

Summary
Variable (V), diversity (D), and joining (J) (V(D)J) recombination is the first determinant of antigen receptor diversity. Understanding how recombination is regulated requires a comprehensive, unbiased readout of V gene usage. We have developed VDJ sequencing (VDJ-seq), a DNA-based next-generation-sequencing technique that quantitatively profiles recombination products. We reveal a 200-fold range of recombination efficiency among recombining V genes in the primary mouse Igh repertoire. We used machine learning to integrate these data with local chromatin profiles to identify combinatorial patterns of epigenetic features that associate with active VH gene recombination. These features localize downstream of VH genes and are excised by recombination, revealing a class of cis-regulatory element that governs recombination, distinct from expression. We detect two mutually exclusive chromatin signatures at these elements, characterized by CTCF/RAD21 and PAX5/IRF4, which segregate with the evolutionary history of associated VH genes. Thus, local chromatin signatures downstream of VH genes provide an essential layer of regulation that determines recombination efficiency.
Graphical Abstract
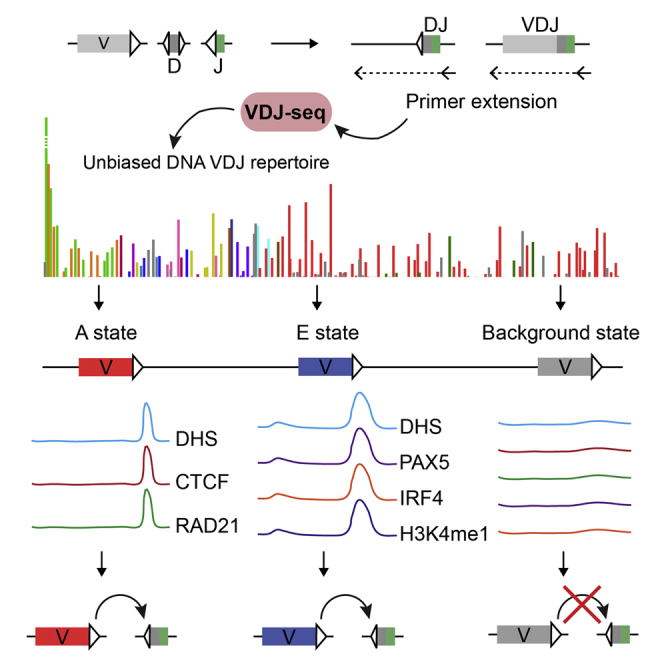
Highlights
-
•
VDJ-seq enables precise quantification of antibody V(D)J recombination products
-
•
Two distinct cis-regulatory designs characterize actively recombining V genes
-
•
Putative recombination regulatory elements map downstream of mouse Igh V genes
-
•
Recombination regulatory architecture reflects the V genes’ evolutionary history
Bolland et al. develop a technique to quantitatively profile antigen receptor diversity. Using VDJ-seq in the mouse Igh locus, they uncover the regulatory logic underlying the highly varying recombination rates of V gene segments, with implications for immune disorders and aberrant recombination in cancer.
Introduction
Variable (V), diversity (D) and joining (J) (V(D)J) recombination of antigen receptor (AgR) loci is the first step in generating the diverse AgR repertoires that enable the adaptive immune system to respond to a vast array of pathogens and regulate tissue homeostasis and surveillance. This process, which occurs at the immunoglobulin (Ig) loci in progenitor B cells and the T cell receptor (TCR) loci in progenitor T cells, involves the sequence-specific cutting and joining of VDJ gene segments to form a functional immunoglobulin (BCR) or TCR gene (Corcoran, 2010, Schatz and Ji, 2011). Failure to generate sufficiently diverse repertoires underpins a wide variety of immunodeficiency diseases and poor immune function in aging (Dunn-Walters and Ademokun, 2010), while inappropriate recombination targeting can lead to genome instability (Teng et al., 2015) and chromosomal translocations in T and B cell leukemias (Marculescu et al., 2002).
The mouse immunoglobulin heavy chain (Igh) locus encompasses 2.8 Mb of chromosome 12 and contains 4 JH genes, 10 DH genes, and 195 VH genes (Johnston et al., 2006, Ye, 2004). The VH genes have been classified into 16 families in three clans, based on sequence similarity and are organized in distinct domains within the VH region (Johnston et al., 2006). The in-frame joining of a VH to a DJH segment to complete the sequence of an IgH polypeptide is the critical event underpinning commitment to the B lineage, since expression of an IgH protein in the pre-B cell receptor is required to switch off Igh recombination (allelic exclusion) and enable progression. Many genetic and epigenetic features have been implicated in the regulation of VH recombination. These include the quality of the downstream Rag recombinase binding sites (recombination signal sequences [RSSs]), sense, and antisense non-coding transcription (Bolland et al., 2004, Yancopoulos and Alt, 1985), modified histones, locus compaction, and chromatin looping (Fuxa et al., 2004, Jhunjhunwala et al., 2008, Sayegh et al., 2005, Stubbington and Corcoran, 2013). Binding of transcription factors including PAX5, YY1, IKAROS, and CTCF, is critical for locus compaction and looping. CTCF promotes local looping in the distal V region, while Pax5 promotes longer-range movement of local distal V domains toward the DJ domain (Degner et al., 2011, Fuxa et al., 2004, Gerasimova et al., 2015, Guo et al., 2011, Liu et al., 2007, Medvedovic et al., 2013, Montefiori et al., 2016, Reynaud et al., 2008). Under the current model, the combined action of these transcription factors brings all V genes into close proximity with the DJ segment, providing equal spatial opportunity for all to participate in V(D)J recombination (Schatz and Ji, 2011).
Despite these advances, it still remains unclear why recombination frequencies of individual VH genes vary enormously (Buchanan et al., 1997, Love et al., 2000, Perlmutter et al., 1985, Yancopoulos et al., 1988). A significant hurdle has been the absence of a comprehensive and quantitative profile of the recombination frequencies of all 195 VH genes. Real-time PCR-based approaches using cocktails of VH primers (Rouaud et al., 2012) and their adaptation for deep sequencing (Georgiou et al., 2014) have increased the throughput of these but remain prone to bias associated with differential primer efficiency and are often incomplete (Kaplinsky et al., 2014). These biases have been mitigated by deep sequencing of the mRNA output of VDJH-recombined products (Choi et al., 2013), but this approach captures only productive recombination events (Eberle et al., 2009) and does not account for varying VH gene promoter activity (Buchanan et al., 1997, Love et al., 2000). In the smaller T cell receptor β locus, these challenges have recently been addressed using DNA-based deep-sequencing, thereby revealing the contributions of key chromatin features to recombination efficiency (Gopalakrishnan et al., 2013), but the methodology used in that study is difficult to adapt to larger AgR loci. Thus, quantitative immunoglobulin repertoire analysis at the DNA level, at which V(D)J recombination occurs, has not been achieved (Benichou et al., 2012).
To address this deficit, we developed VDJ sequencing (VDJ-seq), a quantitative, high-throughput next-generation sequencing assay based on the capture and sequencing of primer extension products of genomic DNA from JH gene oligonucleotides. As each of the four JH genes recombines with the entire spectrum of DH and VH genes, this circumvents the use of multiple V gene primers and enables unbiased detection of DJH and VDJH recombination products. We quantify the primary output of Igh V(D)J recombination by applying VDJ-seq to mouse pro-B cells, in which recombination is ongoing and not yet significantly skewed by downstream processes. We integrate these data with profiles of expression, transcription factor binding, and the chromatin state to reveal two mutually exclusive cis-regulatory signatures at active V genes that localize to the downstream RSS-proximal sequences. These findings establish a paradigm for the regulation of V(D)J recombination that may be widely applicable to other AgR loci.
