

Abstract
Staphylococcus aureus is an important cause of both hospital- and community-associated methicillin-resistant S. aureus (MRSA) infections worldwide. β-Lactam antibiotics are the drugs of choice to treat S. aureus infections, but resistance to these and other antibiotics make treatment problematic. High-level β-lactam resistance of S. aureus has always been attributed to the horizontally acquired penicillin binding protein 2a (PBP 2a) encoded by the mecA gene. Here, we show that S. aureus can also express high-level resistance to β-lactams, including new-generation broad-spectrum cephalosporins that are active against methicillin-resistant strains, through an uncanonical core genome-encoded penicillin binding protein, PBP 4, a nonessential enzyme previously considered not to be important for staphylococcal β-lactam resistance. Our results show that PBP 4 can mediate high-level resistance to β-lactams.
INTRODUCTION
Antibiotic resistance presents an important therapeutic challenge and constitutes a considerable burden medically and economically (1–3). Staphylococcus aureus is an important bacterial pathogen that causes a wide variety of community- and health care-related infections, and widespread β-lactam resistance in S. aureus has been a growing problem worldwide (1, 4, 5).
The antibacterial activity of β-lactams is mediated by covalent binding to penicillin binding proteins (PBPs), thereby inhibiting the transpeptidase (TPase) activity of PBP-mediated bacterial cell wall synthesis (6–9). In S. aureus, class resistance to β-lactams, termed methicillin resistance, is widespread. Class resistance to β-lactams in methicillin-resistant strains of S. aureus (MRSA) is due to expression of PBP 2a or PBP 2a′, which are encoded by horizontally acquired genes (1, 10), mecA and mecC (11), respectively. PBP 2a confers class resistance to β-lactam antibiotics due to its low-affinity binding to β-lactams (8, 12). In addition to PBP 2a, MRSA has four other PBPs (PBPs 1 through 4), which are encoded in the bacterial core genome. PBPs 1 to 4 have not been thought to play a role in high-level β-lactam resistance (7, 9, 13), although PBP 2a-mediated resistance depends on PBP 2 (14).
Ceftaroline and ceftobiprole are broad-spectrum β-lactam antibiotics that are active against MRSA strains, and ceftaroline is FDA approved for treatment of skin and skin structure MRSA infections (15–18) and is clinically effective for other infections, including pneumonia and bacteremia (19). Ceftaroline and ceftobiprole are active against MRSA strains because they effectively bind to and inhibit PBP 2a. Ceftaroline inhibits PBP 2a allosterically by binding to the protein and causing a conformational change, which makes PBP 2a vulnerable to the action of a second molecule of ceftaroline or another β-lactam (20). Ceftobiprole bypasses the resistance mechanism of PBP 2a by having a vinylpyrrolidinone moiety (R2 group) that allows access to the PBP 2a active site (20, 21). MRSA strains that are resistant to ceftaroline and ceftobiprole have already been reported (13, 22–25). High-level resistance to both drugs has been attributed to point mutations in two amino acid residues (Y446 and E447) in the mecA gene (13, 26, 27). Both amino acid residues are located near the transpeptidase active site of PBP 2a, and mutations in them are predicted to interfere with drug binding (20). Moreover, Y466N and E447K mutations have been shown to cause ineffective binding of ceftaroline to PBP 2a (27).
In previous studies, we have shown that a mecA-negative mutant of the archaic S. aureus COL strain, which expresses homogeneous resistance to methicillin and other β-lactams, can express high-level ceftobiprole resistance independently of PBP 2a, indicating alternative mediators of its resistance (26). Genome sequencing of the mecA-negative ceftobiprole-resistant mutant strain (CRB) revealed mutations in three genes: pbp4, gdpP, and acrB (28). The roles of GdpP, a cyclic-di-AMP phosphodiesterase, and AcrB, an efflux pump, in β-lactam resistance are not well delineated. PBP 4 is a low-molecular-weight, core genome-encoded, nonessential PBP implicated in low- to moderate-level β-lactam resistance (29, 30). In this study, we sought to address (i) if passage in another lineage generates mutants that are similar to those found in CRB, (ii) if ceftaroline selects for mutations similar to those selected for by ceftobiprole, and (iii) what is the contribution of PBP 4, as this has not been addressed in previous work. We show that a representative strain of the USA300 community MRSA genotype, which expresses heterogeneous resistance to β-lactams and which is responsible for the majority of MRSA infections in the United States (31), can also exhibit high-level resistance to ceftobiprole, ceftaroline, and other β-lactams in the absence of mecA. Both ceftobiprole- and ceftaroline-resistant strains displayed mutations in pbp4 and gdpP. Finally, we show PBP 4 to be the driving factor responsible for this high-level resistance. These results underscore the potential role of PBP 4 in β-lactam resistance in S. aureus (32).
MATERIALS AND METHODS
Bacterial strains and plasmids.
All the strains were grown in Trypticase soy broth (TSB) with aeration, on Trypticase soy agar (TSA), or on blood agar (Remel) at 37°C. The plasmid pKOR1 was selected in bacterial medium containing ampicillin (100 μg ml−1) or chloramphenicol (10 μg ml−1) for Escherichia coli and S. aureus, respectively. Bacterial medium supplemented with tetracycline (12.5 μg ml−1) was used to select for pTXΔ. The strains used in this study are listed in Table 1. The plasmids and primers used in the study are listed in Table S1 in the supplemental material.
