

Abstract
We synthesized a new series of conjugated hydrazones that were found to be active against malaria parasite in vitro, as well as in vivo in a murine model. These hydrazones concentration-dependently chelated free iron and offered antimalarial activity. Upon screening of the synthesized hydrazones, compound 5f was found to be the most active iron chelator, as well as antiplasmodial. Compound 5f also interacted with free heme (KD [equilibrium dissociation constant] = 1.17 ± 0.8 μM), an iron-containing tetrapyrrole released after hemoglobin digestion by the parasite, and inhibited heme polymerization by parasite lysate. Structure-activity relationship studies indicated that a nitrogen- and sulfur-substituted five-membered aromatic ring present within the benzothiazole hydrazones might be responsible for their antimalarial activity. The dose-dependent antimalarial and heme polymerization inhibitory activities of the lead compound 5f were further validated by following [3H]hypoxanthine incorporation and hemozoin formation in parasite, respectively. It is worth mentioning that compound 5f exhibited antiplasmodial activity in vitro against a chloroquine/pyrimethamine-resistant strain of Plasmodium falciparum (K1). We also evaluated in vivo antimalarial activity of compound 5f in a murine model where a lethal multiple-drug-resistant strain of Plasmodium yoelii was used to infect Swiss albino mice. Compound 5f significantly suppressed the growth of parasite, and the infected mice experienced longer life spans upon treatment with this compound. During in vitro and in vivo toxicity assays, compound 5f showed minimal alteration in biochemical and hematological parameters compared to control. In conclusion, we identified a new class of hydrazone with therapeutic potential against malaria.
INTRODUCTION
Malaria is one of the foremost widespread lethal diseases in the world. Nearly 40% of the global population is affected by malaria, and it has become a major public health concern in the developing countries according to the World Health Organization, with 198 million reported cases and around 584,000 deaths in 2013, most of which comprised of children below 5 years of age. Malaria poses a serious threat to a large chunk of the human population. Malaria has evolved as a gigantic infectious killer, and its control has become a high-precedence assignment globally (1, 2). P. falciparum is the deadliest among the Plasmodium species that causes malaria in humans. With an alarming increase in the emergence of drug-resistant parasites, especially multidrug-resistant (MDR) strains, established antimalarials have become more and more ineffective, thereby instigating active exploration for developing newer substitutes to confront the disease. In this regard, a new generation of in vivo-active antimalarials is needed (3–5). The recognition and design of new chemical scaffolds, particularly those influencing the validated biological targets crucial for parasite survival and disease development, might prove instrumental in achieving these goals (5). With advancement in research on the biology of Plasmodium, many potential drug targets have been identified and evaluated, most of which belong to the asexual blood stages. In this context, iron chelation in parasites has proved promising; however, it is largely underutilized.
To cause infection, nearly all protozoa, fungi, and bacteria obtain growth-essential iron from their hosts. Iron is required for the development of the parasite since many enzymes of the plasmodial metabolic pathways (such as δ-aminolevulinate synthase, responsible for the de novo synthesis of heme, or ribonucleotide reductase, involved in DNA synthesis) depend on the presence of this element (6). Deprivation of utilizable iron by chelation is a proficient approach to arrest parasite growth and associated infection (7). A strong iron chelator, deferoxamine (DFO), reportedly diminishes the activity of Fe(III)-containing enzymes in parasitic protozoa (8–11). Therefore, iron chelators have recently gained importance as potent antimalarial compounds. It is proposed that iron chelator antimalarials mainly arrest iron-dependent ribonucleotide reductase-mediated production of nucleotides, although the precise mechanistic details of antiparasitic action are still unclear (12).
The intraerythrocytic stages during malaria are mainly responsible for the clinical symptoms related to malaria. During that stage, the malaria parasite digests enormous quantities of hemoglobin inside the food vacuole and thereby releasing high quantities of toxin-free heme (13–16). Free heme can change membrane permeability by intercalating into the cell membrane and also may induce oxidative stress (17, 18), which may lead to parasite death. However, by converting heme into the less-toxic hemozoin, parasites cleverly evade the toxicity of free heme (13, 14, 19). Oxidative stress-mediated parasite death through the prevention of hemozoin formation is also an accepted therapeutic strategy against malaria (20). Heme contains an iron atom at the center of its tetrapyrrole ring; therefore, an iron-chelating molecule may be able to interact with free heme through its iron. Furthermore, it is worth mentioning that this sort of heme-interacting molecule may inhibit hemozoin formation to induce parasite death (20, 21). Therefore, a small molecule, which can both chelate free iron and prevent hemozoin formation through free heme binding would be a novel dual target antimalarial agent. Here, we report the designing, synthesis, and antimalarial activities of conjugated hydrazones. These molecules chelated free iron, interacted with free heme, inhibited hemozoin formation, and prevented P. falciparum growth in vitro. Compound 5f (the lead molecule) also showed antimalarial activity in vivo against the MDR strain of P. yoelii in Swiss albino mice.
MATERIALS AND METHODS
Chemicals.
RPMI 1640, hemin, saponin, dimethyl sulfoxide (DMSO), sodium dodecyl sulfate (SDS), sodium bicarbonate, tripyridyl triazine (TPTZ), Saponin, d-glucose, and Giemsa stain were purchased from Sigma (St. Louis, MO). Albumax II, gentamicin, penicillin, streptomycin, and fetal bovine serum were purchased from Gibco/Life Technologies. [3H]hypoxanthine was purchased from Perkin-Elmer. All other chemicals were of analytical-grade purity.
Chemical synthesis.
The general procedure for the synthesis of hydrazone compounds (1, 2, 3, 4, and 5) was as follows. To a solution of the corresponding hydrazine (30 mmol) in methanol (50 ml), 1.1 equivalents of the appropriate aldehyde dissolved in methanol were added at room temperature. After proper mixing of the solution, stirring was continued overnight. The solid products were isolated and purified by column chromatography. The structures of hydrazones were confirmed by nuclear magnetic resonance (NMR) and mass spectral analysis.
Iron-chelating activity in vitro.
The iron-chelating activity of the synthesized compounds was monitored according to a previously described protocol (22). The assay was carried out in optically clear 1-ml quartz cuvette containing 20 mM phosphate-buffered saline (PBS) (pH 7.4) and Fe(II) (10 μM). Compounds were added with increasing concentrations (1 to 20 μM) with the instant addition of tripyridyl triazine (TPTZ; 20 μM) in small volumes to the sample cuvette. The same volume of DMSO was added to the reference cuvette concomitantly (the compounds were dissolved in DMSO). An assay system without a compound was considered a control set. A well-known iron chelator deferoxamine was used as a positive control. The iron-chelating capability of compounds at various concentrations was monitored immediately after each addition by determining the decrease in the absorbance of the Fe(II)-TPTZ complex at 595 nm in a Shimadzu UV/VIS spectrophotometer, and this value was expressed as the concentration required to decrease the original absorbance by 50%.
Parasite culture in vitro and in vivo parasite maintenance.
P. falciparum 3D7 strain (chloroquine sensitive) and K1 strain (chloroquine and pyrimethamine resistant) were cultured as mentioned earlier with some modifications (23, 24). In brief, the parasite culture was maintained in RPMI 1640 medium supplemented with 25 mM HEPES, 1.76 g of sodium bicarbonate per liter, 2 g of d-glucose per liter, 50 μg of gentamicin per ml, 370 μM hypoxanthine, and 0.5% (wt/vol) AlbuMax II at a hematocrit level of 5% in culture flasks inside sealed candle jars at a temperature maintained at 37°C. The culture medium was exchanged with fresh medium at 24-h interval. P. falciparum growth was regularly monitored by Giemsa staining of thin blood smears. In vivo malaria was maintained by intraperitoneal inoculation of P. yoelii (N-67, an MDR strain) in male Swiss albino mice (20 to 25 g) as described earlier (25). Giemsa staining of thin blood smears was used to quantify parasitemia. Animal experiments were conducted in accordance with the institutional guidelines and permission from the animal ethics committee of the Committee for the Purpose of Control and Supervision of Experiments on Animals, New Delhi, India (permit 147/1999/CPCSEA).
In vitro antimalarial activity by SYBR green assay.
