

Abstract
The usefulness of β-lactam antimicrobial agents is threatened as never before by β-lactamase-producing bacteria. For this reason, there has been renewed interest in the development of broad-spectrum β-lactamase inhibitors. Herein we describe the results of dose fractionation and dose-ranging studies carried out using a one-compartment in vitro infection model to determine the exposure measure for CB-618, a novel β-lactamase inhibitor, most predictive of the efficacy when given in combination with meropenem. The challenge panel included Enterobacteriaceae clinical isolates, which collectively produced a wide range of β-lactamase enzymes (KPC-2, KPC-3, FOX-5, OXA-48, SHV-11, SHV-27, and TEM-1). Human concentration-time profiles were simulated for each drug, and samples were collected for drug concentration and bacterial density determinations. Using data from dose fractionation studies and a challenge Klebsiella pneumoniae isolate (CB-618-potentiated meropenem MIC = 1 mg/liter), relationships between change from baseline in log10 CFU/ml at 24 h and each of CB-618 area under the concentration-time curve over 24 h (AUC0–24), maximum concentration (Cmax), and percentage of the dosing interval that CB-618 concentrations remained above a given threshold were evaluated in combination with meropenem at 2 g every 8 h (q8h). The exposure measures most closely associated with CB-618 efficacy in combination with meropenem were the CB-618 AUC0–24 (r2 = 0.835) and Cmax (r2 = 0.826). Using the CB-618 AUC0–24 indexed to the CB-618-potentiated meropenem MIC value, the relationship between change from baseline in log10 CFU/ml at 24 h and CB-618 AUC0–24/MIC ratio in combination with meropenem was evaluated using the pooled data from five challenge isolates; the CB-618 AUC0–24/MIC ratio associated with net bacterial stasis and the 1- and 2-log10 CFU/ml reductions from baseline at 24 h were 27.3, 86.1, and 444.8, respectively. These data provide a pharmacokinetics-pharmacodynamics (PK-PD) basis for evaluating potential CB-618 dosing regimens in combination with meropenem in future studies.
INTRODUCTION
The usefulness of β-lactam antimicrobial agents is threatened as never before by β-lactamase-producing bacteria (1). Today, the β-lactamase enzymes that are of greatest clinical concern include Ambler class A extended-spectrum β-lactamases (ESBL) (e.g., CTX-M), class A serine class carbapenemases (e.g., KPC-type), class B metallo-β-lactamases (e.g., NDM-type and VIM-type), class C cephalosporinases (e.g., AmpC-type), and class D serine oxacillinases (e.g., OXA-type cephalosporinases and OXA-type carbapenemases) (2). Among Enterobacteriaceae, ESBL- and AmpC-type are common β-lactamases (3, 4, 5). The CTX-M-type β-lactamases, which were first reported in the late 1980s (6), have become one of the most globally dominant groups (7, 8). KPC-type β-lactamases were first identified in the United States in 1996 (9) and have since increased in prevalence and become a global concern (10). Increases in the prevalence of OXA-type β-lactamases are also of concern due to variable inhibition of investigational β-lactamase inhibitor agents (2), with OXA-48 being among the most problematic given the rapid recent dissemination observed (11, 12).
Given the above-described diversity of β-lactamases, the increasing number of β-lactamases produced by each pathogen, and the potential for different enzymes to increase in prevalence in different regions, there has been renewed interest in the development of novel broad-spectrum β-lactamase inhibitors (13, 14). CB-618 is a novel non-β-lactam β-lactamase inhibitor that belongs to the diazabicyclooctane class and that is structurally related to avibactam (data on file at Merck & Co., Kenilworth, NJ, USA). Like avibactam, CB-618 inhibits predominantly Ambler class A and C β-lactamases but also select class D β-lactamases (15; data on file at Merck & Co., Kenilworth, NJ, USA).
One-compartment and hollow-fiber in vitro infection models have emerged as important tools for evaluating the adequacy of candidate β-lactam–β-lactamase inhibitor dosing regimens (16, 17, 18, 19, 20). These models allow for evaluation of the relationship between β-lactamase inhibitor exposure in the context of a given β-lactam exposure and change in bacterial burden (17, 18). The resultant exposure-response relationships for both the β-lactamase inhibitor and β-lactam agent can be used as inputs in subsequent analyses to support dose selection.
