

Abstract
Protozoan parasites of the Leishmania donovani complex are the causative agents of visceral leishmaniasis (VL), the most severe form of leishmaniasis, with high rates of mortality if left untreated. Leishmania parasites are transmitted to humans through the bite of infected female sandflies (Diptera: Phlebotominae), and approximately 500,000 new cases of VL are reported each year. In the absence of a safe human vaccine, chemotherapy, along with vector control, is the sole tool with which to fight the disease. Miltefosine (hexadecylphosphatidylcholine [HePC]), an antitumoral drug, is the only successful oral treatment for VL. In the current study, we describe the phenotypic traits of L. donovani clonal lines that have acquired resistance to HePC. We performed whole-genome and RNA sequencing of these resistant lines to provide an inclusive overview of the multifactorial acquisition of experimental HePC resistance, circumventing the challenge of identifying changes in membrane-bound proteins faced by proteomics. This analysis was complemented by assessment of the in vitro infectivity of HePC-resistant parasites. Our work underscores the importance of complementary “omics” to acquire the most comprehensive insight for multifaceted processes, such as HePC resistance.
INTRODUCTION
Visceral leishmaniasis (VL) is the most severe clinical manifestation of leishmaniasis and is caused by several species of the Leishmania donovani complex (1). This protozoan parasite is transmitted to humans through the bite of infected phlebotomine sandflies, and the fatality rate in developing countries can be as high as 100% within 2 years. Ninety percent of VL cases occur in Bangladesh, Brazil, Ethiopia, India, South Sudan, and Sudan, and approximately 500,000 new cases are reported each year (2). The absence of a reliable and safe human vaccine makes chemotherapy, along with vector control, the only tool with which to fight the disease. Additionally, chemotherapy presents several drawbacks in regard to treatment regimes, which are usually species specific, expensive, and associated with high toxicity and require prolonged administration schedules. Pentavalent antimonial compounds have been the mainstay of chemotherapeutic treatment for the last 75 years. The emergence in the parasite of clinical resistance to antimonials has driven the search for new and safer drugs to fight the disease (3). The use of pentamidine, amphotericin B, and paromomycin as alternative treatments is limited by their toxicity and the requirement for parenteral administration by trained medical professionals. Initially introduced as an antitumor drug, the alkyl-lyso-phospholipid analogue hexadecylphosphatidylcholine (HePC), known commercially as miltefosine, is the sole oral drug available to treat VL (4) and was recently approved by the FDA (Impavido) to treat leishmaniasis in the United States. The biochemical properties of this lipid analogue have permitted the generation of soluble and stable formulations, facilitating administration and treatment compliance. One of the major pitfalls in the initial implementation of HePC—uncontrolled access to the drug—has been resolved in recent years through improved regulation of its distribution (5). Unfortunately, however, this unrestricted use has led to a reduction in the effectiveness of HePC, which paralleled the increase in the relapse rate found in a phase III trial conducted during 1999 and 2000 (5). Even though observations of clinical resistance to HePC are scarce, its long half-life (approximately 120 h) and small therapeutic window make the emergence of resistance on a larger scale very likely. Until fully characterized resistant field isolates become available, experimental selection of HePC resistance in the laboratory may offer insight into potential mechanisms of resistance and contribute to design strategies to prevent the emergence and spread of resistance.
Previous findings have demonstrated that HePC internalization depends on a P-type ATPase transporter present in the plasma membrane of the parasite (6, 7). The functional form of the transporter requires the presence of two functional subunits: LdMT and LdRos3. The presence of loss-of-function point mutations in any of the transporter subunits led to reduced HePC intake and parasite survival. These mutations can be easily selected for by exposing parasites to increasing drug concentrations (8). Nevertheless, there is a growing awareness of the multifactorial nature of HePC resistance. Clinical isolates from relapsed VL cases showed lower susceptibility to HePC in the absence of mutations in the transporter or changes in expression of LdMT/LdRos3 genes (9). In another important study of 120 VL patients in Nepal treated with HePC, the susceptibilities of isolates from definite cures were similar to those of isolates from relapses, and thus, drug resistance was not likely involved in treatment failure in Nepal (10). Additionally, increased efflux of the drug as a consequence of the overexpression of ABC transporters has been reported, resulting in reduced HePC susceptibilities (11–13). Furthermore, augmented expression of an L. infantum gene coding for a protein of unknown function conferred resistance not only to HePC, but also to antimonial tartrate (14). Our knowledge of these mechanisms derives primarily from experimental resistance induced in promastigotes, and thus, clinical isolates may indeed display different characteristics. Altogether, a holistic approach is needed in order to better comprehend HePC resistance in Leishmania. We have studied the phenotypic traits of clonal lines of HePC-resistant L. donovani promastigotes following stepwise selection. Whole-genome and RNA sequencing was carried out on HePC-resistant strains, revealing defects in the drug translocation machinery, as well as up- and downregulation of specific genes associated with stress, membrane composition, and amino acid and folate metabolism. The nature of some differentially expressed genes and the interconnection between drug resistance and parasite ecology and biology as a key factor affecting the dissemination of resistant isolates into vertebrate hosts (15, 16) prompted us to study metacyclogenesis and to assess the in vitro infectivity of HePC-resistant lines.
MATERIALS AND METHODS
Cell culture and development of resistant lines.
A wild-type (WT) clone of L. donovani strain 1S2D (MHOM/SD/62/1S-CL2D) and its resulting HePC-resistant line were grown in M199 medium containing 10% fetal calf serum (FCS) at 26°C and pH 7.4. HePC-resistant cultures were generated following a stepwise method. Briefly, proliferation was monitored by microscopy, and cultures were passaged every 4 days from an initial density of 5 × 105 promastigotes/ml. Increasing concentrations of HePC were added to the cultures starting at 2.5 μM (LdR2.5) and sequentially to 5 μM (LdR5), 8 μM (LdR8), 10 μM (LdR10), 15 μM (LdR15), 20 μM (LdR20), 30 μM (LdR30), and 40 μM (LdR40). Each increase in the drug concentration was made when the growth rate of HePC-exposed cultures matched that of the WT. At least two clones of every resistant line (denoted LdR[HePC]0.1 and LdR[HePC]0.2) were generated by plating on M199 plates (17) and were maintained in parallel. The stability of the resistant phenotype was assessed by determination of drug susceptibility at different times after growth of the parasite in the absence of the drug (independent cell lines denoted LdR[HePC]0.1no and LdR[HePC]0.2nob). Parasite stress was monitored by flow cytometry using two fluorescent apoptotic markers. Membrane permeability was monitored via membrane permeability/dead cell apoptosis (Invitrogen), following the manufacturer's instructions. Briefly, samples were pelleted, washed, and resuspended in M199 medium. After the addition of 1 μl of Yo-Pro (Molecular Probes) and propidium iodide, samples were incubated at 4°C for 20 min and analyzed by flow cytometry using a Beckman Coulter FC500 flow cytometer. Furthermore, phosphatidylserine (PS) exposure was studied with annexin V-fluorescein isothiocyanate (FITC) (Miltenyi Biotec) following the manufacturer's instructions. A total of 10,000 events per sample were recorded, and at least two independent experiments were performed. RAW264.7 macrophages were cultured at 37°C with 5% CO2 in RPMI-C (RPMI supplemented with 10% FCS, penicillin-streptomycin, and l-glutamine) and passaged every 2 or 3 days.
Growth curves and drug susceptibility assays.
The growth rates of resistant cultures were measured and compared to that of WT controls. Parasites were seeded in triplicate at an initial concentration of 5 × 105 parasites/ml, and the promastigote density was assessed daily by direct microscopic counting until the parasites reached stationary phase. The leishmanicidal effects of HePC and the reference compounds amphotericin B (AmpB) (Sigma), pentamidine isethionate (PI) (Sigma), paromomycin (Pm) (Sigma), and potassium antimony(III) tartrate (SbIII) (Sigma) were evaluated in the resistant strains and compared to those in WT cultures. Culture viability was measured by using the resazurin-based method (CellTiter-Blue [Promega]) (18). Briefly, 1 × 106 parasites/ml were seeded in a 96-well plate and incubated in the presence of increasing drug concentrations for 48 h at 26°C, along with appropriate solvent controls. Twenty microliters of the reagent was added to 100 μl of culture, and after 4 h at 37°C, the fluorescence was measured (555-nm excitation wavelength [λexc]/580-nm emission wavelength [λem]) using a Typhoon FLA 9500 laser scanner (GE Healthcare) and analyzed with ImageQuant TL software (GE Healthcare). Each assay was performed in triplicate. The half-maximal effective concentrations (EC50s) were calculated by nonlinear regression analysis using SigmaPlot for Windows version 11.0. HePC susceptibility in amastigotes was evaluated via a back-transformation method or by Giemsa staining and light microscopy using RAW264.7 murine macrophage lines and infective-stage resistant (R) and WT promastigotes as previously described (19). Each assay was performed in triplicate. The EC50 values were calculated by nonlinear regression analysis using SigmaPlot for Windows version 11.0.
Determination of HePC accumulation and energy depletion experiments.