The SYBR green assay was performed in 96-well plate, as mentioned earlier (26). In brief, parasite culture with 4% hematocrit and 1% parasitemia in a synchronized ring stage was dispensed into a 96-well plate with increasing concentrations of compounds in three sets and kept in candle jar in a total volume of 100 μl per well. After 48 h, the cells were lysed in 100 μl of lysis buffer (20 mM Tris [pH 7.5], 5 mM EDTA, 0.008% [wt/vol] saponin, 0.08% [vol/vol] Triton X-100, and 2× SYBR green nucleic I acid stain) in each well by pipetting and then incubated in the dark at room temperature for 1 h. Deferoxamine, a standard iron chelator, was used as a positive control. Artesunate, a water-soluble semisynthetic derivative of artemisinin and chloroquine were used as positive controls to check the antimalarial activity. The fluorescence intensities were determined upon excitation at 485 nm and after emission at 530 nm using a Hitachi F-7000 fluorimeter.
Fe(III)-compound 5f complex formation.
Each Fe(III)-compound complex was prepared as described previously (27). Ligand (compound 5f, 2 mmol) was dissolved in 45 ml of dry methanol with subsequent addition of triethylamine (2 mmol) to the solution. After the complete dissolution of the ligand, a methanolic solution of FeCl3·6H2O (2.75 mmol) was added dropwise to the ligand solution, and the resulting mixture was refluxed for 1 h. Progression of the reaction was monitored by thin-layer chromatography. Upon completion of the reaction, the reaction mixture was allowed to come to room temperature, and precipitated product was filtered off and washed with methanol several times, followed by treatment with water and then acetone. The complex obtained was subjected to infrared (IR) spectroscopy for characterization.
Heme interaction studies.
The interaction of hydrazone derivatives with heme was evaluated by differential optical spectroscopy, as mentioned before (28). In brief, assay system was carried out with heme (1 μM) in 100 mM sodium acetate buffer (pH 5.2) in a final volume of 1 ml, and differential optical spectra were recorded using quartz cells with a 1-cm light path in a Shimadzu 15UV/VIS spectrophotometer at 25 ± 1°C. Native heme binding of hydrazones was documented with increasing concentrations (1 to 20 μM) of hydrazones. Differential spectroscopy of heme versus heme-hydrazone interactions was performed by the addition of heme solution (1 μM) in both the reference and the sample cuvettes to obtain a baseline trace at a final volume of 1 ml. This step was followed by the addition of increasing concentrations of hydrazones (1 to 20 μM) to the sample cuvette, along with a concomitant addition of the same volume of DMSO (used to dissolve the hydrazones; 0.5 to 10 μl) to the reference cuvette. The Soret spectrum of thoroughly mixed contents was recorded without hydrazones and after the successive addition of hydrazones immediately. The equilibrium dissociation constant (KD) for heme-hydrazone complex formation was evaluated by an equation described by Schejter et al. (28): 1/ΔA = (KD/ΔA∞)1/S + 1/ΔA∞, where S is the concentration of hydrazones, ΔA is the absorption change at a particular wavelength and at a saturating concentration of the ligand (hydrazones), and absorption changes are expressed as ΔA∞.
Preparation of parasite lysate.
The parasite (P. yoelii) lysate was prepared as described earlier (29, 30). In brief, erythrocytes (from P. yoelii-infected mice blood) were collected and centrifuged at 800 × g for 5 min. The pellet was washed twice and reconstituted in cold PBS. To lyse the erythrocytes, PBS containing an equal volume of 0.5% saponin (final concentration, 0.25%) was mixed well with erythrocyte suspension in PBS and allowed to stand for 15 min in ice. The mixture was then centrifuged at 1,300 × g for 5 min to obtain the parasite pellet. The pellet was rinsed thrice with cold PBS and kept at −80°C for further use. The parasite pellet was then lysed in PBS by mild sonication (a 30-s pulse in water bath-type sonicator) under ice-cold conditions, and the protein content of the lysate was quantified. The lysate obtained was stored at −20°C for further use.
Hemozoin (β-hematin) formation in vitro by parasite lysate and in P. falciparum culture.
Formation of hemozoin (β-hematin) in the presence of parasite lysate was evaluated as described earlier with some modifications (31–33). In brief, the reaction was carried out in the presence of 100 mM hemin, parasite lysate (20 μg), and different concentrations of hydrazones (1 to 100 μM) in a final volume of 1 ml of sodium acetate buffer (100 mM, pH 5.2). The reaction performed in the absence of hydrazones was considered the control. The reaction was carried out at 37°C for 12 h to allow the conversion of hemin into hemozoin. After 12 h, the reaction mixture was centrifuged at 15,000 × g for 15 min at room temperature. The obtained pellet was rinsed three times with Tris buffer (100 mM [pH 7.8] containing 2.5% SDS) and once in bicarbonate buffer (100 mM, pH 9.2). The resultant insoluble pellet (hemozoin) was then dissolved using 100 μl of 2 N NaOH and diluted with 0.9 ml of 2.5% SDS. The absorbance of the solution was recorded at 400 nm using Shimadzu 15UV/VIS spectrophotometer at 25 ± 1°C using quartz cells with a 1-cm light path.
The efficacy of the lead compound was further tested for its activity against hemozoin formation in P. falciparum in culture at various concentrations. Parasite in culture medium was incubated with the compound for 48 h, and then the cells were collected by centrifugation and treated with 0.01% saponin in PBS to release the parasite and lyse the uninfected red blood cells (RBCs). The parasite pellet was washed with PBS three times and lysed by sonication. The total protein content of all of the samples was estimated. Free heme from parasite lysate was removed by washing with 2.5% SDS and 0.1 M sodium bicarbonate (pH 9.2), with subsequent discarding of the supernatant after centrifugation. The remaining pellet was resuspended in 1 ml of 0.2 N NaOH, and the absorbance was measured at 400 nm.
[3H]hypoxanthine incorporation assay to follow antimalarial activity in vitro.
The inhibition of P. falciparum growth by the compound was confirmed by monitoring [3H]hypoxanthine uptake as described previously (20, 34). The synchronized ring stage of the parasite (1% parasitemia) was cultured in multiwell plates (200 μl/well) using different concentrations of the compound in triplicate. Parasite samples with DMSO alone were used as vehicle controls. After 24 h, [3H]hypoxanthine (5 μCi/well) was added to each well, and the plate was kept in an incubator for another 24 h to the allow the incorporation of free [3H]hypoxanthine into parasite nucleic acids. After treatment, the parasite was pelleted and washed three times in PBS. The packed RBC pellet was washed and dissolved in 100 μl of 3 N NaOH and kept at 37°C for 6 h. The lysate content was then dissolved in 10 ml of scintillation fluid (phenyl-oxazole-phenyl [POP], 4 g; phenyl-oxazole-phenyl-oxazole-phenyl [POPOP], 200 mg; naphthalene, 60 g; ethylene glycol, 20 ml; methanol, 100 ml [all in 1 liter of 1,4-dioxane]) and transferred to scintillation vials. The level of incorporated [3H]hypoxanthine was assessed using a Perkin-Elmer Tricarb 2810TR liquid scintillation counter. The radioactivity of the experimental samples was expressed as disintegrations per minute, a value which was proportional to the amount of [3H]hypoxanthine incorporated into the DNA.
Evaluation of antimalarial activity of compound 5f in Swiss albino mice.
The in vivo antimalarial efficacy of compound 5f was assessed in a murine malaria model, as mentioned previously (20). In brief, a MDR strain of P. yoelii (N-67; a strain resistant to chloroquine, mefloquine, and halofantrine) was used to infect Swiss albino mice. Compound 5f was dissolved in alkaline water and administered intraperitoneally at three different doses (10, 25, and 50 mg kg [body weight]−1). For each treatment, a group of six mice (each weighing 25 ± 5 g) was inoculated with ∼106 parasitized RBCs on day 0, and compound 5f was administered from days 4 to 7 postinfection. The effectiveness of compound 5f was evaluated by constant observation of parasitemia and animal survival every day. Giemsa staining of thin blood smears was used to measure the levels of parasitemia in the mice. A drop of blood was extracted from the tail vein of the mice and smeared onto a glass slide to obtain a thin smear. The slide was air dried and fixed in absolute methanol for 20 s. A Giemsa stain working solution was prepared by dilution in PBS (pH 7.2) at a dilution of 1:3. Fixed and air-dried blood smears were stained for 30 min and washed under running tap water. Slides were viewed and pictured at a resolution of ×100 under an oil immersion lens. The mean value of the percent parasitemia observed from an individual set of mice was used to analyze the percent inhibition of parasitemia compared to the vehicle control group. α/β-Arteether (an ethyl ether derivative of artemisinin) was used as a positive control at a dose of 50 mg kg (body weight)−1 day−1.
