hBD2 expression (Schlee et al., 2007; Khan et al., 2008). In contrast, very few studies have been carried out on human intestinal tissue so far. Whereas experiments using human intestinal xenografts in mice have confirmed hBD2 induction by Salmonella typhi (O'Neil et al., 1999), no change in expression was observed after infection with S. flexneri (Sperandio et al., 2008). To the best of our knowledge, this is the first study using IVOC of human intestinal biopsies to investigate the AMP response to bacterial infection.
We further investigated the bacterial factors involved in hBD2 induction and found that EHEC flagellin was both necessary and sufficient to stimulate a response ex vivo and in vitro. This is in agreement with studies on EPEC and Salmonella enterica serotype Enteritidis which also demonstrated hBD2 induction mediated by flagellin (Ogushi et al., 2001; Schlee et al., 2007; Khan et al., 2008). Interestingly, the hBD2 response to flagellin from non-pathogenic E. coli such as ATCC 259922, JM109, and Nissle 1917 is strain-dependent suggesting differences in binding to a potential receptor (Schlee et al., 2007). We further tested which signal transduction pathways were involved in flagellin-mediated hBD2 induction by using chemical inhibitors of NF-κB and MAPKs. These studies showed that flagellin-induced hBD2 expression was dependent on NF-κB, MAPK p38, and JNK but not ERK1/2. NF-κB dependency of hBD2 induction has been described before in LPS-stimulated macrophages (Tsutsumi-Ishii and Nagaoka, 2002) and intestinal epithelial cells treated with flagellin from E. coli Nissle (Wehkamp et al., 2004). The latter study also demonstrated involvement of the MAPKs JNK but not p38 or ERK 1/2. In addition, Bacteroides fragilis enterotoxin induced hBD2 by p38 activation (Yoon et al., 2010), and inhibitors of p38, JNK and ERK 1/2 reduced hBD2 induction by probiotic lactobacilli, although this did not reach significance (Schlee et al., 2008).
In addition to studying the AMP response to EHEC infection, we also examined expression of the pro-inflammatory cytokine IL-8 which acts as a chemoattractant and recruits neutrophils to the site of infection (Baggiolini and Clark-Lewis, 1992). Previous studies using non-polarized cells have shown that EHEC infection induces IL-8 release, and that this is dependent on NF-κB and MAP kinases (Dahan et al., 2002; Gobert et al., 2007). While several bacterial factors including long polar fimbriae, hemorrhagic coli pili, and Shiga toxins (Thorpe et al., 2001; Ledesma et al., 2010; Farfan et al., 2013) have been implicated in this response, many studies suggest that flagellin is the principal inducer of IL-8 production during EHEC infection (Berin et al., 2002; Zhou et al., 2003; Miyamoto et al., 2006). This is confirmed by our results on human colonic biopsies and polarized T84 cells and agrees with earlier ex vivo studies using human colonic xenografts in mice (Miyamoto et al., 2006).
While flagellin provides the initial stimulus for a pro-inflammatory response, it has been shown that T3S effectors encoded outside the locus of enterocyte effacement dampen down NF-κB activation and IL-8 release at later stages of EHEC and EPEC infection (Hauf and Chakraborty, 2003; Nadler et al., 2010; Baruch et al., 2011). In addition, a similar T3S-dependent suppressive effect has been observed in EPEC-dependent hBD2 expression (Khan et al., 2008). Notably, all of these studies have been performed using cervical HeLa cells or undifferentiated Caco-2 cells. Interestingly, we did not detect any significant inhibitory effect of the T3SS on hBD2 and IL-8 expression during EHEC infection of colonic biopsies and polarized T84 cells. A similar observation has been reported by Ruchaud-Sparagano and colleagues who demonstrated T3S-dependent suppression of IL-8 induction in EPEC-infected polarized Caco-2 but not T84 cells (Ruchaud-Sparagano et al., 2007). As fully differentiated Caco-2 cells display a small intestinal rather than colonic phenotype (Engle et al., 1998), these results might suggest a delayed or absent immunosuppressive effect of EHEC infection in human colonic vs. small intestinal epithelium.
