

Abstract
Aims
We aimed to assess the contemporary outcome of Eisenmenger syndrome (ES), delineate the use of disease targeting therapies (DTT) in these patients and to investigate the effect of treatment on outcome in the community.
Methods and results
Patients with ES were systematically identified from the German National Register for Congenital Heart Defects. Data on underlying diagnosis, medical therapy, and survival were collected. The impact of DTT on survival was assessed using time-dependant Cox analysis. Overall, 153 ES patients were included (mean age 34.0 ± 13.3 years, 46% females). Of these, 88 (57.5%) were treated with at least one DTT (76.1% Bosentan, 20.5% Sildenafil) while 17.6% were on dual DTT. In addition, 24.8% of patients received digoxin, 10.5% angiotensin-converting enzyme-inhibitors/angiotensin receptor blockers, and 17.6% β-blockers. Moreover, 17.6% of patients were treated with oral anticoagulants, while 23.5% of patients received Aspirin. The survival rate at 1, 5, and 10 years of follow-up was only 92, 75, and 57% in the entire cohort, and was even worse in treatment naive ES patients (survival rate 86, 60, and 34% at 1, 5, and 10 years). Use of DTT was independently associated with a better survival (hazard ratio 0.42, P= 0.015).
Conclusion
This study illustrates the alarmingly poor survival prospects of Eisenmenger patients by community-based data even in the current era with advanced DTT and in a country with a wealthy health system. Treatment naive ES patients had especially high mortality rates approaching 60–70% at 10 years of follow-up. Treatment with DTT was associated with better survival.
Keywords: Adult congenital heart disease, Eisenmenger syndrome, Advanced therapy, Registry
Introduction
Eisenmenger syndrome (ES) is a multisystem disorder characterized by severely elevated pulmonary arterial pressures and right-to-left shunting with cyanosis.1–4 It is a common complication in patients with large, unrestrictive and unrepaired shunt lesions, especially at arterial or ventricular level. The exact prevalence of the condition in the community is unknown but it has been reported to account for ∼4% of adult patients seen at large tertiary centres for congenital heart disease.5 With increasing availability of timely surgical or interventional repair over the last few decades the number of ES patients in developed countries is likely to decrease. Nevertheless, the condition will continue to occur in patients, unsuitable for early repair, and individuals immigrating from regions with reduced access to high-quality paediatric cardiology and cardiothoracic surgery.
Data on symptoms and survival prospects of ES patients have been published previously based on single, tertiary centre cohorts or consortia of such centers.6–8 Nevertheless, conflicting data exist on the outcome of the condition.9,10 The assumed relatively benign outcome compared with idiopathic pulmonary hypertension has recently been questioned.9,11 In addition, it remains unclear whether the available data, mainly based on cohorts of patients treated at specialized expert centres, is subject to referral bias (favouring patients with more advanced disease) and whether outcomes and treatment regimes are similar in ES patients in the community. Furthermore, only limited data exist on the use of vasoactive disease targeting therapies (DTT) at large and on the effect of this type of medical treatment on survival.12,13 The rather unique data set of the German National Register for Congenital Heart Defects allows to address these questions. Specifically, we aimed to assess the outcome of ES patients in the current era, delineate the use of DTT in this cohort, and investigate the effect of treatment on outcome in the community.
Patients and methods
The German National Register for Congenital Heart Defects provides a nation-wide data base with a uniquely large population of patients with congenital heart disease not primarily gathered by tertiary referral centres but rather representing the community-based population. By now, the register comprises 46 628 patients. All patients included in the Register were screened for the diagnosis of pulmonary arterial hypertension and ES syndrome. Initially, an electronic database search was performed based on the International Paediatric and Congenital Cardiac Code published by the International Society for Nomenclature of Paediatric and Congenital Heart Disease (http://www.ipccc.net).14 The search included the following codes: 10.13.01—PH= pulmonary hypertension; 10.13.02—PH, pulmonary hypertension, primary; 10.13.20—PH, pulmonary hypertension, secondary; 10.13.21—PH, pulmonary hypertension, associated with left-to-right shunts; 10.13.06—PH, pulmonary vascular disease; 10.13.08—PH, irreversible pulmonary vascular disease (Eisenmenger). Subsequently, all electronically identified patients were screened manually to ensure that they had ES syndrome in accordance to current recommendations and as published previously.5
Eisenmenger syndrome was defined as pulmonary hypertension in the presence of a large non-restrictive intracardiac or extracardiac communication. Patients who previously underwent corrective surgery or interventions and did not present with an unrestrictive residual shunt were excluded. In addition, patients with Glenn-type physiology and those with segmental pulmonary hypertension (e.g. pulmonary atresia with ventricular septal defects) were excluded. The diagnosis of congenital heart disease was based on echocardiography, cardiovascular magnetic resonance, or cardiac catheterization data in all patients. Cardiac defects were classified into two categories according to complexity, as simple and complex defects. Simple defects comprised atrial or ventricular septal defects and persistent arterial duct, while complex defects represent combinations of congenital defects as well as atrioventricular septal defects, transposition of the great arteries, and patients with functionally univentricular hearts. Data on age, gender, cardiac and extracardiac diagnosis, NYHA functional class, resting oxygen saturation (SpO2), pulmonary hypertension specific medication, heart failure medication, oral anticoagulation, aspirin, and 6-min walk test (6-MWT) results were extracted from the medical reports. For the scope of the study, follow-up at a larger centre of care was defined as being followed at one of the top four contributing centres to the register.
