

Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that causes considerable morbidity and mortality, specifically during intensive care. Antibiotic-resistant variants of this organism are more difficult to treat and cause substantial extra costs compared to susceptible strains. In the laboratory, P. aeruginosa rapidly developed resistance to five medically relevant antibiotics upon exposure to stepwise increasing concentrations. At several time points during the acquisition of resistance, samples were taken for whole-genome sequencing. The increase in the MIC of ciprofloxacin was linked to specific mutations in gyrA, parC, and gyrB, appearing sequentially. In the case of tobramycin, mutations in fusA, HP02880, rplB, and capD were induced. The MICs of the beta-lactam compounds meropenem and ceftazidime and the combination of piperacillin and tazobactam correlated linearly with beta-lactamase activity but not always with individual mutations. The genes that were mutated during the development of beta-lactam resistance differed for each antibiotic. A quantitative relationship between the frequency of mutations and the increase in resistance could not be established for any of the antibiotics. When the adapted strains are grown in the absence of the antibiotic, some mutations remained and others were reversed, but this reversal did not necessarily lower the MIC. The increased MIC came at the cost of moderately reduced cellular functions or a somewhat lower growth rate. In all cases except ciprofloxacin, the increase in resistance seems to be the result of complex interactions among several cellular systems rather than individual mutations.
INTRODUCTION
The medical consequences of antibiotic resistance, such as fewer options for and increased costs of treating infectious diseases, are well recognized. The pathway to resistance consists of sequential mutations or acquisition of resistance genes driven by the selective pressure caused by antibiotic exposure (1). Once resistance has been acquired, the cell rarely reverses to become sensitive again, compensating for the metabolic costs instead (2, 3). The increased level of resistance caused by an antibiotic treatment typically prescribed by primary care physicians is very noticeable when subsequent further treatment is necessary (4). Hence, in order to limit the development of resistance when antibiotics have to be used, treatment protocols need to be devised to prevent this side effect. Rational design of such protocols requires knowledge of the molecular mechanisms that cause resistance. One of the central questions is whether similar mechanisms are operational for all drugs or whether resistance to each drug is induced in a distinct manner. Other basic questions center on evolutionary pathways to clinically significant resistance and the persistence of molecular changes after treatment.
Molecular changes that cause the development and persistence of drug resistance can be identified by combining experimental evolution and whole-genome sequencing (WGS), provided that the proper controls are used (5, 6). This study used the pathogen Pseudomonas aeruginosa as a model to achieve this goal, as it is an important opportunistic pathogen, for example, in patients suffering from cystic fibrosis (7). Several antibiotics are used as the treatment of choice for intensive care patients infected with P. aeruginosa. The bacteria were adapted to the following five most-often-used drugs: the fluoroquinolone ciprofloxacin, the aminoglycoside tobramycin, and the beta-lactam antibiotics ceftazidime, meropenem, and piperacillin in combination with the beta-lactamase inhibitor tazobactam. This experimental design allows the comparison of three classes of antibiotics and has three members of a single class to document the variability within a class as a biological control for the interclass comparison. The outcome suggests that the de novo buildup of resistance cannot be attributed only to DNA mutations but rather develops as a result of intricate interactions between cellular adaptation and mutations.
MATERIALS AND METHODS
Bacterial strains, growth media, and culture conditions.
The antibiotic-susceptible wild-type strain P. aeruginosa ATCC 27853 was used as the ancestor strain in all resistance evolution experiments. Batch cultures were grown in either rich or defined minimal medium to assess the influence of the growth environment on the development of resistance. The rich medium was cation-adjusted Mueller-Hinton broth (Sigma-Aldrich) autoclaved at 115°C for 10 min. The minimal medium was Evans medium containing 55 mM glucose at pH 6.9 (8). Evans medium was autoclaved for 20 min at 121°C, with the exception of glucose, which was autoclaved for 10 min at 110°C and added afterward. Continuous cultures were performed only with Evans medium with the concentrations of glucose and Na2HPO4 lowered to 5 and 10 mM, respectively.
Precultures for the inoculation of 96-well plates, batch cultures, and continuous cultures were grown overnight in 100-ml flasks shaken at 200 rpm at 37°C. Continuous cultures were carried out in Sixfors fermenter vessels (Infors AG, Bottmingen, Switzerland) consisting of six vessels with a working volume of 250 ml at 37°C and constant stirring at 250 rpm. The pH was maintained at 6.9 by automatically adding sterilized 2 N NaOH. Culture parameters such as pH, temperature, and the stirring rate were monitored continuously. The continuous culture was assumed to have reached a steady state when all of the parameters measured, including cell density and optical density at 600 nm (OD600), remained constant after five to seven volume changes. Samples were taken at every steady state to determine the dry weight and number of cells and the glucose concentration of the culture medium.
Evolution experiments.