Results
The VDJ-Seq Technique
VDJ-seq exploits the fact that every DJH and VDJH recombination event ends with one of only four JH genes and is based on two sequential primer extension and capture steps using biotinylated JH region oligos on sonicated, adaptor-ligated genomic DNA. The first step depletes unrecombined sequences located upstream of each JH, the second captures DJH and VDJH recombined sequences using JH-specific oligos (Figure 1A; Data S1). Captured JH primer-extension products are then PCR-amplified using primers to the VH-end adaptor sequence together with nested JH oligos. The use of only JH primers for both primer extension and PCR steps enables unbiased detection of VDJH/DJH recombined sequences since VH gene primers with their inherent biases are not used at any stage.
Figure 1.

The VDJ-Seq Technique
(A) Genomic DNA from sorted pro-B cells (1) containing unknown VDJH and DJH joins (only VDJH depicted) is sonicated to 500 bp (2), end-repaired, A-tailed, and a custom adaptor ligated (3). Primer-extension is performed with forward and reverse primers that hybridize upstream of each JH gene (4). Following depletion of unrecombined primer-extended DNA with streptavidin beads (4), a second primer-extension is performed extending upstream into VDJH or DJH recombined sequences from biotinylated primers that hybridize downstream of each JH (5). After capture with streptavidin beads, two rounds of PCR generate the sequencing library; the first using adaptor-specific paired-end 1 (PE1) and J-specific paired-end 2 (PE2) primers (6), the second using flow-cell PE1 and PE2 primers (7) to generate the library (8).
(B) Differentiation of early B cell progenitors in bone marrow showing when DH-to-JH and VH-to-DJH joining occur. Rag1−/− mice are incapable of V(D)J recombination.
See also Figures S1 and S3.
We generated two biological replicate libraries from ex vivo flow-sorted wild-type (WT) bone marrow pro-B cells (B220+CD19+CD43+CD25−sIgM−) and one from CD19+ Rag1−/− bone marrow pro-B cells as a negative control. WT pro-B cells have almost completed DH-to-JH recombination (Ehlich et al., 1994, Rumfelt et al., 2006) and are undergoing VH-to-DJH joining, while Rag1−/− cells cannot recombine, so any non-JH region sequences represent background (Figure 1B). V(D)JH recombination generates variability due to combinatorial joining of different VH, DH, and JH gene segments and junctional diversity from nucleotide additions and exonuclease nibbling (Figure S1A). We used this variability in the JH read plus the positional variability of the VH/DH read start locations (from random DNA shearing) to develop a deduplication pipeline to identify unique sequences (Figures S1A and S1B; Supplemental Experimental Procedures). Following deduplication, ∼99% of reads detected in Rag1−/− mapped to unrecombined JH sequences (Figure S1C). In contrast, in WT cells, unrecombined JH sequences constituted ∼20% of reads, while ∼50% of reads mapped to the DH cluster and 25%–30% to the VH region (Figures S1C and S1D). These corresponded to 382,932 and 441,640 DJH joins with 220,363 and 239,090 VDJH recombinants for the two wild-type replicates (Figure S1C), equivalent to 20% detection rate (1.7 million cell equivalents/replicate; 3.4 million alleles; 35% VDJH recombined alleles; see Figure S3).
In addition to these canonical DJH and VDJH joins, we also detected a variety of aberrant products (Figure S2), some of which have been reported previously (Fang et al., 1996, Hu et al., 2015, Sollbach and Wu, 1995). These include the joining of adjacent JH segments, inverted DH-to-JH joining, DH region cryptic recombination, and VH signal sequences joined to DJH segments. While these products were detected at low frequencies (<1%), they were not observed in Rag1−/−, indicating they are genuine products of V(D)J recombination.
We performed extensive quality control of VDJ-seq. Wild-type replicates were highly correlated for both VDJH and DJH recombination (Figures S3A–S3C). Frequencies of DJH and VDJH recombination correlated closely with published VDJ:DJ/GL ratios and with a DNA fluorescence in situ hybridization (FISH) assay of VH-to-DJH joining developed here (Figures S3D–S3G; Supplemental Experimental Procedures). Frequencies of in-frame productive, non-productive, and out-of-frame recombination products analyzed in IMGT HighV-quest (Alamyar et al., 2012) (Figure S3H; Supplemental Experimental Procedures) were close to previous reports, as were complementarity determining region 3 (CDR3) lengths (Figure S3I). Notably, each replicate samples a different part of the highly complex, randomly generated pro-B Igh repertoire (Figure S3J). At the same time, close correlation of VH and DH recombination frequencies within families between replicates indicates that differential gene usage is robust across the entire repertoire (Figures S3B and S3C), and sequencing depth is sufficient for quantitative analysis.
VDJ-Seq Reveals Complete Primary DJH and VDJH Repertoires
For D-to-JH recombination, >98% of the sequences mapping to the DH region originated from the ten canonical C57BL6 DH genes, with DHFL16.1 recombining with the highest frequency, consistent with previous studies (reviewed in Ye, 2004) (Figure 2A).
Figure 2.

VDJ-Seq Reveals Widely Varying DH and VH Gene Recombination
(A) DH gene recombination. Reads per DH gene were counted using Seqmonk for each WT pro-B replicate, normalized to the replicate with the lowest total read count for all DH genes then expressed as a percentage of the total (Data S1). Bars indicate the mean of these values. Replicate values are not shown as DH usage was almost identical between the two replicates. Dotted line, expected value if recombination was equal for all ten DH genes.
(B) Recombination frequencies of the 195 VH genes. Reads for each VH gene were counted for each replicate then normalized to the replicate with the lowest total read count for all VH genes (Data S1) and are shown as open circles. Bars indicate the mean of these values for each VH gene, colored by family. Dotted line, expected frequency if recombination was equal for all VH genes. Individual gene names for actively recombining VH genes (determined by binomial testing) are shown below together with map position on mouse chromosome 12.
See also Figures S2, S4, and S7.
For V-to-DJH recombination, we detected wide variation in usage of V genes across the Igh locus (Figure 2B). To determine which genes were actively undergoing recombination, we used a binomial test (Figure 3A; Supplemental Experimental Procedures). Using a stringent threshold (fdr-adjusted p value <0.01) threshold, we detected 128 recombining and 67 recombinationally silent VH genes and pseudogenes (Data S1) out of the 195 total (Johnston et al., 2006). Recombining VH genes and pseudogenes and recombinationally silent VH genes and pseudogenes are referred to hereafter as “active” and “inactive,” respectively. There was a 200-fold range of recombination efficiencies among the 128 active VH genes (Figures 2B and S4A; Data S1).
Figure 3.

Random Forest Classification Identifies RSS as a Binary Switch for VH Gene Recombination but with No Predictive Value for Recombination Frequency
(A) Frequency distribution of VDJ-seq read counts for 195 VH genes, color-coded as active (recombining) or inactive (non-recombining) using a binomial test to gauge the significance of the recombination level. Red: active fdr-adjusted (p value <0.01); blue: inactive fdr-adjusted (p value ≥0.01).
(B) Average of out-of-bag variable importance (the gini impurity) in predicting active genes from a Random Forest classifier applied on 18 factors. Error bars show the SEs from a 10-fold cross validation procedure to further control for overfitting. Variables with high gini importance were consistent with the permutation importance measure (data not shown).
(C) Distribution of area under curve (AUC) scores from 2,048 RF models derived from all possible combinations of the 11 most important factors from the RF classifier.
(D) RSS RIC scores of active versus inactive VH genes.
(E) RSS RIC scores at active genes show little correlation with rates of recombination of individual VH genes.