TABLE 1.
Strains used in the study
| Strain | MecA | Passage in: | Notes | Reference |
|---|---|---|---|---|
| E. coli | Invitrogen | |||
| RN4220 | − | Laboratory S. aureus strain | BEI Resources | |
| COLnex | − | 13 | ||
| COLnex Δpbp4 | − | pbp4 deletion mutant in COLnex | This study | |
| SF8300 | + | USA300 MRSA clinical isolate | 29 | |
| SF8300ex | − | SF8300 strain with mecA excised | 29 | |
| SF8300ex Δpbp4 | − | pbp4 deletion mutant in SF8300ex | This study | |
| SRT | − | Ceftaroline | This study | |
| SRB | − | Ceftobiprole | This study | |
| Sgap | − | SF8300ex strain in which pbp4 (E183A, F241R), gdpP (N182K), and acrB (I960V) mutations analogous to those in the CRB strain (28) were introduced | This study | |
| SgapT | − | Ceftaroline | This study | |
| Sp | − | SF8300ex strain in which pbp4 (E183A, F241R) mutations analogous to those in the CRB strain (28) were introduced | This study | |
| SpT | − | Ceftaroline | This study | |
| SRB Δpbp4 | − | pbp4 deletion mutant in SRB | This study | |
| SRT Δpbp4 | − | pbp4 deletion mutant in SRT | This study | |
| SgapT Δpbp4 | − | pbp4 deletion mutant in SgapT | This study | |
| SpT Δpbp4 | − | pbp4 deletion mutant in SpT | This study | |
| SRB Δpbp4 [empty plasmid] | − | SRB Δpbp4 with empty plasmid pTXΔ | This study | |
| SRB Δpbp4 [pbp4 (SF8300)] | − | SRB Δpbp4 complemented with pTXΔ containing pbp4 from SF8300 | This study | |
| SRB Δpbp4 [pbp4 (SRB)] | − | SRB Δpbp4 complemented with pTXΔ containing pbp4 from SRB | This study | |
| SRT Δpbp4 [empty plasmid] | − | SRT Δpbp4 with empty plasmid pTXΔ | This study | |
| SRT Δpbp4 [pbp4 (SF8300)] | − | SRT Δpbp4 complemented with pTXΔ containing pbp4 from SF8300 | This study | |
| SRT Δpbp4 [pbp4 (SRT)] | − | SRT Δpbp4 complemented with pTXΔ containing pbp4 from SRT | This study |
a +, present; −, absent.
Antibiotics.
Ceftobiprole solution was prepared fresh daily from ceftobiprole powder (provided by Johnson and Johnson Pharmaceutical Research and Development) at a concentration of 2 mg ml−1. Ceftaroline solution was prepared from ceftaroline active powder (provided by Forest Laboratories) at a concentration of 1 mg ml−1. All other antibiotics were purchased from Sigma-Aldrich.
Multipassage selection.
SF8300ex was serially passaged in ceftobiprole as previously described (26) to obtain strain SRB. Briefly, 10-ml preparations of TSB containing various concentrations of ceftobiprole were inoculated at a 1:100 dilution with overnight cultures containing 109 CFU ml−1. The ceftobiprole concentration was doubled at each passage, as tolerated. The strains SF8300ex, Sp, and Sgap were serially passaged in ceftaroline in a similar manner to obtain strains SRT, SpT, and SgapT, respectively (Table 1). The strains mentioned above were passaged until bacterial growth was observed in at least 128 μg ml−1 of the drug specified (Fig. 1). Two to four single colonies were isolated from each passaging study. MIC tests revealed that each of the colonies was similarly resistant to the respective antibiotic that was used in the passaging study (data not shown). A single colony was selected from each passaged strains and used throughout the study.
FIG 1.

Serial passaging of bacteria in ceftobiprole and ceftaroline. Bacteria were passaged every day in ceftobiprole or ceftaroline as described in Materials and Methods. (A) Strain SF8300ex passage in ceftobiprole (SRB) and ceftaroline (SRT). (B) Strain Sgap and Sp passage in ceftaroline.
Serial passaging of COLnex, SF8300ex, and their isogenic Δpbp4 strains was carried out in nafcillin as described above, except that they were grown in 50 ml TSB medium. The passaging experiment for COLnex and SF8300ex was terminated when bacterial growth was observed at 256 μg ml−1 of nafcillin (see Fig. 5).
FIG 5.
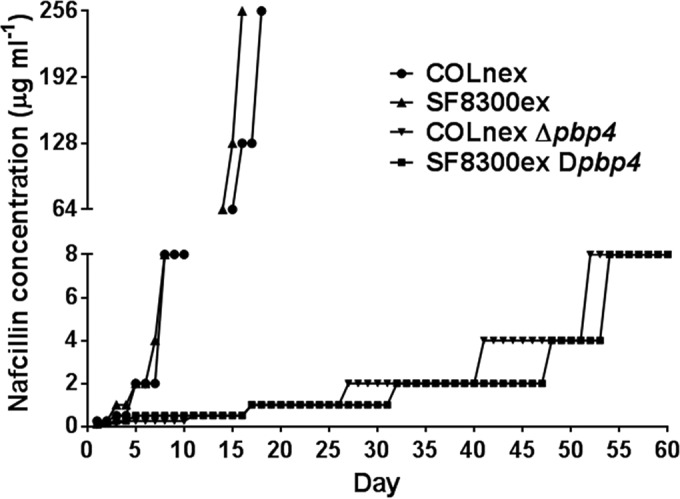
Serial passaging of bacteria in nafcillin. Bacteria were passaged every day in nafcillin as described in Materials and Methods. Each day, the drug concentration at which visible bacterial growth was observed was plotted.