Log-phase parasites (5 × 105) were washed with HPMI buffer (20 mM HEPES, 132 mM NaCl, 3.5 mM KCl, 0.5 mM MgCl2, 1 mM CaCl2, [pH 7.4]) and preincubated in the same buffer for 30 min at 26°C with 1 mM phenylmethylsulfonyl fluoride (to block lipid catabolism) and then labeled with 5 μM HePC–boron-dipyrromethene (BODIPY) (20) for 45 min at 26°C in the dark. The fluorescent analogue was added directly from an ethanol stock solution. The parasites were washed with HPMI supplemented with 4% bovine serum albumin (BSA) to remove the noninternalized probe (back exchange), resuspended in HPMI, and maintained on ice. Cellular fluorescence was measured by flow cytometry using a Beckman Coulter FC500 flow cytometer. Propidium iodide was added to monitor cell viability, and 10,000 events per sample were recorded. Solvent controls were included, and the experiment was performed in triplicate. Energy depletion studies were carried out by incorporating 10 mM KCN into the samples before the addition of the fluorescent analogue for 30 min at 26°C in HPMI buffer.
HePC efflux studies.
Log-phase parasites (L. donovani promastigotes; 5 × 105) were incubated with 5 μM HePC-BODIPY for different times up to 120 min at 26°C in M199 medium, washed twice with 4% (wt/vol) BSA–phosphate-buffered saline (PBS), and resuspended in PBS at 1 × 106 cells/ml. Samples were analyzed by flow cytometry in a Beckman Coulter FC500 flow cytometer.
Confocal microscopy.
Cells were incubated with 2.5 μM HePC-BODIPY for 1 h at 26°C and washed twice with 1% (wt/vol) BSA-PBS. Living cells were immobilized on Hydrogel Cygel (Biostatus Ltd.) according to the instructions issued by the supplier and were subsequently imaged on a Leica TCS-SP2-AOBS-UV ultraspectral confocal microscope (Leica Microsystems). The fluorescence settings for HePC-BODIPY were 488-nm λexc/520-nm λem.
RNA extraction and real-time PCR analysis (quantitative real-time [qRT]-PCR).
Total RNA was isolated from logarithmic- and stationary-phase promastigotes using TRIzol reagent (Invitrogen). The RNA was reverse transcribed with SuperScript II reverse transcriptase (Invitrogen) after DNase I treatment with the Turbo DNA-free kit (Ambion, Invitrogen). At least three biological samples were included per experiment. All real-time PCRs were performed in triplicate in 10-μl volumes using the SYBR green fluorescence quantification system (Invitrogen) in a 7500 Fast real-time PCR system (Applied Biosystems). The ΔΔCT method was used to calculate relative changes in gene expression (21). The data are presented as the fold change in expression of the target gene in resistant L. donovani cultures normalized to the internal-control (glyceraldehyde-3-phosphate dehydrogenase [GAPDH] and superoxide dismutase [SOD]) genes and relative to the L. donovani WT reference strain. The standard PCR conditions were 95°C (10 min) and 40 cycles of 94°C (1 min), 60°C (1 min), and 72°C (2 min), followed by the denaturation curve. Primer designs were based on the nucleotide sequences of L. infantum genes coding for LdMT (GenBank accession number AY321297.1), LdRos3 (GenBank accession number DQ205096.1), SHERP (GenBank accession number XM_001683391), GAPDH (GenBank accession number XP_001467145.1), and SOD (GenBank accession number XP_001467867.1). Differences in the transcription levels were compared using Student's t test. The significance level (P value) was determined with a confidence interval of 95% in a two-tailed distribution. Three biological replicates were assayed, and experiments were done in triplicate.
Whole-genome sequencing (WGS).
The genomic-DNA concentration was determined using the Qubit 2.0 fluorometer and broad-range DNA assay (Life Technologies Corp., Carlsbad, CA). Libraries were constructed using the TruSeq Nano DNA LT library preparation kit and reference guide 15041110 D (Illumina, Inc., San Diego, CA) following the manufacturer's protocol for the 550-bp insert size. To calculate the final library concentration, the quantity and average fragment length for each library were measured using the Agilent Bioanalyzer DNA 7500 assay (Agilent Technologies, Santa Clara, CA) and the Qubit DNA High Sensitivity assay (Life Technologies Corp). Libraries were normalized to 10 nM in buffer EB (Qiagen, Santa Clarita, CA) and combined in equal molar amounts. Sequencing was performed on the Illumina MiSeq platform (Illumina, Inc.) using the MiSeq reagent kit v3 (600 cycles) according to the manufacturer's instructions. A total of 13.12 Gb of data were obtained using 301-bp paired-end reads. Changes in the number of individual chromosomes were calculated using the script “find_copy_number.pl” (22). The median coverage for each of the chromosomes was calculated, followed by the determination of all chromosomal medians. This value was divided by 2, which represents the median coverage for a haploid allele of a chromosome. The median coverage of each chromosome was then divided by the haploid chromosome coverage to obtain the somy of individual chromosomes.
RNA sequencing.
Total RNA was extracted from three biological replicates of resistant (R30.1) and WT logarithmic-phase promastigotes using RNeasy minikit extraction according to the manufacturer's recommendations (Qiagen). The initial RNA concentration was determined using the Qubit 2.0 fluorometer and the RNA Broad Range assay (Life Technologies Corp.). Total RNA integrity was assessed using the Agilent 2100 Bioanalyzer, along with the RNA Nano kit (Agilent Technologies). The sequencing library was constructed using the TruSeq RNA sample preparation kit and companion protocol (Illumina). Briefly, poly(A)-containing mRNA was purified from 3 μg total RNA using oligo poly(T) attached to magnetic beads and then heat fragmented at 94°C for 4 min. First-strand cDNA was synthesized using reverse transcriptase and random hexamers, followed by second-strand synthesis. The ends of the double-stranded cDNA were polished, 3′ adenylated, and then ligated with indexing adapters to prepare them for hybridization on the flow cell. The library was then PCR amplified to enrich for fragments properly ligated and purified using AMPure XP beads (Beckman Coulter, Inc.). The final library integrity and size distribution were assessed using the Agilent Bioanalyzer 2100, along with the DNA 7500 assay (Agilent Technologies). The Qubit 2.0 fluorometer, along with the High Sensitivity DNA assay (Life Technologies Corp.), was used to determine the final library concentration.
Libraries were normalized to 10 nM in buffer EB (Qiagen) and combined for multiplexing. To prepare the library for sequencing, 4 nM the pooled library was denatured by addition of an equal volume of fresh 0.2 N NaOH. Chilled HT1 buffer (Illumina) was used to dilute the library to a final loading concentration of 6 pM. Paired-end sequencing of 2 × 79 bp was performed using the Illumina MiSeq platform and MiSeq reagent kit v3 (150 cycles; Illumina). A total of 68.1 million raw paired-end sequencing reads were obtained, and 55.5 million were retained after quality filtering.
Bioinformatics.
For bioinformatics analysis, raw sequences were trimmed of adapters with Trimmomatic version 0.32 and assessed for quality with FastQC version 0.11.2. The trimmed sequences were aligned with the L. infantum JPCM5 reference genome (23) with TopHat version 2.0.11 using Bowtie 2 version 2.2.2. The choice of the L. infantum genome was due to its superior annotation compared with the L. donovani BPK282A1 genome, which is still in version 1 and not well annotated. The sorting of corresponding alignments was completed with SAMtools version 0.1.19, and read counts were generated with HTSeq-count version 0.6.1. Statistical analysis was completed in R version 3.1.0 implementing the EdgeR library. The Benjamini-Hochberg method (24) was implemented to determine the significance of expression and to adjust for multiple testing. Gene names and gene ontology (GO) terms were identified using the TriTryp database (http://www.tritrypdb.org). Extended functional annotation of sequence data was performed with Blast2GO (version 3.0.9) using standard parameters (25). The FASTA sequences of proteins were downloaded from the TriTrypDB.org database and subsequently subjected to a BLAST search to find homologous sequences, mapped to collect GO terms associated with BLAST hits, and finally, annotated to assign information to query sequences.
For whole-genome sequencing, trimmed sequences (with an average 58× coverage) were aligned to the L. infantum JPCM5 reference genome (23) using the MEM algorithm of BWA (26) version 0.7.8. Further processing of mapped reads was conducted with SAMtools version 0.1.19 and Picard Tools (Broad Institute) version 1.119 according to the Genome Analysis Toolkit (GATK) best practices recommendations (27). Single nucleotide polymorphism (SNP) calling was performed using the haplotype caller walker of GATK (28) version v3.1-1. Further filtering on variants was performed, based on changes in the wild-type sample, changes occurring within a gene region, and having quality scores greater than 30.
Metacyclogenesis and macrophage infections.
A Ficoll 400 (Sigma) gradient was set up using 4 ml of 20% Ficoll (bottom layer) overlaid by 4 ml of 10% Ficoll in M199 medium without FCS and 4 ml of day 4 stationary-phase culture (29). The gradient was centrifuged for 10 min at 1,300 × g at room temperature. Metacyclic parasites were recovered from the upper interface. The number of parasites was determined by counting with a microscope before and after the procedure to determine the percentage of metacyclic parasites. Infection assays were performed using the RAW264.7 murine macrophage lines and 106 infective-stage metacyclic promastigotes isolated as described above. Infection was performed for 8 h in RPMI medium at a ratio of 10 metacyclic parasites per macrophage (10:1). Free parasites were removed by one wash with RPMI without FCS. The number of intracellular parasites in 100 macrophages was monitored at 0, 12, and 18 h postinfection by DiffQuick staining of cytospin whole-cell preparations and visualization by light microscopy. Experiments were done in triplicate, and appropriate controls were included for each period.
Statistics.
Significance was determined by P values calculated from a two-tailed Student's t test in GraphPad Prism 6.0 unless otherwise stated.
RESULTS
Phenotypic characterization of HePC-resistant L. donovani promastigotes generated in the study.