As expected, the majority of functional (protein-coding, pre-BCR pairing) genes (99/103) were active. Notably, 29/92 pseudogenes also recombined, albeit at lower frequencies. All of these had RSSs predicted as recombinogenic for Igh V gene RSSs (recombination information content [RIC] score >−58.45) (Cowell et al., 2002, Lee et al., 2003). Separation of the 128 active VH genes and pseudogenes into the 15 VH gene families plus one pseudogene class revealed large differences in recombination frequencies both across and within the families (Figure S4B). Thus, with VDJ-seq, we have quantified the full range of VH genes and pseudogenes and identified those that do and do not participate in Igh V(D)J recombination. The wide range of recombination frequencies suggests that complex regulatory mechanisms are at play.
Factors Predictive of Active VH Gene Recombination
We set out to identify the genetic and epigenetic features that associate with active VH recombination. We integrated our VDJ-seq data with the profiles of transcription factor binding and histone modifications and the quality of the V genes’ RSSs. To determine the individual and combinatorial roles of these factors in V to DJ recombination, we used them as predictors in a Random Forest (RF) classifier trained to distinguish active from inactive VH genes (Supplemental Experimental Procedures). We first trained a RF model using 18 features as predictors including RSS RIC score, chromatin immunoprecipitation sequencing (ChIP-seq) signals for histone marks and transcription factors (TF), and sense and antisense RNA levels (Data S1). Features were assessed in 2.5 kb windows, spanning the VH gene itself, including 1 kb upstream to incorporate the promoter and 1 kb downstream, including the RSS. Classification based on these 18 factors showed a very high predictive power (Figures 3A–3C), indicating that they effectively distinguish active from inactive VH genes. Significant predictors of active recombination included a high RIC score, DNase hypersensitivity (DHS), chromatin marks associated with active chromatin states (H3K4me3 and H3K4me1), and several architectural and transcription factors. Some of these (CTCF, RAD21, PAX5, and YY1) are known to be required for Igh recombination (Degner et al., 2011, Fuxa et al., 2004, Liu et al., 2007), while others (PU.1, P300, MED1, and IRF4) have not been previously reported to function in this capacity. In contrast, local sense germline transcription, long implicated as a requirement for Igh recombination (Bolland et al., 2004, Yancopoulos and Alt, 1985), had a weak predictive power in our analysis (Figure 3B). Our strand-specific nuclear RNA sequencing (RNA-seq) dataset aimed to enrich for non-coding transcripts, but sense V gene transcripts were infrequent (Figure S4C). Antisense transcription was also not a robust predictor, which was expected as it localizes to a small number of discrete domains across the locus (Figure S4C). A previous study (Choi et al., 2013) had also failed to find a strong correlation between transcription and recombination, both according to the authors and in our reanalysis (Figure S4D). Thus, while the importance of transcription cannot be excluded, the available data do not support a predictive role in active recombination.
We then evaluated the classification rate of all possible combinations of the top 11 predictors reported by the RF classifier (2048 RF models; Figure 3C). In all cases, the classification rate differed considerably depending on whether or not the RIC score was included, indicating a strong association of the Rag-binding RSS sequence with active recombination, consistent with previous findings (Choi et al., 2013). The highest classification rates (>95%) were obtained when RIC scores together with DHS, H3K4me1, CTCF, and RAD21 were used as predictors. RSS quality was nevertheless not a sufficient predictor, since 34% of inactive genes had RIC scores above the predicted functional cut-off (Figure 3D). Importantly, within the group of active genes, the RSS scores, although generally elevated (Figure 3D), showed no further correlation with individual rates of VH recombination (Figure 3E). Moreover, models excluding RSS as a predictor also produced high classification rates (>80%). Together, these data suggest that the other factors above determine individual V gene recombination capacity.
Consistent with these findings, DHS, H3K4me1, CTCF, RAD21, IRF4, PAX5, and MED1 significantly co-localized with active VH genes, while PU.1 did not (Figure 4A). For example, CTCF binding sites were found adjacent to 34 VH genes, of which 31 were active. In contrast, some features, including H3K9 acetylation, did not co-localize with VH genes (Figure S5). Notably, active VH genes with none of the above chromatin marks or TF binding events in the vicinity had much lower recombination rates than those with chromatin marks, independent of their RSS quality (Figure 4B). Conversely, a subset of active VH genes with poor RIC scores, but six or more adjacent binding events, showed comparable recombination frequencies to subsets with good RIC scores (Figure 4B), suggesting that cooperative TF binding may partially compensate for a poor Rag binding context.
Figure 4.

Epigenetic Factors Co-localize with VH Genes
(A) P values from a χ2 test to gauge the significance level of the observed number of actively recombining genes that co-localize with eight factors compared to the number expected if co-localization was randomly distributed between active and inactive V genes. A value of 1.3 (log10 of 0.05) or above indicates significant association with active V genes. The remaining factors are not co-localized (Figure S5A).
(B) Relationship between the number of factor peaks associated with VH genes and recombination frequency (upper panel) and RSS RIC scores (lower panel). Dashed red lines indicate the threshold VDJ-seq read count for active VH genes (upper panel) and the pass/fail RSS RIC score threshold (lower panel). Numbers of VH genes in each group are shown above.
(C) Distances from VH genes to the nearest peaks for CTCF, RAD21, PAX5, and IRF4 exhibit bimodality (see also Figure S5B). The subset of VH genes with nearby peaks (≤1 kb, light shading) was enriched for active genes (red). The curves in the left hand y axis illustrate the frequency of genes in each group. Chromosomal position and location within the VH region are shown below.
See also Figure S5.
The absence of a quantitative link between RSS quality and recombination frequency of active VH genes, together with the requirement for other factors suggest that the RSSs serve as enabling genetic “binary switches” of recombination at each VH gene, while other features control the efficiency of this mechanism.
Two Distinct cis-Regulatory Designs at RSS Regions Associate with Active Recombination
The transcription factors and histone marks identified as predictive of active recombination (Figure 4A) bind heterogeneously throughout the Igh V region. We asked how their binding sites are distributed across the region. First, we analyzed the distance between active VH genes and the nearest ChIP-seq peak summits of these factors. As expected, CTCF, RAD21, IRF4, PAX5, DHS, H3K4me1, MED1, and PU.1 localized very close to subsets of active genes (light shaded panels in Figures 4C, S5A, and S5B; active genes are red). Importantly, patterns of factors co-localizing with specific VH genes appeared mutually exclusive. In particular, CTCF and RAD21 preferentially associated with JH-proximal active VH genes, while PAX5 and IRF4 commonly co-localized with active genes in the middle and distal regions (Figure 4C).
To further investigate this, we used ChromHMM (Ernst and Kellis, 2012) to partition the VH region into discrete states based on the chromatin marks and TFs used in the RF classification (Supplemental Experimental Procedures). ChromHMM has recently been used AgR-wide to identify putative enhancer regions (Predeus et al., 2014). Surprisingly, despite the complexity of the locus and the large number of factors included, we observed that low within-class heterogeneity and a high between-state separation could be achieved with just three chromatin states: two highly distinctive “regulatory” states and a “background” (Bg) state depleted of all of these factors (Figures 5A, 5B, and S6A). The first regulatory state (termed “A”) was characterized by the binding of “architectural proteins” CTCF and RAD21 (Phillips-Cremins et al., 2013). The second state was best characterized by the binding of PAX5, IRF4, and YY1, and the enrichment of “active” chromatin marks H3K4me1, H3K4me2, H3K4me3, and H3K9ac (Figures 5A, 5C, and S6A). It thus had features associated with hematopoietic transcriptional regulatory elements (Lelli et al., 2012), and we refer to it as the “E” state. DHS, PU.1, and MED1 were enriched in both states.
Figure 5.

Identification of Chromatin States across the VH Locus
(A) ChromHMM emission probability (composition) of 12 epigenetic factors in three chromatin states: background (Bg), architectural (A), and enhancer (E). Range, zero (white) to 1 (dark blue).