Allelic exchange and complementation.
The plasmid pKOR1 was used to conduct allelic exchanges as previously described (33). Briefly, constructs for allelic exchange were created via splice overlap extension (SOE) PCR. The PCR fragments generated with the primers listed in Table S1 in the supplemental material were cloned into the plasmid pKOR1 using BP Clonase II. The constructs were transformed into E. coli Top10 cells. Purified plasmids from E. coli Top10 cells were electroporated into competent S. aureus RN4220. The plasmids were either transformed or transduced with phage ϕ11 from RN4220 to host S. aureus strains of choice. The strains were grown at 30°C and 42°C for allelic exchange. Cultures were plated onto 1 μg ml−1 anhydrotetracycline plates for counterselection.
Complementation was carried out with the constitutively expressed plasmid pTXΔ (34). The pbp4 gene was amplified with the primers specified in Table S1 in the supplemental material by standard PCR, digested with the restriction enzymes BamHI and MluI, and ligated to the empty pTXΔ plasmid digested with the same enzymes prior to the ligation step. The resultant plasmids were first selected in strain RN4220, followed by transformation or transduction to the strain of choice as stated.
Antibiotic resistance measurement.
MICs were determined by broth microdilution. Briefly, 1 × 105 CFU were incubated for 24 or 48 h at 37°C in 0.2 ml cation-adjusted Mueller-Hinton broth (CAMHB) containing increasing concentrations of antibiotic. MIC levels were recorded as the lowest concentration without growth.
Population analyses were done by the agar method, as previously described (26). Tetracycline (12.5 μg ml−1) was added to the agar plates to select for the plasmid pTXΔ whenever stated. A 10-μl volume of serially diluted culture was spotted onto agar plates containing various concentrations of antibiotic. The plates were incubated at 37°C for 72 h. The plates were read, and the results were expressed as CFU per milliliter. Both MIC and population analyses were repeated at least twice, and the data from one experiment are presented.
Sanger sequencing.
Mutations (see Table 3), including point mutations introduced in strains Sgap and Sp, were verified by Sanger sequencing. pbp4 knockout strains and complemented constructs were verified by Sanger sequencing.
TABLE 3.
Point mutations detected in S. aureus PBPs, GdpP, and AcrB
| Strain | Mutation(s)a |
|||||
|---|---|---|---|---|---|---|
| PBP1 | PBP2 | PBP3 | PBP4 | GdpP | AcrB | |
| SRB | H499R; E567K | Y437C; V445L; Q453R; M559I | W228X | E183V; F241R | T509A | |
| SRT | N138K; H270L | Y306X | ||||
| Sgap | E183Ab; F241Rb | N182Kb | I960Vb | |||
| SgapT | H499R | N138I; E183Ab; R200L; F241Rb | N182Kb | I960Vb | ||
| Sp | E183Ab; F241Rb | |||||
| SpT | G581D | N138I; E183Vb,c T201A; F241Rb | N214del | |||
X, stop codon.
Mutation was introduced by allelic replacement.
E183A was spontaneously mutated to E183V during the construction of SpT.
Bioinformatic and statistical analyses.
Statistical analyses were performed using GraphPad Prism 5.04. Comparisons between groups were analyzed using two-way analysis of variance (ANOVA) whenever stated. DNA sequence analysis was performed using DNAStar. In silico analysis of the mutations on the cefotaxime-bound PBP 4 crystal structure (Protein Data Bank [PDB] 3HUM) was carried out with UCSF Chimera (35).
RESULTS
A USA300 strain of S. aureus can attain high-level resistance to ceftobiprole and ceftaroline in the absence of mecA.
A SF8300 strain with mecA excised (SF8300ex) (36) was highly susceptible to a wide range of β-lactams compared to the isogenic wild-type strain, SF8300 (Table 2), underscoring the importance of mecA in β-lactam resistance in an S. aureus USA300 background. In contrast, both strains SF8300 and SF8300ex showed susceptibility to ceftaroline and ceftobiprole, confirming the activity of these drugs against the mecA-positive S. aureus USA300 background strain (Table 2).
TABLE 2.