As described herein, a one-compartment in vitro infection model was used to simulate a clinically applicable meropenem dosing regimen administered in combination with a range of CB-618 dosing regimens to characterize the pharmacokinetics-pharmacodynamics (PK-PD) relationship for CB-618 efficacy against β-lactamase-producing Enterobacteriaceae. The specific objectives were 2-fold. The first objective was to identify the CB-618 exposure measure most closely associated with CB-618 efficacy. The second objective was to determine the magnitude of the CB-618 PK-PD index associated with net bacterial stasis and 1- and 2-log10 CFU/ml reductions from baseline at 24 h.
MATERIALS AND METHODS
Bacteria, antimicrobial, and β-lactamase inhibitor.
A total of five serine carbapenemase-producing clinical isolates, one Escherichia coli isolate and four Klebsiella pneumoniae isolates, were utilized in these studies (JMI Laboratories, North Liberty, IA). These isolates were selected based upon their elevated MIC values to meropenem alone and meropenem in the presence of CB-618 at a fixed concentration of 4 mg/liter (i.e., the CB-618-potentiated meropenem MIC values), both of which were required to span the upper margin of their respective MIC distributions. The presence of β-lactamases was confirmed by JMI Laboratories using PCR, sequencing, and isoelectric focusing using standard methodologies. Meropenem and CB-618 were provided by Cubist Pharmaceuticals (Lexington, MA).
Media and in vitro susceptibility studies.
Susceptibility studies were completed in triplicate over a 2-day period in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines (21). The susceptibility of each isolate to meropenem and CB-618 was determined by broth microdilution methodology using cation-adjusted Mueller-Hinton growth medium (BD Laboratories, Franklin Lakes, NJ). The susceptibilities of each clinical isolate to meropenem and CB-618 alone and meropenem in combination with CB-618 using a fixed CB-618 concentration (4 mg/liter), following CLSI recommendations for the beta-lactamase inhibitor tazobactam (21), were determined. The modal MIC values were utilized in all analyses.
PK-PD in vitro model and sample processing.
A previously described one-compartment PK-PD in vitro infection model was utilized in the single-isolate dose-ranging, dose fractionation, and multiple-isolate dose-ranging studies (17, 18). The in vitro model consisted of a central infection compartment containing a suspension of the challenge isolate in Mueller-Hinton II broth which was set upon a magnetic stir plate. Within the central infection compartment, the challenge isolate was exposed to meropenem and CB-618. Through the use of computer-controlled syringe pumps, selected free-drug concentration-time profiles were simulated for each study drug. The PK-PD in vitro model was placed within a temperature- and humidity-controlled incubator at 35°C. Specimens for determination of bacterial density and drug concentration were collected from the central infection compartment at predetermined time points.
In these experiments, bacterial suspensions of 1.0 × 106 CFU/ml were prepared for each challenge isolate. These suspensions were generated from a culture grown overnight on Trypticase soy agar supplemented with 5% sheep blood (BD Laboratories). Isolated colonies were taken from the overnight cultures and grown to mid-logarithmic phase in Mueller-Hinton broth at 35°C and 125 rotations per minute in a shaking water bath. The bacterial concentration within the flask was determined by optical density for each challenge isolate. Bacteria were then exposed to changing free-drug concentrations of meropenem and CB-618, simulating human half-lives of 1 h for meropenem (22) and 1.8 h for CB-618 (data on file at Merck & Co., Kenilworth, NJ, USA) using computer-controlled syringe pumps. At baseline and 2, 4, 8, 12, and 24 h after therapy initiation, 1-ml samples were collected for bacterial density determination. Each sample was centrifuged, washed, and resuspended with sterile normal saline twice to prevent drug carryover. The washed samples were serially diluted in sterile normal saline and cultured onto drug-free Trypticase soy agar enriched with 5% sheep blood. Plated samples were placed in a humidified incubator at 35°C for 24 h, and subsequently, the bacterial density was determined. In addition, 1-ml samples were collected for drug concentration determination at 1, 3, 5, 7, 9, 11, 13, and 23 h after initiation of therapy for each study. All samples collected for drug concentration determination were immediately frozen at −80°C until assayed using liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Single-isolate dose-ranging studies.