(B) Comparison of the significance of the read counts for CTCF, RAD21, PAX5, and IRF4 in the three states (Figure S6A; remaining eight factors).
(C) Examples of recombining genes in the A (enriched for CTCF and RAD21) and E (enriched for DHS, PAX5, YY1, H3K4me1, MED1, PU.1, and IRF4) states.
(D and E) Comparison of VDJ-seq read counts (D) and RSS RIC scores (E) for active genes in each of the three states. P values are driven by Wilcoxon test.
See also Figure S6.
VH genes associated with either A or E states recombined significantly more than Bg state genes (Figure 5D). Consistent with this, 76% of active VH genes (97/128) associated with either one of the two “regulatory” states (state A: 33 genes; state E: 78 genes; Bg state: 84; Data S1). The remaining 24% of active VH genes (31/128) associated with the Bg state all recombined poorly, despite having higher RIC scores than the average for A state genes (Figure 5E). Conversely, 80% of inactive genes (53/67; Fisher’s exact test p = 6.9e-13 versus random expectation) were associated with the Bg state. Importantly, this included most (18/23) of the inactive genes with functional RSSs (23/67). This is strikingly illustrated within the large J558 family. When the active VH genes therein are separated into high and low recombiners, there is no difference between RIC scores of the two subgroups, but marked differences in the chromatin state (Figure S6B). Together, these results indicate that the Bg chromatin state is refractory to recombination, while state A and state E represent two distinct regulatory architectures associated with active recombination.
ChromHMM analysis of JH and DH genes revealed that all four JH genes overlap with hallmarks of state E (Pax5, IRF4) and lack state A CTCF and Rad21 binding (Figure S6C). In turn, the two most 3′ DH genes (DQ52 and DST4) and the most 5′ DH gene (DFL16.1) (Figure 2) overlap with the E state (Figure S6C). The six DSP genes all overlap with the Bg state, in agreement with previous reports of repressive chromatin marks at these genes (Chakraborty et al., 2007), despite significant antisense transcription (Bolland et al., 2007) and frequent recombination (Figure 2).
Fine-scale analysis of ChIP-seq signals revealed that CTCF and RAD21 were enriched specifically over the RSS of A state genes (Figures 6A and 6B), consistent with previous reports for some A state genes (Choi et al., 2013, Lucas et al., 2011). Surprisingly, the E state-associated transcription factors PAX5 and IRF4 were also enriched close to the RSS region of their respective genes and not at VH promoter regions (Figures 6A and 6B), along with the high DNase hypersensitivity common to both signatures. Our analyses, therefore, suggest the RSS region as a key cis-regulatory region for enhancement of recombination conforming to either of two distinctive cis-regulatory designs.
Figure 6.

Epigenetic Factors Are Specifically Enriched at the VH RSS
(A) Aligned region plot for the 2 kb centered on the RSS for VH genes in the two active states and the VH3609 outlier family. This shows enrichment for DHS, CTCF, and RAD21 for the A state and DHS, IRF4, and PAX5 for the E state. Enrichment is localized close to the RSS in all cases.
(B) Line graph of average relative enrichment for each factor for genes in the A and E states.
(C) Signal intensity at genome-wide Rag1-ChIP peaks. Signal intensity of five features (chosen to represent both regulatory states) and Rag2 at 3388 Rag1 peaks (Teng et al., 2015). Density defined as normalized log-based read counts in 2 kb regions centered at the summit of peaks.
To determine whether these two cis-regulatory designs have a more wide-spread role in determining recombination-permissive sites, we examined the >3,000 Rag1 binding sites across the genome that colocalize with Rag2 binding and H3K4me3 enrichment (Teng et al., 2015). We found that CTCF, PAX5, and IRF4 binding profiles all align closely with Rag1 peaks (Figure 6C), suggesting that state A- and E-specific signatures provide a more focused set of candidate sites for off-target Rag recombination.
cis-Regulatory Architecture of Active VH Genes Reflects Their Evolutionary History
Generally, the A state was more abundant at the JH-proximal end, while the E state was more enriched in the distal domain of the Igh locus (Figure S6D). However, there were numerous exceptions, suggesting that genomic position is not the primary determinant of VH gene regulatory architecture. We asked whether the distribution of the regulatory states across VH genes could be explained by their evolutionary history. The 16 VH gene families have evolved into three separate clans—a large clan 1 and the more closely related smaller clans 2 and 3 (Figure 7A), based on conservation of VH gene framework 1 (FR1) sequences (Schroeder et al., 1990, Kirkham et al., 1992) and overall DNA sequence (Johnston et al., 2006). We found the A state (CTCF/RAD21) exclusively at VH gene families from clans 2 and 3, while the E state was almost exclusively associated with VH genes of clan 1, with the exceptional inclusion of the VH3609 family forming an outlier group in clan 2 (Schroeder et al., 1990) (Figure 7B). The association of different clans with each of the two states is particularly striking in the poorly understood “middle families” region, which contains a frequently “oscillating” mixture of clan 1 and 3 genes and undergoes a consistent frequent “switching” of states A and E across the region (Figures 7C and S6D). The segregation of regulatory states with evolutionary clans was observed even for the minority of inactive genes (14/67) that associated with a regulatory state (Data S1). Collectively, these data demonstrate that the recombination-regulatory states reflect the evolutionary history of their respective VH gene clans.
Figure 7.

Regulatory States Co-segregate with Evolutionary VH Clans
(A) Evolutionary organization of V gene families in clans.
(B) Relationship between regulatory states and evolutionary VH clans.
(C) Geographical position of actively recombining VH genes overlapping the three states (top) and VH clans (bottom) across the mouse VH region. Clans 2 and 3 are colored the same (red) to reflect their overlap in state classification (A). The VH3609 family is colored green to denote its outlier status as an E state, but clan 2, family. Co-switching of the A and E states with clans 2+3 and 1, respectively, can be seen for the middle families.
See also Figure S6D.
Clan-Specific Differences in Expression of the Recombined VDJ Repertoire
We observed an appreciable correlation between the DNA-based VDJ-seq and the outputs of the expressed Igh repertoire (Choi et al., 2013) (Figure S7A). However, there were also significant differences in individual genes, and expression from 14 active genes was undetectable (Figure S7B). Segregation of the data into clans demonstrated that the RNA-based data deviated from VDJ-seq in a clan-specific fashion (Figure S7C). Specifically, while clan 1 showed comparable median output between the two assays, clan 2 and particularly clan 3 genes were generally under-represented in the RNA-based assay. This comparison contributes to our understanding of the modulation of the expressed repertoire by downstream factors including promoter strength (Love et al., 2000), mRNA stability, and productivity, while highlighting the limitations of RNA-based approaches for quantification of primary recombination events.
Local Regulatory Events at V Genes Occur in Addition to Global Locus Looping
We asked whether recombination frequency reflects the topological localization of the V genes. A recent 5C-based study (Montefiori et al., 2016) identified six sites in the V region that potentially provide a structural “backbone” of independent chromatin subdomains, within which more local interactions occur. However, we did not observe selective clustering of highly recombining V genes at these sites based on either VDJ-seq (Figure 7D) or expression (Choi et al., 2013; not shown). Active V genes also did not consistently co-localize with the DJ-interacting domains in the V region defined by 4C-seq (Medvedovic et al., 2013). Together, these results suggest that the looping events detected by these studies act in combination with local regulatory mechanisms to shape the V(D)J repertoire.
Discussion
The DNA-based VDJ-seq technique established here has enabled a quantitative assessment of the recombination frequency of all VH genes in the mouse Igh locus and led to detection of cis-regulatory signatures associated with active recombination. These signatures explain the enormous variation in recombination efficiency throughout the VH region, including at geographically neighboring V genes. Therefore, in addition to the large-scale looping mechanisms necessary to bring distal V genes close to DJ segments (Chaumeil and Skok, 2012, Fuxa et al., 2004, Gerasimova et al., 2015, Guo et al., 2011, Medvedovic et al., 2013), local chromatin regulation of V genes likely plays a pivotal role in recombination outcome.