For experiments documenting the development of resistance, cultures were initially grown at the maximum antibiotic concentration that allowed growth. Whenever normal or approximately normal growth (OD600 at >75% of the OD600 for normal growth) occurred, a small aliquot of the culture was used to start two more incubations, one at the same concentration and the other at double the concentration (9). The stepwise increasing exposure to an antibiotic was stopped when the saturation level of the antibiotic was reached or continued for 30 days at most. After the adaptation, cultures were grown in fresh medium without drugs for 15 days to observe the sustainability of the acquired resistance after treatment. Independent duplicates were performed with each antibiotic. The MIC was determined every day for both duplicates. Daily samples were preserved at −80°C for further tests, including WGS, beta-lactamase activity measurement, and fitness evaluation. These tests were done with samples revived at the concentration of antibiotic to which they were adapted.
Antibiotics and MIC measurement.
Five antibiotics were tested in this study, i.e., three beta-lactam antibiotics, ceftazidime, meropenem, and piperacillin, combined with the beta-lactamase inhibitor tazobactam; the aminoglycoside tobramycin; and the fluoroquinolone ciprofloxacin. The 10-mg/ml stock solutions of ceftazidime (Fresenius Kabi), meropenem (Fresenius Kabi), piperacillin-tazobactam (Fresenius Kabi), and ciprofloxacin (Fluka) were filter sterilized (0.2 μm) and preserved in a freezer at −20°C. Each stock solution of these drugs was used only once and remade freshly every week. The tobramycin was purchased in a solution of 80 mg/2 ml (Obracin), stored according to the manufacturer's instructions, and used before the expiration date.
MICs were measured by monitoring the growth of cells exposed to antibiotic concentrations increasing by factors of 2 in 96-well plates as described previously (10). The ranges of antibiotic concentrations were adjusted according to the expected resistance level of the sample tested. All measurements were performed in duplicate. The starting OD600 was 0.05. The MIC was defined as the minimal concentration of antibiotic that limited growth to an OD600 of ≤0.2 after 23 h.
De novo sequencing and annotation of the reference strain.
The culture's genome was isolated with the DNeasy blood and tissue kit (Qiagen). De novo sequencing of the reference strain was performed by using the Illumina and PacBio platforms at BaseClear B.V. (Leiden, The Netherlands). For Illumina sequencing, high-molecular-weight genomic DNA (gDNA) was used as the input for library preparation with the Illumina Nextera XT library preparation kit (Illumina). Briefly, the gDNA was fragmented by random transposon integration, DNA adapters with sample-specific bar codes were added, and the library was amplified by PCR. The resulting Illumina library was checked on a Bioanalyzer (Agilent) and quantified. The library was multiplexed, clustered, and sequenced on an Illumina HiSeq 2500 by a paired-end 125-cycle protocol. For PacBio sequencing, high-molecular-weight gDNA was sheared to about 10-kb lengths with g-TUBES (Covaris) and further processed into a PacBio sequencing library by standard protocols (Pacific Biosciences). The resulting PacBio library was checked on a Bioanalyzer (Agilent), quantified, and sequenced on a PacBio RSII.
The quality of the Illumina FASTQ sequences was enhanced by trimming off low-quality bases with the program bbduk, which is part of the BBMap suite, version 34.46. The quality-filtered sequence reads were assembled into a number of contig sequences with ABySS version 1.5.1 (11). These contigs were then linked and placed into superscaffolds based on the alignment of the PacBio CLR reads with BLASR (12). From the alignment, the orientation, order, and distance between the contigs were estimated. This analysis was performed with the SSPACE-LongRead scaffolder, version 1.0 (13). The gapped regions within the superscaffolds were (partially) closed in an automated manner with GapFiller, version 1.10 (14). The complete sequence was reached in one scaffold with a total size of 6,827,737 bp.
Genome annotation of the assembled contig or scaffold sequences was performed with the BaseClear (BaseClear B.V., Leiden, The Netherlands) annotation pipeline, which is based on the Prokka Prokaryotic Genome Annotation System (Victorian Bioinformatics Consortium, Melbourne, Australia).
WGS and data analysis.
WGS was used at those points where the MIC significantly increased in response to drug exposure at the end of the drug exposure period and after 15 days of continued growth in the absence of the drug. Strains grown in either Mueller-Hinton broth or mineral medium for 30 days served as controls for mutations occurring during growth in the absence of antibiotics. DNA was also collected with the DNeasy blood and tissue kit (Qiagen). gDNA libraries were generated according to the manufacturer's protocols with the Ion Xpress Plus gDNA Fragment Library Preparations (Life Technologies). Shearing of 100 ng of gDNA was performed with the Covaris M220 Focused-ultrasonicator in accordance with the 200-bp protocol provided by Life Technologies. Bar-coded libraries were prepared with the Ion Plus fragment library kit (Life Technologies) and the Ion Xpress DNA bar-coding kit (Life Technologies) according to the instructions of the manufacturer of the 200-base-read Ion Proton libraries. The size distribution and yield of the bar-coded libraries were assessed with the 2200 Tapestation System by using Agilent High Sensitivity D1000 ScreenTapes (Agilent Technologies). Sequencing templates were prepared with the Ion PI Template OT2 200 kit v3 on an Ion OneTouch 2 system and enriched on an Ion OneTouch ES system (Life Technologies). Sequencing was performed with the Ion Proton system with the Ion PI Chip v2 and the Ion PI Sequencing 200 kit v3 (Life Technologies) according to the manufacturer's protocols.