As it was technically not feasible to accurately quantify epithelial IL-8 secretion in colonic biopsies due to the presence and varying thickness of the submucosal tissue, polarized T84 cells were employed for this part of the study. Unexpectedly, we found that IL-8 secretion was vectorial and depended on the direction of the stimulus: Whereas, apical exposure to EHEC or flagellin induced apical IL-8 release, basolateral exposure resulted in IL-8 secretion to the basolateral side. Whilst EHEC-induced IL-8 secretion has not been examined in polarized T84 cells, studies on EPEC and EPEC flagellin have reported increased basolateral IL-8 secretion after basolateral but not apical stimulation (Zhou et al., 2003; Ruchaud-Sparagano et al., 2007). Very few studies have examined apical IL-8 secretion as it is generally assumed that IL-8 is mainly secreted basolaterally to mediate neutrophil recruitment to the epithelium. In line with this theory, predominantly basolateral IL-8 secretion has been shown in polarized HCA-7 cells stimulated with EHEC flagellin and polarized Caco-2 cells apically exposed to EPEC (Berin et al., 2002; Ruchaud-Sparagano et al., 2007). However, similar to our results, stimulation of polarized HT 29/19A or Caco-2 cells with IL-1 or TNF-α resulted in polarized IL-8 secretion dependent on the side of exposure (Lammers et al., 1994; Sonnier et al., 2010). In addition, vectorial IL-8 release has been shown in flagellin-stimulated polarized Caco-2 cells (Rossi et al., 2013). While luminal IL-8 could aid neutrophil transmigration across the epithelium as demonstrated in urinary tract infections (Godaly et al., 2001), autocrine epithelial signaling via apically expressed IL-8 receptors (CXCR1) has also been suggested (Rossi et al., 2013).
Taken together, our study demonstrates that colonic EHEC infection elicits a weak innate immune response with induction of hBD2 but no other AMPs and low levels of IL-8. These results suggest that inflammation plays a minor role in intestinal EHEC pathogenesis.
Author contributions
SL and SS designed the study, analyzed the data, and prepared the manuscript. SL and SE performed the experimental work. AP, VC, SC, and WG selected suitable patients, obtained informed consent, and provided human biopsy samples.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Grégory Jubelin and Christine Martin (INRA CR Clermont-Ferrand/Theix, France) for strain EDL933 and ΔfliC and Δstx mutants, Amin Tahoun and David Gally (Roslin Institute, Edinburgh) for purified H7 flagellin, Jorge Girón (University of Florida, US) for strains EDL933 ΔescN and 85–170 ΔhcpA, Alfredo Torres (University of Texas, US) for mutant 85–170 ΔlpfA1, Roberto La Ragione (University of Surrey) for strain Walla-1, and Geraldine Smith (Public Health England) for isolate H0-7184-0336. We are grateful to Nigel Belshaw and Giles Elliott (Institute of Food Research, Norwich) for support with qPCR, Bertrand Lézé (University of East Anglia, Norwich) for help with electron microscopy and Mark Lucas (University College London) for helpful discussions. This work was supported by the University of East Anglia (FMH studentship to SL), the BBSRC (DTP studentship to SE), the NIHR (SC), and the MRC (MR/J002062/1, SS).
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2016.00068
Kinetics of hBD2 and IL-8 gene expression during EHEC infection. Polarized T84 cells were apically infected with EHEC strain EDL933 for 3–12 h, or left NI for 12 h. hBD2 and IL-8 mRNA levels were quantified by qPCR and expressed as fold changes relative to NI controls. Data represent means ± SEM of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NI control.
Influence of different treatments on T84 cell barrier function. Polarized T84 cells were apically inoculated with EHEC wild-type EDL933 or 85–170 (WT), deletion mutants (ΔfliC, escN, stx, hcp, or lpfA1), purified flagellin (1 μg/ml) or left non-infected (NI) for 9 h (A) or incubated with EDL933, IL-1β (10 ng/ml), flagellin (1 μg/ml), or left NI on the apical (AP) or basolateral side (BL) for 24 h (B). TER after treatment is expressed as percentage of TER before treatment. Data are shown as means ± SEM of three independent experiments performed in duplicate.