This was a retrospective study. The starting point for analysis was the time of inclusion in the register for all patients. Survival status and time of death were ascertained from medical records and through inquiries at the local registration office for all patients. Approval by the appropriate Ethics Committee was obtained.
Statistical analysis
Data are presented as numbers (percentage) for categorical variables or mean ± standard deviation or median (25th to 75th percentile) for continuous variables, as appropriate. The relation between parameters and all-cause mortality was assessed with the use of uni- and multivariable time-dependent Cox proportional hazards regression analysis, with the date of inclusion in the register or start/escalation date of therapy used as start date. The proportional hazards assumption was verified for each time-dependent variable using generalized linear regression analysis, thus testing for a non-zero slope of the scaled Schoenfeld residuals on functions of time in addition to visual inspections of the graphs of the regression. Relevant clinical parameters were tested for significance on univariable analysis. Parameters significantly predicting prognosis on univariable analysis were subsequently included in multivariable time-dependent models. Interaction terms were used to explore the association between DTT and outcome with respect to centre size. For all analyses, a two-tailed P-value of <0.05 was used as the criterion for statistical significance. Analyses were performed with the use of R version 3.1.0 (R Foundation for Statistical Computing).
Results
Overall, 153 ES patients were identified and included in the current analysis. Of these, 83 (54%) presented with simple lesions (five of them with atrial septal defects) and 70 had complex congenital heart lesions. The included patients were under follow-up at one of 21 different institutions in Germany. The included patients per institution ranged from 1 to 68 patients. As illustrated in Table 1, patients with complex defects were significantly younger and had lower resting oxygen saturations compared with patients with simple defects. There were no significant differences with regard to height, weight, functional capacity, and medication between the groups.
Table 1.
Baseline characteristics, diagnoses, and therapy of the included patients
| Parameter | All | Simple defects | Complex defects | P-value |
|---|---|---|---|---|
| n | 153 | 83 | 70 | |
| Age | 34.0 ± 13.3 | 36.6 ± 14.2 | 30.9 ± 11.4 | 0.007 |
| Gender (female, %) | 45.8 | 60.2 | 55.7 | 0.69 |
| Down syndrome | 50 (32.7%) | 22 (26.5%) | 28 (40.0%) | 0.11 |
| Diagnoses | ||||
| ASD | 5 | 5 | ||
| VSD | 66 | 66 | ||
| PDA | 12 | 12 | ||
| AVSD | 35 | 35 | ||
| APW/TAC | 11 | 11 | ||
| TGA | 1 | 1 | ||
| Univentricular physiology | 23 | 23 | ||
| Weight (kg) | 57.0 ± 14.5 | 57.5 ± 15.2 | 56.4 ± 13.9 | 0.68 |
| Height (cm) | 159.5 ± 16.7 | 159.4 ± 15.4 | 159.5 ± 18.2 | 0.98 |
| Resting SpO2 (%) | 81.2 ± 8.9 | 83.8 ± 8.6 | 78.3 ± 8.3 | <0.0001 |
| NYHA class (I/II/III/IV) | 0/66/77/10 | 0/41/39/3 | 0/25/38/7 | 0.11 |
| 6-MWT distance (m) | 380.1 ± 108.3 | 395.8 ± 122.8 | 360.5 ± 85.2 | 0.24 |
| DTT (%) | 57.5 | 60.2 | 54.3 | 0.56 |
| Diuretics (%) | 35.3 | 28.9 | 42.9 | 0.10 |
| Digoxin (%) | 24.8 | 21.7 | 28.6 | 0.43 |
| ACE-i/ARBs (%) | 10.5 | 6.0 | 15.7 | 0.09 |
| β-Blocker (%) | 17.6 | 15.7 | 20.0 | 0.63 |
| Oral anticoagulation (%) | 17.6 | 20.5 | 14.3 | 0.43 |
| Aspirin (%) | 23.5 | 25.3 | 21.4 | 0.71 |
P-values compare patients with simple defects with those having complex underlying heart conditions.