The FASTQ files were subjected to quality control procedures. The quality of individual samples was assessed with fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). In addition, several quality metrics (sequencing depths, read length distributions, read quality distributions, mean read quality along the read, base frequencies at each read position) were compared across the samples, in relation to the experimental factors, with in-house software based on samtools (15) and R statistical software (https://www.r-project.org/). Tmap (16) was used to map all accepted reads from all samples to the P. aeruginosa ATCC 27853 reference genome. The Ion Proton system generates sequencing reads of variable lengths, and Tmap combines a short-read algorithm (17) and long-read algorithms (18, 19) in a multistage mapping approach. The average sequencing depth was 220, with a range of 185 to 253. Deviations from the reference genome, such as single-nucleotide variants, insertions, and deletions, were identified with the standalone Torrent Variant Caller, v4.2. (Thermo Fisher Scientific, Waltham, MA). The abundance of the mutations is presented as a mutation frequency calculated as the ratio of the number of reads containing a genetic variation to the overall read number.
Possible gene duplication was searched for by aligning reads of experimental samples with the reference genome and calculating the number of reads that map for every gene. Both forward and reverse complemented reads were counted in nonoverlapping “windows” of 100 nucleotides. Counts were normalized and copy numbers were estimated with haplocn.mops, which is especially designed for haploid organisms (http://www.bioinf.jku.at/research/ehec/ehec.html) (20). Pairwise comparisons of the control and experimental samples for every gene were performed in the R programming language to calculate differences as follows: diff = log2 sample count − log2 control count.
Genes were plotted in the order of the genome and assessed for individual genes or cluster of genes that gained a copy or was deleted. A gene was considered to have been duplicated if the ratio exceeded 1, representing a single duplication, and it occurred in more than one experimental sample. Applying these standards, no gene was found to have been duplicated.
To distinguish mutations caused by sequencing errors or selected for by the growth medium, the detected genetic variations were excluded in the final analysis when one of the following conditions applied: (i) the Phred quality score was <20, (ii) the depth was <100, (iii) mutations appeared only once and at a frequency of <10%, or (iv) mutations also occurred in cells growing in the absence of drugs for 30 days. Mutations with Phred quality scores between 10 and 20 and/or sequencing depths between 40 and 100 were included when they were found more than once.
Reproducibility of mutations detected.
WGS of one of the duplicate strains was performed, and PCR was used to ascertain the presence of these mutations in the other replicate at the same time points. The Primers used are shown in Table 1. The PCR products of six colonies for each combination of antibiotic and time point were purified with the MSB Spin PCRapace kit (Invitek) and sequenced by Macrogen Europe by Sanger sequencing with an ABI 3730XL DNA analyzer.
TABLE 1.
Sequences of the primers used for PCR of the areas of interest within the genes indicated
| Gene | Primer sequence (5′-3′) |
|
|---|---|---|
| Forward | Reverse | |
| ampD | GTAGACCACCACCAGAAG | AATACCTTTCCTCGACGC |
| dacB | ATCGGGCCTGGAGAAT | TTCGCGTGATGTCCGT |
| yerD | GACATGAAAAAGCCGGAG | CGAAGAAGGTGACTACCA |
| hfq | CCCTTCCAGATGCACCA | TTGTCCGTCTGTTTCCG |
| prkC | GAA AAC CAG GAC GC | GTC TTT CTG CCC CGT |
| HP06356 | AGC AAT GTT GTG CCG AT | CTT TGC CGT AAC TTT CAT TC |
| oprD | CTGCGTGCTATAAGTTAG | CTACGCCCTTCCTTTATA |
| mexR | AAGCGGATACCTGAAACG | AAGCCTCGCGTGAAAACA |
| mexB | TCGAGGTGAAGACCGT | GTCGATCCTCAAGCATCG |
| NC3312 | TCA CCC TGA TCG CTC | TTT CCT GGG TTG ATT GAT CG |
| ampC | GCT CAT GGC ACC ATC ATA G | GGG GCG GTT TCT CAT |
| NC3725 | ACC GTG TTC GAG AAA GG | TCG ACC TCC TCC AAC |
| gyrA | CGT TGA TTT GTA GTG AGT TGG | ACT CTC GCT ATA GGT AGG G |
| gyrB | ACCCGCAACTATTGAAAG | CAAGCTACAGGCAAA |
| parC | GGGAGACGACATTC | GCTACTCGGCATG |
| CPA_2 | CCATCGGTAGCGAA | CGT CTA CGC ATT GTC |
| fusA | CAA GAA GCG TGA AGA | AGCCACGACAGTAAA |
| UK02280 | CCCGTCCGTTTT | GCGTTATACTCGCC |
| rplB | GGGCAAGCGTAA | CTTGCGGTCGTT |
| capD | TATTTGCCAGACCAA | AAA GCA GTC GCT TC |
Beta-lactamase assay.
Beta-lactamase activity was measured with an assay based on the chromogenic substrate nitrocefin (21) Briefly, 1 ml of culture was harvested and washed in buffer (pH 7.0). The cells were lysed in 1% Triton X-100, and the lysates were centrifuged. Beta-lactamase activity was determined as the rate of nitrocefin hydrolysis with protein as the normalization factor.