The suppressive activity of compound 5f against P. yoelii was also evaluated in vivo by applying Peter's 4-day test (35) in Swiss albino mice with slight modifications. In brief, blood from an infected donor mouse was diluted with isotonic saline to yield an inoculum containing 2 × 107 infected erythrocytes per ml. On the first day (D0), 30 mice were inoculated intraperitoneally with 0.2 ml of the inoculum before being randomly divided into five treatment groups of six mice each. At 3 h postinoculation, three experimental groups of mice were treated with compound 5f intraperitoneally, at three different doses (10, 25, or 50 mg kg−1 day−1) for 4 days (D0 to D3), respectively. The fourth group was treated with α/β-arteether, which was used as a positive control, at a dose of 50 mg kg−1 day−1. A fifth group was considered the control. On day 5 (D4), at 24 h after the last treatment, a thin smear was prepared from the tail blood of each mouse and stained with Giemsa stain to determine the percent parasitemia (by determining the number of parasitized erythrocytes per 300 erythrocytes in random fields). Parasitemia was determined by light microscopy using a ×100 objective lens. The suppression of the average percent parasitemia was calculated as follows: % parasitemia = [(A − B)/A] × 100, where A is the average percent parasitemia in the negative-control group, and B is the average percent parasitemia in the test group.
In vitro hemolysis assay.
The cytotoxic effect of compound 5f on RBCs was evaluated by hemolysis assay, as described earlier, with some modifications (36). The freshly obtained RBCs were washed with 3 volumes of PBS (pH 7.4) three times. Hemolysis assay was performed at 2% hematocrit with different concentrations of drug in a final volume of 0.3 ml, followed by incubation at 37°C for 2 h and 12 h. Saponin (0.05% in PBS) was used as a positive control, whereas 0.2% DMSO was used as a vehicle control. After incubation, the tubes were centrifuged at 1,000 × g for 10 min, and the absorbance of the supernatants was determined over a wavelength range of 300 to 700 nm. The absorbance of the supernatants was also measured at 540 nm in an enzyme-linked immunosorbent assay plate reader (μQuant; BioTek) to measure the amount of hemoglobin released upon RBC lysis. The experiment was performed in triplicate.
In vitro cytotoxic effect of compound 5f against mammalian cells.
The cytotoxicity of compound 5f was further assessed using a human liver cell line (HepG2) and human embryonic kidney 293 (HEK293) cells cultured in RPMI 1640 and Dulbecco modified Eagle medium, respectively, supplemented with 10% heat-inactivated fetal bovine serum and antibiotics, in a 5% CO2 atmosphere at 37°C. When confluent, the cells were washed with respective culture media, treated with trypsin, and replated in flat-bottom 24-well plates (5 × 104 cells/well) for the determination of cell adherence, as described earlier (37). Compound 5f was added to each well at various concentrations (1 to 50 μM), and the plate was further incubated for 24 h. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (1 mg/ml in PBS; 500 μl/well) was added, followed by incubation for 3 h under the same culture conditions. The supernatant was carefully removed, and 500 μl of DMSO was added with mixing to dissolve the formazan crystals formed. The optical density was determined at 570 nm. Cell viability was expressed as the percent viability relative to the control.
Evaluation of in vivo toxicity of compound 5f according to biochemical and hematological parameters.
To check for any toxic effects of compound 5f on the liver, the serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), and bilirubin (both conjugated and unconjugated) were measured using kits from Span Diagnostics Limited (Surat, India) (38). To check for any toxic effects of compound 5f on the kidneys, the serum urea and creatinine levels were analyzed using kits from Span Diagnostics (38). To monitor the effect of compound 5f on other blood parameters, the hemoglobin content was measured, along with a total count of blood cells and a differential count of white blood cells from mice blood exposed to compound 5f (50 mg kg−1 day−1) for 4 successive days. Six animals were taken in each set, and all of the animals from all of the sets were sacrificed on day 5. The packed cell volume was determined using a conventional method by microcapillary centrifugation.
Statistical analysis.
To determine the 50% inhibitory concentration (IC50), each experiment was performed three times in triplicate. The data are expressed as the means of the three experiments and the corresponding standard deviations (SD). Statistically significant differences between groups were determined by a Student t test using GraphPad Prism 4. P values of <0.005 were considered significant.
RESULTS
Chemistry.
The hydrazones were synthesized via conjugation of differentially substituted aldehydes and hydrazines (Fig. 1). Synthesized compounds were purified by column chromatography using the ethyl acetate in hexane solvent gradient. All the mass, 1H and 13C NMR spectra of benzothiazole hydrazones (BHs) were consistent with the anticipated product structure (data not shown). Other compounds were characterized by the mass, 1H and 13C NMR spectroscopies (data not shown). All the 30 compounds were synthesized by following a similar reaction condition as described (Fig. 1). The percent yield of the synthesized compounds was given in parentheses (Fig. 1). All of these synthesized compounds were then screened to explore whether they can chelate free iron and exhibit any antiplasmodial activity.
FIG 1.

Synthesis of conjugated arylhydrazones. Hydrazones were synthesized using methanol at room temperature. Different substitutions with the percent yields of the respective compounds are mentioned in parentheses.
Iron chelation and antimalarial activity of synthesized hydrazones.
Synthesized hydrazones were screened for Fe(II) chelating activity by measuring the decrease of Fe(II)-TPTZ color in the presence of increasing concentrations of compounds at 595 nm. Absorbance was decreased due to Fe(II)-hydrazone interaction which minimizes the Fe(II)-TPTZ complexation, thus inhibiting color formation. All of the synthesized compounds showed iron-chelating activity (Table 1). The data revealed that BHs exhibited better iron-chelating activity compared to other synthesized series of compounds (compounds 1, 2, 3, and 4) (Table 1). A comparison of the iron-chelating activity in terms of a 50% decrease in the absorbance at 595 nm of synthesized compounds showed that methoxylated hydrazones had less binding affinity compared to hydrazones containing free hydroxyl groups (Table 1). Deferoxamine was used as a reference compound (Table 1).
TABLE 1.
Iron-chelating and in vitro antimalarial activities of synthesized conjugated hydrazone derivatives
| Compound | Activity (mean concn [μM] ± SEM)a |
|
|---|---|---|
| Iron-chelating activity | In vitro antimalarial activity | |
| 1a | 83.86 ± 5.30 | NA |
| 1b | 75.24 ± 8.57 | NA |
| 1c | >100 | NA |
| 1d | >100 | 59.48 ± 9.55 |
| 1e | 11.73 ± 2.73 | 71.33 ± 6.93 |
| 1f | 9.73 ± 1.89 | 42.96 ± 3.63 |
| 2a | 83.66 ± 9.33 | NA |
| 2b | >100 | NA |
| 2c | >100 | NA |
| 2d | >100 | 62.16 ± 8.54 |
| 2e | 13.90 ± 2.82 | 78.85 ± 7.22 |
| 2f | 15.12 ± 4.15 | 65.56 ± 4.91 |
| 3a | 65.24 ± 10.95 | NA |
| 3b | 53.58 ± 7.30 | NA |
| 3c | >100 | NA |
| 3d | >100 | >100 |
| 3e | 8.94 ± 3.14 | >100 |
| 3f | 12.26 ± 4.63 | 89.44 ± 10.74 |
| 4a | 21.37 ± 4.15 | NA |
| 4b | 12.48 ± 1.68 | NA |
| 4c | >100 | NA |
| 4d | >100 | 92.98 ± 6.93 |
| 4e | 2.45 ± 0.99 | >100 |
| 4f | 2.51 ± 1.97 | 76.55 ± 9.16 |
| 5a | 8.28 ± 3.22 | >100 |
| 5b | 6.10 ± 1.81 | NA |
| 5c | 43.73 ± 5.89 | NA |
| 5d | 78.11 ± 8.25 | 27.11 ± 2.87 |
| 5e | 2.35 ± 0.35 | 53.87 ± 4.32 |
| 5f | 1.87 ± 0.24 | 16.95 ± 1.19 |
| Deferoxamine | 0.72 ± 0.19 | 27.13 ± 2.03 |
| Chloroquine | ND | 0.020 ± 0.0012 |
| Artesunate | ND | 0.015 ± 0.0009 |
Values indicate the means (± the standard errors of the means, where applicable) of a single parallel determination performed in triplicate. Iron-chelating activity was determined as a 50% decrease in TPTZ-Fe(II) absorbance. In vitro antimalarial activity was determined using a SYBR green assay and is expressed as the IC50. NA, not applicable; ND, not determined.