MICs for antibiotic-treated strainsa
| Strain | MIC (μg ml−1)a |
||||||
|---|---|---|---|---|---|---|---|
| NAF | AMP | CTX | FOX | CFZ | BPR | CPT | |
| SF8300 | 32 | 128 | >256 | 32 | 256 | 1 | 0.5 |
| SF8300ex | 0.5 | 0.25 | 4 | 4 | 1 | 0.5 | 0.25 |
| SRB | 8 | 4 | 32 | 1 | 16 | >64 | 4 |
| SRT | 64 | >256 | >256 | 8 | >256 | 8 | >64 |
| Sgap | 0.5 | <0.25 | 4 | 4 | 0.5 | 1 | <0.25 |
| SgapT | >256 | >256 | 256 | 8 | 256 | >64 | 64 |
| Sp | 1 | 0.25 | 4 | 4 | 0.5 | 0.5 | 0.25 |
| SpT | >256 | >256 | >256 | 8 | >256 | >64 | >64 |
NAF, nafcillin; AMP, ampicillin; CTX, ceftriaxone; FOX, cefoxitin; CFZ, cefazolin; BPR, ceftobiprole; and CPT, ceftaroline. The CLSI breakpoint for resistance is ≥4 μg ml−1 (45).
To investigate if a mecA-negative strain can attain resistance to ceftobiprole and ceftaroline, we serially passaged strain SF8300ex in increasing concentrations of each drug. Strain SF8300ex attained growth in 256 μg ml−1 of ceftaroline within 15 days; the mutant strain selected for study is designated SRT. Strain SF8300ex grew in 128 μg ml−1 of ceftobiprole after 43 days of passage; the mutant strain selected for study is designated SRB (Fig. 1A; see Fig. S1 in the supplemental material). These data show that a representative USA300 strain can adapt to attain high-level resistance to either ceftobiprole or ceftaroline in the absence of mecA.
As described previously, COLnex, an archaic MRSA isolate of non-USA300 lineage and lacking the mecA gene, when passaged in ceftobiprole resulted in a highly broad-spectrum β-lactam-resistant derivative, strain, CRB (26). Whole-genome sequencing of CRB revealed point mutations in 3 genes, pbp4, gdpP, and acrB, that led to amino acid substitutions (28). To determine whether mutations detected in CRB play a role in β-lactam resistance in S. aureus USA300 background strains, we introduced mutations (N182K in GdpP, I960V in AcrB, and E183A and F241R in PBP 4) in strain SF8300ex to create strain Sgap and E183A and F241R in PBP 4 to create strain Sp (Table 1). Strains Sgap and Sp showed no detectable resistance in MIC tests performed using β-lactam drugs, including ceftaroline and ceftobiprole (Table 2). Thus, mutations associated with resistance in strain CRB were not able to confer β-lactam resistance in the USA300 background strain, probably due to marked differences in background between strains USA300 and COL (the parent of CRB).
Next, we tested if the mutations in strains Sgap and Sp can confer earlier resistance to ceftaroline by passaging both strains in increasing concentrations of ceftaroline, creating strains SgapT and SpT, respectively. Both SgapT and SpT grew at 256 μg ml−1 of ceftaroline within 15 days at a pace similar to that of strain SRT (Fig. 1A and B; see Fig. S1 in the supplemental material), suggesting that the mutations that were introduced in Sgap and Sp did not facilitate earlier development of resistance to ceftaroline.
The new-generation broad-spectrum cephalosporin-resistant isolates show broad-spectrum β-lactam resistance.
The strains passaged in ceftobiprole and ceftaroline showed high-level stable resistance against the drugs in which they were passaged. SRB (passaged in ceftobiprole) and SRT, SgapT, and SpT (passaged in ceftaroline) were highly resistant (MICs > 64 μg ml−1) compared to their parental strain (MICs ≤ 1 μg ml−1) (Table 2). Strains SRB and SRT showed lower levels of resistance to ceftaroline (MIC = 4 μg ml−1) and ceftobiprole (8 MIC = μg ml−1), respectively, supporting different modes of action for these drugs, as described previously (Table 2) (20, 21). On the other hand, both strains SpT and SgapT (passaged in ceftaroline) showed high-level cross-resistance to ceftobiprole (MICs ≥ 64 μg ml−1), suggesting the mutations introduced prior to passaging (N182K in GdpP, I960V in AcrB, and E183A and F241R in Pbp4 in Sgap and E183A and F241R in PBP 4 in Sp) might have contributed to the cross-resistance postpassaging (Table 2).
In addition, all the passaged strains showed across-the-board moderate to very high-level β-lactam resistance (nafcillin, ≥8; ampicillin, ≥4; ceftriaxone, ≥32; and cefazoline, ≥16 μg ml−1) compared to their parental strain, SF8300ex (Table 2).
The resistant isolates revealed multiple mutations in genes known to confer β-lactam resistance.
To investigate the underlying cause of resistance to ceftobiprole and ceftaroline in the passaged isolates, we sequenced all the core genome-encoded penicillin binding proteins (encoded by pbp1 through pbp4). In addition, we also sequenced the gdpP and acrB genes, in which mutations were detected in the ceftobiprole-resistant passaged strain CRB (28). Sanger sequencing of the above-mentioned candidate genes revealed that our passaged isolates primarily contained mutations in all pbp and gdpP genes (Table 3). None of the passaged isolates revealed any mutations in acrB except SgapT, in which I960V in acrB was introduced during the creation of strain Sgap (see Materials and Methods).