Assessment of fitness cost.
To determine maximum growth rates (μmax) the growth of batch cultures was monitored for 23 h by measuring OD600. The μmax was calculated as the average growth rate of four independent replicates during exponential growth. Dry weight was calculated as the added weight on a preweighed filter dried overnight at 110°C. Cell number was quantified by counting colonies on antibiotic-free agar plates after dilution. Glucose concentrations were determined enzymatically.
RESULTS
To explore the role of mutations in the de novo development of resistance by P. aeruginosa, cells were made resistant to medically relevant antibiotics and WGS was performed at several time points during this process. Resistance to three beta-lactam antibiotics, ceftazidime, meropenem, and piperacillin, in combination with the beta-lactamase inhibitor tazobactam; to the aminoglycoside tobramycin; and to the fluoroquinolone ciprofloxacin was induced. Identification of the mutations that accompany the acquisition of resistance allowed a comparison of the potential differences within the beta-lactam class of antibiotics and the expected differences between the representatives of separate classes. Since preliminary experiments showed that the acquisition of resistance differs when P. aeruginosa is grown in mineral or rich medium, both media were used.
No or barely any resistance to the beta-lactam antibiotics developed during growth and exposure in mineral medium (Fig. 1). There was no meaningful difference in the development of resistance to tobramycin and ciprofloxacin in mineral or rich medium (compare Fig. 1 and 2). In rich medium, the MIC of a drug for P. aeruginosa increased within 20 to 30 days by 7 to 11 2-fold steps, depending on the drug (Fig. 2). Once the drug was removed and growth continued in its absence, the MIC decreased slightly or not at all. The difference between the replicates in the acquisition of resistance was meaningful only in the case of the 8-fold lower MIC of piperacillin-tazobactam for one replicate than for the other. The MIC of ceftazidime decreased for one replicate after exposure was ended but not for the other.
FIG 1.
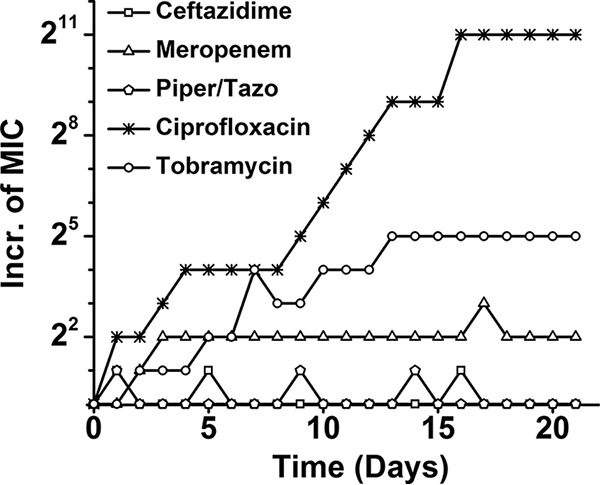
Antibiotic resistance development by P. aeruginosa expressed as 2-fold increases (Incr.) in MICs as a function of time (days) in minimal medium with stepwise increasing concentrations of the antibiotics indicated. Piper, piperacillin; Tazo, tazobactam.
FIG 2.

Increase (Incr.) in the MIC by factors of 2 and frequencies of mutations observed in the replicates on which WGS was performed as a function of time (days) in the presence of the antibiotic indicated and after its removal from the growth medium. All mutations found at frequencies exceeding 0.5 were also discovered in the second replicate by PCR, except for the parC mutations in response to ciprofloxacin, where P85L in the first was replaced by S87N in the second. Piper, piperacillin; Tazo, tazobactam.
At four or five time points (indicated with red arrows in Fig. 2), WGS of one of the replicates was performed with a sequencing depth of roughly 200 times. The hypothesis was that mutations would appear at a low frequency in the beginning and over time become dominant. The actual outcome differed from that expected to various degrees (Fig. 2). In the figures, mutations are omitted that appeared only once and at a frequency of <10%. The presence of the same mutations in the replicate cultures was checked by sequencing PCR products of the DNA in the region around the mutation. Unless specifically mentioned, the first and second replicates had the same mutations. The theoretical possibility that additional mutations had occurred in the replicate strain was not verified experimentally.
The accumulation of mutations was comparatively straightforward when cells were exposed to ciprofloxacin. After five transfers, the usual T83I gyrA mutation for fluoroquinolones (22) appeared, accompanied by a mutation in CPA2 (Fig. 2A). After another seven transfers, the CPA2 mutation disappeared entirely and two parC mutations emerged in less than half of the population. Analysis of the individual reads, averaging 200 bases, suggests that the neighboring parC mutations never occurred in a single chromosome. In the next stage, when the highest MIC had been reached, only one of the two parC mutations, P85L in the first replicate and S87N in the second, remained and a gyrB mutation appeared in the whole population. Even though these mutations remained, the MIC was slightly reduced during growth in the absence of ciprofloxacin. Exposure to tobramycin immediately yielded a mutation in fusA in the whole population and a low frequency of mutation in HP02280 (Fig. 2B). Both were found at a frequency of one a few transfers later, accompanied by a mutation in rpB. When the highest MIC was reached, a new capD mutation attained a frequency of almost one as well. After 15 transfers in drug-free medium, the HP02280 mutation disappeared and the MIC was considerably reduced.