VH Gene Downstream Sites: Dual-Function Recombination Regulatory Elements?
Integrating VDJ-seq data with genetic and epigenetic annotations, we have produced a functional view of the factors associated with recombination of individual active V genes. Consistent with previous studies, we find that the stringency of the Rag-binding RSSs does not fully explain the differential recombination efficiency across the Igh locus (Choi et al., 2013, Merelli et al., 2010). Nevertheless, our results suggest that regions proximal to the RSSs regulate VH gene usage via combinatorial transcription factor recruitment. These elements therefore exquisitely juxtapose the two requirements for recombination: a functional RSS and a permissive chromatin state likely generated by key transcription factors. Importantly, these downstream elements localize on the opposite sides of the VH genes from their promoters and are excised during recombination of their associated VH gene. Therefore, the regulatory mechanisms underpinning efficient recombination are both spatially and temporally separated from those driving the expression of recombined VH genes.
We further reveal two mutually exclusive designs at these recombination-regulatory sites, implicating CTCF/RAD21 and PAX5/IRF4 in the local regulation of VH gene recombination efficiency in cis. It was previously hypothesized that CTCF forms the base of local loops at D-proximal V genes, thus impacting on recombination efficiency (Lucas et al., 2011). Here, we show that both CTCF and RAD21 have a strong association with active VH genes in clans 2 and 3. RAD21 binding is developmentally restricted to pro-B cells undergoing V-to-DJ recombination (Degner et al., 2009) and may have a more stage-specific local role in activating recombination.
PAX5 association with recombination-regulatory elements is consistent with a previous model, in which Pax5 recruits Rag to individual V genes (Zhang et al., 2006). Pax5 was postulated to enable recombination of distal VH genes by promoting DNA looping (Fuxa et al., 2004). However, its local role revealed here may provide an additional explanation of the reduced recombination of state-E VH genes in a PAX5 mutant, particularly those that are not distally located such as the VGam 3.8 family (Fuxa et al., 2004).
Finally, we provide evidence that IRF4 is associated with recombination of Igh state E VH genes. IRF4 was previously shown to regulate Igκ recombination (Johnson et al., 2008) potentially by promoting accessibility of Igκ RSSs (Bevington and Boyes, 2013). IRF4 is a known target of PAX5 (Revilla-I-Domingo et al., 2012) and its colocalization with PAX5 around VH RSSs suggests a feed-forward loop (Palomero et al., 2006).
The Distribution of Regulatory States across the Igh Locus
Detailed analysis of interspersed V gene families led us to the discovery that the chromatin states were based on clan evolution and not on geographical location. We note that our conclusions differ from those of Choi et al. (2013), who proposed that the V region is divided into four chromatin states in a geographical manner, each with characteristics that favor recombination and/or compensate for unfavorable factors. We attribute the different conclusions in part to greater sequencing depth and the ability of VDJ-seq to report on DNA rather than downstream RNA expression and detect non-productive recombination events and recombining pseudogenes. Inclusion of a wider range of relevant transcription factors, including Pax5, IRF4, YY1, and PU.1 enabled us to resolve V genes into just two active chromatin states.
Recombination and Transcription Mediated by Spatially Separate Elements
The discovery of germline transcription from Igh V gene promoters before V to DJ recombination prompted the accessibility hypothesis, which proposed that V(D)J recombination is regulated by controlling access of the RAG enzymes to the antigen receptor loci (Yancopoulos and Alt, 1985). However, subsequent studies have produced conflicting findings both in vitro and in vivo (Baumann et al., 2003, Bevington and Boyes, 2013, Buchanan et al., 1997, Du et al., 2008, Ji et al., 2010, Kondilis-Mangum et al., 2010, Love et al., 2000). While we cannot reconcile the debate fully, our evidence supports the notion that neither promoter activity nor transcription discriminate between active and inactive genes. First, we did not find sense non-coding RNA transcription over unrecombined VH genes to be a strong predictor of active recombination. In part, this reflects the very low sense transcript levels detected by RNA-seq. Neither did we find a correlation between transcription factor binding and antisense transcription at RSSs. Second, fine-scale mapping of ChIP-seq datasets around state A and E genes did not reveal any recombination-associated patterns at the promoters. Rather, the canonical promoter-associated mark H3K4me3 was enriched around the RSSs of state E genes, consistent with its additional role in facilitating Rag2 binding (Matthews et al., 2007). Although germline transcription does not discriminate between actively recombining versus inactive VH genes, it may still be required for recombination, for example, by providing the first level of chromatin “priming.” It is clear that V gene promoter activity plays an important role in shaping the expressed repertoire post-recombination, as we show by comparing DNA-based (VDJ-seq) and RNA-based (Choi et al., 2013) outputs.
Implications for the Human IGH Locus
In humans, VH genes maintain evolutionarily conserved clan identities, but have a very different geographical organization to the mouse, with interspersed gene families and no polarity of clan position (Das et al., 2008, de Bono et al., 2004, Schroeder et al., 1990). The human locus is also much smaller than the mouse (1 Mb versus 2.5 Mb), and the role of looping in its regulation is unknown. Despite these differences and consistent with our findings in mouse, CTCF is associated with 90% of clan 2/3 genes in a human lymphoblastoid cell line (GM12878) (ENCODE Project Consortium, 2012), supporting the evolutionary conservation of downstream recombination-regulatory sites. Accordingly, an attractive hypothesis is that the segregating local chromatin states and their potential role in active recombination reported here also apply to the human IGH locus.
Implications for Other Antigen Receptor Loci
RSS-mediated recombination is common to all AgR loci. Thus, it will be important to determine whether other AgRs have a similar cis-regulatory organization. For instance, CTCF and RAD21 may play a role in local Igκ or TCR gene recombination, and indeed CTCF regulates Igk recombination (Ribeiro de Almeida et al., 2011), while CTCF and Rad21 regulate TCRα and TCRβ locus conformation and recombination (Chen et al., 2015, Seitan et al., 2011, Shih et al., 2012). Some of the key factors for Igh are only expressed in B cells (PAX5, IRF4) and thus may be relevant for Igκ and Igl, but not for TCRs. Indeed, IRF4 and IRF8 are implicated in Igk recombination (Johnson et al., 2008). T cell-specific factors implicated in TCR recombination, including RUNX1 may also have local as well as the long-range looping roles (Cieslak et al., 2014).
Insights into Inappropriate Rag-Mediated Recombination
Rag recombinases mediate aberrant chromosomal translocations and deletions (Helmink and Sleckman, 2012, Hu et al., 2015), but the determinants of their mis-localization are not fully understood. While cryptic RSSs are extremely frequent throughout the genome, RSS quality is a weak predictor of aberrant recombination (Zhang and Swanson, 2008). Permissive chromatin structure has been implicated (Shimazaki et al., 2012), and indeed, Rag-mediated DNA breaks in acute lymphoblastic leukemias, while poorly predictable by RSS quality, are enriched at active promoters and enhancers (Papaemmanuil et al., 2014). Consistent with this, recent studies have revealed thousands of Rag1 binding sites at promoters and enhancers (Teng et al., 2015). Here, we show that signature chromatin states at canonical RSS sites are critical features associated with their recombination potential. Notably, state E genes are enriched in H3K4me3, H3K4me1, canonical promoter, and enhancer marks that provide docking sites for Rag1 and Rag2 throughout the genome. However, state A genes are depleted of these marks suggesting the promoter/enhancer signature is not a universal requirement for RSS-mediated recombination, and an additional class of CTCF/Rad21-associated signatures warrants investigation for cryptic RSS cleavage. In support of this hypothesis, the 3,000 Rag1/Rag2 bound sites in the genome were enriched for markers of state A and state E. Combining RSS quality with identification of these chromatin structures may provide a more in-depth set of rearrangement-prone sites, which, as for canonical sites, may differ in different lymphocyte subpopulations.