Duplicate CB-618 dose-ranging studies, for which 0.03 to 2 g of CB-618 was administered every 8 h (q8h) for 24 h, were conducted using the PK-PD one-compartment in vitro infection model, a single KPC-3-producing challenge isolate (K. pneumoniae 79), and a fixed meropenem dosing regimen (2 g q8h). The activities of all treatment regimens were compared to that of the no-treatment control group.
Dose fractionation studies.
The CB-618 dosing regimens selected for use in the 24-h dose fractionation PK-PD in vitro infection model studies were guided by results of the single-isolate dose-ranging studies. The CB-618 dosing regimens selected were those which bracketed and included one-half the maximal effect seen in the dose-ranging study. In these studies, the same KPC-3-producing challenge isolate as evaluated for the single-isolate dose-ranging studies (K. pneumoniae 79) was utilized. The same total daily CB-618 dose was fractionated and administered as equal divided doses every 6, 8, or 12 h in combination with a fixed meropenem dosing regimen (2 g q8h). All studies were completed in duplicate and included a no-treatment control group.
Multiple-isolate dose-ranging studies.
After the dose fractionation studies, 24-h CB-618 dose-ranging studies for which 0.008 to 3 g of CB-618 was administered q8h in the context of a fixed meropenem dosing regimen (1 or 2 g q8h) were conducted utilizing an expanded carbapenemase-producing clinical isolate challenge panel. The expanded challenge panel included one E. coli isolate and three additional K. pneumoniae isolates (Table 1). All studies were completed in duplicate and included a no-treatment control group.
TABLE 1.
Known β-lactamase profiles and susceptibility results for the five Enterobacteriaceae isolates utilized in the PK-PD in vitro infection model studies
| Isolate | β-Lactamase(s)a | MIC (mg/liter) |
||
|---|---|---|---|---|
| Meropenem | CB-618 | Meropenem + CB-618 at 4 mg/liter | ||
| K. pneumoniae 79 | KPC-3, SHV-11, TEM-1, FOX-5 | 128 | 256 | 1 |
| K. pneumoniae 908 | KPC-2, SHV-27, TEM-1 | 64 | 256 | 0.5 |
| K. pneumoniae 6501 | KPC-3, SHV-11, TEM-1 | 512 | 256 | 2 |
| K. pneumoniae 4192 | OXA-48 | 64 | >512 | 0.25 |
| E. coli 4189 | OXA-48 | 16 | >512 | 0.5 |
Boldface indicates carbapenemase enzymes.
PK-PD analysis.
Data from the dose fractionation studies were evaluated using Hill-type models and nonlinear least-squares regression. Data were weighted by the inverse of the estimated measurement variance. Relationships between change from baseline in log10 CFU/ml at 24 h and the ratio of the CB-618 area under the concentration-time curve over 24 h (AUC0–24), maximum CB-618 concentration (Cmax), and percentage of the dosing interval that CB-618 concentrations remained above a given threshold (%Time>threshold) were characterized. The CB-618 concentration threshold was identified through an iterative process previously described (16). In brief, a range of candidate CB-618 concentration thresholds (0.5 to 4 mg/liter) were evaluated. Discrimination of candidate concentration thresholds was accomplished by evaluating the dispersion of data along the %Time>threshold axis and optimizing r2 values for the relationship between change from baseline in log10 CFU/ml at 24 h and %Time>threshold.
Using a Hill-type model, nonlinear least-squares regression, the data from the four clinical isolates utilized in the multiple-isolate dose-ranging studies, and those from the single isolate used in dose fractionation studies, the relationship between change from baseline in log10 CFU/ml at 24 h and the CB-618 PK-PD index most associated with efficacy based on the dose fractionation studies was evaluated. The CB-618 AUC0–24/MIC ratio and Cmax/MIC ratio were determined by indexing each exposure measure to the CB-618-potentiated meropenem MIC value for each challenge isolate. Data were weighted by the inverse of the estimated measurement variance. Using this relationship, the magnitudes of the CB-618 PK-PD index in combination with meropenem that was associated with net bacterial stasis and 1- and 2-log10 CFU/ml reductions from baseline at 24 h were determined.