The picture for the three beta-lactam antibiotics was considerably more complex. No persistent mutations were observed in the initial stages, when considerable resistance had already developed. When cells were already almost completely resistant to the combination of piperacillin and tazobactam, two mutations were observed, the usual one in ampC and an unknown single-nucleotide polymorphism in a noncoding region (Fig. 2C). Both remained when the antibiotic pressure was removed, while the MIC remained unchanged. Growth in the presence of meropenem initially resulted in one synonymous mutation with a frequency of one in a hypothetical protein (Fig. 2D). This mutation was no longer observed afterward. Instead, a low-frequency mutation appeared in oprD, which codes for a membrane channel known to be involved in meropenem resistance (23). In the next stage, at an intermediate MIC, the frequency of none of the six observed mutations exceeded 0.5. The mutations occurred three times in oprD, twice in mexR, and once in a noncoding region. None of the different mutations detected in a specific gene occurred together on the chromosome of a single cell. In cells with the highest MIC, mutations in oprD, mexR, mexB, and a noncoding region had taken over the entire population and attained a frequency of one. After removal of the drug, the MIC remained unchanged at high levels, but two new mutations were detected, while the earlier oprD mutation decreased to a very low frequency. The exact same synonymous mutation that was observed at the start of the meropenem experiment also appeared upon exposure to ceftazidime (Fig. 2E). While the MIC increased steadily, low-frequency mutations appeared and disappeared. Only at the very end, when the MIC had decreased because of removal of the drug, was a mutation in prkC, which codes for a serine/threonine-protein kinase, detected in the entire population.
The experimental samples were searched for evidence of gene duplication or deletion, but none was found, indicating that under these experimental conditions, only point mutations occurred.
Strains adapted to ceftazidime maintained their beta-lactamase activity when grown in the absence of a drug, but meropenem- and piperacillin-tazobactam-adapted cells did not (Fig. 3A). Since the MICs of the latter two antibiotics did not decrease during growth in their absence, beta-lactamase activity cannot be the sole factor determining the MICs. Still, there is a considerable correlation between the measured beta-lactamase activity and the increase in the MICs of the three beta-lactam antibiotics (ceftazidime, R2 = 0.926; piperacillin-tazobactam, R2 = 0.921; meropenem, R2 = 0.635) (Fig. 3B). This correlation does not necessarily mean that beta-lactamase activity determines the MIC of this class of antibiotics. The positive cross-resistance of only two to four 2-fold steps (Table 2) suggests that a common mechanism is merely part of the total beta-lactam resistance system. Cross-resistance with the antibiotics of the other classes is either absent or negative. The negative cross-resistance of tobramycin-adapted cells indicates that becoming resistant to this antibiotic increases sensitivity to the others.
FIG 3.

(A) β-Lactamase activities measured in cells adapted to the beta-lactam antibiotics indicated and after subsequent growth for 15 days in the absence of the drugs. The unit of β-lactamase activity is nanomoles of nitrocefin hydrolyzed per minute per milligram of protein. (B) Increase (Incr.) in the MIC as a function of β-lactamase activity. The correlation between β-lactamase activity and MIC is statistically significant for the two antibiotics indicated with asterisks. Piper, piperacillin; tazo, tazobactam.
TABLE 2.
Cross-resistance after drug exposure/removala
| Drug(s) | Ceftazidime | Meropenem | Piperacillin-tazobactam | Tobramycin | Ciprofloxacin |
|---|---|---|---|---|---|
| Ceftazidime | +27/25 | +23/+22 | +23/+22 | –b | – |
| Meropenem | +24/– | +29/+29 | +22/– | – | – |
| Piperacillin-tazobactam | +24/+22 | +22/– | +27/+27 | – | – |
| Tobramycin | −22/−24 | – | −22/−23 | +28/+24 | −23/−24 |
| Ciprofloxacin | – | – | – | – | +27/+25 |
Shown are the increases (+) or decreases (−) in the MICs of the antibiotics at the top upon acquisition of resistance to the antibiotics on the left. After P. aeruginosa became resistant to tobramycin, it was more sensitive to the other antibiotics.
–, no change in the MIC.
The costs of resistance for the cell were measured as a decrease in μmax (Fig. 4A) and as maintenance energy, defined as energy metabolism devoted to purposes other than growth (Fig. 4B). Resistance to all antibiotics caused some decrease in μmax. This decrease was statistically significant in cells adapted to ceftazidime, meropenem, and tobramycin. Ceftazidime-adapted cells had the lowest μmax of 0.85 h−1, compared to 1.15 for the wild type. Maintenance energy could only be measured in cells adapted to ciprofloxacin and tobramycin, because only these grew in mineral medium. In rich medium, too much glucose and other potential carbon and energy sources are available to use the carbon and energy source as a growth rate-limiting factor. The results indicate an increase in the maintenance energy by up to 0.3 mmol of glucose/1010 cells/h when cells become resistant to ciprofloxacin and roughly half of that amount in the case of tobramycin. Taken together, these observations indicate that P. aeruginosa pays a modest metabolic price for becoming resistant to the antibiotics tested.