Experimental Procedures
All mice were maintained in accordance with Babraham Institute Animal Welfare and Ethical Review Body and Home office rules and ARRIVE guidelines under Project Licence 80/2529. Detailed methods are in the Supplemental Information available online.
VDJ-Seq
VDJ-seq is the capture and amplification for Illumina sequencing of Igh DJ and VDJ recombined genes from genomic DNA by primer extension from reverse-oriented biotinylated J gene oligonucleotides. A flow chart of the VDJ-seq assay is provided in Figure 1. Oligonucleotide sequences are provided in Data S1.
VDJ-Seq Pipeline: Babraham LinkON
A bioinformatic pipeline was developed to process raw sequences. True J gene containing reads were identified and deduplicated to identify unique V-J read pairs based on the J sequence and the V read start position. Only J sequences with different V read start positions were called as unique VDJ joins. Flow chart of Babraham LinkON is provided in Figure S1A. We used Seqmonk (Babraham Bioinformatics), a freely available java-based tool, to visualize and quantify unique DJ and VDJ sequences in mapped sequencing data.
VDJ-Seq IMGT HighV-Quest Pipeline
IMGT HighV-quest is a high-throughput web tool for the analysis of VDJ junctions providing data including V, D, and J genes used, coding potential, P and N nucleotides, and CDR3 AA sequence. Analysis of VDJ-seq data using IMGT HighV-quest required a pipeline to link V and J reads. See the details in the Supplemental Information.
Computational and Statistical Approaches
Random Forest Analysis
Recombining active VH genes were defined as those enriched for VDJ-seq reads compared with the V region as a whole, ascertained using a binomial test (padj < 0.01) for each gene. The not-significantly recombining genes were defined as inactive. These binary recombination classes were used as response variables in a Random Forest classifier model (Liaw and Wiener, 2002). Predictors for the model were signal intensities from DHS-seq, ChIP data, and RNA-seq data extracted from 2.5 kb surrounding each V gene. The model was evaluated in a 10-fold cross-validation to prevent over-fitting.
Co-localization
ChIP peaks (including DHS) were called using MACS2 (Zhang et al., 2008). The distance between the RSS and the summit of nearest peak was measured for each dataset. The significance of co-localization (versus active and inactive genes) was assessed using a χ2 test.
Chromatin Segmentation
The Igh locus was split into 200 bp bins. Each bin overlapping with ChIP or DHS peaks was assigned 1, otherwise 0. The resulting binary matrix was used as input for the chromatin segmentation algorithm chromHMM (Ernst and Kellis, 2012). The significance of association between chromatin states and V gene recombination classes was assessed by a Fisher’s exact test.
Author Contributions
A.L.W. conceived the first version of VDJ-seq. D.J.B. developed the final version reported here and generated VDJ-seq data. K.T. sequenced Illumina libraries. F.K. and S.R.A. developed the Babraham LinkON pipeline. L.M., M.J.T.S., A.B.-E., P.C., and B.A.S. refined the VDJ-seq protocol and analysis pipeline. L.M. generated RNA-seq and ChIP-seq datasets. H.K. pre-processed and Q.C. checked all NGS datasets and conceived and performed machine learning analysis. D.J.B. and H.K. visualized the data and prepared the figures. D.J.B., H.K., M.S., and A.E.C. interpreted the results and wrote the manuscript.
Conflicts of Interest
D.J.B., A.L.W., L.S.M., and A.E.C. are named inventors on a patent filed, “Covering the VDJ-seq technique: Method of identifying VDJ recombination products.” (UK Patent Application No. GB1203720.6, filed March 2, 2012; PCT Patent Applic No. PCT/GB2013/05056, published September 6, 2013. National applications filed Europe, USA, Japan. US Publication number: 20150031042, publication date January 29, 2015.)
Acknowledgments
We thank Peter Fraser and Martin Turner for critical reading of the manuscript. This work was supported by the Biotechnology and Biological Sciences Research Council UK (BBSRC) (D.J.B., A.L.W., H.K., L.M., F.K., M.J.T.S., P.C., K.T., S.R.A., M.S., and A.E.C.) and the Medical Research Council UK (A.B.-E. and B.A.S.).
Published: June 2, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.05.020.
Contributor Information
Mikhail Spivakov, Email: mikhail.spivakov@babraham.ac.uk.
Anne E. Corcoran, Email: anne.corcoran@babraham.ac.uk.
Accession Numbers
The accession number for VDJ-seq, H3K4me3 ChIP-seq, and nuclear RNA-seq datasets reported in this paper is GEO: GSE80155.
Supplemental Information
References
- Alamyar E., Giudicelli V., Li S., Duroux P., Lefranc M.-P. IMGT/HighV-QUEST: the IMGT web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res. 2012;8:26. [Google Scholar]
- Baumann M., Mamais A., McBlane F., Xiao H., Boyes J. Regulation of V(D)J recombination by nucleosome positioning at recombination signal sequences. EMBO J. 2003;22:5197–5207. doi: 10.1093/emboj/cdg487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benichou J., Ben-Hamo R., Louzoun Y., Efroni S. Rep-Seq: uncovering the immunological repertoire through next-generation sequencing. Immunology. 2012;135:183–191. doi: 10.1111/j.1365-2567.2011.03527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevington S., Boyes J. Transcription-coupled eviction of histones H2A/H2B governs V(D)J recombination. EMBO J. 2013;32:1381–1392. doi: 10.1038/emboj.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolland D.J., Wood A.L., Johnston C.M., Bunting S.F., Morgan G., Chakalova L., Fraser P.J., Corcoran A.E. Antisense intergenic transcription in V(D)J recombination. Nat. Immunol. 2004;5:630–637. doi: 10.1038/ni1068. [DOI] [PubMed] [Google Scholar]
- Bolland D.J., Wood A.L., Afshar R., Featherstone K., Oltz E.M., Corcoran A.E. Antisense intergenic transcription precedes Igh D-to-J recombination and is controlled by the intronic enhancer Emu. Mol. Cell. Biol. 2007;27:5523–5533. doi: 10.1128/MCB.02407-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan K.L., Smith E.A., Dou S., Corcoran L.M., Webb C.F. Family-specific differences in transcription efficiency of Ig heavy chain promoters. J. Immunol. 1997;159:1247–1254. [PubMed] [Google Scholar]
- Chakraborty T., Chowdhury D., Keyes A., Jani A., Subrahmanyam R., Ivanova I., Sen R. Repeat organization and epigenetic regulation of the DH-Cmu domain of the immunoglobulin heavy-chain gene locus. Mol. Cell. 2007;27:842–850. doi: 10.1016/j.molcel.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Chaumeil J., Skok J.A. The role of CTCF in regulating V(D)J recombination. Curr. Opin. Immunol. 2012;24:153–159. doi: 10.1016/j.coi.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Carico Z., Shih H.-Y., Krangel M.S. A discrete chromatin loop in the mouse Tcra-Tcrd locus shapes the TCRδ and TCRα repertoires. Nat. Immunol. 2015;16:1085–1093. doi: 10.1038/ni.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N.M., Loguercio S., Verma-Gaur J., Degner S.C., Torkamani A., Su A.I., Oltz E.M., Artyomov M., Feeney A.J. Deep sequencing of the murine IgH repertoire reveals complex regulation of nonrandom V gene rearrangement frequencies. J. Immunol. 2013;191:2393–2402. doi: 10.4049/jimmunol.1301279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieslak A., Le Noir S., Trinquand A., Lhermitte L., Franchini D.M., Villarese P., Gon S., Bond J., Simonin M., Vanhille L. RUNX1-dependent RAG1 deposition instigates human TCR-δ locus rearrangement. J. Exp. Med. 2014;211:1821–1832. doi: 10.1084/jem.20132585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran A.E. The epigenetic role of non-coding RNA transcription and nuclear organization in immunoglobulin repertoire generation. Semin. Immunol. 2010;22:353–361. doi: 10.1016/j.smim.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Cowell L.G., Davila M., Kepler T.B., Kelsoe G. Identification and utilization of arbitrary correlations in models of recombination signal sequences. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0072. research0072.1–research0072.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Nozawa M., Klein J., Nei M. Evolutionary dynamics of the immunoglobulin heavy chain variable region genes in vertebrates. Immunogenetics. 2008;60:47–55. doi: 10.1007/s00251-007-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bono B., Madera M., Chothia C. VH gene segments in the mouse and human genomes. J. Mol. Biol. 2004;342:131–143. doi: 10.1016/j.jmb.2004.06.055. [DOI] [PubMed] [Google Scholar]