We were then interested in evaluating the effect of these compounds on the growth of P. falciparum by using a SYBR green assay. This dye binds to DNA and shows fluorescence depending on the number of parasites present in the culture. The data indicated that among the synthesized hydrazones, free hydroxyl groups containing hydrazones inhibited SYBR green fluorescence significantly, indicating the inhibition of P. falciparum growth in culture, whereas methoxylated hydrazones did not inhibit parasite growth (Table 1). The IC50 data indicated that BHs (compounds 5a to 5f) were more efficient at inhibiting P. falciparum growth than were other synthesized compounds (compounds 1, 2, 3, and 4). Furthermore, among the BH compounds, 5f was found to be the most effective antiplasmodial (Table 1). Deferoxamine, a standard iron chelator, also exhibited antiplasmodial activity in vitro (Table 1). Known antimalarials, chloroquine and artesunate, were used as positive controls (Table 1).
Structure-activity relationship study.
Based on the antimalarial activity of synthesized hydrazones, we attempted to search for the active functional group behind their activity. Previous results showed that among the synthesized molecules, BHs were more effective at chelating iron and against P. falciparum growth compared to other hydrazone derivatives (compounds 1, 2, 3, and 4). Various hydrazones derived from aromatic hydrazines bearing differential substitutions such as chloro-fluoro (1a-f), bromo (2a-f), cyano (3a-f), and hydroxyl (4a-f) could not enhance their antimalarial activity. Hence, extraconjugation of an electron-withdrawing group (−CN) or an electron-donating group (−OH), along with the substitution of halogen groups on the aromatic ring, did not result in any enhanced hydrazone activity. In contrast, BH-derived molecules showed both higher antimalarial and iron-chelating activity (Table 1). Conjugation of the simple aromatic system fused with a nitrogen- and sulfur-substituted five-membered ring might be the reason for the improved activity of the BHs. Among the BHs, the methoxy-substituted aldehydes were ineffective, based on the data regarding iron chelation, heme binding, and in vitro antimalarial activities (Tables 1 and 2). 3,5- and 3,4-hydroxy-substituted BHs were found to be the more effective, whereas only 4-hydroxy-substituted BH was futile against P. falciparum (Table 1). When iron-chelating and antiplasmodial activities were compared, compound 5f (3,4-hydroxy substituted benzothiazole) was found to be the most efficient.
TABLE 2.
Heme-binding activity of benzothiazole hydrazonesa
| Compound | Heme binding (mean KD [μM] ± SD) |
|---|---|
| 5a | 9.69 ± 1.8 |
| 5b | 9.23 ± 2.3 |
| 5c | 11.19 ± 4.1 |
| 5d | 5.58 ± 1.2 |
| 5e | 3.03 ± 0.9 |
| 5f | 1.17 ± 0.8 |
Differential optical Soret spectroscopy was performed in order to determine benzothiazole hydrazone compound-heme interaction, and the KD values were calculated from 1/λmax(nm) versus 1/[compounds 5a to 5f] (Fig. 3). KD, dissociation constant for compound-heme interaction. All experiments were performed in triplicate.
Compound 5f interacts with Fe(III).
To further understand the interaction of hydrazone with Fe(III), we performed IR spectroscopy of the compound 5f alone upon forming its complex with Fe(III). The IR spectrum of compound 5f exhibited prominent peaks corresponding to the two O-H groups (the peak corresponding to the bond marked “d”) at 3,466.41 cm−1 (stretch) and one hydrazone N-H group (the peak corresponding to the bond marked “b”) at 3,245.81 cm−1 (stretch) (Fig. 2A). A higher-frequency stretching of the aromatic 5-membered C=N group (the peak corresponding to the bond marked “a”) appeared at 1,602.60 cm−1 (stretch) and of hydrazone C=N group (the peak corresponding to the bond marked “c”) appeared at 1,562.03 cm−1 (stretch) (Fig. 2A). In contrast, the complex exhibited a broad peak at 3,418.17 cm−1 instead of two distinct peaks of O-H and N-H present in compound 5f. The IR peak corresponding to bond “b” (N-H stretching) at 3,245.81 cm−1 totally disappeared in the complex. The position of the peaks of compound 5f at 1,602.60 cm−1, 1,513.32 cm−1, and 1,458.23 cm−1 were shifted in the compound 5f-Fe(III) complex at 1,627.21 cm−1, 1,532.94 cm−1, and 1,467.74 cm−1, respectively, indicating the involvement of these specific bonds for iron complexation (Fig. 2B). The stretching frequency at 1,562.03 cm−1 (the peak corresponding to the bond marked “c”) in the parent compound disappeared in the Fe(III)-compound 5f complex, indicating the alteration of the hydrazone C=N group (Fig. 2B).
FIG 2.

IR spectra of compound 5f and its iron complex. IR spectra of compound 5f alone (A) and compound 5f plus Fe(III) (B). The structure of compound 5f with the bonds responsible for the IR peaks is assigned using alphabetical notation in the inset in panel A. Arrows indicate the change of IR peaks due to chelation.
BHs interact with heme.
Heme is an iron-containing tetrapyrrole and, because hydrazones can interact with iron, we were interested in testing the heme-interacting property of BHs. The binding of BHs to heme was evaluated by differential optical spectroscopy and, subsequently, binding constants were determined (Fig. 3 and Table 2). The approximate KD values for the binding of BHs to heme were analyzed from the plot of 1/Δλmax against 1/[BHs] (Fig. 3, insets). Compound 5f exhibited the highest affinity to heme (KD = 1.17 ± 0.8 μM), as evidenced by a comparative analysis of the equilibrium dissociation constant (KD) of the heme interaction of various BHs (Table 2).
FIG 3.

Interaction of BHs with heme. (A to F) The interaction of BHs (compounds 5a to 5f) with heme was studied by using optical-differential Soret spectroscopy at concentrations from 1 to 20 μM (i, 1 μM; f, 20 μM). Compound names are indicated in brackets. The inset shows the plot of 1/λmax(nm) versus 1/[compounds 5a to 5f] used to calculate the KD values of BH-heme interaction.
BHs inhibit hemozoin (β-hematin) formation by parasite lysate in vitro.
Heme-interacting molecule may inhibit hemozoin formation (39, 40). Thus, to check the effect of these molecules on hemozoin formation, we measured hemozoin formation in vitro by parasite lysate in presence or absence of different BHs. The data revealed that BHs in a concentration-dependent manner inhibited hemozoin formation (Fig. 4). Dose-response studies indicated that among BHs, compound 5f, followed by compounds 5d and 5e, showed potent activity against hemozoin formation (Fig. 4).
FIG 4.
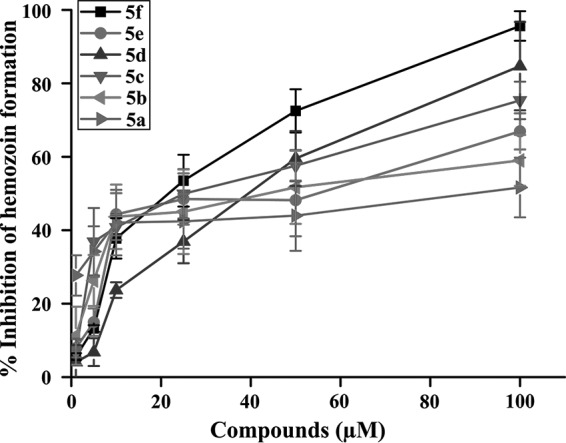
Inhibition of hemozoin formation by BHs. Compounds 5a to 5f inhibit hemozoin formation and favor the accumulation of free heme in the parasite. Hemozoin formation was measured in the presence or absence of BHs as described in Materials and Methods. Values are normalized by using the upper and lower plateaus of the best-fit curve as 100 and 0% inhibitions, respectively, of hemozoin formation. Each experiment was performed in triplicate. The data are shown as means ± the SD.
Compound 5f inhibits hemozoin (β-hematin) formation and [3H]hypoxanthine uptake in P. falciparum.
Since compound 5f is the most active compound among the BHs, its efficacy in inhibition of hemozoin formation was further evaluated in parasites in culture. P. falciparum was treated with increasing concentrations of compound 5f and kept for 48 h. The data indicated that inhibition of hemozoin formation in P. falciparum was significantly increased by increasing concentrations of compound 5f (Fig. 5A). Compound 5f, concentration dependently, not only inhibited hemozoin formation but also P. falciparum growth, as evidenced by [3H]hypoxanthine incorporation into parasitic DNA (Fig. 5B). Treatment with vehicle (DMSO) did not show any effect on parasite growth (data not shown).
FIG 5.