References
- Baggiolini M., Clark-Lewis I. (1992). Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 307, 97–101. 10.1016/0014-5793(92)80909-Z [DOI] [PubMed] [Google Scholar]
- Baruch K., Gur-Arie L., Nadler C., Koby S., Yerushalmi G., Ben-Neriah Y., et al. (2011). Metalloprotease type III effectors that specifically cleave JNK and NF-kappaB. EMBO J. 30, 221–231. 10.1038/emboj.2010.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berin M. C., Darfeuille-Michaud A., Egan L. J., Miyamoto Y., Kagnoff M. F. (2002). Role of EHEC O157:H7 virulence factors in the activation of intestinal epithelial cell NF-kappaB and MAP kinase pathways and the upregulated expression of interleukin 8. Cell. Microbiol. 4, 635–648. 10.1046/j.1462-5822.2002.00218.x [DOI] [PubMed] [Google Scholar]
- Croxen M. A., Finlay B. B. (2010). Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 8, 26–38. 10.1038/nrmicro2265 [DOI] [PubMed] [Google Scholar]
- Croxen M. A., Law R. J., Scholz R., Keeney K. M., Wlodarska M., Finlay B. B. (2013). Recent advances in understanding enteric pathogenic Escherichia coli. Clin. Microbiol. Rev. 26, 822–880. 10.1128/CMR.00022-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan S., Busuttil V., Imbert V., Peyron J. F., Rampal P., Czerucka D. (2002). Enterohemorrhagic Escherichia coli infection induces interleukin-8 production via activation of mitogen-activated protein kinases and the transcription factors NF-kappaB and AP-1 in T84 cells. Infect. Immun. 70, 2304–2310. 10.1128/IAI.70.5.2304-2310.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann L., Kagnoff M. F. (2005). Intestinal mucosal responses to microbial infection. Springer Semin. Immunopathol. 27, 181–196. 10.1007/s00281-005-0207-5 [DOI] [PubMed] [Google Scholar]
- Engle M. J., Goetz G. S., Alpers D. H. (1998). Caco-2 cells express a combination of colonocyte and enterocyte phenotypes. J. Cell. Physiol. 174, 362–369. [DOI] [PubMed] [Google Scholar]
- Estrela A. B., Rohde M., Gutierrez M. G., Molinari G., Abraham W. R. (2013). Human beta-defensin 2 induces extracellular accumulation of adenosine in Escherichia coli. Antimicrob. Agents Chemother. 57, 4387–4393. 10.1128/AAC.00820-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren A., Hammarström S., Danielsson A., Hammarström M. L. (2003). Increased expression of antimicrobial peptides and lysozyme in colonic epithelial cells of patients with ulcerative colitis. Clin. Exp. Immunol. 131, 90–101. 10.1046/j.1365-2249.2003.02035.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren A., Hammarström S., Danielsson A., Hammarström M. L. (2004). beta-Defensin-3 and -4 in intestinal epithelial cells display increased mRNA expression in ulcerative colitis. Clin. Exp. Immunol. 137, 379–385. 10.1111/j.1365-2249.2004.02543.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfan M. J., Cantero L., Vergara A., Vidal R., Torres A. G. (2013). The long polar fimbriae of STEC O157:H7 induce expression of pro-inflammatory markers by intestinal epithelial cells. Vet. Immunol. Immunopathol. 152, 126–131. 10.1016/j.vetimm.2012.09.017 [DOI] [PubMed] [Google Scholar]
- Fitzhenry R., Dahan S., Torres A. G., Chong Y., Heuschkel R., Murch S. H., et al. (2006). Long polar fimbriae and tissue tropism in Escherichia coli O157:H7. Microbes. Infect. 8, 1741–1749. 10.1016/j.micinf.2006.02.012 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick M. M., Shah V., Trompeter R. S., Dillon M. J., Barratt T. M. (1992). Interleukin-8 and polymorphoneutrophil leucocyte activation in hemolytic uremic syndrome of childhood. Kidney Int. 42, 951–956. 10.1038/ki.1992.372 [DOI] [PubMed] [Google Scholar]
- Gobert A. P., Coste A., Guzman C. A., Vareille M., Hindré T., De Sablet T., et al. (2008). Modulation of chemokine gene expression by Shiga-toxin producing Escherichia coli belonging to various origins and serotypes. Microbes Infect. 10, 159–165. 10.1016/j.micinf.2007.10.018 [DOI] [PubMed] [Google Scholar]
- Gobert A. P., Vareille M., Glasser A. L., Hindré T., De S. T., Martin C. (2007). Shiga toxin produced by enterohemorrhagic Escherichia coli inhibits PI3K/NF-kappaB signaling pathway in globotriaosylceramide-3-negative human intestinal epithelial cells. J. Immunol. 178, 8168–8174. 10.4049/jimmunol.178.12.8168 [DOI] [PubMed] [Google Scholar]