- Degner S.C., Wong T.P., Jankevicius G., Feeney A.J. Cutting edge: developmental stage-specific recruitment of cohesin to CTCF sites throughout immunoglobulin loci during B lymphocyte development. J. Immunol. 2009;182:44–48. doi: 10.4049/jimmunol.182.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner S.C., Verma-Gaur J., Wong T.P., Bossen C., Iverson G.M., Torkamani A., Vettermann C., Lin Y.C., Ju Z., Schulz D. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc. Natl. Acad. Sci. USA. 2011;108:9566–9571. doi: 10.1073/pnas.1019391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H., Ishii H., Pazin M.J., Sen R. Activation of 12/23-RSS-dependent RAG cleavage by hSWI/SNF complex in the absence of transcription. Mol. Cell. 2008;31:641–649. doi: 10.1016/j.molcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn-Walters D.K., Ademokun A.A. B cell repertoire and ageing. Curr. Opin. Immunol. 2010;22:514–520. doi: 10.1016/j.coi.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Eberle A.B., Herrmann K., Jäck H.-M., Mühlemann O. Equal transcription rates of productively and nonproductively rearranged immunoglobulin mu heavy chain alleles in a pro-B cell line. RNA. 2009;15:1021–1028. doi: 10.1261/rna.1516409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlich A., Martin V., Müller W., Rajewsky K. Analysis of the B-cell progenitor compartment at the level of single cells. Curr. Biol. 1994;4:573–583. doi: 10.1016/s0960-9822(00)00129-9. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W., Mueller D.L., Pennell C.A., Rivard J.J., Li Y.S., Hardy R.R., Schlissel M.S., Behrens T.W. Frequent aberrant immunoglobulin gene rearrangements in pro-B cells revealed by a bcl-xL transgene. Immunity. 1996;4:291–299. doi: 10.1016/s1074-7613(00)80437-9. [DOI] [PubMed] [Google Scholar]
- Fuxa M., Skok J., Souabni A., Salvagiotto G., Roldan E., Busslinger M. Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev. 2004;18:411–422. doi: 10.1101/gad.291504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou G., Ippolito G.C., Beausang J., Busse C.E., Wardemann H., Quake S.R. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 2014;32:158–168. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova T., Guo C., Ghosh A., Qiu X., Montefiori L., Verma-Gaur J., Choi N.M., Feeney A.J., Sen R. A structural hierarchy mediated by multiple nuclear factors establishes IgH locus conformation. Genes Dev. 2015;29:1683–1695. doi: 10.1101/gad.263871.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan S., Majumder K., Predeus A., Huang Y., Koues O.I., Verma-Gaur J., Loguercio S., Su A.I., Feeney A.J., Artyomov M.N., Oltz E.M. Unifying model for molecular determinants of the preselection Vβ repertoire. Proc. Natl. Acad. Sci. USA. 2013;110:E3206–E3215. doi: 10.1073/pnas.1304048110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Gerasimova T., Hao H., Ivanova I., Chakraborty T., Selimyan R., Oltz E.M., Sen R. Two forms of loops generate the chromatin conformation of the immunoglobulin heavy-chain gene locus. Cell. 2011;147:332–343. doi: 10.1016/j.cell.2011.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink B.A., Sleckman B.P. The response to and repair of RAG-mediated DNA double-stranded breaks. Annu. Rev. Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Zhang Y., Zhao L., Frock R.L., Du Z., Meyers R.M., Meng F.-L., Schatz D.G., Alt F.W. Chromosomal loop domains direct the recombination of antigen receptor genes. Cell. 2015;163:947–959. doi: 10.1016/j.cell.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhunjhunwala S., van Zelm M.C., Peak M.M., Cutchin S., Riblet R., van Dongen J.J.M., Grosveld F.G., Knoch T.A., Murre C. The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell. 2008;133:265–279. doi: 10.1016/j.cell.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y., Little A.J., Banerjee J.K., Hao B., Oltz E.M., Krangel M.S., Schatz D.G. Promoters, enhancers, and transcription target RAG1 binding during V(D)J recombination. J. Exp. Med. 2010;207:2809–2816. doi: 10.1084/jem.20101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K., Hashimshony T., Sawai C.M., Pongubala J.M.R., Skok J.A., Aifantis I., Singh H. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- Johnston C.M., Wood A.L., Bolland D.J., Corcoran A.E. Complete sequence assembly and characterization of the C57BL/6 mouse Ig heavy chain V region. J. Immunol. 2006;176:4221–4234. doi: 10.4049/jimmunol.176.7.4221. [DOI] [PubMed] [Google Scholar]
- Kaplinsky J., Li A., Sun A., Coffre M., Koralov S.B., Arnaout R. Antibody repertoire deep sequencing reveals antigen-independent selection in maturing B cells. Proc. Natl. Acad. Sci. USA. 2014;111:E2622–E2629. doi: 10.1073/pnas.1403278111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham P.M., Mortari F., Newton J.A., Schroeder H.W., Jr. Immunoglobulin VH clan and family identity predicts variable domain structure and may influence antigen binding. EMBO J. 1992;11:603–609. doi: 10.1002/j.1460-2075.1992.tb05092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondilis-Mangum H.D., Cobb R.M., Osipovich O., Srivatsan S., Oltz E.M., Krangel M.S. Transcription-dependent mobilization of nucleosomes at accessible TCR gene segments in vivo. J. Immunol. 2010;184:6970–6977. doi: 10.4049/jimmunol.0903923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.I., Fugmann S.D., Cowell L.G., Ptaszek L.M., Kelsoe G., Schatz D.G. A functional analysis of the spacer of V(D)J recombination signal sequences. PLoS Biol. 2003;1:E1. doi: 10.1371/journal.pbio.0000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli K.M., Slattery M., Mann R.S. Disentangling the many layers of eukaryotic transcriptional regulation. Annu. Rev. Genet. 2012;46:43–68. doi: 10.1146/annurev-genet-110711-155437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. Classification and regression by randomForest. R. News. 2002;2:18–22. [Google Scholar]
- Liu H., Schmidt-Supprian M., Shi Y., Hobeika E., Barteneva N., Jumaa H., Pelanda R., Reth M., Skok J., Rajewsky K., Shi Y. Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 2007;21:1179–1189. doi: 10.1101/gad.1529307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love V.A., Lugo G., Merz D., Feeney A.J. Individual V(H) promoters vary in strength, but the frequency of rearrangement of those V(H) genes does not correlate with promoter strength nor enhancer-independence. Mol. Immunol. 2000;37:29–39. doi: 10.1016/s0161-5890(00)00023-7. [DOI] [PubMed] [Google Scholar]