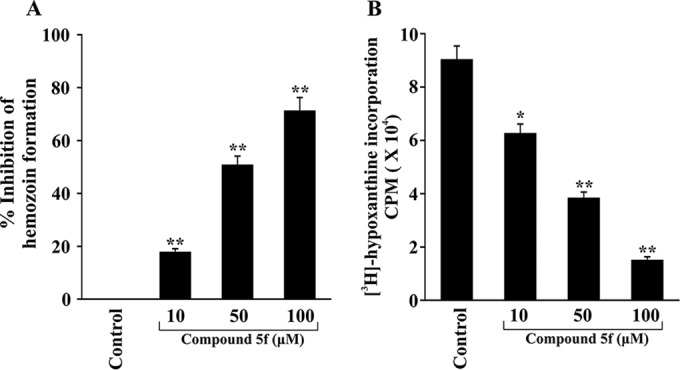
Dose-dependent inhibition of hemozoin formation and [3H]hypoxanthine uptake by compound 5f. (A) Hemozoin formation was measured from a P. falciparum culture with increasing doses of compound 5f. The data are presented as the percent inhibition of hemozoin formation compared to the control. Data are the means ± the SD from three independent experiments (**, P < 0.05 versus control). (B) P. falciparum growth was monitored by measuring the incorporation of [3H]hypoxanthine into parasite DNA and is expressed as the average radioactive counts/minute (cpm) in a liquid scintillation counter. Data are the means ± the SD from three independent experiments (*, P < 0.5; **, P < 0.05 [versus the control]).
Iron supplementation inhibits the antiplasmodial activity of compound 5f in vitro in P. falciparum culture.
Next, we were interested in determining the effect of iron supplementation on the antiplasmodial activity of compound 5f in vitro in a P. falciparum culture. We performed an iron supplementation assay with various concentrations of FeCl3 (1 to 100 μM) against a fixed concentration of compound 5f (25 μM, in which an ∼75% inhibition of parasite growth was observed). Iron supplementation offered significant, although not complete, protection against the antiplasmodial effect of compound 5f. The data indicated that the antiplasmodial activity of compound 5f gradually decreased in the presence of increasing concentrations of FeCl3 (Fig. 6). No significant change in the parasite growth was observed when iron is supplemented in the absence of compound 5f (data not shown).
FIG 6.
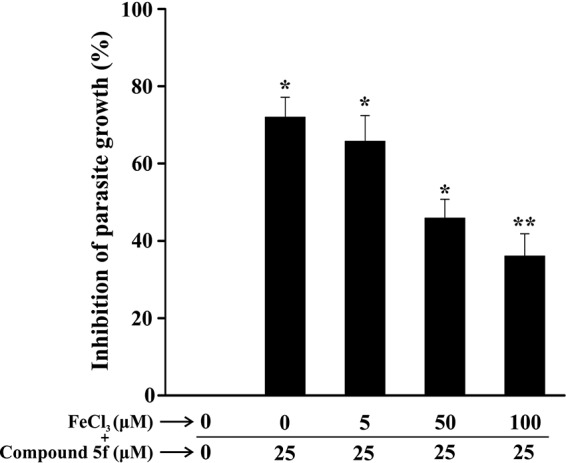
Effect of iron supplementation on the antimalarial activity of compound 5f in vitro. The effect of compound 5f (25 μM) on P. falciparum growth was monitored in the presence of increasing concentrations of FeCl3 (0 to 100 μM). After 48 h of treatment, the parasites were lysed in 100 μl of lysis buffer containing 2× SYBR green nucleic I acid stain, followed by incubation for 1 h. Data were collected upon excitation at 485 nm and after emission at 530 nm in terms of the fluorescence intensities. The experiment was performed in triplicate. Data are shown as means ± the SD (*, P < 0.5; **, P < 0.05 [versus the control]).
In vitro antimalarial activity of compound 5f against chloroquine-resistant P. falciparum K1.
We were interested in evaluating the antimalarial activity of compound 5f against a chloroquine- and pyrimethamine-resistant strain of P. falciparum (K1) in vitro. Interestingly, compound 5f offered significant antiplasmodial activity in a concentration-dependent fashion (Fig. 7). The IC50 of compound 5f was found to be 14.17 ± 1.34 μM. Both artesunate and chloroquine were used as reference drugs to validate the resistant strain of P. falciparum. Although artesunate (100 nM) inhibited the growth of parasite, chloroquine (100 nM), which was effective against 3D7 strain, failed to inhibit the growth of this parasite (Fig. 7).
FIG 7.
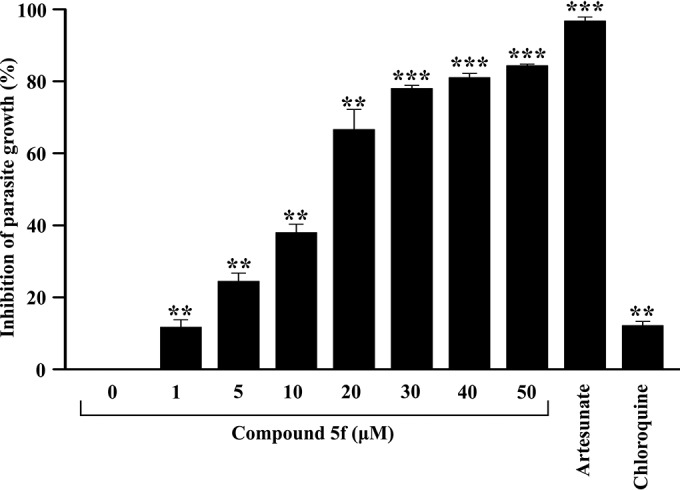
Antimalarial activity of compound 5f in the chloroquine- and pyrimethamine-resistant P. falciparum K1 strain. The growth of a chloroquine- and pyrimethamine-resistant P. falciparum K1 strain was evaluated in the presence of different concentrations of compound 5f (1 to 50 μM). Artesunate (100 nM) and chloroquine (100 nM) were used as a positive and negative controls, respectively. The experiment was performed in triplicate. Data are shown as means ± the SD (**, P < 0.05; ***, P < 0.005 [versus the control]).
In vivo antimalarial activity of compound 5f against an MDR P. yoelii malaria parasite.
We sought to determine the in vivo antimalarial activity of the lead compound 5f against a lethal MDR P. yoelii strain in a murine model. Postinfection administration of compound 5f (from days 4 to 7) showed dose-dependent antimalarial activity in vivo (Table 3). Compound 5f not only suppressed the mean parasitemia by 31, 57, and 74% at dosages of 10, 25, and 50 mg kg−1, respectively, but also significantly increased the survival of infected mice, as evidenced by the number of living mice in treated groups compared to infected controls (Table 3). The administration of compound 5f did not lead to any mortality among uninfected mice at the highest concentration (50 mg kg−1) we used for antimalarial evaluation (data not shown). The suppression of parasite growth by compound 5f in Swiss albino mice was evaluated by determining the parasitemia levels on the slides of thin blood smears under a light microscope during infection. The occurrence of lethal P. yoelii parasites on day 7 was measured in infected controls, the α/β-arteether-treated group (50 mg kg−1), and the experimental group treated with increasing concentrations of compound 5f (Table 3). Compound 5f showed antimalarial activity against the MDR P. yoelii strain, where existing halofantrine, mefloquine, and chloroquine were ineffective, as described earlier (20). At a dose of 50 mg kg−1, α/β-arteether resulted in 100% parasite clearance, whereas compound 5f showed parasite suppression in the range of 60 to 90% (Table 3). Antimalarial activity of compound 5f against P. yoelii was also evaluated in vivo by monitoring parasite growth suppression after performing Peter's four-day test. The administration of compound 5f (intraperitoneal) produced dose-dependent chemosuppression (Table 4). A significant reduction in parasitemia was observed in treated mice compared to vehicle-treated controls. α/β-Arteether treatment was used as a positive control (Table 4).
TABLE 3.
Antimalarial activity of compound 5f against an MDR P. yoelii strain in vivo in a mouse modela
| Compound | Dose (mg/kg) | Mean % parasitemia ± SD |
Mean % parasitemia suppression ± SDb | Survival rate | |||
|---|---|---|---|---|---|---|---|
| Day 2 | Day 4 | Day 6 | Day 7 | ||||
| Control | 2.5 ± 0.3 | 13.3 ± 2.2 | 42.5 ± 4.5 | 58.6 ± 9.5 | 0 | 0/6 | |
| 5f | 10 | 3.5 ± 1.1 | 15.8 ± 0.9 | 28.7 ± 2.9 | 40.3 ± 3.3 | 31.2 ± 3.6 | 0/6 |
| 25 | 2.8 ± 0.8 | 14.9 ± 1.9 | 18.6 ± 3.1 | 24.8 ± 2.6 | 57.7 ± 6.4 | 2/6 | |
| 50 | 2.6 ± 1.3 | 14.1 ± 1.4 | 12.6 ± 3.1 | 14.8 ± 2.6 | 74.7 ± 8.3 | 5/6 | |
| α/β arteether | 3.2 ± 0.4 | 14.2 ± 1.4 | 6.5 ± 0.7 | 0 | 100 | 6/6 | |
Swiss albino mice (six mice in each group) were infected by an MDR P. yoelii strain (i.e., resistant to chloroquine, mefloquine, and halofantrine) and treated with compound 5f (at 10, 25, and 50 mg/kg [body weight]) from days 4 to 7 intraperitoneally, and the parasitemia was monitored over time.