Among mutations found in PBP 1, H499R was detected in strains SRB and SgapT. PBP 1 is an essential PBP and primarily functions as a transpeptidase in S. aureus (37). Several mutations were detected in pbp2 in strains SRB and SpT. PBP 2 is the only bifunctional PBP in S. aureus, displaying both transglycosylase and transpeptidase activities (14). Interestingly, all the mutations detected in pbp2 were located in the transpeptidase domain. A nonsense mutation was detected in pbp3 of strain SRB that led to a premature stop codon, abolishing the entire C-terminal transpeptidase domain of PBP 3 (38). All of our passaged strains showed at least two point mutations in PBP 4, making it a highly mutated candidate in our study (Table 3). PBP 4 is a supplementary PBP known to function as a transpeptidase and a carboxypeptidase (CPase) (14, 39). Thus far, PBP 4 has been associated only with low to moderate β-lactam resistance in S. aureus (30, 32). The PBP 4 mutations detected in this study clustered near its active site (Fig. 2). Additionally, each mutation detected in PBP 4 brought a change to the chemical properties of the amino acids. N138K/I and T201A brought in an electrically charged or hydrophobic amino acid in place of a polar amino acid. The E183V, R200L, and H270L mutations changed an electrically charged amino acid to a hydrophobic amino acid. F241R caused a change of a hydrophobic amino acid to an electrically charged one. All these amino acid changes suggest a significant change in enzyme activity for the mutant PBP 4. Furthermore, accumulation of all the PBP mutations in the transpeptidase domain affected the β-lactam target site, suggesting a common feature among the detected mutations.
FIG 2.

Point mutations mapped on the PBP 4-cefotaxime complex. PBP 4 point mutations (yellow) detected in this study are depicted near the active site of cefotaxime-bound PBP 4 (PDB 3HUM).
Our study also shows several point mutations in GdpP, an enzyme crucial for maintaining cyclic-di-AMP (CDA) balance in bacteria. CDA is a recently discovered second messenger known to influence several life processes in bacteria, including virulence, ion export, and biofilm formation. Recent studies have suggested a role for CDA in the β-lactam resistance of S. aureus (40, 41). Mutations in GdpP in strains SRT, SRB, and SpT (Table 3) (only SgapT had a gdpP mutation introduced by allelic exchange), particularly the Y306Stop and T509A mutations (in SRT and SRB, respectively), were found adjacent to the DHH domain, crucial for the phosphodiesterase activity of GdpP (40). These mutations may alter the function of GdpP and could help mediate bacterial resistance or survival in our passaged isolates. However, further studies are needed to support this hypothesis.
PBP 4 mediates high-level class resistance to β-lactams in the resistant isolates.
To determine if pbp4 is the driving factor for high-level β-lactam resistance, we deleted the pbp4 gene in the passaged isolates (Table 1). MIC assays performed for a variety of β-lactam drugs, including ceftobiprole and ceftaroline, revealed that the strains were fully susceptible to the entire class of β-lactams tested. MIC tests with cefoxitin, a known semiselective inhibitor of PBP 4 (42), showed only moderate decreases (2- to 4-fold) in the MIC for the Δpbp4 knockout strains (Table 4). Population analysis of the passaged isolates and their pbp4 knockout strains for nafcillin also showed complete susceptibility of the knockouts compared to their isogenic wild-type strains, indicating that the resistant phenotype of the passaged strain was dependent on pbp4 (Fig. 3A to D).
TABLE 4.
MICs of passaged strains and their isogenic pbp4 deletion knockout strains
| Strain | MIC (μg ml−1)a |
||||||
|---|---|---|---|---|---|---|---|
| NAF | AMP | CTX | FOX | CFZ | BPR | CPT | |
| SRB | 8 | 4 | 32 | 1 | 16 | >64 | 4 |
| SRB Δpbp4 | 0.25 | 0.25 | 0.25 | 0.5 | 0.25 | 4 | 0.125 |
| SRT | 64 | >256 | >256 | 8 | >256 | 8 | >64 |
| SRT Δpbp4 | 0.25 | 0.25 | 2 | 4 | 0.25 | 0.5 | 0.125 |
| SgapT | >256 | >256 | 256 | 8 | 256 | >64 | 64 |
| SgapT Δpbp4 | 0.25 | 0.25 | 1 | 2 | 0.25 | 0.25 | 0.125 |
| SpT | >256 | >256 | >256 | 8 | >256 | >64 | >64 |
| SpT Δpbp4 | 0.25 | 0.25 | 2 | 4 | 0.25 | 0.5 | 0.125 |
NAF, nafcillin; AMP, ampicillin; CTX, ceftriaxone; FOX, cefoxitin; CFZ, cefazolin; BPR, ceftobiprole; and CPT, ceftaroline. The CLSI breakpoint for resistance is ≥4 μg ml−1 (45).
FIG 3.

Population analysis of the passaged strains and their isogenic Δpbp4 knockout strains in nafcillin. (A to D) Ten microliters of serially diluted bacterial overnight cultures was spotted onto agar plates containing various concentrations of nafcillin, as indicated. Bacterial colonies were enumerated (CFU ml−1) after 72 h of incubation at 37°C.