Following stepwise selection of each resistant line in the presence or absence of increasing drug pressure, proliferation rates were assessed by counting on a microscope. HePC-resistant promastigotes proliferate as WT parasites, suggesting that promastigote viability was not affected by HePC selection (see Fig. S1 in the supplemental material). In order to fully determine drug susceptibility, an EC50 assay was performed using the resazurin-based CellTiter-Blue method (Promega) (18). As expected, cultures grown in the presence of higher HePC concentrations displayed reduced susceptibilities (2.4- to 7.7-fold more resistant) to the drug, in contrast to cultures grown under lower HePC concentrations (Fig. 1). All cell lines selected under HePC pressure and grown further in the absence of the drug showed EC50s higher than the WT value. Remarkably, clones LdR40.1no and LdR40.2no, cultured without HePC for at least 100 passages (2 passages per week; ca. 12-month period) presented EC50s similar to those of parasites maintained in the presence of HePC (Fig. 1). For reference, HePC susceptibility was measured in intracellular amastigotes for the WT and R30.1 (EC50s, 7.77 ± 0.60 and 43.77 ± 2.05 μM, respectively). Two fluorescence-activated cell sorting (FACS)-based fluorescent apoptotic markers were used to investigate the stress and fitness levels of the parasites growing in HePC-containing medium: (i) Yo-Pro and (ii) annexin V. Figure S2 in the supplemental material shows the results obtained with the WT and R30 lines: LdR30.1 (grown in 30 μM HePC) and LdR30.1no and LdR30no.1b (both lines that were obtained independently from LdR30.1 and maintained without drug after the initial selection process). Morphometric profiles, as well as histograms, corresponding to the Yo-Pro and annexin V experiments suggested a minimal stress level compared to controls. As a positive control, WT parasites were treated with 20 μM HePC for 24 h (see Fig. S1 and S3 in the supplemental material). Similar results were obtained with all the populations generated for this study (data not shown).
FIG 1.
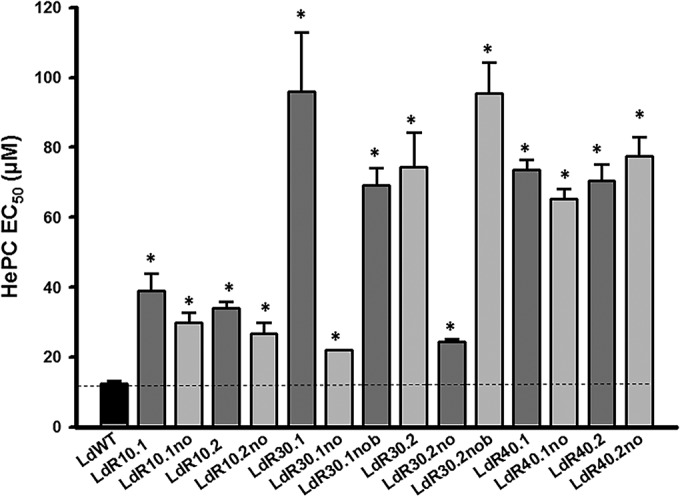
HePC susceptibility of HePC-resistant L. donovani populations determined by EC50 assays. The parasite lines showed increasing degrees of resistance to HePC as the selection pressure increased. All the populations were analyzed in triplicate, and the error bars indicate standard deviations. *, P < 0.05.
HePC-resistant populations do not show cross-resistance to other antileishmanial drugs.
Previous reports have shown partial cross-resistance of both laboratory and clinical HePC-treated isolates with some alternative leishmanicidal compounds (3, 30, 31). Cross-resistance was evaluated by comparison of the EC50s of different drugs in the resistant lines. Figure S4 in the supplemental material summarizes the results obtained for the responses to AmpB, PI, Pm, and SbIII.
Accumulation of HePC in resistant lines.
The uptake of HePC-BODIPY in the WT, LdR30.1, LdR30.1no, and LdR30.1nob lines was assessed by flow cytometry, as well as confocal microscopy. Experiments were carried out at 26°C, and the noninternalized HePC was removed by back exchange (6, 7, 12, 32–34). Under these conditions, WT and LdR30.1no promastigotes efficiently internalized HePC-BODIPY. Both LdR30.1 and LdR30.1nob showed a reduction (>85%) in their HePC-BODIPY uptake from the medium (Fig. 2A and C). To determine whether the reduction observed was the result of increased HePC-BODIPY efflux to the extracellular medium, parasites were loaded with HePC, and the amount of fluorescence retained was measured at different time points (Fig. 2B). No significant differences between the tested populations were reported, indicating that variations in HePC accumulation were not the result of increased drug efflux but rather a consequence of deficient drug intake. As judged from our data, an association between a higher tolerance for HePC treatment and impaired drug accumulation was experimentally sustained. To support our argument, the LdR30.1 and LdR30.1nob lines exhibited 7.75- and 5.5-fold increases, respectively, in their EC50s compared to the WT. Notably, HePC uptake was strongly reduced in the two lines. In contrast, the resistant LdR30.1no line, showing a 2.15-fold increase in resistance, displayed drug accumulation similar to that of WT parasites. The defective HePC intake may indicate that the observed phenotype was the result of a modulation or a defect in the transporter expression. To explore this possibility, qRT-PCR was performed on WT (control) and LdR30.1 parasites (Fig. 3A). The expression of the LdMT and LdRos3 genes was evaluated and normalized to the expression of two housekeeping genes: SOD and GAPDH genes. The results shown are expressed as the fold change in gene expression compared to WT levels. We also included in this study the analysis of two ABC transporters, MDR1 and ABCG4, whose overexpression has been previously linked to HePC-increased extrusion and therefore increased HePC tolerance (11, 12). Our data indicated that the expression of both the HePC transporter and its subunit is downregulated in resistant parasites. The expression of the ABC transporters tested was also reduced compared to control parasites (Fig. 3A), and drug efflux (Fig. 2B) was not increased in resistant strains. The translocation of phospholipids by the transporter from the exoplasmic monolayer of the plasma membrane into the cytoplasmic monolayer requires ATP hydrolysis (35). To rule out any nonspecific incorporation of HePC-BODIPY into the parasites, WT and resistant parasites were pretreated with 10 mM KCN to abolish ATP production (Fig. 3B). In both cases, a 1-log-unit reduction was obtained for both strains, exemplifying that most of the HePC is incorporated in an energy-dependent fashion.
FIG 2.

HePC uptake by Leishmania promastigotes. (A) Confocal fluorescence images were obtained from WT and resistant parasites (R30 line) incubated with 2.5 μM HePC-BODIPY (green fluorescence). The cells were stained with 10 μg/ml DAPI (4′,6-diamidino-2-phenylindole) for the nuclei and kinetoplast for 5 min before sample observation. Fluorescence settings were as follows: λexc, 488 nm, and λem, 520 nm for HePC and λexc, 350 nm, and λem, 460 nm for DAPI. (B) Time dependence of HePC-BODIPY efflux. Promastigotes of L. donovani were incubated with 5 μM HePC-BODIPY for different times up to 120 min at 26°C, washed with 4% (wt/vol) BSA-PBS, and analyzed by flow cytometry. Excitation was set at 488 nm, and emission was detected in the FL1 channel. The efflux scale was adapted to stress changes in the different lines. The data are expressed as arbitrary units of fluorescence (AU) and represent means and standard errors of the results of three independent experiments. (C) HePC-BODIPY intracellular accumulation in R30 parasite strains. Cells were incubated with 2.5 μM HePC-BODIPY, washed twice with 4% (wt/vol) BSA in HPMI, and analyzed by flow cytometry. *, P < 0.05.
FIG 3.
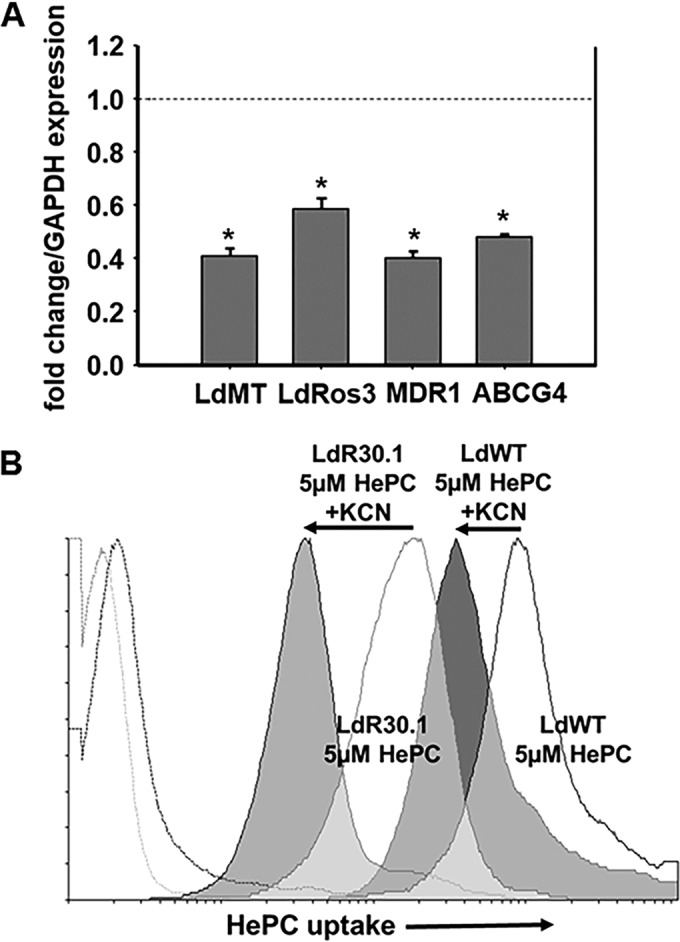
mRNA levels for selected genes associated with HePC resistance and effect of energy depletion on HePC uptake. (A) qRT-PCR experiments carried out on LdR30.1 parasites showed decreased HePC transporter expression (LdMT/LdRos3 subunits), as well as reduced transcription of the ABC transporters MDR1 and ABCG4, compared to the WT strain, using the GAPDH gene as a housekeeping gene. Similar results were obtained when using the SOD gene. Samples were analyzed in triplicate, and the error bars indicate standard deviations (*, P < 0.05). (B) LdR30.1 and WT parasites were incubated with 5 μM HePC-BODIPY with (solid histograms) or without (open histograms) KCN to deplete intracellular ATP pools. Drug intake was followed by flow cytometry. The excitation wavelength was set at 488 nm, and emission was detected in the FL1 channel. Parasite autofluorescence (broken profiles on the left) is also shown. A total of 10,000 cells were counted for each histogram. Experiments were done in triplicate with profiles similar to those shown.