The percent parasitemia suppression at day 7 was calculated as follows: [(C − T)/C] × 100, where C is the parasitemia in the control group and T is the parasitemia in the treated group. The data represent the means ± SD (n = 6). The survival number was determined at day 10 (in the control group, all of the mice were dead at day 7).
TABLE 4.
Four-day parasite-suppressive activity of compound 5f against an MDR P. yoelii strain in mice
| Compound | Dose (mg/kg) | Mean % ± SDa |
|
|---|---|---|---|
| Parasitemia | Suppression | ||
| Control | 58.65 ± 0.3 | 0 | |
| 5f | 10 | 41.91 ± 1.1* | 28.48 ± 1.26 |
| 25 | 24.73 ± 0.8* | 57.79 ± 4.43 | |
| 50 | 11.32 ± 1.3* | 80.69 ± 3.59 | |
| α/β arteether | 50 | 0** | 100 |
*, P < 0.05; **, P < 0.005 (compared to the control).
Cytotoxicity analysis of compound 5f.
After evaluating the antimalarial activity of compound 5f, we sought to determine the cytotoxic activity of compound 5f in the host. Therefore, we monitored hemolysis by measuring the release of hemoglobin from RBCs in presence or absence of various concentrations of compound 5f. The data showed that compound 5f did not produce any significant hemolysis at concentrations of 0 to 100 μM (Fig. 8A). Saponin treatment was used as a positive control. We also checked the release of hemoglobin in presence or absence of compound 5f at 100 μM by Soret spectroscopy (Fig. 8A, inset). Spectral analysis revealed that the compound did not cause significant hemolysis (Fig. 8A, inset) compared to saponin. We also assessed the cytotoxic effect of compound 5f in mammalian HepG2 and HEK293 cells by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] reduction assay. The data indicated that compound 5f did not produce any significant toxicity to HepG2 cells, as well as HEK293 cells, at concentrations even higher than the concentration required for exhibiting in vitro antimalarial activity (Fig. 8B).
FIG 8.

Compound 5f did not show cytotoxic effects in vitro. (A) RBC lysis was measured in the presence of compound 5f at various concentrations (0 to 100 μM) at 2 and 12 h. (Inset) Soret spectra of supernatant of untreated RBCs and RBCs incubated with saponin/compound 5f (100 μM) at 2 and 12 h provided in the range of 300 to 700 nm. (B) The viability of HepG2 and HEK293 cells was assessed by an MTT assay of triplicate cultures, expressed as the percentage of untreated control. Data represent means ± the SD (**, P < 0.05 [versus 0 μM in HepG2 cells]; ##, P < 0.05 [versus 0 μM in HEK293 cells]).
Biochemical and hematological evaluation.
Having performed all of the in vitro toxicity assays, we next checked any probable in vivo toxicity of compound 5f by measuring the biochemical and hematological parameters in mice after compound 5f treatment for four successive days. The data revealed that compound 5f did not alter the levels of ALT, AST, GGT, total bilirubin, conjugated bilirubin, and unconjugated bilirubin, indicating no hepatotoxicity upon treatment with an effective dose of compound 5f (Table 5). The serum level of both creatinine and urea in the presence of compound 5f was found to be close to the range of the normal control level (Table 5), indicating that this compound did not significantly affect renal functions at the effective dose. The hematological parameters were also assessed for the possible toxicity of the compound 5f in vivo. Compound 5f did not produce any significant changes in the hemograms of treated mice compared to controls (Table 6).
TABLE 5.
Biochemical analysis of serum of mice treated with compound 5f
| Parameter | Mean ± SD |
|
|---|---|---|
| Control | Compound 5f (50 mg/kg)a | |
| AST (U/liter) | 62.33 ± 3.32 | 68.94 ± 2.59 |
| ALT (U/liter) | 41.00 ± 2.64 | 43.58 ± 1.70 |
| ALP (U/liter) | 89.00 ± 4.58 | 74.22 ± 5.50 |
| GGT (U/liter) | 9.66 ± 1.52 | 7.35 ± 2.51 |
| Total bilirubin (mg/dl) | 0.22 ± 0.13 | 0.26 ± 0.13 |
| Conjugated bilirubin (mg/dl) | 0.07 ± 0.04 | 0.13 ± 0.05 |
| Unconjugated bilirubin (mg/dl) | 0.15 ± 0.08 | 0.13 ± 0.05 |
| Creatinine (mg/dl) | 0.30 ± 0.04 | 0.24 ± 0.12 |
| Urea (mg/dl) | 38.33 ± 3.73 | 40.33 ± 2.14 |
P < 0.05 (compared to the control) for all of the values in this column.
TABLE 6.
Hemogram of mice treated with compound 5f
| Parameter | Control | Compound 5f (50 mg/kg) |
|---|---|---|
| Hemoglobin (g%) | 12.80 | 11.8 |
| Neutrophil (%) | 31 | 21 |
| Lymphocyte (%) | 68 | 79 |
| Monocyte (%) | 0 | 0 |
| Eosinophil (%) | 1 | 0 |
| Basophil (%) | 0 | 0 |
| Packed cell vol (%) | 42.00 | 38.00 |
DISCUSSION
In this study, we report the synthesis of a series of hydrazone derivatives that exhibited iron-chelating and antiplasmodial activities in vitro. We also provide evidence that compound 5f, a lead molecule, exhibited a high free heme-binding property and significantly inhibited hemozoin formation in P. falciparum. Moreover, compound 5f showed antimalarial activity in vivo against a lethal MDR strain of P. yoelii in a mouse model.
Iron is crucial for many biochemical reactions involved in the growth and multiplication of the malaria parasite that can be repressed by iron chelators. A number of biochemical studies have indicated that deprivation of iron would adversely affect the activity of ribonucleotide reductase, cytochrome, and other vital cellular functions, which in turn inhibit the growth of the malaria parasite (16, 21). Parasite digests host hemoglobin and as a consequence free heme is released, which is very toxic to the parasite (3, 14, 41). However, parasite converts toxic free heme into a less-toxic hemozoin for its own survival (42). Iron chelators may interact with free heme through the iron present at its center. This kind of interaction may subsequently hinder hemozoin formation from free heme, which in turn may lead to the inhibition of parasite growth. Among the synthesized molecules, hydroxylated hydrazones showed more free iron-chelating activity compared to the methoxylated hydrazones. Hydroxyl group contains a labile hydrogen atom, which may be replaced or can take part in conjugation during chelation with iron, whereas in the case of the methoxy group, the C-O bond is too strong to be replaced in the normal physiological condition. We found that the introduction of polar or ionizable groups within compounds rendered them more active for free iron chelation. To determine the outcome of iron chelation on parasite health, the antimalarial activity of synthesized hydrazones was screened by a SYBR green assay. The highest inhibition was obtained for hydroxylated compounds of heterocyclic BHs. The data further suggested the importance of ionizable hydroxyl groups on iron chelation and its consecutive effect on parasite growth. Among the synthesized hydrazones, compound 5f was found to be the most active compound. Interaction of compound 5f with Fe(III) was further evaluated by complex formation and subsequent identification by IR spectroscopy. IR spectroscopy is a spectroscopic tool that provides valuable information on alterations in bond vibration when a compound binds to a metal compared to the original compound. The omission and shifting of specific stretching frequencies corresponding to particular bonds in complexes provides an indication of their interaction with metal. We evaluated the alterations in bond vibration to explore the involvement of specific bonds of the lead molecule (compound 5f) when it forms a complex with Fe(III). Owing to a structural similarity of BHs, they are expected to interact with Fe(III) in the similar way, reducing the need for testing compound-Fe(III) complex formation with other BHs. Thus, spectral changes in the Fe(III)-compound 5f complex occurred as a consequence of the reorientation of bonds in compound 5f in the presence of Fe(III).