In total, 10.5% of patients were treated with angiotensin-converting enzyme (ACE)-inhibitors or angiotensin receptor blockers (ARBs). Especially in ES patients with complex defects, a considerable proportion (16%) of patients received these drugs. In addition, 25% of patients were treated with digoxin and 18% with β-blockers. Oral anticoagulation was administered in 18% of patients and aspirin in 23%.
As illustrated in Figure 1, 57.5% of patients were treated with at least one DTT, while 17.6% of patients were treated with a combination of two drugs. The majority of them were on Bosentan (76.1% of patients on monotherapy). In addition, this drug was used as a second line medication in 44.4% of patients treated primarily with Sildenafil. The median time to escalation of therapy was 2.6 years (interquartile range, IQR, 0.9–5.2 years). A greater proportion of patients without Down syndrome was treated with DTT compared with those with Down syndrome (62 vs. 48%); however, this difference did not reach significance (P= 0.14).
Figure 1.

Distribution of therapy. The Figure illustrates that overall 57.5% of patients were treated with at least one disease targeting therapy drug. Of those patients treated with disease targeting therapies, over 95% of patients were on Bosentan or Sildenafil.
Over a median follow-up period of 5.0 years (IQR 1.4–9.3 years), 47 patients (30%) died. Survival rates at 1, 5, and 10 years of follow-up were 92.1% [95% confidence interval, CI, 87.8–96.7%], 74.9% [95% CI 67.6–83.1%], and 57.2% [95% CI 47.6–68.8%], respectively (Figure 2). The leading causes of death were heart failure, sudden cardiac death, malignant disease, perioperative mortality, and infections as illustrated in Supplementary material online, Figure SA1. No difference in survival was observed between males and females (log-rank P= 0.92), between patients with or without Down syndrome (P= 0.31) or patients with simple or complex underlying heart defects (P= 0.48). Table 2 shows the results of the Cox proportional hazards analysis. On univariable Cox analysis, resting oxygen saturation, follow-up at a larger centre and use of DTT emerged as the only significant predictors of mortality. On stepwise multivariable analysis, only oxygen saturation (hazard ratio, HR, 0.952 [95% CI 0.922–0.983], P= 0.0024) and use of DTT (HR 0.436 [95% CI 0.198–0.959], P= 0.04), were independently related to outcome. In fact, ES patients at larger centres were more likely to be treated with DTT (62.7 vs. 47.1% on DTT, P= 0.08) and there was a statistically significant interaction between centre size and use of DTT on Cox analysis (P= 0.0004).
Figure 2.

Kaplan–Meier survival function of all Eisenmenger patients included in the current study. (A) Time scale based on follow-up time in the National Register. (B) Time scale based on patients' age.
Table 2.
Results of the univariate Cox analysis
| Parameter | HR (95% CI) | P-value |
|---|---|---|
| Age (years) | 0.99 (0.97–1.02) | 0.54 |
| Female gender | 0.97 (0.54–1.74) | 0.92 |
| Complex lesion | 0.81 (0.46–1.44) | 0.48 |
| Down syndrome | 0.72 (0.39–1.35) | 0.31 |
| Weight (kg) | 0.98 (0.96–1.01) | 0.23 |
| Height (cm) | 0.98 (0.96–1.00) | 0.054 |
| Resting SpO2 (%) | 0.95 (0.92–0.98) | 0.0015 |
| NYHA Class > II | 0.75 (0.42–1.33) | 0.33 |
| 6-MWT distance (m) | 1.00 (0.99–1.00) | 0.14 |
| Larger centre of carea | 0.53 (0.30–0.97) | 0.04 |
| DTT | 0.30 (0.17–0.54) | 0.0001 |
| Diuretics | 1.01 (0.55–1.85) | 0.97 |
| Digoxin | 1.59 (0.85–2.98) | 0.14 |
| ACE-i/ARBs | 1.30 (0.52–3.29) | 0.58 |
| β-Blocker | 0.35 (0.13–1.00) | 0.051 |
| Oral anticoagulation | 1.07 (0.52–2.22) | 0.86 |
| Aspirin | 0.93 (0.47–1.82) | 0.83 |
DTT, disease targeting therapies.
aLarger centre of care was defined as the top four contributors of patients to the register.