Whole-genome sequencing of resistant L. donovani lines.
We carried out a more exhaustive analysis of point mutations and/or indels in the four lines mentioned above. LdR30.1 (resistant [R]), LdR30.1no (susceptible [S]), and LdR30.1nob (R) are clonal lines derived from the parent WT (S). Only polymorphisms that were selected for under HePC pressure were considered (Table 1). Our analysis did not reveal any of the previously described point mutations in LdMT and LdRos3 associated with experimental resistance to HePC in vitro (6, 8) (two nonsense [M1* and W210*] and two missense [T421N and L856P] mutations) but a novel substitution in the LdMT (LinJ.13.1590) gene in position 620293, generating a stop codon in R30.1. Interestingly, this homozygous variant was absent from the sensitive R30.1no clone and was heterozygous in R30.1nob, which showed a resistance phenotype similar to that of R30.1, suggesting that in the absence of HePC pressure, the mutation is reversible. Surprisingly, no variants were found in the LdRos3 gene. Our analysis also revealed other homozygous mutations in the proteophosphoglycan 4 (LinJ.35.0520) and the folate-biopterine transporter (LinJ.10.0400) genes in the two resistant phenotypes, R30.1 and R30.1nob. A complete list of SNPs and indels is shown in Table S1 in the supplemental material. We also determined chromosome somy that may account for the differences in resistance. As shown in Fig. S6 in the supplemental material, most of the chromosomes were disomic in WT and resistant populations, except chromosome 31, which is tetrasomic, as previously shown for L. donovani (36), and chromosomes 5, 21 to 23, and 26, which appear to be trisomic.
TABLE 1.
Gene mutations identified in L. donovani miltefosine-resistant mutantsa
| Gene product (GeneID) | SNP/indel | Nucleotide position | Amino acid change | Mutationc |
||
|---|---|---|---|---|---|---|
| R30.1 | R30.1no | R30.1nob | ||||
| Proteophosphoglycan 4 (LinJ.35.0520) | G→C | 206838 | V→L | Hom | .:.:.:.:. | Hom |
| Deletion | 210357 | CVVCTV and Qxb | Hom | .:.:.:.:. | .:.:.:.:. | |
| G→A | 210393 | V→I | Hom | .:.:.:.:. | .:.:.:.:. | |
| G→A | 210798 | V→I | Hom | .:.:.:.:. | .:.:.:.:. | |
| Deletion | 214074 | VVCAVQQQLRAVGVLVVCAVQQQLRAVGVL | Het | .:.:.:.:. | .:.:.:.:. | |
| AG→A | 214111 | RA→HT | Het | .:.:.:.:. | .:.:.:.:. | |
| Deletion | 214114 | Het | .:.:.:.:. | .:.:.:.:. | ||
| G→A | 214438 | RA→HT | Hom | .:.:.:.:. | Hom | |
| G→A | 214440 | Hom | .:.:.:.:. | Hom | ||
| S-Adenosylmethionine transporter (LinJ.10.0370) | G→C | 151608 | G→A | Het | .:.:.:.:. | .:.:.:.:. |
| C→A | 151609 | G→A | Het | .:.:.:.:. | .:.:.:.:. | |
| C→G | 151616 | Q→D | Het | .:.:.:.:. | .:.:.:.:. | |
| G→T | 151618 | Het | .:.:.:.:. | .:.:.:.:. | ||
| C→A | 151619 | Q→K | Het | .:.:.:.:. | .:.:.:.:. | |
| Folate-biopterin transporter (LinJ.10.0390) | A→G | 163647 | H→R | Hom | .:.:.:.:. | .:.:.:.:. |
| Folate-biopterin transporter (LinJ.10.0400) | Insertion | 171999 | A→R | Het | .:.:.:.:. | .:.:.:.:. |
| Deletion | 172001 | A→S | Het | .:.:.:.:. | .:.:.:.:. | |
| A→G | 172184 | N→S | Hom | .:.:.:.:. | Hom | |
| Insertion | 172186 | T→S | Hom | .:.:.:.:. | Hom | |
| Deletion | 172188 | I→A | Hom | .:.:.:.:. | Hom | |
| GP63 (LinJ.10.0520) | Deletion | 216455 | Deletions shift ORF and generate truncated proteinb | .:.:.:.:. | .:.:.:.:. | Hom |
| Deletion | 216461 | Hom | .:.:.:.:. | .:.:.:.:. | ||
| Deletion | 216554 | .:.:.:.:. | .:.:.:.:. | Het | ||
| C→T | 216928 | Het | .:.:.:.:. | Het | ||
| Phosphoglycan beta galactosyltransferase (LinJ.31.3330) | A→G | 1462550 | T→A | Het | .:.:.:.:. | .:.:.:.:. |
| G→T | 1462552 | Het | .:.:.:.:. | .:.:.:.:. | ||
| G→T | 1462557 | R→L | Het | .:.:.:.:. | .:.:.:.:. | |
| A→G | 1462560 | K→R | Het | .:.:.:.:. | .:.:.:.:. | |
| C→A | 1462567 | T→V | Het | .:.:.:.:. | .:.:.:.:. | |
| Deletion | 1462570 | K→T | Het | .:.:.:.:. | .:.:.:.:. | |
| A→C | 1462577 | Kxb | Het | .:.:.:.:. | .:.:.:.:. | |
| Beta fructofuranosidase (LinJ.04.0310) | Insertion | 98726 | N→b | Hom | .:.:.:.:. | .:.:.:.:. |
| Phosphoglycan beta galactosyltransferase (LinJ.02.0170) | G→A | 90694 | A→V | Het | .:.:.:.:. | .:.:.:.:. |
| G→A | 90781 | A→V | Hom | .:.:.:.:. | .:.:.:.:. | |
| LdMT (LinJ.13.1590) | G→T | 620293 | Sxb | Hom | .:.:.:.:. | Het |
| P-glycoprotein (MDR1) (LinJ.34.1060) | G→C | 439247 | R→S | Het | .:.:.:.:. | Het |
| G→A | 440541 | D→N | Het | .:.:.:.:. | Het | |
| C→T | 441121 | A→V | Het | .:.:.:.:. | Het | |
| mTOR kinase (LinJ.34.3750) | G→A | 1511126 | R→H | Het | .:.:.:.:. | Het |
| Kinesin K39 (LinJ.14.1600) | C→T | 104163 | A→T | Hom | .:.:.:.:. | .:.:.:.:. |
Summary of mutations (SNPs and indels) identified in miltefosine-resistant clones derived from the parent WT L. donovani. Heterozygous (Het) and homozygous (Hom) mutations are indicated compared to the WT strain. ORF, open reading frame.
Stop codon.
The dots indicate that mutations (SNP/indels) were not found.
High-throughput RNA sequencing (RNA-seq) of HePC-resistant lines.
RNAs isolated from three biological replicates of resistant (R30.1) and WT logarithmic-phase promastigotes were sequenced on an Illumina MiSeq platform. The sequencing library was constructed using the TruSeq RNA sample preparation kit (Illumina), where poly(A)-containing mRNA was purified from total RNA using poly(T) oligo-attached magnetic beads. We generated a total of 68.1 million raw paired-end sequence reads and retained 55.5 million paired-end reads after quality filtering. The raw data can be found in Table S2 in the supplemental material. In Fig. 4, the log fold change (logFC), i.e., the log ratio of normalized expression levels between R30.1 (R) and WT parasites, was plotted against the log counts per million (logCPM). The genes selected as differentially expressed are shown. A short list of the top genes that were up- or downregulated in resistant parasites was added to the plot. A more comprehensive list of differentially expressed genes (the logFC R/WT ratio was less than −1.5 or more than 1.5) is presented in Table 2.
FIG 4.
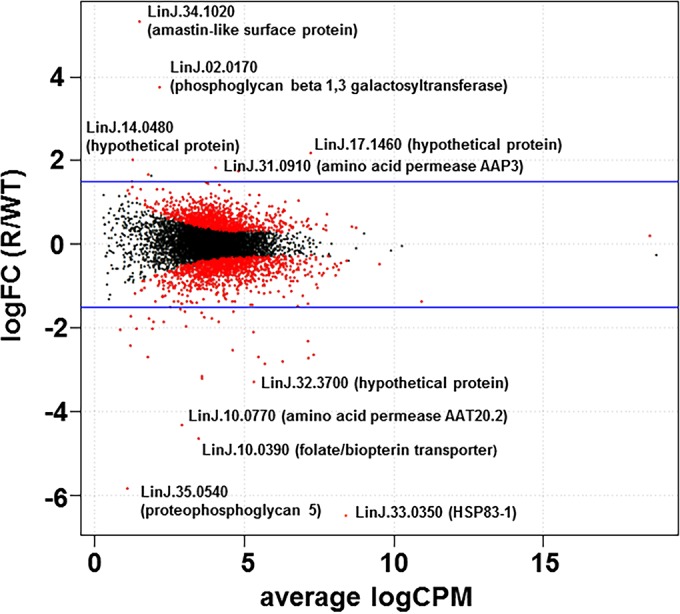
RNA-seq. Shown are differentially expressed mRNAs in resistant and control L. donovani strains. The logFC of normalized expression levels between R30.1 (R) and the WT were plotted against the logCPM. Genes selected as differentially expressed are shown as red dots. The blue lines indicate 1.5-fold changes.