Analytical method.
All samples were assayed by liquid chromatography-mass spectrometry (AB Sciex, Framingham, MA). Standard curves ranged from 0.025 to 50 mg/liter for meropenem and CB-618. The meropenem and CB-618 standard curves were linear over their respective ranges (r2 = 0.994 and 0.998, respectively). The lower limit of quantification was 0.025 mg/liter for both compounds, and the average interday coefficients of variation were 4.4 and 4.7% for the meropenem and CB-618 standard curves, respectively (data on file at Merck & Co., Kenilworth, NJ, USA).
RESULTS
In vitro susceptibility testing.
The MIC values for meropenem and CB-618 alone and in combination are presented in Table 1 for the five clinical Enterobacteriaceae isolates evaluated. Collectively, these clinical isolates produced a wide range of β-lactamase enzymes, including carbapenemases (KPC-2, KPC-3, and OXA-48) as well as other β-lactamases (FOX 5, SHV 11, SHV 27, and TEM 1), with corresponding meropenem MIC values varying from 16 to 512 mg/liter. However, when meropenem was potentiated with 4 mg/liter of CB-618, the MIC values decreased markedly (0.25 to 2 mg/liter). As evidenced by MIC values of 256 or >512 mg/liter, CB-618 displayed a minimal antibacterial effect alone.
PK.
The simulated meropenem and CB-618 pharmacokinetic (PK) profiles matched the targeted profiles well in the PK-PD in vitro infection model for all dosing regimens evaluated (Fig. 1), as evidenced by the agreement between observed and predicted concentrations of each drug (for meropenem, r2 = 0.957, slope = 0.933, and intercept = 0.4368; for CB-618, r2 = 0.9616, slope = 1.004, and intercept = 0.0284).
FIG 1.
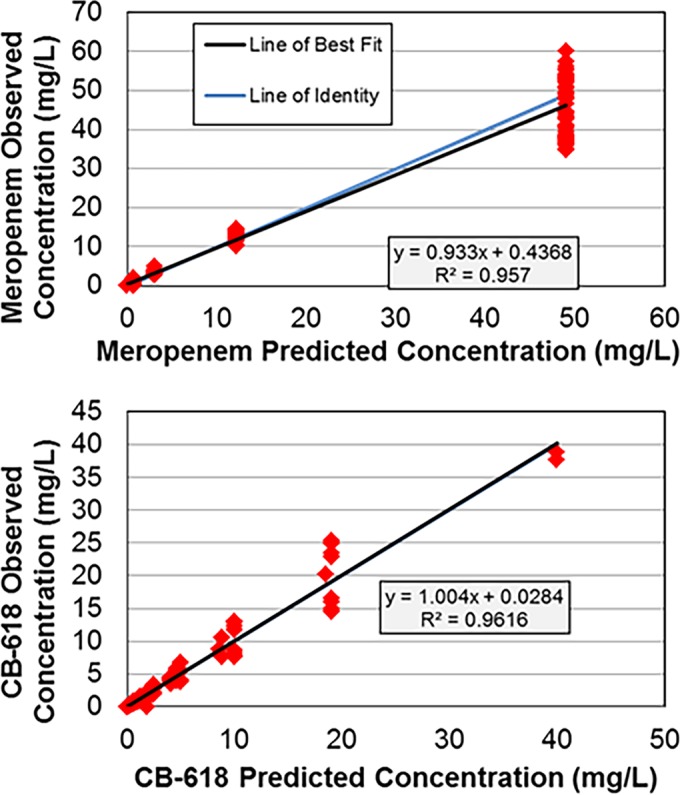
Relationships between observed and predicted meropenem and CB-618 concentrations simulated within the one-compartment PK-PD in vitro infection models.