Survival at 5 years of follow-up was 60.3% [95% CI 48.2–75.5%] in treatment naive patients and 83.0% [95% CI 72.6–94.9%] in those with DTT. This is illustrated in Figure 3, based on the results of the Cox proportional-hazard analysis. In addition, Supplementary material online, Figure SA2 illustrates the association between DTT and outcome using patient age as a time scale variable. No difference could be found between survival in patients on mono- vs. dual therapy on time-dependant Cox analysis (Figure 4). No significant association between conventional medical treatment (Diuretics, Digoxin, ACE-inhibitors/ARBs, etc.) and outcome could be established, although a trend between β-blocker therapy and better survival was observed on this analysis. Similarly, oral anticoagulation or aspirin use was not significantly related to outcome (see Supplementary material online, Figure SA3; Table 2).
Figure 3.
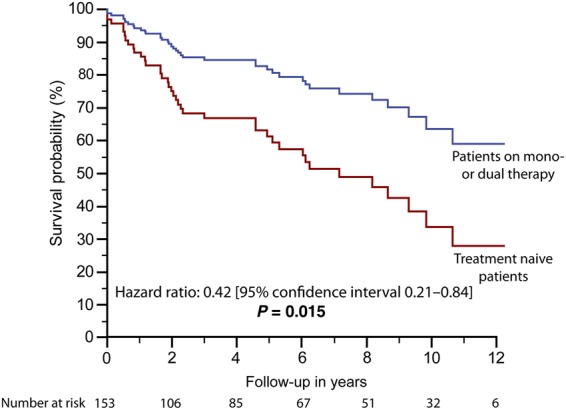
Survival of treatment naïve patients compared with those treated with at least one disease targeting drug modelled based on the results of time-dependant Cox analysis.
Figure 4.
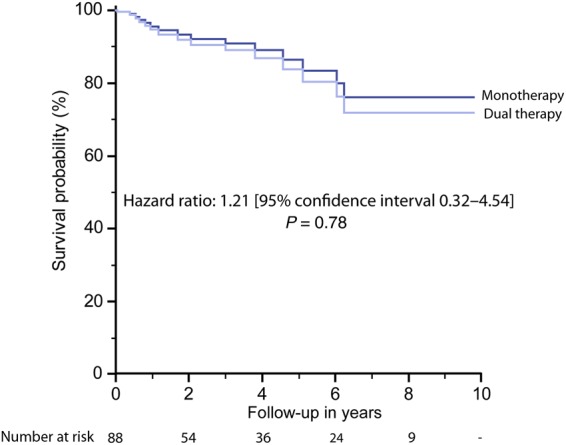
Survival of patients on disease targeting monotherapy compared with those treated with dual therapy modelled based on the results of time-dependant Cox analysis.
Discussion
The data from the German National Register for Congenital Heart Defects highlights poor survival prospects of ES patients, worse than in previous reports.6–8,10 In addition, our results suggest that patients at larger centres and especially those treated with DTT fared better compared with the remaining patients. This is in agreement with a recent report suggesting improved survival of adult congenital heart disease patients, in general, after the establishment of large tertiary centres in Canada, but is a new finding specifically for ES patients.15 Interestingly, the association between centre size and outcome appears to be mainly mediated via the more frequent use of DTT at these larger centres. This observation is consistent with a recent report showing no clear indication that survival prospects of treatment naive ES patients have improved since the 1960s.
The relatively poor survival prospects of Eisenmenger patients observed in the current study are of particular relevance when considering that the study comprises a relatively contemporary cohort of patients treated in a well-developed, wealthy health system with universal access to paediatric and adult cardiology services. Unlike, previous studies, the current report is not based on data from a single tertiary institution or groups of expert centres specialized in adult congenital heart defects but rather reflects the treatment reality and fate of ES patients at large.6–8 We have recently reviewed the literature on survival prospects of ES patients and found that, after accounting for confounding statistical effects, the 10-year mortality rate is ∼30–40% in treatment naive individuals.9 In contrast, the current study—using a broader cohort of patients, not all of whom were attending specialized centres—revealed a 10-year mortality rate approaching 60–70%. The reasons for this discrepancy are unclear, especially given that this still represents a selected cohort of patients registered in a National database for congenital heart disease. Although, enrolment in the Register is patient driven, it is generally prompted by an according recommendation of the treating physician. In addition, ours is a relatively contemporary cohort (registration date between 2001 and 2012) and, thus, should supposedly not fare worse compared with published historical cohorts.