TABLE 2.
Genes differentially expressed (resistant/WT)
| GeneIDa | Log fold change (R/WT) | P value | Product description | GO description |
|---|---|---|---|---|
| LinJ.34.1020 | 5.318479787 | 8.16E−16 | Amastin-like surface protein, putative | NAb |
| LinJ.02.0170 | 3.74401895 | 1.42E−19 | SCGR3 | Membrane; galactosyltransferase activity; protein glycosylation |
| LinJ.14.0480 | 2.008543142 | 1.44E−05 | Hypothetical protein, conserved | Metabolic process; transferase activity, transferring hexosyl groups |
| LinJ.17.1460 | 1.822730736 | 1.43E−20 | Hypothetical protein, conserved | Heme binding |
| LinJ.31.0910 | 1.741031952 | 3.30E−36 | Amino acid permease 3 (AAP3) | Integral component of membrane; arginine transport |
| LinJ.28.2960 | 1.650656184 | 6.26E−02 | Heat shock protein hsp70, putative | ATP binding; response to stress |
| LinJ.29.1850 | −1.500403343 | 2.74E−10 | Histone H2A, putative | Nucleus; DNA binding; nucleosome; protein heterodimerization activity |
| LinJ.18.0270 | −1.545833842 | 5.54E−11 | Glycogen synthase kinase 3 (GSK3) | Protein phosphorylation; ATP binding; protein kinase activity |
| LinJ.32.3330 | −1.608152327 | 1.50E−11 | Ribosomal protein L3, putative | Translation; ribosome; structural constituent of ribosome; ribosome biogenesis |
| LinJ.14.0490 | −1.61880324 | 1.81E−15 | Hypothetical protein, unknown function | NA |
| LinJ.35.3720 | −1.642687961 | 2.90E−21 | Hypothetical protein, conserved | Mitochondrion; NADH dehydrogenase (ubiquinone) activity; mitochondrial electron transport, NADH to ubiquinone; ubiquinone-biosynthetic process; sodium ion transport; proton transport |
| LinJ.15.0140 | −1.717620556 | 1.96E−05 | Zinc finger (CCCH type) protein, putative | Metal ion binding |
| LinJ.14.1600 | −1.755225783 | 2.17E−15 | Kinesin K39, putative | Protein kinase activity; ATP binding; protein phosphorylation |
| LinJ.35.1850 | −1.762112566 | 1.77E−08 | Protein kinase-like protein | Protein kinase activity; protein phosphorylation; ATP binding |
| LinJ.29.2940 | −1.766012806 | 7.17E−27 | Hypothetical protein, conserved | Nucleic acid binding; nucleotide binding |
| LinJ.22.1540 | −1.842571349 | 2.14E−10 | Metallopeptidase, clan MA(E), Family M3, putative, partial | Metalloendopeptidase activity; Proteolysis |
| LinJ.24.1640 | −1.855177439 | 1.38E−07 | Hypothetical protein, conserved | Nucleic acid binding; nucleotide binding |
| LinJ.04.0160 | −1.859253913 | 6.30E−35 | Hypothetical protein, conserved in leishmania | Zinc ion binding |
| LinJ.04.0170 | −1.961165045 | 2.52E−20 | Surface antigen-like protein | Protein binding |
| LinJ.18.1680 | −2.012242952 | 5.92E−10 | Hypothetical protein, conserved | Integral component of membrane; transport |
| LinJ.34.2740 | −2.027394855 | 1.36E−07 | Hypothetical protein, conserved | Lipid metabolic process |
| LinJ.34.2310 | −2.030919495 | 1.25E−04 | Protein phosphatase 2C-like protein | Protein serine/threonine phosphatase activity; metal ion binding; protein dephosphorylation; protein serine/threonine phosphatase complex |
| LinJ.25.2560 | −2.107183485 | 7.62E−52 | Histone H4 | Nucleosome; nucleus; DNA binding; protein heterodimerization activity; nucleosome assembly |
| LinJ.10.0400 | −2.411193056 | 6.67E−09 | Folate/biopterin transporter, putative (FT1) | Transport; integral component of membrane |
| LinJ.21.0020 | −2.532488862 | 1.01E−68 | Histone H4 | Nucleosome; nucleus; DNA binding; protein heterodimerization activity; nucleosome assembly |
| LinJ.22.0012 | −2.68327233 | 1.42E−11 | Hypothetical protein, conserved | Mitochondrion |
| LinJ.32.3320 | −2.715526764 | 6.20E−74 | Ribosomal protein L3, putative | Ribosome; structural constituent of ribosome; translation; ribosome biogenesis |
| LinJ.33.0860 | −2.793529841 | 1.64E−95 | Beta-tubulin | Cytoplasm; microtubule; GTPase activity; structural constituent of cytoskeleton; GTP binding; GTP catabolic process; microtubule-based process; protein polymerization |
| LinJ.32.3700 | −3.282578343 | 8.04E−75 | Hypothetical protein, conserved | NA |
| LinJ.10.0770 | −4.321242608 | 1.26E−42 | Amino acid permease, putative (AAT20.2) | Plasma membrane; integral component of membrane; l-proline transmembrane transporter activity; drug transmembrane transporter activity; alanine transmembrane transporter activity; regulation of l-glutamate transport; drug transmembrane transport; hypotonic response; proline transmembrane transport; regulation of proline transport; glutamate homeostasis; arginine homeostasis |
| LinJ.10.0390 | −4.636169845 | 3.76E−97 | Folate/biopterin transporter, putative | Transport; integral component of membrane |
| LinJ.35.0540 | −5.821310277 | 8.91E−18 | Proteophosphoglycan 5 | Myosin complex; motor activity; ATP binding; metabolic process |
| LinJ.33.0350 | −7.333460885 | 1.01E−276 | Heat shock protein 83, heat shock protein 83-1 (HSP83-1) | Cytoplasm; ATP binding; unfolded protein binding; protein folding; response to stress |
Only genes showing a logFC R/WT ratio above 1.5 (overexpressed) or less than −1.5 (underexpressed) are shown.
NA, not applicable.
Metacyclogenesis.
WGS and RNA-seq revealed variants and differential expression of phosphoglycan beta-1,3-galactosyltransferase (SCGR3) and proteophosphoglycan in HePC-resistant parasites and thus prompted us to measure changes in metacyclogenesis. The examination of WT and LdR30.1 stationary-phase cultures enriched for metacyclic promastigotes via a Ficoll 400 step gradient revealed that metacyclogenesis was unusually impaired in the resistant line (Fig. 5, left). A qRT-PCR assay was performed to amplify the metacyclic marker SHERP, which is highly and almost exclusively expressed in infective and nonreplicative stages of the parasite (29, 37). As shown in Fig. 5 (right), SHERP mRNA expression was considerably reduced in LdR30.1 parasites (P < 0.05), confirming the results obtained via Ficoll enrichment.
FIG 5.
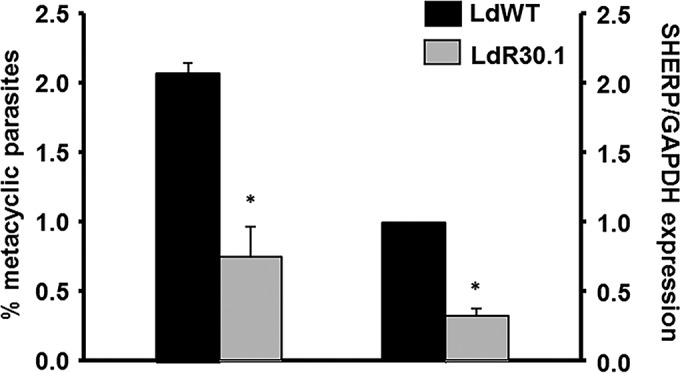
Abundances of metacyclic promastigotes in different lines and their association with HePC resistance. Promastigotes resistant to HePC exhibit decreased metacyclogenesis, as determined by Ficoll enrichment (left) and qRT-PCR of SHERP expression using GAPDH as an internal expression control (right). Populations were analyzed in triplicate, and the error bars indicate standard deviations (*, P < 0.05).
In vitro macrophage infection.
Metacyclogenesis is often associated with the parasite's ability to infect and survive inside the host. RAW264.7 murine macrophages were infected with 106 Ficoll-enriched parasites. The results were contrasted with macrophage cultures infected with an equal number of (normalized) WT metacyclics. No significant differences from controls in parasite infectivity (see Fig. S5A in the supplemental material) or intracellular replication (see Fig. S5B and C in the supplemental material) were observed.
DISCUSSION
Previous studies found that the EC50s for L. donovani HePC-resistant lines at any given concentration were maintained after they were cultured in the absence of the drug for 12 weeks (38). We, too, observed this occurrence only with our 40 μM HePC-resistant parasites after long-term passage (1 year). In addition, most of the parasite populations assayed did not display a change in susceptibility to any other drugs tested. This suggests that the HePC resistance mechanism is exclusive to the compound and likely is not due to the induction of a generalized resistance pattern. LdR30.1nob was the sole exception, and susceptibility was observed only with SbIII, with a 4.25-fold reduction in the EC50 compared to the WT line.