Given that meropenem MIC values for the challenge isolates ranged from 16 to 512 mg/liter, the percentage of the dosing interval that meropenem concentrations remained above the MIC (%T>MIC) for meropenem active control regimens were low (0 to 29.5%). For the meropenem/CB-618 combination regimens and the meropenem MIC values of the challenge isolates, which were determined in the presence of 4 mg/liter of CB-618 and which ranged from 0.25 to 2 mg/liter, the meropenem %T>MIC ranged from 69.9 to 100% across challenge isolates.
Single-isolate dose-ranging studies.
Bacteria in the no-treatment control groups grew well, reaching a bacterial density of greater than 1.0 × 108 CFU/ml by 8 h (Fig. 2). The range of CB-618 exposures, used in combination with the fixed meropenem regimen of 2 g q8h, provided a full spectrum of effect. Low CB-618 exposures (31.3 to 62.5 mg) demonstrated magnitudes of bacterial burden relatively similar to that of the no-treatment control group by 24 h, while high exposures (500 mg and greater) resulted in ≥1-log10 CFU/ml reductions from baseline at 24 h. Intermediate exposures (125 to 250 mg) resulted in net bacterial stasis at 24 h. The results of these single-isolate dose-ranging studies were sufficient to select exposures for use in the dose fractionation studies. The total daily CB-618 exposures selected for the dose fractionation studies were 0.188, 1.5, and 6 g.
FIG 2.

Single-isolate dose-ranging study results for meropenem at 2 g q8h in combination with CB-618 exposures ranging from 31.3 mg to 2 g q8h.
PK-PD analyses.
The relationships between change from baseline in log10 CFU/ml at 24 h and the CB-618 AUC0–24, Cmax, and %Time>threshold based on the data from the dose fractionation studies for the challenge isolate (K. pneumoniae 79) are presented in Fig. 3. As demonstrated by the dispersion of the data around the fitted functions based on the Hill-type models for each exposure measure, the CB-618 AUC0–24 (r2 = 0.835) and Cmax (r2 = 0.826) were most closely associated with CB-618 efficacy in combination with meropenem. It should be noted that while the association for %Time>threshold using a CB-618 concentration threshold of 1 mg/liter was reasonably high (r2 = 0.792) compared to that for the other thresholds evaluated (0.5, 2, and 4 mg/liter), this exposure measure was not selected for further evaluation using the data from the multiple-isolate dose-ranging studies due the variability in effect (approximately a 3-log10 CFU/ml range) at the 100% Time>threshold.
FIG 3.

Relationships between CB-618 AUC0–24, Cmax, and %Time>threshold and the change from baseline in log10 CFU/ml of K. pneumoniae 79 at 24 h (CB-618-potentiated meropenem MIC value = 1 mg/liter). The colors of the symbols represent the different dosing intervals.
Figure 4 shows the relationships between change from baseline in log10 CFU/ml at 24 h and the CB-618 AUC0–24/MIC ratio across all five clinical isolates. As demonstrated by the dispersion of the data around the fitted function, the CB-618 AUC0–24/MIC ratio (r2 = 0.721) described the relationship between drug exposure and response well. The CB-618 AUC0–24/MIC ratio in combination with meropenem that was associated with net bacterial stasis and 1- and 2-log10 CFU/ml reductions from baseline at 24 h were 27.3, 86.1, and 444.8, respectively. The parameter estimates (standard error) for the Hill-type model describing the relationship between change from baseline in log10 CFU/ml at 24 h and CB-618 AUC0–24/MIC shown in Fig. 4 were 3.3 (1.0), 6.12 (1.77), 0.61 (0.26), and 21.2 (9.4) for change from baseline in log10 CFU/ml at 24 h in the absence of drug (E0), the maximum change in log10 CFU/ml from E0 (Emax), Hill coefficient, and the CB-618 AUC0–24/MIC ratio associated with half-maximal effect (EC50), respectively.
FIG 4.