FIG 4.

μmax and maintenance energy of strains adapted to the antibiotics indicated immediately after growth in the presence of the drugs (after exposure) and after 14 days of subsequent growth in their absence (after removal). An asterisk indicates that the growth rate was significantly lower than that of the wild type, while double asterisks indicate that the growth rates before and after growth in the absence of the antibiotic differed significantly. Panels: A, μmax in duplications (Piper, piperacillin; tazo, tazobactam); B, estimation of the maintenance energy by extrapolation to D (specific growth rate in duplication per h) = 0 of the specific glucose (gluc) consumption measured at several dilution rates; C, μmax of wild-type P. aeruginosa and cells adapted to tobramycin (TBM) in the presence or absence of the drug as a function of pH.
Induced resistance in Escherichia coli came at the price of a reduced ability to grow under less-than-optimal conditions (24). Therefore, growth at nonoptimal pHs and salt concentrations was measured in P. aeruginosa as well. Only cells adapted to tobramycin had a reduced pH range (Fig. 4C), indicating that regulation of the internal pH interfered with counteraction of the drug. In all other cases, growth rates at different pHs and salt concentrations were similar to those of the wild type (data not shown).
DISCUSSION
The outcome of this study suggests that P. aeruginosa acquires resistance to the five antibiotics tested by different genetic mechanisms. WGS did not reveal mutations common to all five types of resistance, not even when limited to the three beta-lactam antibiotics. Some of the genes that were mutated were identified as resistance genes in P. aeruginosa before (25, 26). There was no clear correlation between the frequency of specific mutations and the increase in the MICs of any of the antibiotics tested, though for fluoroquinolone resistance, the gyrA mutation is always required. Below, we will argue that all of the data combined indicate that, as shown for E. coli (22, 24), mutations alone cannot explain the development of resistance but that an intricate interaction between the adaptation of cellular systems and the DNA mutation level leads to higher levels of resistance.
Intrinsic resistance of P. aeruginosa to fluoroquinolones is due to four RND-type drug efflux pumps of the Mex-Opr families (27), which were not affected during de novo development of ciprofloxacin resistance in this study. In a hospital setting, fluoroquinolone use did not correlate with the occurrence of the gyrA and parC mutations found in this study (28). Therefore, it seems that several distinct mechanisms can neutralize the effects of fluoroquinolones on P. aeruginosa. The role of the initial CPA2 mutation is unclear, but mutations often co-occur without an obvious functional relationship during the adaptation of P. aeruginosa to changing environmental conditions (29).
Cell elongation and clustering were shown to be essential steps during the initial development of resistance in P. aeruginosa (30). Therefore, the role of fusA, which codes for an elongation factor, in the initial protection against tobramycin can be understood in terms of its function in biofilm formation by Pseudomonas chlororaphis (31). The rplB gene codes for a 50S ribosome-associated L2 protein that is known to be involved in bactobolin resistance (32). The effect of the rplB mutation may be exerted through interaction with the 30S target protein of aminoglycosides (27). The CapD protein has a function in type 1 capsular polysaccharide biosynthesis (33) and therefore can have an indirect effect on the cellular access of aminoglycosides.
The ceftolozane-tazobactam combination caused several mutations during the development of resistance in P. aeruginosa (34), but the ampC mutation was the only functional one observed when piperacillin-tazobactam was used in this study. Since this mutation appeared at the very last MIC increase stages, its contribution may not have been very important. Possibly, the induction of increased beta-lactamase activity did not involve DNA mutations but was the result of increased expression that remained at higher levels even after many generations, as found in E. coli (24). In contrast, the highest levels of meropenem resistance correlated with mutations in the well-known resistance genes of the oprD and mex families (35). There are, however, several other genes involved in resistance to beta-lactam antibiotics (36, 37) that did not show mutations in this study. The dacB mutations observed at low frequencies during the development of resistance to ceftazidime influence the expression of AmpC beta-lactamases (38). The ampD mutations observed after the growth of cells made resistant to ceftazidime in antibiotic-free medium fulfill a role in peptidoglycan synthesis (39). The mechanism by which the two mutations in yerD, which codes for the large subunit of glutamate synthase, interact with ceftazidime resistance is unclear. It is possible that genetic hitchhiking (40), rather than a functional relationship, is the cause of these mutations.
While development of resistance to tobramycin and ciprofloxacin seems to be a more or less linear process with two unsuccessful mutations in the initial stages in the case of ciprofloxacin, the process is less straightforward in the case of beta-lactam antibiotics. In the latter case, the diverse-community model (41, 42) seems more applicable, in particular in the case of ceftazidime. In the early stages of development of resistance to meropenem and ceftazidime, multiple mutations in different genes are observed at frequencies of less than one and sometimes even at very low frequencies. Either the mutated cells also provide neighboring cells with protection against the antibiotic (43) or it represents evolution by trial and error. Alternatively, the well-documented regulation at the level of cellular processes (24, 44) may be the origin of the initial increase in the MIC observed in a similar manner in the case of all five antibiotics tested.