Next, the SRB Δpbp4 and SRT Δpbp4 knockout strains were complemented in trans with pbp4 that originated either from the wild-type strain SF8300 or from the passaged strain SRB or SRT. Population analysis for nafcillin showed that PBP 4s originating either from SRB or from SRT provided significantly (P < 0.0001) higher resistance than the wild-type pbp4, clearly underscoring the importance of the mutations in PBP 4s in conferring elevated resistance (Fig. 4A and B). The wild-type pbp4 from strain SF8300 provided significantly (P < 0.0001) higher resistance to SRB than the empty-vector control. In contrast, wild-type pbp4 failed to confer resistance on SRT (Fig. 4A and B). These results suggest cooperation between PBP 4 and the other PBPs, all of which had the mutations in SRB and none of which had mutations in SRT. However, other, unknown candidate genes could also account for the resistant phenotype in the SRB strain.
FIG 4.

Population analysis of the complemented strains in nafcillin. (A and B) Ten microliters of serially diluted bacterial overnight cultures, as indicated, were spotted onto agar plates containing tetracycline (12.5 μg ml−1) and various concentrations of nafcillin, as indicated. Bacterial colonies were enumerated (CFU ml−1) after 72 h of incubation at 37°C. pbp4 (SF8300), pbp4 (SRB), and pbp4 (SRT) represent the pbp4 genes from strains SF8300, SRB, and SRT, respectively, that were cloned into pTXΔ and introduced into the indicated strains. Two-way ANOVA of the data revealed significant differences (P < 0.0001) between the strains.
PBP 4 is essential for conferring high-level β-lactam resistance on S. aureus.
Cross-linking of muropeptides during S. aureus cell wall synthesis is thought to be coordinated by multiple PBPs. It has been proposed that PBP 2, in particular, cooperates with PBP 4 to mediate proper cell wall synthesis (14). To determine if high-level β-lactam resistance can occur in the absence of pbp4, we serially passaged strains COLnex and SF8300ex, along with their isogenic Δpbp4 strains, in nafcillin. Nafcillin was used as a prototype β-lactam for this experiment due to its easy commercial availability. While strains COLnex and SF8300ex attained growth in 256 μg ml−1 of nafcillin within 18 days, their isogenic Δpbp4 strains did not attain growth beyond 8 μg ml−1 of nafcillin even after 60 days of our passaging experiment (Fig. 5). These results together further underline the critical role played by PBP 4 in the emergence of high-level β-lactam resistance.
DISCUSSION
Widespread antimicrobial resistance among bacterial pathogens, including S. aureus, has spurred renewed interest in searching for new and better drugs. The search will be facilitated by a detailed understanding of the mechanisms that lead to susceptibility and resistance to antibiotics (43; http://www.cdc.gov/drugresistance/). We show that S. aureus strain USA300 can attain high-level resistance to β-lactams, including those active against MRSA, in the absence of mecA. Our observation supports previous work showing that a mecA-negative COLn strain was able to develop high-level β-lactam resistance (26). We have extended those results by demonstrating that an uncanonical member of the penicillin binding protein family, PBP 4, can mediate resistance in the community MRSA USA300 strain background (Fig. 3A to D and Table 4). In S. aureus, pbp4 codes for a low-molecular-weight penicillin binding protein that is believed to function primarily as a transpeptidase and carboxypeptidase to perform the penultimate stages of bacterial cell wall synthesis (39). Although pbp4 is conserved among all S. aureus strains, its function is generally considered supplementary and nonessential for both bacterial cell wall synthesis and high-level β-lactam resistance (30). Earlier work suggested that PBP 4 is responsible for forming higher cross-linked peptidoglycan chains and can be associated with moderate-level β-lactam resistance in CA-MRSA strains (14, 32). Our work supports these data and, in addition, indicates that PBP 4 can be associated with very high-level β-lactam resistance if needed.
Our study identified several point mutations in PBP 4 that were shown to provide elevated resistance (Fig. 4A and B). Mapping of these mutations to the recently crystalized PBP 4 identified all of them in close proximity to the active site of PBP 4 (Fig. 2). Interestingly, the E183A mutation that was introduced into strain Sp was mutated to E183V postpassaging (Table 3). Although our study does not address the biochemical basis of the altered/superior function of the mutated PBP 4, we hypothesize the following scenarios: (i) the mutations may alter the binding affinity of PBP 4 for the β-lactams and/or (ii) the mutations may cause a gain of function, thereby turning the protein into a major TPase and CPase to allow bacterial cell wall synthesis. Experiments are ongoing to address these possibilities.
In addition to PBP 4, several point mutations were also detected in other PBPs (PBPs 1 to 3) in our study. Interestingly, all these mutations were present near the transpeptidase active site of the PBPs, the primary target of β-lactam drugs. The β-lactam susceptibility in our pbp4 knockout strains to the β-lactam drugs tested here (Fig. 3A to D and Table 4), along with the inability of the pbp4-negative strains to display high-level resistance upon passaging in nafcillin (Fig. 5), confirm the key role of PBP 4 for the emergence and expression of resistance in our passaged isolates. It also seems likely that mutations in PBPs 1 to 3 have some supplementary role(s) in providing resistance to our isolates.
Our study also detected mutations in the gdpP gene in each of the passaged isolates examined. Recently, several points of evidence have associated GdpP with β-lactam resistance (44). Although a direct role of GdpP in S. aureus β-lactam resistance has yet to be identified, our study suggests that GdpP may provide some fitness to the bacteria to survive the β-lactam challenge. Finally, additional mutations that are not described in this study are likely to be present. Genome-sequencing studies are under way to provide further details of our finding on a genome-wide basis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carolina Roberte de Oliveira for technical help and Michael Otto for providing the plasmid pTXΔ.