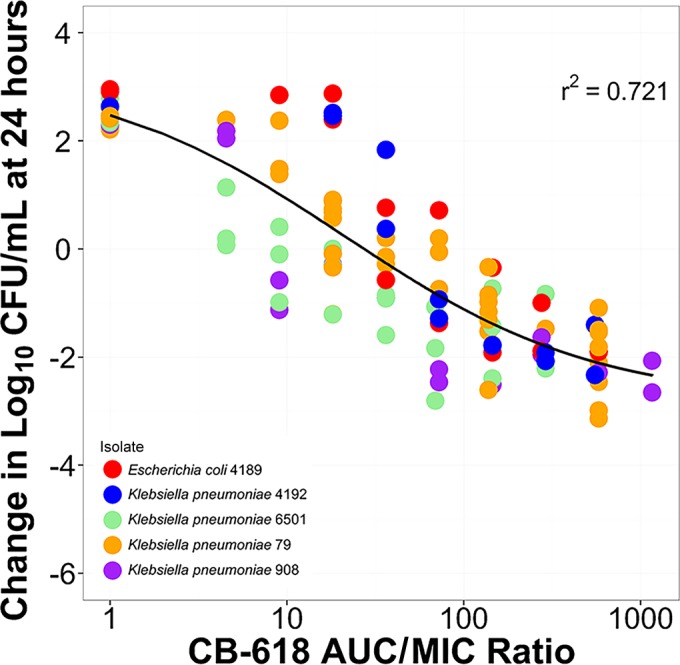
Relationship between CB-618 AUC0–24/MIC ratio and the change from baseline in log10 CFU/ml at 24 h based on data from the Enterobacteriaceae challenge panel. The different-colored circles represent each of the five different clinical isolates, with CB-618-potentiated meropenem MIC values that ranged from 0.25 to 2 mg/liter.
DISCUSSION
There were two objectives of these studies; the first was to identify the CB-618 exposure measure in combination with meropenem that was most closely associated with efficacy, and the second was to determine the magnitude of the CB-618 PK-PD index in combination with meropenem associated with net bacterial stasis and 1- and 2-log10 CFU/ml reductions from baseline at 24 h.
The results of dose fractionation studies determined the CB-618 AUC0–24 to be the exposure measure that was most closely associated with CB-618 efficacy. Although the associations for each CB-618 exposure measure were high, we emphasized the CB-618 AUC0–24/MIC ratio over the CB-618 Cmax/MIC ratio due to the transient nature of Cmax and the CB-618 AUC0–24/MIC ratio over the CB-618%Time>threshold due the variability in effect (approximately a 3-log10 CFU/ml range) at 100% Time>threshold.
Based on data from dose-ranging studies, the next step was to index the CB-618 exposure measures to the CB-618-potentiated meropenem MIC values for each of the five carbapenemase-producing clinical isolates evaluated and determine the magnitude of the CB-618 AUC0–24/MIC ratio associated with efficacy. This was accomplished by evaluating the relationship between change from baseline in log10 CFU/ml at 24 h and the CB-618 AUC0–24/MIC ratio using the pooled data from these isolates. Based on the Hill-type model, which described these data well, the magnitudes of the CB-618 AUC0–24/MIC ratio in combination with meropenem associated with net bacterial stasis and 1- and 2-log10 CFU/ml reductions from baseline at 24 h were 27.3, 86.1, and 444.8, respectively.
It is apparent that the exposure measure associated with β-lactamase inhibitor efficacy can vary among agents. Previously, we identified the %Time>threshold as the tazobactam PK-PD index most closely associated with efficacy in combination with ceftolozane, piperacillin, or cefepime (18, 23, 24), while herein we identified the AUC0–24 as the exposure measure most associated with efficacy for CB-618 when used in combination with meropenem. The basis for this observation is unknown. One possible explanation involves differences in the stability of the β-lactamase inhibitor–β-lactamase complex among β-lactamase inhibitors. While both tazobactam and CB-618 bind covalently to the β-lactamase target, tazobactam binds irreversibly (25) and CB-618 binds reversibly in a manner similar to that for avibactam (26; data on file at Merck & Co., Kenilworth, NJ).
There are a number of limitations to the studies described herein. First, five clinical isolates (4 K. pneumoniae and 1 E. coli), producing a small subset of clinically relevant β-lactamase enzymes, were studied. Thus, the results may not be reflective of the entirety of the Enterobacteriaceae family. However, the challenge organisms represented clinical isolates producing serine carbapenemase enzymes relevant to carbapenem therapy and are likely representative of isolates encountered in clinical settings.