- Godaly G., Bergsten G., Hang L., Fischer H., Frendeus B., Lundstedt A. C., et al. (2001). Neutrophil recruitment, chemokine receptors, and resistance to mucosal infection. J. Leukoc. Biol. 69, 899–906. [PubMed] [Google Scholar]
- Golan L., Gonen E., Yagel S., Rosenshine I., Shpigel N. Y. (2011). Enterohemorrhagic Escherichia coli induce attaching and effacing lesions and hemorrhagic colitis in human and bovine intestinal xenograft models. Dis. Model. Mech. 4, 86–94. 10.1242/dmm.005777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin P. M., Olmstead L. C., Petras R. E. (1990). Escherichia coli O157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology 99, 142–149. [DOI] [PubMed] [Google Scholar]
- Hase K., Eckmann L., Leopard J. D., Varki N., Kagnoff M. F. (2002). Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect. Immun. 70, 953–963. 10.1128/IAI.70.2.953-963.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf N., Chakraborty T. (2003). Suppression of NF-kappa B activation and proinflammatory cytokine expression by Shiga toxin-producing Escherichia coli. J. Immunol. 170, 2074–2082. 10.4049/jimmunol.170.4.2074 [DOI] [PubMed] [Google Scholar]
- Islam D., Bandholtz L., Nilsson J., Wigzell H., Christensson B., Agerberth B., et al. (2001). Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 7, 180–185. 10.1038/84627 [DOI] [PubMed] [Google Scholar]
- Jarvis K. G., Kaper J. B. (1996). Secretion of extracellular proteins by enterohemorrhagic Escherichia coli via a putative type III secretion system. Infect. Immun. 64, 4826–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J., Oryshak A., Wenetsek M., Grabiec J., Handy S. (1990). The colonic pathology of Escherichia coli O157:H7 infection. Am. J. Surg. Pathol. 14, 87–92. 10.1097/00000478-199001000-00010 [DOI] [PubMed] [Google Scholar]
- Khan M. A., Bouzari S., Ma C., Rosenberger C. M., Bergstrom K. S., Gibson D. L., et al. (2008). Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect. Immun. 76, 1410–1422. 10.1128/IAI.01141-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers K. M., Jansen J., Bijlsma P. B., Ceska M., Tytgat G. N., Laboisse C. L., et al. (1994). Polarised interleukin 8 secretion by HT 29/19A cells. Gut 35, 338–342. 10.1136/gut.35.3.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma M. A., Ochoa S. A., Cruz A., Rocha-Ramírez L. M., Mas-Oliva J., Eslava C. A., et al. (2010). The hemorrhagic coli pilus (HCP) of Escherichia coli O157:H7 is an inducer of proinflammatory cytokine secretion in intestinal epithelial cells. PLoS ONE 5:e12127. 10.1371/journal.pone.0012127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S. B., Cook V., Tighe R., Schüller S. (2015). Enterohemorrhagic Escherichia coli colonization of human colonic epithelium in vitro and ex vivo. Infect. Immun. 83, 942–949. 10.1128/IAI.02928-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara J. L., Stafford J., Dharmsathaphorn K., Carlson S. (1987). Structural analysis of a human intestinal epithelial cell line. Gastroenterology 92, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y., Iimura M., Kaper J. B., Torres A. G., Kagnoff M. F. (2006). Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell. Microbiol. 8, 869–879. 10.1111/j.1462-5822.2005.00673.x [DOI] [PubMed] [Google Scholar]
- Muniz L. R., Knosp C., Yeretssian G. (2012). Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front. Immunol. 3:310. 10.3389/fimmu.2012.00310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata A., Shimazu T., Yamamoto T., Taenaka N., Nagayama K., Honda T., et al. (1998). Profiles of circulating inflammatory- and anti-inflammatory cytokines in patients with hemolytic uremic syndrome due to E. coli O157 infection. Cytokine 10, 544–548. 10.1006/cyto.1997.0329 [DOI] [PubMed] [Google Scholar]
- Nadler C., Baruch K., Kobi S., Mills E., Haviv G., Farago M., et al. (2010). The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 6:e1000743. 10.1371/journal.ppat.1000743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrig T. G., Karpman D. (2012). Shiga toxin pathogenesis: kidney complications and renal failure. Curr. Top. Microbiol. Immunol. 357, 105–136. 10.1007/82_2011_172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogushi K., Wada A., Niidome T., Mori N., Oishi K., Nagatake T., et al. (2001). Salmonella enteritidis FliC (flagella filament protein) induces human beta-defensin-2 mRNA production by Caco-2 cells. J. Biol. Chem. 276, 30521–30526. 10.1074/jbc.M011618200 [DOI] [PubMed] [Google Scholar]