The variation in drug uptake detected in the HePC-resistant parasite populations generated for this work may indicate that the observed phenotype was a result of a modulation in the transporter expression or a defect of the translocation machinery. WGS analysis of R30.1 revealed a homozygous variant in the LdMT (LinJ.13.1590) gene at position 620293, generating a stop codon and inactivation of the protein. R30.1no, a clone derived from the resistant line and cultured in the absence of HePC, reversed the mutation, while a second clonal line, R30.1.nob, with a resistant phenotype in the absence of drug, displayed a heterozygous variant of the mutation and should still produce approximately 50% of the functional transporter.
It has been established that the membrane composition and fluidity are different in parasites with reduced susceptibility to HePC and that these differences may influence drug-membrane interactions (39). The plasma membrane of HePC-resistant parasites is characterized by reduced fluidity as a result of a decrease in the percentage of unsaturated alkyl chains of the phospholipid. Rakotomanga et al. (39) were able to correlate decreased fluidity with reduced HePC-external phospholipid monolayer interactions. Interestingly, changes in membrane fluidity in Leishmania have been ascribed to antimonial (40) or amphotericin B (41) resistance. The BSA used in our experiments to remove surface-adsorbed HePC-BODIPY did not remove HePC inserted in the internal membrane leaflet; thus, our uptake fraction may have included both inserted and incorporated HePC-BODIPY.
Next-generation sequencing (NGS) technologies have become more accessible in recent years, and their application to drug resistance in Leishmania is starting to gain momentum (reviewed in reference 42). For example, whole-genome sequencing was used to study experimental HePC resistance in L. major (43), and distinct mutations were identified, with a potential role in the mechanism of resistance. NGS also encompasses the study of the entire transcriptome of an organism via RNA-seq, with the advantage of quantifying the most significant changes in gene expression. We applied this technology to better understand HePC resistance in Leishmania. The most upregulated gene (logFC, 5.31) in the resistant line is LinJ.34.1020, encoding an amastin-like protein comprising the eukaryotic surface glycoprotein amastin. These genes have been prominent in screens for vaccine candidates in L. donovani, and transcripts from some subfamilies are developmentally restricted to the amastigote stage (44). LinJ.02.0170 (logFC, 3.74) is an SCGR3 that is potentially implicated in the biosynthetic pathway to create the structural phosphoglycan repertoire. A homozygous variant was found at nucleotide position 90781. Phosphoglycans are usually attached to the cell surface through phosphatidylinositol anchors or are secreted as protein-containing glycoconjugates. In L. donovani, metacyclogenesis is associated with the expression of longer lipophosphoglycans on the surface, thus rendering the parasite less susceptible to the innate defenses of the host (45). LinJ.14.0480 (logFC, 2.00) and LinJ.17.1460 (logFC, 1.82) are hypothetical (uncharacterized) proteins. LinJ.31.0910 (logFC, 1.74) is an amino acid permease 3 (AAP3) gene. It codes for a high-affinity arginine transporter, and its transport was not inhibited by other amino acids or arginine-related compounds. It has been suggested that this transporter might play a role in host-parasite interaction (46), where its upregulation may lead to increased levels of the amino acid, as well as to greater sequestration of the arginine, preventing the formation of nitric oxide by the host cell. Using a multianalytical platform, Canuto et al. measured significant elevation of arginine levels in L. donovani strains that were experimentally resistant to HePC (47). This led to the conclusion that HePC-resistant lines may undergo a metabolic adaptation to preserve their fitness inside the macrophage's parasitophorous vacuole. LinJ.33.0350 (logFC, −7.33), a heat shock protein (HSP83), is prominently downregulated in resistant lines. HSP83 is constitutively expressed, and its transcription is not induced by heat shock (48). HSP83 expression is regulated by posttranscriptional control (49) and posttranslational modifications (50). It has also been shown that inhibition of HSP83 in L. donovani induces differentiation from the promastigote to the amastigote parasitic stage (51). In the context of drug resistance, HSP83 was shown to be overexpressed after in vitro selection under antimonials, with the conclusion of the study being that parasites that overexpressed HSP83 were better protected against drug-induced programmed cell death (PCD) (52). We can only speculate that decreased levels of HSP83 (if correlated at the protein level) may confer on HePC-resistant parasites an adaptation to avoid PCD in a novel and unknown fashion. LinJ.35.0540 (logFC, −5.82) is annotated in the genome as a proteophosphoglycan (PPG5). PPG has been shown to be the key molecule conferring resistance to midgut digestive enzymes during sandfly infection (53). It would be interesting to test whether the survival rates of HePC-resistant parasites after artificial feeding of competent sandflies were affected as a consequence of PPG downregulation. LinJ.10.0390 (logFC, −4.63) is a folate/biopterin transporter and an integral component of the membrane. Leishmania parasites acquire folates via active transport, the mechanism of which was first identified by functional studies of methotrexate-resistant mutants, where the folate transporter is strongly downregulated (54, 55). Although the data are preliminary, inhibitors of the folate pathway may not be appropriate for combined therapy with HePC, as HePC-resistant parasites will likely present cross-resistance. WGS data revealed SNPs and indels in the transporter. Interestingly, some of the variants were found in both resistant clones (R30.1 and R30.1nob), although their implication in HePC resistance will require additional studies. LinJ.10.0770 (logFC, −4.32) is an amino acid permease (AAT20.2) downregulated in resistant lines. This neutral amino acid transporter translocates proline and alanine across the L. donovani plasma membrane and plays multiple roles (56). Metabolomics studies have identified increased amounts of proline in HePC-resistant parasites (47). It remains to be established whether this accumulation is a consequence o (i) a lower rate of proline metabolism, (ii) a lack of correlation of RNA and protein expression levels, or (iii) its connection with arginine levels. Interestingly, higher proline levels were associated with playing a protective role against antimonial-induced, but not HePC-induced, oxidative stress (57).
Metacyclogenesis accounts for the transformation of poorly infective procyclic promastigotes into highly infective metacyclic promastigotes. In L. donovani, metacyclogenesis is associated with the expression of longer lipophosphoglycans on the surface, rendering the parasite less susceptible to the innate defenses of the host. This event takes place naturally in the insect vector and is often associated with an increased ability to infect and survive inside the host (58). Metacyclogenesis, therefore, becomes one of the principal contributors to parasite fitness. Stationary cultures can mimic the natural process occurring in the sandfly midgut, leading to in vitro induction of metacyclogenesis (59). By Ficoll enrichment and qRT-PCR experiments, we were able to determine that the metacyclogenesis event takes place in the resistant parasites to a lesser extent than in WT cells. Conversely, a previous report associated increased metacyclogenesis with antimonial resistance in L. donovani (60), and our own results in L. major populations resistant to HePC have led us to link reduced HePC susceptibility to augmented metacyclogenesis rates (61). Altogether, these results strongly suggest that the specific genetic background of the parasite has a key role in determining the parasite response to resistance emergence, not only among species, but also among species-specific strains. To further study virulence and parasite survival, and thus the fitness of resistant lines, an in vitro macrophage infection assay was performed. Our results indicated that although resistant parasites present an impaired ability to transform and become highly infective (metacyclogenesis), their capacity to successfully invade host cells and replicate as amastigotes is not affected.
At this point, it is risky to extrapolate our results to field isolates, as they may indeed behave differently due to additional pressure from the immune response of the host that may crucially shape the resulting resistant phenotype. In fact, the higher metacyclogenesis levels in clinical isolates of L. donovani resistant to antimony have been shown to correlate with greater success of in vitro infection (60). Additionally, it is worth noting that differences in phenotypic outcome are possible when drug selection, e.g., paromomycin, is performed in axenic promastigotes or intracellular amastigotes (19, 62).
In the current work, we approached HePC resistance using NGS technologies, such as WGS and RNA sequencing. A feature of the Leishmania genome (and those of other trypanosomatids) is the unusual nature of transcription and RNA processing. Protein-coding genes are organized into long clusters that are transcribed as polycistronic RNAs, which are posttranscriptionally processed into mature mRNAs by concomitant transsplicing and polyadenylation (63). Traditionally, one of the challenges faced by proteomics is attaining a significant detection threshold for changes in membrane-bound proteins that are difficult to resolve by mass spectrometry proteomics, since the technologies are biased toward soluble hydrophilic peptides (64). In fact, of the comparative proteomics studies addressing drug resistance (65–68), only one stressed the importance of PgP (a membrane glycoprotein) expression as a beacon for HePC resistance, underscoring the importance of complementary omics to acquire the most comprehensive insight for multifaceted processes, such as HePC resistance. In this regard, other complementary proteomic approaches concerning the lines described in this study are in progress.
Supplementary Material
ACKNOWLEDGMENTS
We thank William Archer and Tatyana Orlova (Notre Dame Integrated Imaging Facility) for technical assistance with scanning electron microscopy (SEM). We thank Peter Myler (Seattle Biomedical Research Institute, Seattle, WA) for his guidance and expertise in RNA-seq experiments and Rachel Wiltshire for reading the manuscript.
P.V., L.R., and M.A.M. designed the research; P.V., B.N.-M., M.A.A., and M.T.S. performed the research; P.V., J.S., M.E.P., L.R., and M.A.M. analyzed the data; M.A.M. and L.R. wrote the paper.