- Lucas J.S., Bossen C., Murre C. Transcription and recombination factories: common features? Curr. Opin. Cell Biol. 2011;23:318–324. doi: 10.1016/j.ceb.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marculescu R., Le T., Simon P., Jaeger U., Nadel B. V(D)J-mediated translocations in lymphoid neoplasms: a functional assessment of genomic instability by cryptic sites. J. Exp. Med. 2002;195:85–98. doi: 10.1084/jem.20011578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews A.G.W., Kuo A.J., Ramón-Maiques S., Han S., Champagne K.S., Ivanov D., Gallardo M., Carney D., Cheung P., Ciccone D.N. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J., Ebert A., Tagoh H., Tamir I.M., Schwickert T.A., Novatchkova M., Sun Q., Huis In ’t Veld P.J., Guo C., Yoon H.S. Flexible long-range loops in the VH gene region of the Igh locus facilitate the generation of a diverse antibody repertoire. Immunity. 2013;39:229–244. doi: 10.1016/j.immuni.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merelli I., Guffanti A., Fabbri M., Cocito A., Furia L., Grazini U., Bonnal R.J., Milanesi L., McBlane F. RSSsite: a reference database and prediction tool for the identification of cryptic Recombination Signal Sequences in human and murine genomes. Nucleic Acids Res. 2010;38:W262–W267. doi: 10.1093/nar/gkq391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori L., Wuerffel R., Roqueiro D., Lajoie B., Guo C., Gerasimova T., De S., Wood W., Becker K.G., Dekker J. Extremely Long-Range Chromatin Loops Link Topological Domains to Facilitate a Diverse Antibody Repertoire. Cell Rep. 2016;14:896–906. doi: 10.1016/j.celrep.2015.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomero T., Lim W.K., Odom D.T., Sulis M.L., Real P.J., Margolin A., Barnes K.C., O’Neil J., Neuberg D., Weng A.P. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA. 2006;103:18261–18266. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E., Rapado I., Li Y., Potter N.E., Wedge D.C., Tubio J., Alexandrov L.B., Van Loo P., Cooke S.L., Marshall J. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014;46:116–125. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter R.M., Kearney J.F., Chang S.P., Hood L.E. Developmentally controlled expression of immunoglobulin VH genes. Science. 1985;227:1597–1601. doi: 10.1126/science.3975629. [DOI] [PubMed] [Google Scholar]
- Phillips-Cremins J.E., Sauria M.E.G., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S.K., Ong C.-T., Hookway T.A., Guo C., Sun Y. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predeus A.V., Gopalakrishnan S., Huang Y., Tang J., Feeney A.J., Oltz E.M., Artyomov M.N. Targeted chromatin profiling reveals novel enhancers in Ig H and Ig L chain Loci. J. Immunol. 2014;192:1064–1070. doi: 10.4049/jimmunol.1302800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revilla-I-Domingo R., Bilic I., Vilagos B., Tagoh H., Ebert A., Tamir I.M., Smeenk L., Trupke J., Sommer A., Jaritz M., Busslinger M. The B-cell identity factor Pax5 regulates distinct transcriptional programmes in early and late B lymphopoiesis. EMBO J. 2012;31:3130–3146. doi: 10.1038/emboj.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud D., Demarco I.A., Reddy K.L., Schjerven H., Bertolino E., Chen Z., Smale S.T., Winandy S., Singh H. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat. Immunol. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro de Almeida C., Stadhouders R., de Bruijn M.J.W., Bergen I.M., Thongjuea S., Lenhard B., van Ijcken W., Grosveld F., Galjart N., Soler E., Hendriks R.W. The DNA-binding protein CTCF limits proximal Vκ recombination and restricts κ enhancer interactions to the immunoglobulin κ light chain locus. Immunity. 2011;35:501–513. doi: 10.1016/j.immuni.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Rouaud P., Vincent-Fabert C., Fiancette R., Cogné M., Pinaud E., Denizot Y. Enhancers located in heavy chain regulatory region (hs3a, hs1,2, hs3b, and hs4) are dispensable for diversity of VDJ recombination. J. Biol. Chem. 2012;287:8356–8360. doi: 10.1074/jbc.M112.341024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumfelt L.L., Zhou Y., Rowley B.M., Shinton S.A., Hardy R.R. Lineage specification and plasticity in CD19- early B cell precursors. J. Exp. Med. 2006;203:675–687. doi: 10.1084/jem.20052444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh C.E., Jhunjhunwala S., Riblet R., Murre C. Visualization of looping involving the immunoglobulin heavy-chain locus in developing B cells. Genes Dev. 2005;19:322–327. doi: 10.1101/gad.1254305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz D.G., Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011;11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- Schroeder H.W., Jr., Hillson J.L., Perlmutter R.M. Structure and evolution of mammalian VH families. Int. Immunol. 1990;2:41–50. doi: 10.1093/intimm/2.1.41. [DOI] [PubMed] [Google Scholar]
- Seitan V.C., Hao B., Tachibana-Konwalski K., Lavagnolli T., Mira-Bontenbal H., Brown K.E., Teng G., Carroll T., Terry A., Horan K. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature. 2011;476:467–471. doi: 10.1038/nature10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H.-Y., Verma-Gaur J., Torkamani A., Feeney A.J., Galjart N., Krangel M.S. Tcra gene recombination is supported by a Tcra enhancer- and CTCF-dependent chromatin hub. Proc. Natl. Acad. Sci. USA. 2012;109:E3493–E3502. doi: 10.1073/pnas.1214131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki N., Askary A., Swanson P.C., Lieber M.R. Mechanistic basis for RAG discrimination between recombination sites and the off-target sites of human lymphomas. Mol. Cell. Biol. 2012;32:365–375. doi: 10.1128/MCB.06187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollbach A.E., Wu G.E. Inversions produced during V(D)J rearrangement at IgH, the immunoglobulin heavy-chain locus. Mol. Cell. Biol. 1995;15:671–681. doi: 10.1128/mcb.15.2.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington M.J.T., Corcoran A.E. Non-coding transcription and large-scale nuclear organisation of immunoglobulin recombination. Curr. Opin. Genet. Dev. 2013;23:81–88. doi: 10.1016/j.gde.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Teng G., Maman Y., Resch W., Kim M., Yamane A., Qian J., Kieffer-Kwon K.-R., Mandal M., Ji Y., Meffre E. RAG represents a widespread threat to the lymphocyte genome. Cell. 2015;162:751–765. doi: 10.1016/j.cell.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos G.D., Alt F.W. Developmentally controlled and tissue-specific expression of unrearranged VH gene segments. Cell. 1985;40:271–281. doi: 10.1016/0092-8674(85)90141-2. [DOI] [PubMed] [Google Scholar]
- Yancopoulos G.D., Malynn B.A., Alt F.W. Developmentally regulated and strain-specific expression of murine VH gene families. J. Exp. Med. 1988;168:417–435. doi: 10.1084/jem.168.1.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J. The immunoglobulin IGHD gene locus in C57BL/6 mice. Immunogenetics. 2004;56:399–404. doi: 10.1007/s00251-004-0712-z. [DOI] [PubMed] [Google Scholar]
- Zhang M., Swanson P.C. V(D)J recombinase binding and cleavage of cryptic recombination signal sequences identified from lymphoid malignancies. J. Biol. Chem. 2008;283:6717–6727. doi: 10.1074/jbc.M710301200. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Espinoza C.R., Yu Z., Stephan R., He T., Williams G.S., Burrows P.D., Hagman J., Feeney A.J., Cooper M.D. Transcription factor Pax5 (BSAP) transactivates the RAG-mediated V(H)-to-DJ(H) rearrangement of immunoglobulin genes. Nat. Immunol. 2006;7:616–624. doi: 10.1038/ni1339. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.