This work was funded in part by NIH grant R01-AI100291 (to H.F.C.). T.M.D.C. is sponsored by CNPq-Brazil.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00358-16.
REFERENCES
- 1.Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee SS, Otto M. 2013. Improved understanding of factors driving methicillin-resistant Staphylococcus aureus epidemic waves. Clin Epidemiol 5:205–217. doi: 10.2147/CLEP.S37071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 4.Bush K. 2012. Antimicrobial agents targeting bacterial cell walls and cell membranes. Rev Sci Tech 31:43–56. [DOI] [PubMed] [Google Scholar]
- 5.Miller EL. 2002. The penicillins: a review and update. J Midwifery Womens Health 47:426–434. doi: 10.1016/S1526-9523(02)00330-6. [DOI] [PubMed] [Google Scholar]
- 6.Frere JM, Page MG. 2014. Penicillin-binding proteins: evergreen drug targets. Curr Opin Pharmacol 18:112–119. doi: 10.1016/j.coph.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 7.Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. 2006. Penicillin binding proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol Rev 30:673–691. doi: 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 8.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 9.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and β-lactam resistance. FEMS Microbiol Rev 32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 10.Berger-Bachi B. 1999. Genetic basis of methicillin resistance in Staphylococcus aureus. Cell Mol Life Sci 56:764–770. doi: 10.1007/s000180050023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laurent F, Chardon H, Haenni M, Bes M, Reverdy ME, Madec JY, Lagier E, Vandenesch F, Tristan A. 2012. MRSA harboring mecA variant gene mecC, France. Emerg Infect Dis 18:1465–1467. doi: 10.3201/eid1809.111920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livermore DM. 2006. Can β-lactams be re-engineered to beat MRSA? Clin Microbiol Infect 12(Suppl 2):S11–S16. [DOI] [PubMed] [Google Scholar]
- 13.Chan LC, Basuino L, Diep B, Hamilton S, Chatterjee SS, Chambers HF. 2015. Ceftobiprole- and ceftaroline-resistant methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 59:2960–2963. doi: 10.1128/AAC.05004-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leski TA, Tomasz A. 2005. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J Bacteriol 187:1815–1824. doi: 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson SD, Gums JG. 2008. Ceftobiprole: an extended-spectrum anti-methicillin-resistant Staphylococcus aureus cephalosporin. Ann Pharmacother 42:806–816. doi: 10.1345/aph.1L016. [DOI] [PubMed] [Google Scholar]
- 16.Barbour A, Derendorf H. 2010. Resistance and the management of complicated skin and skin structure infections: the role of ceftobiprole. Ther Clin Risk Manag 6:485–495. doi: 10.2147/TCRM.S5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambers HF. 2006. Ceftobiprole: in vivo profile of a bactericidal cephalosporin. Clin Microbiol Infect 12(Suppl 2):S17–S22. [DOI] [PubMed] [Google Scholar]
- 18.File TM Jr, Wilcox MH, Stein GE. 2012. Summary of ceftaroline fosamil clinical trial studies and clinical safety. Clin Infect Dis 55(Suppl 3):S173–S180. doi: 10.1093/cid/cis559. [DOI] [PubMed] [Google Scholar]
- 19.Casapao AM, Davis SL, Barr VO, Klinker KP, Goff DA, Barber KE, Kaye KS, Mynatt RP, Molloy LM, Pogue JM, Rybak MJ. 2014. Large retrospective evaluation of the effectiveness and safety of ceftaroline fosamil therapy. Antimicrob Agents Chemother 58:2541–2546. doi: 10.1128/AAC.02371-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otero LH, Rojas-Altuve A, Llarrull LI, Carrasco-Lopez C, Kumarasiri M, Lastochkin E, Fishovitz J, Dawley M, Hesek D, Lee M, Johnson JW, Fisher JF, Chang M, Mobashery S, Hermoso JA. 2013. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc Natl Acad Sci U S A 110:16808–16813. doi: 10.1073/pnas.1300118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lovering AL, Gretes MC, Safadi SS, Danel F, de Castro L, Page MG, Strynadka NC. 2012. Structural insights into the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of ceftobiprole. J Biol Chem 287:32096–32102. doi: 10.1074/jbc.M112.355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez R, Paz LI, Rosato RR, Rosato AE. 2014. Ceftaroline is active against heteroresistant methicillin-resistant Staphylococcus aureus clinical strains despite associated mutational mechanisms and intermediate levels of resistance. Antimicrob Agents Chemother 58:5736–5746. doi: 10.1128/AAC.03019-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley WL, Jousselin A, Barras C, Lelong E, Renzoni A. 2015. Missense mutations in PBP2A affecting ceftaroline susceptibility detected in epidemic hospital-acquired methicillin-resistant Staphylococcus aureus clonotypes ST228 and ST247 in Western Switzerland archived since 1998. Antimicrob Agents Chemother 59:1922–1930. doi: 10.1128/AAC.04068-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long SW, Olsen RJ, Mehta SC, Palzkill T, Cernoch PL, Perez