Second, CB-618 was studied in combination with a single β-lactam, meropenem, which is intrinsically stable to all of the beta-lactamase enzymes produced by these isolates other than carbapenemase enzymes KPC-2 and KPC-3. While it is likely that the CB-618 exposure measure that is most closely associated with efficacy would be consistent across β-lactam partner agents as well as other β-lactamases, the magnitude of the PK-PD index necessary for a given level of bactericidal activity may vary among agents and across clinically relevant β-lactam partner agent dosing regimens. Additional studies are required to address these limitations.
In conclusion, we successfully identified the CB-618 exposure measure in combination with meropenem that was most closely associated with CB-618 efficacy and determined the magnitude of the CB-618 PK-PD index associated with net bacterial stasis and 1- and 2-log10 CFU/ml reductions from baseline at 24 h. The PK-PD index most closely associated with CB-618 efficacy in combination with meropenem was the CB-618 AUC0–24/MIC ratio. These findings demonstrated a PK-PD index associated with efficacy that differed from that identified for tazobactam in combination with either piperacillin, ceftolozane, or cefepime. These data provide a PK-PD basis for evaluating potential CB-618 dosing regimens in combination with meropenem in future studies.
ACKNOWLEDGMENTS
We thank Kim A. Charpentier from the Institute for Clinical Pharmacodynamics for assistance with the manuscript and for technical support.
This study was sponsored by Merck & Co., Kenilworth, NJ, USA. The Institute for Clinical Pharmacodynamics (B.D.V., S.M.B., J.M., H.C., and P.G.A.) has received research support from Achaogen, Astellas, AstraZeneca, Basilea Pharmaceutica, Bayer HealthCare, Bristol-Meyers Squibb, Cempra Pharmaceuticals, Cerexa, Cellceutix Corporation, Cubist Pharmaceuticals, Durata Pharmaceuticals, Fedora Pharmaceuticals, Forest Research Institute, Furiex Pharmaceuticals, GlaxoSmithKline, Meiji Seika Pharma, Melinta Therapeutics, Merck & Co., Nabriva Therapeutics, Nimbus, Pfizer, Roche Bioscience, Rock Therapeutics, Tetraphase Pharmaceuticals, and the Medicines Company.
REFERENCES
- 1.Bush K. 2010. Bench-to-bedside review: the role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit Care 14:224. doi: 10.1186/cc8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biedenbach D, Bouchillon S, Hackel M, Hoban D, Kazmierczak K, Hawser S, Badal R. 2015. Dissemination of NDM metallo-β-lactamase genes among clinical isolates of Enterobacteriaceae collected during the SMART Global Surveillance Study from 2008 to 2012. Antimicrob Agents Chemother 59:826–830. doi: 10.1128/AAC.03938-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacoby GA. 2009. AmpC beta-lactamases. Clin Microbiol Rev 22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Castanheira M, Farrell SE, Deshpande LM, Mendes RE, Jones RN. 2013. Prevalence of β-lactamase-encoding genes among Enterobacteriaceae bacteremia isolates collected in 26 U.S. hospitals: report from the SENTRY Antimicrobial Surveillance Program (2010). Antimicrob Agents Chemother 57:3012–3020. doi: 10.1128/AAC.02252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauernfeind A, Stemplinger I, Jungwirth R, Ernst S, Casellas JM. 1996. Sequences of β-lactamase genes encoding CTX-M-1 (MEN-1) and CTX-M-2 and relationship of their amino acid sequences with those of other β-lactamases. Antimicrob Agents Chemother 40:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Andrea MM, Arena F, Pallecchi L, Rossolini GA. 2013. CTX-M-type β-lactamases: a successful story of antibiotic resistance. Int J Med Microbiol 303:305–317. doi: 10.1016/j.ijmm.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Cantón R, González-Alba JM, Galán JC. 2012. CTX-M enzymes: origin and diffusion. Front Microbiol 3:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, Alberti S, Bush K, Tenover FC. 2001. Novel carbapenem-hydrolyzing β-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother 45:1151–1161. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantón R, Coque TM. 2006. The CTX-M β-lactamase pandemic. Curr Opin Microbiol 9:466–475. doi: 10.1016/j.mib.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 11.Tängdén T, Giske CG. 2015. Global dissemination of extensively drug-resistant carbapenemase-producing Enterobacteriaceae: clinical perspectives on detection, treatment and infection control. J Intern Med 277:501–512. doi: 10.1111/joim.12342. [DOI] [PubMed] [Google Scholar]