A compound which interacts with or chelates iron may also interact with iron present in free heme, and inhibition of hemozoin formation by a novel heme-interacting molecule is considered to be an effective way to prevent the growth of malarial parasites (43, 44). Hence, synthesized active BHs were screened against heme-binding activity by differential spectroscopy. Compound 5f showed the lowest dissociation constant (KD) for heme interaction, as well as the maximum inhibitory power against hemozoin formation. To determine the impact of the inhibition of hemozoin formation in parasite growth, we concurrently measured the content of hemozoin in parasites and the growth of parasites at different concentrations of compound 5f. Concentration-dependent inhibition of both hemozoin formation and parasite growth was observed with compound 5f. At this point, we were interested to determine the effect of iron supplementation on the antimalarial activity of compound 5f. Iron supplementation was supposed to produce a free iron-compound 5f complex, decreasing the affinity of compound 5f for free cellular iron. Iron supplementation significantly reduced the antimalarial activity of compound 5f. However, the inhibition of antimalarial activity upon iron supplementation was not absolute because compound 5f might have display other mechanisms that inhibit parasite growth. The inhibition of hemozoin formation may be one of the ways compound 5f exhibited a significant inhibition of hemozoin formation. We observed that deferoxamine showed higher iron chelation activity but less antimalarial activity than did compound 5f. The reason for this may be the inability of deferoxamine to inhibit hemozoin formation (45). We further checked the antimalarial activity of compound 5f against the resistant P. falciparum K1 strain. Compound 5f significantly inhibited P. falciparum growth both in chloroquine-sensitive and in chloroquine/pyrimethamine-resistant strains. In vitro-active antiplasmodial molecules may be active or inactive in vivo because, under in vivo conditions, there are many other factors that may play a significant part in deciding drug activity. The drug may encounter an absorption problem, or it may undergo degradation by host cells. Moreover, a drug must be specific in targeting parasite without harming host cells. The advantage of small molecules as the drug is their lower probability of degradation under physiological conditions. In vivo drug testing is important because it shows the effects of a new compound on a living subject. On the other hand, the development of resistance in parasites against the most effective drugs in use is a serious issue and demands immediate attention (46). Therefore, after confirming the activity of compound 5f in chloroquine-sensitive and -resistant P. falciparum strains, we used an MDR P. yoelii parasite for in vivo drug testing. We performed animal drug testing under highly parasitemic conditions to determine the drug efficacy when the number of parasites is high enough to cause physiological stress in the host (47). Moreover, under such conditions, we were able to compare the lead molecule with the frontline antimalarial α/β-arteether that is known to be effective in a similar situation (47). Compound 5f significantly suppressed the parasite growth. The activity of this molecule against drug-resistant parasites highlighted its efficacy in avoiding common mechanisms of drug resistance present in malarial parasites. After evaluation of the antimalarial activity of compound 5f, we measured both the in vitro and the in vivo toxicity of this compound before establishing its therapeutic potential. Hemolysis of host RBCs by any compound may also lead to the death of the malaria parasite. To determine whether compound 5f has any hemolytic activity, we performed a hemolysis assay. The data indicated that compound 5f was nonhemolytic; thus, the antimalarial activity is not due to RBC lysis. Furthermore, we evaluated the cytotoxicity of the compound 5f in HepG2 and HEK293 cells, where no significant change in cell viability was observed, indicating the nontoxic nature of compound 5f in vitro against mammalian cells. Since xenobiotic metabolism is mostly attributed to liver and kidney, the evaluation of liver and kidney functions was extremely relevant in order to check any potential in vivo toxicity of compound 5f. Therefore, mice were treated for four successive days with compound 5f, whereupon the serum levels of AST, ALT, ALP, GGT, bilirubin, creatinine, and urea, along with the hematological parameters, were stringently measured. All of the biochemicals, as well as the hematological parameters, were found to be comparable to the control (healthy) mice, indicating the negligible or nontoxic nature of compound 5f. Thus, we identified the lead compound 5f, which is active in vivo against resistant and highly lethal P. yoelii and may be developed as a novel antimalarial drug.
The effectiveness of current antimalarial treatments is threatened by the naturally occurring evolution of the parasites that allows the selection of resistant strains. Existing antimalarial drugs mostly contain a few basic chemotypes, against which malarial parasites have become resistant (48). As the threat of antimalarial drug resistance grows, there is growing pressure for the discovery of new antimalarial drugs that are able to overcome these issues. Here, we identified BHs as a new chemotype that can prevent the growth of malarial parasites. This chemotype may be used as a scaffold to design next-generation antimalarial drugs. Proper investigation of its structure activity relationship and its suitable optimization by side chain modifications may result in better bioavailability and improved efficacy. Thus, compound 5f is a lead chemotype that has shown antimalarial activity both in vitro and in vivo against resistant parasites and can be modified into effective compound by fragment-based modification or combination therapy with artemisinin and other well-known antimalarial agents.
ACKNOWLEDGMENTS
This study was supported by Council of Scientific and Industrial Research, New Delhi, India, through grants (SPlenDID, BSC 0104) and a fellowship to S.S. and A.A.S. Financial support from the Department of Science & Technology, Government of India (J. C. Bose Fellowship), is also acknowledged.
REFERENCES
- 1.Goldberg DE. 1993. Hemoglobin degradation in Plasmodium-infected red blood cells. Semin Cell Biol 4:355–361. doi: 10.1006/scel.1993.1042. [DOI] [PubMed] [Google Scholar]
- 2.Hill AV. 2011. Vaccines against malaria. Philos Trans R Soc Lond B Biol Sci 366:2806–2814. doi: 10.1098/rstb.2011.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alam A, Goyal M, Iqbal MS, Pal C, Dey S, Bindu S, Maity P, Bandyopadhyay U. 2009. Novel antimalarial drug targets: hope for new antimalarial drugs. Expert Rev Clin Pharmacol 2:469–489. doi: 10.1586/ecp.09.28. [DOI] [PubMed] [Google Scholar]
- 4.White NJ. 2004. Antimalarial drug resistance. J Clin Invest 113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gelb MH, Van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. 2003. Protein farnesyl and N-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol Biochem Parasitol 126:155–163. doi: 10.1016/S0166-6851(02)00282-7. [DOI] [PubMed] [Google Scholar]
- 6.Weinberg ED, Moon J. 2009. Malaria and iron: history and review. Drug Metab Rev 41:644–662. doi: 10.1080/03602530903178905. [DOI] [PubMed] [Google Scholar]
- 7.Taylor MC, Kelly JM. 2010. Iron metabolism in trypanosomatids, and its crucial role in infection. Parasitology 137:899–917. doi: 10.1017/S0031182009991880. [DOI] [PubMed] [Google Scholar]
- 8.Atkinson CT, Bayne MT, Gordeuk VR, Brittenham GM, Aikawa M. 1991. Stage-specific ultrastructural effects of desferrioxamine on Plasmodium falciparum in vitro. Am J Trop Med Hyg 45:593–601. [DOI] [PubMed] [Google Scholar]
- 9.Breidbach T, Scory S, Krauth-Siegel RL, Steverding D. 2002. Growth inhibition of bloodstream forms of Trypanosoma brucei by the iron chelator deferoxamine. Int J Parasitol 32:473–479. doi: 10.1016/S0020-7519(01)00310-1. [DOI] [PubMed] [Google Scholar]
- 10.Merschjohann K, Steverding D. 2006. In vitro growth inhibition of bloodstream forms of Trypanosoma brucei and Trypanosoma congolense by iron chelators. Kinetoplastid Biol Dis 5:3. doi: 10.1186/1475-9292-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lytton SD, Mester B, Libman J, Shanzer A, Cabantchik ZI. 1994. Mode of action of iron(III) chelators as antimalarials: II. Evidence for differential effects on parasite iron-dependent nucleic acid synthesis. Blood 84:910–915. [PubMed] [Google Scholar]