- O'Neil D. A., Porter E. M., Elewaut D., Anderson G. M., Eckmann L., Ganz T., et al. (1999). Expression and regulation of the human beta-defensins hBD-1 and hBD-2 in intestinal epithelium. J. Immunol. 163, 6718–6724. [PubMed] [Google Scholar]
- Ostroff S. M., Griffin P. M., Tauxe R. V., Shipman L. D., Greene K. D., Wells J. G., et al. (1990). A statewide outbreak of Escherichia coli O157:H7 infections in Washington State. Am. J. Epidemiol. 132, 239–247. [DOI] [PubMed] [Google Scholar]
- Ou G., Baranov V., Lundmark E., Hammarström S., Hammarström M. L. (2009). Contribution of intestinal epithelial cells to innate immunity of the human gut-studies on polarized monolayers of colon carcinoma cells. Scand. J. Immunol. 69, 150–161. 10.1111/j.1365-3083.2008.02208.x [DOI] [PubMed] [Google Scholar]
- Riley L. W., Remis R. S., Helgerson S. D., McGee H. B., Wells J. G., Davis B. R., et al. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685. 10.1056/NEJM198303243081203 [DOI] [PubMed] [Google Scholar]
- Rossi O., Karczewski J., Stolte E. H., Brummer R. J., Van Nieuwenhoven M. A., Meijerink M., et al. (2013). Vectorial secretion of interleukin-8 mediates autocrine signalling in intestinal epithelial cells via apically located CXCR1. BMC Res. Notes 6:431. 10.1186/1756-0500-6-431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchaud-Sparagano M. H., Maresca M., Kenny B. (2007). Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell. Microbiol. 9, 1909–1921. 10.1111/j.1462-5822.2007.00923.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Harder J., Koten B., Stange E. F., Wehkamp J., Fellermann K. (2008). Probiotic lactobacilli and VSL#3 induce enterocyte beta-defensin 2. Clin. Exp. Immunol. 151, 528–535. 10.1111/j.1365-2249.2007.03587.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Wehkamp J., Altenhoefer A., Oelschlaeger T. A., Stange E. F., Fellermann K. (2007). Induction of human beta-defensin 2 by the probiotic Escherichia coli Nissle 1917 is mediated through flagellin. Infect. Immun. 75, 2399–2407. 10.1128/IAI.01563-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller S. (2011). Shiga toxin interaction with human intestinal epithelium. Toxins (Basel) 3, 626–639. 10.3390/toxins3060626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüller S., Frankel G., Phillips A. D. (2004). Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell. Microbiol. 6, 289–301. 10.1046/j.1462-5822.2004.00370.x [DOI] [PubMed] [Google Scholar]
- Schüller S., Lucas M., Kaper J. B., Girón J. A., Phillips A. D. (2009). The ex vivo response of human intestinal mucosa to enteropathogenic Escherichia coli infection. Cell. Microbiol. 11, 521–530. 10.1111/j.1462-5822.2008.01275.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham H. P., Shames S. R., Croxen M. A., Ma C., Chan J. M., Khan M. A., et al. (2011). Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-kappaB and p38 mitogen-activated protein kinase activation. Infect. Immun. 79, 3552–3562. 10.1128/IAI.05033-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R., Tesfay S., Tomson F. L., Kanteti R. P., Viswanathan V. K., Hecht G. (2006). Balance of bacterial pro- and anti-inflammatory mediators dictates net effect of enteropathogenic Escherichia coli on intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G685–G694. 10.1152/ajpgi.00404.2005 [DOI] [PubMed] [Google Scholar]
- Sonnier D. I., Bailey S. R., Schuster R. M., Lentsch A. B., Pritts T. A. (2010). TNF-alpha induces vectorial secretion of IL-8 in Caco-2 cells. J. Gastrointest. Surg. 14, 1592–1599. 10.1007/s11605-010-1321-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio B., Regnault B., Guo J., Zhang Z., Stanley S. L., Jr., Sansonetti P. J., et al. (2008). Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J. Exp. Med. 205, 1121–1132. 10.1084/jem.20071698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr P. I., Gordon C. A., Chandler W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086. 10.1016/s0140-6736(05)71144-2 [DOI] [PubMed] [Google Scholar]