The fitness costs of resistance can come as a reduced growth rate, lower pathogenicity (2, 45), or a reduced ecological range (24). The μmax of P. aeruginosa was affected less than its maintenance energy, indicating that the operation of, for example, efflux pumps requires additional energy (46). The increased maintenance energy was observed in the absence of antibiotics, indicating a more or less permanent change in metabolism. The moderate effect on μmax is in agreement with observations on clinical strains (47). Whether the acquisition of resistance in vitro also influences pathogenicity, as in these clinical strains, is unclear. No mutations were observed in known pathogenicity genes, but expression levels may have been affected.
The overall conclusion from the comparison of the development of resistance to five antibiotics by P. aeruginosa is that no common mutations or mechanisms can be discerned. Only resistance to ciprofloxacin is built up in a straightforward manner by mutations in the genes coding for the target proteins. The genes affected are different for each of the three beta-lactam antibiotics. While this does not exclude the possibility of a common mechanism regulating the acquisition of resistance, as would be expected, assuming a common killing mechanism for bactericidal antibiotics (48, 49), it does not support that notion either. The lack of correlation between the persistence of mutations and the MIC after removal of the antibiotic from the growth medium indicates that these mutations are not the sole factor determining the MIC.
ACKNOWLEDGMENTS
We thank C. Schultz, C. J. Hodiamont, and R. M. Van Hest for stimulating discussions.
This study was financed by the Netherlands Food and Consumer Product Safety Authority.
REFERENCES
- 1.Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. 2012. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet 44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 3.Gifford DR, MacLean RC. 2013. Evolutionary reversals of antibiotic resistance in experimental populations of Pseudomonas aeruginosa. Evolution 67:2973–2981. doi: 10.1111/evo.12158. [DOI] [PubMed] [Google Scholar]
- 4.Feng Y, Händel N, De Groot MHP, Brul S, Schultsz C, Ter Kuile BH. 2014. Experimental simulation of the effects of an initial antibiotic treatment on a subsequent treatment after initial therapy failure. Antibiotics 3:49–63. doi: 10.3390/antibiotics3010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawecki TJ, Lenski RE, Ebert D, Hollis B, Olivieri I, Whitlock MC. 2012. Experimental evolution. Trends Ecol Evol 27:547–560. doi: 10.1016/j.tree.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Spagnolo F, Rinaldi C, Sajorda DR, Dykhuizen DE. 2015. Evolution of resistance to continuously increasing streptomycin concentrations in populations of Escherichia coli. Antimicrob Agents Chemother 60:1336–1342. doi: 10.1128/AAC.01359-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tümmler B, Wiehlmann L, Klockgether J, Cramer N. 2014. Advances in understanding Pseudomonas. F1000Prime Rep 6:9. doi: 10.12703/P6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans CGT, Herbert D, Tempest DW. 1970. The continuous culture of microorganisms, p 209–344. In Norris JR, Ribbons DW (ed), Methods in microbiology, vol 2 Academic Press, Ltd, London, United Kingdom. [Google Scholar]
- 9.van der Horst MA, Schuurmans JM, Smid MC, Koenders BB, Ter Kuile BH. 2011. De novo acquisition of resistance to three antibiotics by Escherichia coli. Microb Drug Resist 17:141–147. doi: 10.1089/mdr.2010.0101. [DOI] [PubMed] [Google Scholar]
- 10.Schuurmans JM, Nuri Hayali AS, Koenders BB, ter Kuile BH. 2009. Variations in MIC value caused by differences in experimental protocol. J Microbiol Methods 79:44–47. doi: 10.1016/j.mimet.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boetzer M, Pirovano W. 2014. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15:211. doi: 10.1186/1471-2105-15-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol 13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caboche S, Audebert C, Lemoine Y, Hot D. 2014. Comparison of mapping algorithms used in high-throughput sequencing: application to Ion Torrent data. BMC Genomics 15:264. doi: 10.1186/1471-2164-15-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ning Z, Cox AJ, Mullikin JC. 2001. SSAHA: a fast search method for large DNA databases. Genome Res 11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, Hochreiter S. 2012. cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res 40:e69. doi: 10.1093/nar/gks003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai X, Xiang S, Li J, Gao Q, Yang K. 2012. Development of a colorimetric assay for rapid quantitative measurement of clavulanic acid in microbial samples. Sci China Life Sci 55:158–163. doi: 10.1007/s11427-012-4287-x. [DOI] [PubMed] [Google Scholar]