- 12.Mabeza GF, Loyevsky M, Gordeuk VR, Weiss G. 1999. Iron chelation therapy for malaria: a review. Pharmacol Ther 81:53–75. doi: 10.1016/S0163-7258(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 13.Goyal M, Alam A, Bandyopadhyay U. 2012. Redox regulation in malaria: current concepts and pharmacotherapeutic implications. Curr Med Chem 19:1475–1503. doi: 10.2174/092986712799828328. [DOI] [PubMed] [Google Scholar]
- 14.Kumar S, Guha M, Choubey V, Maity P, Bandyopadhyay U. 2007. Antimalarial drugs inhibiting hemozoin (beta-hematin) formation: a mechanistic update. Life Sci 80:813–828. doi: 10.1016/j.lfs.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Haldar K, Murphy SC, Milner DA, Taylor TE. 2007. Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu Rev Pathol 2:217–249. doi: 10.1146/annurev.pathol.2.010506.091913. [DOI] [PubMed] [Google Scholar]
- 16.Kumar S, Bandyopadhyay U. 2005. Free heme toxicity and its detoxification systems in human. Toxicol Lett 157:175–188. doi: 10.1016/j.toxlet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Ryter SW, Tyrrell RM. 2000. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med 28:289–309. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt TH, Frezzatti WA Jr, Schreier S. 1993. Hemin-induced lipid membrane disorder and increased permeability: a molecular model for the mechanism of cell lysis. Arch Biochem Biophys 307:96–103. doi: 10.1006/abbi.1993.1566. [DOI] [PubMed] [Google Scholar]
- 19.Bandyopadhyay U, Dey S. 2011. Antimalarial drugs and molecules inhibiting hemozoin formation, p 205–234. In Apicomplexan parasites. Wiley-VCH Verlag GmbH, Berlin, Germany. [Google Scholar]
- 20.Goyal M, Singh P, Alam A, Das SK, Iqbal MS, Dey S, Bindu S, Pal C, Panda G, Bandyopadhyay U. 2012. Aryl aryl methyl thio arenes prevent multidrug-resistant malaria in mouse by promoting oxidative stress in parasites. Free Radic Biol Med 53:129–142. doi: 10.1016/j.freeradbiomed.2012.04.028. [DOI] [PubMed] [Google Scholar]
- 21.Kumar S, Das SK, Dey S, Maity P, Guha M, Choubey V, Panda G, Bandyopadhyay U. 2008. Antiplasmodial activity of [(aryl)arylsulfanylmethyl]pyridine. Antimicrob Agents Chemother 52:705–715. doi: 10.1128/AAC.00898-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal C, Sarkar S, Mazumder S, Adhikari S, Bandyopadhyay U. 2013. Synthesis and biological evaluation of primaquine-chloroquine twin drug: a novel heme-interacting molecule prevents free heme and hydroxyl radical-mediated protein degradation. MedChemComm 4:731–736. doi: 10.1039/c3md00019b. [DOI] [Google Scholar]
- 23.Abiodun OO, Brun R, Wittlin S. 2013. In vitro interaction of artemisinin derivatives or the fully synthetic peroxidic anti-malarial OZ277 with thapsigargin in Plasmodium falciparum strains. Malar J 12:43. doi: 10.1186/1475-2875-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Guha M, Choubey V, Maity P, Srivastava K, Puri SK, Bandyopadhyay U. 2008. Bilirubin inhibits Plasmodium falciparum growth through the generation of reactive oxygen species. Free Radic Biol Med 44:602–613. doi: 10.1016/j.freeradbiomed.2007.10.057. [DOI] [PubMed] [Google Scholar]
- 26.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. 2004. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernhardt PV, Chin P, Sharpe PC, Wang JY, Richardson DR. 2005. Novel diaroylhydrazine ligands as iron chelators: coordination chemistry and biological activity. J Biol Inorg Chem 10:761–777. doi: 10.1007/s00775-005-0018-0. [DOI] [PubMed] [Google Scholar]
- 28.Pal C, Kundu MK, Bandyopadhyay U, Adhikari S. 2011. Synthesis of novel heme-interacting acridone derivatives to prevent free heme-mediated protein oxidation and degradation. Bioorg Med Chem Lett 21:3563–3567. doi: 10.1016/j.bmcl.2011.04.127. [DOI] [PubMed] [Google Scholar]
- 29.Choubey V, Guha M, Maity P, Kumar S, Raghunandan R, Maulik PR, Mitra K, Halder UC, Bandyopadhyay U. 2006. Molecular characterization and localization of Plasmodium falciparum choline kinase. Biochim Biophys Acta 1760:1027–1038. doi: 10.1016/j.bbagen.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Choubey V, Maity P, Guha M, Kumar S, Srivastava K, Puri SK, Bandyopadhyay U. 2007. Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: a possible antimalarial mechanism. Antimicrob Agents Chemother 51:696–706. doi: 10.1128/AAC.00919-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey AV, Singh N, Tekwani BL, Puri SK, Chauhan VS. 1999. Assay of beta-hematin formation by malaria parasite. J Pharm Biomed Anal 20:203–207. doi: 10.1016/S0731-7085(99)00021-7. [DOI] [PubMed] [Google Scholar]
- 32.Sullivan DJ Jr, Gluzman IY, Goldberg DE. 1996. Plasmodium hemozoin formation mediated by histidine-rich proteins. Science 271:219–222. doi: 10.1126/science.271.5246.219. [DOI] [PubMed] [Google Scholar]
- 33.Trivedi V, Chand P, Maulik PR, Bandyopadhyay U. 2005. Mechanism of horseradish peroxidase-catalyzed heme oxidation and polymerization (beta-hematin formation). Biochim Biophys Acta 1723:221–228. doi: 10.1016/j.bbagen.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16:710–718. doi: 10.1128/AAC.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knight DJ, Peters W. 1980. The antimalarial activity of N-benzyloxydihydrotriazines. I. The activity of clociguanil (BRL 50216) against rodent malaria, and studies on its mode of action. Ann Trop Med Parasitol 74:393–404. [PubMed] [Google Scholar]
- 36.Ajdacic V, Senerovic L, Vranic M, Pekmezovic M, Arsic-Arsnijevic V, Veselinovic A, Veselinovic J, Solaja BA, Nikodinovic-Runic J, Opsenica IM. 2016. Synthesis and evaluation of thiophene-based guanylhydrazones (iminoguanidines) efficient against panel of voriconazole-resistant fungal isolates. Bioorg Med Chem 24:1277–1291. doi: 10.1016/j.bmc.2016.01.058. [DOI] [PubMed] [Google Scholar]
- 37.Kulikova V, Shabalin K, Nerinovski K, Dolle C, Niere M, Yakimov A, Redpath P, Khodorkovskiy M, Migaud ME, Ziegler M, Nikiforov A. 2015. Generation, release and uptake of the NAD precursor nicotinic acid riboside by human cells. J Biol Chem 290:27124–27137. doi: 10.1074/jbc.M115.664458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banerjee A, De M, Ali N. 2011. Combination therapy with paromomycin-associated stearylamine-bearing liposomes cures experimental visceral leishmaniasis through Th1-biased immunomodulation. Antimicrob Agents Chemother 55:1661–1670. doi: 10.1128/AAC.00524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziegler J, Linck R, Wright DW. 2001. Heme aggregation inhibitors: antimalarial drugs targeting an essential biomineralization process. Curr Med Chem 8:171–189. doi: 10.2174/0929867013373840. [DOI] [PubMed] [Google Scholar]
- 40.Coronado LM, Nadovich CT, Spadafora C. 2014. Malarial hemozoin: from target to tool. Biochim Biophys Acta 1840:2032–2041. doi: 10.1016/j.bbagen.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dey S, Bindu S, Goyal M, Pal C, Alam A, Iqbal MS, Kumar R, Sarkar S, Bandyopadhyay U. 2012. Impact of intravascular hemolysis in malaria on liver dysfunction: involvement of hepatic free heme overload, NF-κB activation, and neutrophil infiltration. J Biol Chem 287:26630–26646. doi: 10.1074/jbc.M112.341255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Francis SE, Sullivan DJ Jr, Goldberg DE. 1997. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu Rev Microbiol 51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 43.Sandlin RD, Fong KY, Wicht KJ, Carrell HM, Egan TJ, Wright DW. 2014. Identification of beta-hematin inhibitors in a high-throughput screening effort reveals scaffolds with in vitro antimalarial activity. Int J Parasitol Drugs Drug Resist 4:316–325. doi: 10.1016/j.ijpddr.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorka AP, Alumasa JN, Sherlach KS, Jacobs LM, Nickley KB, Brower JP, de Dios AC, Roepe PD. 2013. Cytostatic versus cytocidal activities of chloroquine analogues and inhibition of hemozoin crystal growth. Antimicrob Agents Chemother 57:356–364. doi: 10.1128/AAC.01709-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sonnet P, Mullie C. 2011. In vitro antimalarial activity of ICL670: a further proof of the correlation between inhibition of beta-hematin formation and of peroxidative degradation of hemin. Exp Parasitol 128:26–31. doi: 10.1016/j.exppara.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 46.Mita T, Tanabe K, Kita K. 2009. Spread and evolution of Plasmodium falciparum drug resistance. Parasitol Int 58:201–209. doi: 10.1016/j.parint.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Dey S, Mazumder S, Siddiqui AA, Iqbal MS, Banerjee C, Sarkar S, De R, Goyal M, Bindu S, Bandyopadhyay U. 2014. Association of heme oxygenase 1 with the restoration of liver function after damage in murine malaria by Plasmodium yoelii. Infect Immun 82:3113–3126. doi: 10.1128/IAI.01598-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calderon F, Wilson DM, Gamo FJ. 2013. Antimalarial drug discovery: recent progress and future directions. Prog Med Chem 52:97–151. doi: 10.1016/B978-0-444-62652-3.00003-X. [DOI] [PubMed] [Google Scholar]