Funding Statement
This work was supported by the Eck Institute of Global Health and partially by the Indiana Clinical and Translational Sciences Institute, funded in part by grant number UL1TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. M.A.M. was supported by capitalization funds from the University of Notre Dame. L.R. was supported by VI PN de I+D+I 2008-2011 FIS PS12-02706, ISCIII Subdirección General de Redes y Proyectos Cooperativos-FEDER RD 12/0018/0007, and CSIC PIE-201020E054. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00478-16.
REFERENCES
- 1.Lukes J, Mauricio IL, Schonian G, Dujardin JC, Soteriadou K, Dedet JP, Kuhls K, Tintaya KW, Jirku M, Chocholova E, Haralambous C, Pratlong F, Obornik M, Horak A, Ayala FJ, Miles MA. 2007. Evolutionary and geographical history of the Leishmania donovani complex with a revision of current taxonomy. Proc Natl Acad Sci U S A 104:9375–9380. doi: 10.1073/pnas.0703678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2010. Control of the leishmaniases. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Kumar D, Kulshrestha A, Singh R, Salotra P. 2009. In vitro susceptibility of field isolates of Leishmania donovani to miltefosine and amphotericin B: correlation with sodium antimony gluconate susceptibility and implications for treatment in areas of endemicity. Antimicrob Agents Chemother 53:835–838. doi: 10.1128/AAC.01233-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sundar S, Makharia A, More DK, Agrawal G, Voss A, Fischer C, Bachmann P, Murray HW. 2000. Short-course of oral miltefosine for treatment of visceral leishmaniasis. Clin Infect Dis 31:1110–1113. doi: 10.1086/318122. [DOI] [PubMed] [Google Scholar]
- 5.Sundar S, Singh A, Rai M, Prajapati VK, Singh AK, Ostyn B, Boelaert M, Dujardin JC, Chakravarty J. 2012. Efficacy of miltefosine in the treatment of visceral leishmaniasis in India after a decade of use. Clin Infect Dis 55:543–550. doi: 10.1093/cid/cis474. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Victoria FJ, Castanys S, Gamarro F. 2003. Leishmania donovani resistance to miltefosine involves a defective inward translocation of the drug. Antimicrob Agents Chemother 47:2397–2403. doi: 10.1128/AAC.47.8.2397-2403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Victoria FJ, Gamarro F, Ouellette M, Castanys S. 2003. Functional cloning of the miltefosine transporter. A novel P-type phospholipid translocase from Leishmania involved in drug resistance. J Biol Chem 278:49965–49971. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Victoria FJ, Sanchez-Canete MP, Castanys S, Gamarro F. 2006. Phospholipid translocation and miltefosine potency require both L. donovani miltefosine transporter and the new protein LdRos3 in Leishmania parasites. J Biol Chem 281:23766–23775. doi: 10.1074/jbc.M605214200. [DOI] [PubMed] [Google Scholar]
- 9.Bhandari V, Kulshrestha A, Deep DK, Stark O, Prajapati VK, Ramesh V, Sundar S, Schonian G, Dujardin JC, Salotra P. 2012. Drug susceptibility in Leishmania isolates following miltefosine treatment in cases of visceral leishmaniasis and post kala-azar dermal leishmaniasis. PLoS Negl Trop Dis 6:e1657. doi: 10.1371/journal.pntd.0001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rijal S, Ostyn B, Uranw S, Rai K, Bhattarai NR, Dorlo TP, Beijnen JH, Vanaerschot M, Decuypere S, Dhakal SS, Das ML, Karki P, Singh R, Boelaert M, Dujardin JC. 2013. Increasing failure of miltefosine in the treatment of Kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin Infect Dis 56:1530–1538. doi: 10.1093/cid/cit102. [DOI] [PubMed] [Google Scholar]
- 11.Perez-Victoria JM, Perez-Victoria FJ, Parodi-Talice A, Jimenez IA, Ravelo AG, Castanys S, Gamarro F. 2001. Alkyl-lysophospholipid resistance in multidrug-resistant Leishmania tropica and chemosensitization by a novel P-glycoprotein-like transporter modulator. Antimicrob Agents Chemother 45:2468–2474. doi: 10.1128/AAC.45.9.2468-2474.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castanys-Munoz E, Alder-Baerens N, Pomorski T, Gamarro F, Castanys S. 2007. A novel ATP-binding cassette transporter from Leishmania is involved in transport of phosphatidylcholine analogues and resistance to alkyl-phospholipids. Mol Microbiol 64:1141–1153. doi: 10.1111/j.1365-2958.2007.05653.x. [DOI] [PubMed] [Google Scholar]
- 13.BoseDasgupta S, Ganguly A, Roy A, Mukherjee T, Majumder HK. 2008. A novel ATP-binding cassette transporter, ABCG6 is involved in chemoresistance of Leishmania. Mol Biochem Parasitol 158:176–188. doi: 10.1016/j.molbiopara.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Choudhury K, Zander D, Kube M, Reinhardt R, Clos J. 2008. Identification of a Leishmania infantum gene mediating resistance to miltefosine and SbIII. Int J Parasitol 38:1411–1423. doi: 10.1016/j.ijpara.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Vanaerschot M, Dumetz F, Roy S, Ponte-Sucre A, Arevalo J, Dujardin JC. 2014. Treatment failure in leishmaniasis: drug-resistance or another (epi-) phenotype? Expert Rev Anti Infect Ther 12:937–946. doi: 10.1586/14787210.2014.916614. [DOI] [PubMed] [Google Scholar]
- 16.Vanaerschot M, Huijben S, Van den Broeck F, Dujardin JC. 2014. Drug resistance in vectorborne parasites: multiple actors and scenarios for an evolutionary arms race. FEMS Microbiol Rev 38:41–55. doi: 10.1111/1574-6976.12032. [DOI] [PubMed] [Google Scholar]
- 17.LeBowitz JH. 1994. Transfection experiments with Leishmania. Methods Cell Biol 45:65–78. [DOI] [PubMed] [Google Scholar]
- 18.Vacchina P, Morales MA. 2014. In vitro screening test using Leishmania promastigotes stably expressing mCherry protein. Antimicrob Agents Chemother 58:1825–1828. doi: 10.1128/AAC.02224-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hendrickx S, Boulet G, Mondelaers A, Dujardin JC, Rijal S, Lachaud L, Cos P, Delputte P, Maes L. 2014. Experimental selection of paromomycin and miltefosine resistance in intracellular amastigotes of Leishmania donovani and L. infantum. Parasitol Res 113:1875–1881. doi: 10.1007/s00436-014-3835-7. [DOI] [PubMed] [Google Scholar]
- 20.Hornillos V, Carrillo E, Rivas L, Amat-Guerri F, Acuna AU. 2008. Synthesis of BODIPY-labeled alkylphosphocholines with leishmanicidal activity, as fluorescent analogues of miltefosine. Bioorg Med Chem Lett 18:6336–6339. doi: 10.1016/j.bmcl.2008.10.089. [DOI] [PubMed] [Google Scholar]
- 21.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. 2006. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol Chapter 15:Unit 15.18. [DOI] [PubMed] [Google Scholar]
- 22.Zhang WW, Ramasamy G, McCall LI, Haydock A, Ranasinghe S, Abeygunasekara P, Sirimanna G, Wickremasinghe R, Myler P, Matlashewski G. 2014. Genetic analysis of Leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog 10:e1004244. doi: 10.1371/journal.ppat.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ Jr, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H. 2010. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
- 25.Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. 2008. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spath GF, Beverley SM. 2001. A lipophosphoglycan-independent method for isolation of infective Leishmania metacyclic promastigotes by density gradient centrifugation. Exp Parasitol 99:97–103. doi: 10.1006/expr.2001.4656. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Victoria JM, Bavchvarov BI, Torrecillas IR, Martinez-Garcia M, Lopez-Martin C, Campillo M, Castanys S, Gamarro F. 2011. Sitamaquine overcomes ABC-mediated resistance to miltefosine and antimony in Leishmania. Antimicrob Agents Chemother 55:3838–3844. doi: 10.1128/AAC.00065-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreira W, Leprohon P, Ouellette M. 2011. Tolerance to drug-induced cell death favours the acquisition of multidrug resistance in Leishmania. Cell Death Dis 2:e201. doi: 10.1038/cddis.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weingartner A, Drobot B, Herrmann A, Sanchez-Canete MP, Gamarro F, Castanys S, Gunther Pomorski T. 2010. Disruption of the lipid-transporting LdMT-LdRos3 complex in Leishmania donovani affects membrane lipid asymmetry but not host cell invasion. PLoS One 5:e12443. doi: 10.1371/journal.pone.0012443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araujo-Santos JM, Gamarro F, Castanys S, Herrmann A, Pomorski T. 2003. Rapid transport of phospholipids across the plasma membrane of Leishmania infantum. Biochem Biophys Res Commun 306:250–255. doi: 10.1016/S0006-291X(03)00947-1. [DOI] [PubMed] [Google Scholar]
- 34.Castanys-Munoz E, Perez-Victoria JM, Gamarro F, Castanys S. 2008. Characterization of an ABCG-like transporter from the protozoan parasite Leishmania with a role in drug resistance and transbilayer lipid movement. Antimicrob Agents Chemother 52:3573–3579. doi: 10.1128/AAC.00587-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JC. 2003. Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell 14:1240–1254. doi: 10.1091/mbc.E02-08-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schonian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin JC, Berriman M. 2011. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res 21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knuepfer E, Stierhof YD, McKean PG, Smith DF. 2001. Characterization of a differentially expressed protein that shows an unusual localization to intracellular membranes in Leishmania major. Biochem J 356:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seifert K, Matu S, Javier Perez-Victoria F, Castanys S, Gamarro F, Croft SL. 2003. Characterisation of Leishmania donovani promastigotes resistant to hexadecylphosphocholine (miltefosine). Int J Antimicrob Agents 22:380–387. doi: 10.1016/S0924-8579(03)00125-0. [DOI] [PubMed] [Google Scholar]