- 22.Händel N, Schuurmans JM, Feng Y, Brul S, Ter Kuile BH. 2014. Interaction between mutations and regulation of gene expression during development of de novo antibiotic resistance. Antimicrob Agents Chemother 58:4371–4379. doi: 10.1128/AAC.02892-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dreier J, Ruggerone P. 2015. Interaction of antibacterial compounds with RND efflux pumps in Pseudomonas aeruginosa. Front Microbiol 6:660. doi: 10.3389/fmicb.2015.00660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Händel N, Schuurmans JM, Brul S, Ter Kuile BH. 2013. Compensation of the metabolic costs of antibiotic resistance by physiological adaptation in Escherichia coli. Antimicrob Agents Chemother 57:3752–3762. doi: 10.1128/AAC.02096-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cabot G, Zamorano L, Moyà B, Juan C, Navas A, Blázquez J, Oliver A. 4 January 2016. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation supply rates. Antimicrob Agents Chemother. doi: 10.1128/AAC.02676-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2:65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morita Y, Tomida J, Kawamura Y. 2014. Responses of Pseudomonas aeruginosa to antimicrobials. Front Microbiol 4:422. doi: 10.3389/fmicb.2013.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cluzet VC, Lautenbach E, Nachamkin I, Cary MS, Fishman NO, Shih NN, Morales KH, Linkin DR. 2015. Risk factors for gyrA and parC mutations in Pseudomonas aeruginosa. Infect Control Hosp Epidemiol 36:387–393. doi: 10.1017/ice.2014.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong A, Rodrigue N, Kassen R. 2012. Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet 8:e1002928. doi: 10.1371/journal.pgen.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng Y, Hodiamont CJ, van Hest RM, Brul S, Schultsz C, Ter Kuile BH. 2016. Development of antibiotic resistance during simulated treatment of Pseudomonas aeruginosa in chemostats. PLoS One 11:e0149310. doi: 10.1371/journal.pone.0149310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D, Dorosky RJ, Han CS, Lo CC, Dichosa AE, Chain PS, Yu JM, Pierson LS III, Pierson EA. 2015. Adaptation genomics of a small-colony variant in a Pseudomonas chlororaphis 30-84 biofilm. Appl Environ Microbiol 81:890–899. doi: 10.1128/AEM.02617-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandler JR, Truong TT, Silva PM, Seyedsayamdost MR, Carr G, Radey M, Jacobs MA, Sims EH, Clardy J, Greenberg EP. 2012. Bactobolin resistance is conferred by mutations in the L2 ribosomal protein. mBio 3:e00499-12. doi: 10.1128/mBio.00499-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Ulm H, Rausch M, Li X, O'Riordan K, Lee JC, Schneider T, Muller CE. 2014. Analysis of the Staphylococcus aureus capsule biosynthesis pathway in vitro: characterization of the UDP-GlcNAc C6 dehydratases CapD and CapE and identification of enzyme inhibitors. Int J Med Microbiol 304:958–969. doi: 10.1016/j.ijmm.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Cabot G, Bruchmann S, Mulet X, Zamorano L, Moya B, Juan C, Haussler S, Oliver A. 2014. Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of AmpC. Antimicrob Agents Chemother 58:3091–3099. doi: 10.1128/AAC.02462-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pai H, Kim J, Kim J, Lee JH, Choe KW, Gotoh N. 2001. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother 45:480–484. doi: 10.1128/AAC.45.2.480-484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bratu S, Landman D, Gupta J, Quale J. 2007. Role of AmpD, OprF and penicillin-binding proteins in beta-lactam resistance in clinical isolates of Pseudomonas aeruginosa. J Med Microbiol 56:809–814. doi: 10.1099/jmm.0.47019-0. [DOI] [PubMed] [Google Scholar]
- 37.Moya B, Dotsch A, Juan C, Blazquez J, Zamorano L, Haussler S, Oliver A. 2009. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog 5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vestergaard M, Paulander W, Marvig RL, Clasen J, Jochumsen N, Molin S, Jelsbak L, Ingmer H, Folkesson A. 2016. Antibiotic combination therapy can select for broad-spectrum multidrug resistance in Pseudomonas aeruginosa. Int J Antimicrob Agents 47:48–55. doi: 10.1016/j.ijantimicag.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W, Lee M, Hesek D, Lastochkin E, Boggess B, Mobashery S. 2013. Reactions of the three AmpD enzymes of Pseudomonas aeruginosa. J Am Chem Soc 135:4950–4953. doi: 10.1021/ja400970n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM. 2013. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500:571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, Kishony R. 2014. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet 46:82–87. doi: 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen D, Singh PK. 2006. Evolving stealth: genetic adaptation of Pseudomonas aeruginosa during cystic fibrosis infections. Proc Natl Acad Sci U S A 103:8305–8306. doi: 10.1073/pnas.0602526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee HH, Molla MN, Cantor CR, Collins JJ. 2010. Bacterial charity work leads to population-wide resistance. Nature 467:82–85. doi: 10.1038/nature09354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernández L, Breidenstein EB, Hancock RE. 2011. Creeping baselines and adaptive resistance to antibiotics. Drug Resist Updat 14:1–21. doi: 10.1016/j.drup.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Andersson DI. 2006. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol 9:461–465. doi: 10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 46.Russell JB, Cook GM. 1995. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol Rev 59:48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roux D, Danilchanka O, Guillard T, Cattoir V, Aschard H, Fu Y, Angoulvant F, Messika J, Ricard JD, Mekalanos JJ, Lory S, Pier GB, Skurnik D. 2015. Fitness cost of antibiotic susceptibility during bacterial infection. Sci Transl Med 7:297ra114. doi: 10.1126/scitranslmed.aab1621. [DOI] [PubMed] [Google Scholar]
- 48.Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol 8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]