- Thorpe C. M., Smith W. E., Hurley B. P., Acheson D. W. (2001). Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 69, 6140–6147. 10.1128/IAI.69.10.6140-6147.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe T., Beatson S. A., Taniguchi H., Abe H., Bailey C. M., Fivian A., et al. (2006). An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U.S.A. 103, 14941–14946. 10.1073/pnas.0604891103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi-Ishii Y., Nagaoka I. (2002). NF-kappa B-mediated transcriptional regulation of human beta-defensin-2 gene following lipopolysaccharide stimulation. J. Leukoc. Biol. 71, 154–162. [PubMed] [Google Scholar]
- Tzipori S., Karch H., Wachsmuth I. K., Robins-Browne R. M., O'brien A. D., Lior H., et al. (1987). Role of a 60-megadalton plasmid and shiga-like toxins in the pathogenesis of enterohaemorrhagic Escherichia coli O157:H7 in gnotobiotic piglets. Infect. Immun. 55, 3117–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan V. K., Hodges K., Hecht G. (2009). Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat. Rev. Microbiol. 7, 110–119. 10.1038/nrmicro2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassing G. M., Bergman P., Lindbom L., Van Der Does A. M. (2014). Complexity of antimicrobial peptide regulation during pathogen-host interactions. Int. J. Antimicrob. Agents 45, 447–454. 10.1016/j.ijantimicag.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Wehkamp J., Harder J., Wehkamp K., Wehkamp-Von Meissner B., Schlee M., Enders C., et al. (2004). NF-kappaB- and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic bacterium. Infect. Immun. 72, 5750–5758. 10.1128/IAI.72.10.5750-5758.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xicohtencatl-Cortes J., Monteiro-Neto V., Ledesma M. A., Jordan D. M., Francetic O., Kaper J. B., et al. (2007). Intestinal adherence associated with type IV pili of enterohemorrhagic Escherichia coli O157:H7. J. Clin. Invest. 117, 3519–3529. 10.1172/JCI30727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y. M., Lee J. Y., Yoo D., Sim Y. S., Kim Y. J., Oh Y. K., et al. (2010). Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I kappaB kinase/NF-kappaB-dependent pathway. Infect. Immun. 78, 2024–2033. 10.1128/IAI.00118-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Girón J. A., Torres A. G., Crawford J. A., Negrete E., Vogel S. N., et al. (2003). Flagellin of enteropathogenic Escherichia coli stimulates interleukin-8 production in T84 cells. Infect. Immun. 71, 2120–2129. 10.1128/IAI.71.4.2120-2129.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilbauer M., Dorrell N., Boughan P. K., Harris A., Wren B. W., Klein N. J., et al. (2005). Intestinal innate immunity to Campylobacter jejuni results in induction of bactericidal human beta-defensins 2 and 3. Infect. Immun. 73, 7281–7289. 10.1128/IAI.73.11.7281-7289.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilbauer M., Jenke A., Wenzel G., Postberg J., Heusch A., Phillips A. D., et al. (2010). Expression of human beta-defensins in children with chronic inflammatory bowel disease. PLoS ONE 5:e15389. 10.1371/journal.pone.0015389 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kinetics of hBD2 and IL-8 gene expression during EHEC infection. Polarized T84 cells were apically infected with EHEC strain EDL933 for 3–12 h, or left NI for 12 h. hBD2 and IL-8 mRNA levels were quantified by qPCR and expressed as fold changes relative to NI controls. Data represent means ± SEM of two independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NI control.
Influence of different treatments on T84 cell barrier function. Polarized T84 cells were apically inoculated with EHEC wild-type EDL933 or 85–170 (WT), deletion mutants (ΔfliC, escN, stx, hcp, or lpfA1), purified flagellin (1 μg/ml) or left non-infected (NI) for 9 h (A) or incubated with EDL933, IL-1β (10 ng/ml), flagellin (1 μg/ml), or left NI on the apical (AP) or basolateral side (BL) for 24 h (B). TER after treatment is expressed as percentage of TER before treatment. Data are shown as means ± SEM of three independent experiments performed in duplicate.