- 39.Rakotomanga M, Saint-Pierre-Chazalet M, Loiseau PM. 2005. Alteration of fatty acid and sterol metabolism in miltefosine-resistant Leishmania donovani promastigotes and consequences for drug-membrane interactions. Antimicrob Agents Chemother 49:2677–2686. doi: 10.1128/AAC.49.7.2677-2686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berg M, Vanaerschot M, Jankevics A, Cuypers B, Maes I, Mukherjee S, Khanal B, Rijal S, Roy S, Opperdoes F, Breitling R, Dujardin JC. 2013. Metabolic adaptations of Leishmania donovani in relation to differentiation, drug resistance, and drug pressure. Mol Microbiol 90:428–442. doi: 10.1111/mmi.12374. [DOI] [PubMed] [Google Scholar]
- 41.Purkait B, Kumar A, Nandi N, Sardar AH, Das S, Kumar S, Pandey K, Ravidas V, Kumar M, De T, Singh D, Das P. 2012. Mechanism of amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother 56:1031–1041. doi: 10.1128/AAC.00030-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leprohon P, Fernandez-Prada C, Gazanion E, Monte-Neto R, Ouellette M. 2015. Drug resistance analysis by next generation sequencing in Leishmania. Int J Parasitol Drugs Drug Resist 5:26–35. doi: 10.1016/j.ijpddr.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coelho AC, Boisvert S, Mukherjee A, Leprohon P, Corbeil J, Ouellette M. 2012. Multiple mutations in heterogeneous miltefosine-resistant Leishmania major population as determined by whole genome sequencing. PLoS Negl Trop Dis 6:e1512. doi: 10.1371/journal.pntd.0001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson AP. 2010. The evolution of amastin surface glycoproteins in trypanosomatid parasites. Mol Biol Evol 27:33–45. doi: 10.1093/molbev/msp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sacks DL, Modi G, Rowton E, Spath G, Epstein L, Turco SJ, Beverley SM. 2000. The role of phosphoglycans in Leishmania-sand fly interactions. Proc Natl Acad Sci U S A 97:406–411. doi: 10.1073/pnas.97.1.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaked-Mishan P, Suter-Grotemeyer M, Yoel-Almagor T, Holland N, Zilberstein D, Rentsch D. 2006. A novel high-affinity arginine transporter from the human parasitic protozoan Leishmania donovani. Mol Microbiol 60:30–38. doi: 10.1111/j.1365-2958.2006.05060.x. [DOI] [PubMed] [Google Scholar]
- 47.Canuto GA, Castilho-Martins EA, Tavares MF, Rivas L, Barbas C, Lopez-Gonzalvez A. 2014. Multi-analytical platform metabolomic approach to study miltefosine mechanism of action and resistance in Leishmania. Anal Bioanal Chem 406:3459–3476. doi: 10.1007/s00216-014-7772-1. [DOI] [PubMed] [Google Scholar]
- 48.Brandau S, Dresel A, Clos J. 1995. High constitutive levels of heat-shock proteins in human-pathogenic parasites of the genus Leishmania. Biochem J 310:225–232. doi: 10.1042/bj3100225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larreta R, Soto M, Quijada L, Folgueira C, Abanades DR, Alonso C, Requena JM. 2004. The expression of HSP83 genes in Leishmania infantum is affected by temperature and by stage-differentiation and is regulated at the levels of mRNA stability and translation. BMC Mol Biol 5:3. doi: 10.1186/1471-2199-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morales MA, Watanabe R, Dacher M, Chafey P, Osorio y Fortea J, Scott DA, Beverley SM, Ommen G, Clos J, Hem S, Lenormand P, Rousselle JC, Namane A, Spath GF. 2010. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc Natl Acad Sci U S A 107:8381–8386. doi: 10.1073/pnas.0914768107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiesgigl M, Clos J. 2001. Heat shock protein 90 homeostasis controls stage differentiation in Leishmania donovani. Mol Biol Cell 12:3307–3316. doi: 10.1091/mbc.12.11.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vergnes B, Gourbal B, Girard I, Sundar S, Drummelsmith J, Ouellette M. 2007. A proteomics screen implicates HSP83 and a small kinetoplastid calpain-related protein in drug resistance in Leishmania donovani clinical field isolates by modulating drug-induced programmed cell death. Mol Cell Proteomics 6:88–101. [DOI] [PubMed] [Google Scholar]
- 53.Secundino N, Kimblin N, Peters NC, Lawyer P, Capul AA, Beverley SM, Turco SJ, Sacks D. 2010. Proteophosphoglycan confers resistance of Leishmania major to midgut digestive enzymes induced by blood feeding in vector sand flies. Cell Microbiol 12:906–918. doi: 10.1111/j.1462-5822.2010.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ouellette M, Drummelsmith J, El-Fadili A, Kundig C, Richard D, Roy G. 2002. Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int J Parasitol 32:385–398. doi: 10.1016/S0020-7519(01)00346-0. [DOI] [PubMed] [Google Scholar]
- 55.Kundig C, Haimeur A, Legare D, Papadopoulou B, Ouellette M. 1999. Increased transport of pteridines compensates for mutations in the high affinity folate transporter and contributes to methotrexate resistance in the protozoan parasite Leishmania tarentolae. EMBO J 18:2342–2351. doi: 10.1093/emboj/18.9.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inbar E, Schlisselberg D, Suter Grotemeyer M, Rentsch D, Zilberstein D. 2013. A versatile proline/alanine transporter in the unicellular pathogen Leishmania donovani regulates amino acid homoeostasis and osmotic stress responses. Biochem J 449:555–566. doi: 10.1042/BJ20121262. [DOI] [PubMed] [Google Scholar]
- 57.Berg M, Garcia-Hernandez R, Cuypers B, Vanaerschot M, Manzano JI, Poveda JA, Ferragut JA, Castanys S, Dujardin JC, Gamarro F. 2015. Experimental resistance to drug combinations in Leishmania donovani: metabolic and phenotypic adaptations. Antimicrob Agents Chemother 59:2242–2255. doi: 10.1128/AAC.04231-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.da Silva R, Sacks DL. 1987. Metacyclogenesis is a major determinant of Leishmania promastigote virulence and attenuation. Infect Immun 55:2802–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sacks DL, Perkins PV. 1984. Identification of an infective stage of Leishmania promastigotes. Science 223:1417–1419. doi: 10.1126/science.6701528. [DOI] [PubMed] [Google Scholar]
- 60.Ouakad M, Vanaerschot M, Rijal S, Sundar S, Speybroeck N, Kestens L, Boel L, De Doncker S, Maes I, Decuypere S, Dujardin JC. 2011. Increased metacyclogenesis of antimony-resistant Leishmania donovani clinical lines. Parasitology 138:1392–1399. doi: 10.1017/S0031182011001120. [DOI] [PubMed] [Google Scholar]
- 61.Turner KG, Vacchina P, Robles-Murguia M, Wadsworth M, McDowell MA, Morales MA. 2015. Fitness and phenotypic characterization of miltefosine-resistant Leishmania major. PLoS Negl Trop Dis 9:e0003948. doi: 10.1371/journal.pntd.0003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hendrickx S, Inocencio da Luz RA, Bhandari V, Kuypers K, Shaw CD, Lonchamp J, Salotra P, Carter K, Sundar S, Rijal S, Dujardin JC, Cos P, Maes L. 2012. Experimental induction of paromomycin resistance in antimony-resistant strains of L. donovani: outcome dependent on in vitro selection protocol. PLoS Negl Trop Dis 6:e1664. doi: 10.1371/journal.pntd.0001664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clayton C, Shapira M. 2007. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol Biochem Parasitol 156:93–101. doi: 10.1016/j.molbiopara.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 64.Whitelegge JP. 2013. Integral membrane proteins and bilayer proteomics. Anal Chem 85:2558–2568. doi: 10.1021/ac303064a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carnielli JB, de Andrade HM, Pires SF, Chapeaurouge AD, Perales J, Monti-Rocha R, Carvalho SF, Ribeiro LP, Dietze R, Figueiredo SG, Lemos EM. 2014. Proteomic analysis of the soluble proteomes of miltefosine-sensitive and -resistant Leishmania infantum chagasi isolates obtained from Brazilian patients with different treatment outcomes. J Proteomics 108:198–208. doi: 10.1016/j.jprot.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 66.Singh G, Chavan HD, Dey CS. 2008. Proteomic analysis of miltefosine-resistant Leishmania reveals the possible involvement of eukaryotic initiation factor 4A (eIF4A). Int J Antimicrob Agents 31:584–586. doi: 10.1016/j.ijantimicag.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 67.Messaritakis I, Christodoulou V, Mazeris A, Koutala E, Vlahou A, Papadogiorgaki S, Antoniou M. 2013. Drug resistance in natural isolates of Leishmania donovani s.l. promastigotes is dependent of Pgp170 expression. PLoS One 8:e65467. doi: 10.1371/journal.pone.0065467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Das S, Shah P, Baharia RK, Tandon R, Khare P, Sundar S, Sahasrabuddhe AA, Siddiqi MI, Dube A. 2013. Over-expression of 60s ribosomal L23a is associated with cellular proliferation in SAG resistant clinical isolates of Leishmania donovani. PLoS Negl Trop Dis 7:e2527. doi: 10.1371/journal.pntd.0002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.