- 12.Nordmann P, Poirel L. 2014. The difficult-to-control spread of carbapenemase producers among Enterobacteriaceae worldwide. Clin Microbiol Infect 20:821–831. doi: 10.1111/1469-0691.12719. [DOI] [PubMed] [Google Scholar]
- 13.Shlaes DM. 2013. New β-lactam-β-lactamase inhibitor combinations in clinical development. Ann N Y Acad Sci 1277:105–114. doi: 10.1111/nyas.12010. [DOI] [PubMed] [Google Scholar]
- 14.Bush K. 2015. A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int J Antimicrob Agents 46:483–493. doi: 10.1016/j.ijantimicag.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Zhanel GG, Lawson CD, Adam H, Schweizer F, Zelenitsky S, Lagacé-Wiens PR, Denisuik A, Rubinstein E, Gin AS, Hoban DJ, Lynch JP III, Karlowsky JA. 2013. Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs 73:159–177. doi: 10.1007/s40265-013-0013-7. [DOI] [PubMed] [Google Scholar]
- 16.Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, VanScoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob Agents Chemother 56:258–270. doi: 10.1128/AAC.05005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanScoy B, Mendes RE, Nicasio AM, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacokinetics-pharmacodynamics of tazobactam in combination with ceftolozane in an in vitro infection model. Antimicrob Agents Chemother 57:2809–2814. doi: 10.1128/AAC.02513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.VanScoy B, Mendes RE, McCauley J, Bhavnani SM, Bulik CC, Okusanya OO, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2013. Pharmacological basis of ß-lactamase inhibitor therapeutics: tazobactam in combination with ceftolozane. Antimicrob Agents Chemother 57:5924–5930. doi: 10.1128/AAC.00656-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.VanScoy BD, Mendes RE, Castanheira M, McCauley J, Bhavnani SM, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2014. Relationship between ceftolozane-tazobactam exposure and selection for Pseudomonas aeruginosa resistance in a hollow-fiber infection model. Antimicrob Agents Chemother 58:6024–6031. doi: 10.1128/AAC.02310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.VanScoy B, Mendes RE, Castanheira M, McCauley J, Bhavnani SM, Forrest A, Jones RN, Okusanya OO, Friedrich LV, Steenbergen J, Ambrose PG. 2013. Relationship between ceftolozane-tazobactam exposure and drug resistance amplification in a hollow-fiber infection model. Antimicrob Agents Chemother 57:4134–4138. doi: 10.1128/AAC.00461-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard, 9th ed CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 22.AstraZeneca. 2015. Merrem (meropenem for injection) package insert. AstraZeneca. Wilmington, DE. [Google Scholar]
- 23.Nicasio AM, VanScoy BD, Mendes RE, Castanheira M, Bulik CC, Okusanya OO, Bhavnani SM, Forrest A, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2016. Pharmacokinetics-pharmacodynamics of tazobactam in combination with piperacillin in an in vitro infection model. Antimicrob Agents Chemother 60:2075–2080. doi: 10.1128/AAC.02747-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.VanScoy BD, Tenero D, Turner S, Livermore DM, McCauley J, Conde H, Mendes R, Bhavnani SM, Rubino CM, Ambrose PG. 2015. Pharmacokinetics-pharmacodynamics of tazobactam in combination with cefepime in an in vitro infection model, abstr A-499 Abstr 55th Intersci Conf Antimicrob Agents Chemother, San Diego, CA, 17 to 21 September 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bush K. 1986. Evaluation of enzyme inhibition data in screening for new drugs. Drugs Exp Clin Res 12:565–576. [PubMed] [Google Scholar]
- 26.Ehmann DE, Jahić H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]