KK, Musick WL, Rosato AE, Musser JM. 2014. PBP2a mutations causing high-level ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother 58:6668–6674. doi: 10.1128/AAC.03622-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biedenbach DJ, Alm RA, Lahiri SD, Reiszner E, Hoban DJ, Sahm DF, Bouchillon SK, Ambler JE. 2015. In vitro activity of ceftaroline against Staphylococcus aureus isolated in 2012 from Asia-Pacific countries as part of the AWARE Surveillance Program. Antimicrob Agents Chemother 60:343–347. doi: 10.1128/AAC.01867-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banerjee R, Gretes M, Basuino L, Strynadka N, Chambers HF. 2008. In vitro selection and characterization of ceftobiprole-resistant methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 52:2089–2096. doi: 10.1128/AAC.01403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long SW, Beres SB, Olsen RJ, Musser JM. 2014. Absence of patient-to-patient intrahospital transmission of Staphylococcus aureus as determined by whole-genome sequencing. mBio 5:e01692–01614. doi: 10.1128/mBio.01692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee R, Gretes M, Harlem C, Basuino L, Chambers HF. 2010. A mecA-negative strain of methicillin-resistant Staphylococcus aureus with high-level β-lactam resistance contains mutations in three genes. Antimicrob Agents Chemother 54:4900–4902. doi: 10.1128/AAC.00594-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henze UU, Berger-Bachi B. 1995. Staphylococcus aureus penicillin-binding protein 4 and intrinsic β-lactam resistance. Antimicrob Agents Chemother 39:2415–2422. doi: 10.1128/AAC.39.11.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katayama Y, Zhang HZ, Chambers HF. 2003. Effect of disruption of Staphylococcus aureus PBP4 gene on resistance to β-lactam antibiotics. Microb Drug Resist 9:329–336. doi: 10.1089/107662903322762752. [DOI] [PubMed] [Google Scholar]
- 31.Talan DA, Krishnadasan A, Gorwitz RJ, Fosheim GE, Limbago B, Albrecht V, Moran GJ, EMERGEncy ID Net Study Group. 2011. Comparison of Staphylococcus aureus from skin and soft-tissue infections in US emergency department patients, 2004 and 2008. Clin Infect Dis 53:144–149. doi: 10.1093/cid/cir308. [DOI] [PubMed] [Google Scholar]
- 32.Memmi G, Filipe SR, Pinho MG, Fu Z, Cheung A. 2008. Staphylococcus aureus PBP4 is essential for β-lactam resistance in community-acquired methicillin-resistant strains. Antimicrob Agents Chemother 52:3955–3966. doi: 10.1128/AAC.00049-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 34.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 35.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 36.Diep BA, Stone GG, Basuino L, Graber CJ, Miller A, des Etages SA, Jones A, Palazzolo-Ballance AM, Perdreau-Remington F, Sensabaugh GF, DeLeo FR, Chambers HF. 2008. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J Infect Dis 197:1523–1530. doi: 10.1086/587907. [DOI] [PubMed] [Google Scholar]
- 37.Pereira SF, Henriques AO, Pinho MG, de Lencastre H, Tomasz A. 2007. Role of PBP1 in cell division of Staphylococcus aureus. J Bacteriol 189:3525–3531. doi: 10.1128/JB.00044-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida H, Kawai F, Obayashi E, Akashi S, Roper DI, Tame JR, Park SY. 2012. Crystal structures of penicillin-binding protein 3 (PBP3) from methicillin-resistant Staphylococcus aureus in the apo and cefotaxime-bound forms. J Mol Biol 423:351–364. doi: 10.1016/j.jmb.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Navratna V, Nadig S, Sood V, Prasad K, Arakere G, Gopal B. 2010. Molecular basis for the role of Staphylococcus aureus penicillin binding protein 4 in antimicrobial resistance. J Bacteriol 192:134–144. doi: 10.1128/JB.00822-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corrigan RM, Abbott JC, Burhenne H, Kaever V, Grundling A. 2011. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog 7:e1002217. doi: 10.1371/journal.ppat.1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corrigan RM, Grundling A. 2013. Cyclic di-AMP: another second messenger enters the fray. Nat Rev Microbiol 11:513–524. doi: 10.1038/nrmicro3069. [DOI] [PubMed] [Google Scholar]
- 42.Wyke AW, Ward JB, Hayes MV, Curtis NA. 1981. A role in vivo for penicillin-binding protein-4 of Staphylococcus aureus. Eur J Biochem 119:389–393. doi: 10.1111/j.1432-1033.1981.tb05620.x. [DOI] [PubMed] [Google Scholar]
- 43.WHO. 2014. Antimicrobial resistance: global report on surveillance. WHO, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/112647/1/WHO_HSE_PED_AIP_2014.2_eng.pdf?ua=1. [Google Scholar]
- 44.Corrigan RM, Bowman L, Willis AR, Kaever V, Grundling A. 2015. Cross-talk between two nucleotide-signaling pathways in Staphylococcus aureus. J Biol Chem 290:5826–5839. doi: 10.1074/jbc.M114.598300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.CLSI. 2013. Performance standards for antimicrobial susceptibility testing; 23rd informational supplement; CLSI document M100-S23. CLSI, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.