Our analysis illustrates that despite absent scientific data and guideline recommendations many ES patients are still treated with Digoxin, standard heart failure medication, and subjected to oral anticoagulation or aspirin treatment. The use of Digoxin is concerning as it has been demonstrated in other cardiovascular settings to increase mortality10,16–18 and a similar tendency was seen in this study (HR 1.6, P= 0.14). Other standard heart failure drugs, namely ACE-inhibitors and ARBs may not share the problem of increasing mortality in their own right, but they have a hypotensive effect and may preclude the use of DTT in adequate doses and especially combination therapy. Therefore, we believe that the use of these drugs should be very restrictive in the absence of guideline recommendations and positive scientific data. The issue of anticoagulation remains controversial. While a recent study found no evidence of a specific survival benefit in congenital pulmonary hypertension patients19 and current guidelines discourage the routine use of oral anticoagulation in ES patients in the absence of additional indications,20 selected patients with large pulmonary thrombi may benefit from this therapy.21 It is likely, however, that a proportion of patients are given oral anticoagulation based on recommendations extrapolated from—but specifically intended for—patients with idiopathic pulmonary hypertension.22 Furthermore, no data on the beneficial effects of aspirin are available—to our knowledge—and the use of this drug is likely to represent the pragmatism of some colleagues who aim to offer some protection against thrombosis, while fearing the potential complications of anticoagulation.
While anticoagulation, aspirin treatment, and heart failure medication were not related to outcome, we found a pronounced association between DTT and all-cause mortality. This is in agreement with a previous study by Dimopoulos et al. reporting an association between use of DTT and lower mortality in ES patients.12 Comparing the current study with this previous report reveals some interesting findings. Firstly, the proportion of ES patients treated with DTT was 57% in the current analysis compared with only 30% in the former study. This likely reflects the increasing uptake of DTT in the setting of ES syndrome by the medical profession. Of those patients treated, a similar percentage was treated with endothelin receptor antagonists (79.5% in our study vs. 73.5% in the study by Dimopoulos). Comparing the two studies also reveals a higher proportion of patients on dual therapy in the current study (27/153 vs. 6/299), likely also related to a more proactive use of DTT driven by an increased experience with the medication and also supported by the positive results of previous studies.
We found a hazard ratio of 0.42 for mortality in treated patients compared with treatment naive individuals as opposed to lower values reported by Dimopoulos et al. (0.21 and 0.16 based on unadjusted and adjusted Cox analyses). This suggests the impact of DTT on survival may be lower in a community setting compared with the results that can be achieved at tertiary institutions but still is substantial as it indicated that the hazard of death is 2.4-times higher (i.e. 1/0.42) in untreated compared with treated ES patients.
Despite the use of a monotherapy-first approach, with escalation of therapy after a median of 2.6 years, we found no evidence of worse survival of patients on dual therapy. As the general approach is to start ES patients on DTT monotherapy and to only escalate therapy when patients deteriorate or fail to achieve a pre-specified therapy goal or an acceptable clinical status, patients on dual therapy represent a sicker patient population with more advance symptoms. The lack of a difference in survival between patients on mono and those on dual DTT should therefore not be interpreted as an argument against escalation of therapy. Rather, we contend that this finding suggests that beyond the recognized impact on symptoms and exercise capacity,23–26 escalation of therapy may be associated with clinical stabilization and may aid to preserve the positive impact of DTT on survival over time. This observation is consistent with findings reporting haemodynamic improvements with dual therapy in ES patients,27 which may provide the pathophysiologic rationale for the observed clinical stabilization.
The borderline association between β-blocker therapy and improved outcome of ES patients in the current study deserves future attention. A potential role for β-blocker therapy to protect right ventricular function has been advocated by some,28,29 and these drugs are currently undergoing clinical trials, albeit in the setting of idiopathic pulmonary hypertension.30
Study limitations
We accept that this study has limitations related to the nature of the data employed. Unlike previous single centre studies, we did not have access to an entire hospital medical database but had to rely on information available from medical reports and clinic letters. We cannot, therefore, exclude the possibility that the association between DTT and outcome was mediated by unobserved variables. However, even after adjusting for other significant determinants of outcome we found that DTT use was associated with better survival compared with treatment naive patients. We believe that this, together with the observation that none of the other conventional drugs showed an effect similar to DTT, suggests that DTT may be genuinely associated with reduced mortality in the setting of ES. One hundred and two (67%) of the included patients were under follow-up at the four largest contributors of patients to the current study. Therefore, it is likely that the patients included here represent a mixture of Eisenmenger patients in the community and those followed at specialized centres, with the latter cohort potentially dominating numerically. Nevertheless, it appears that the superior outcome of patients at larger centres was chiefly mediated by a more frequent use of DTT there, thus supporting the main results of the study. Furthermore, patients with learning difficulties or disabilities may be underrepresented in the study and although the percentage of patients with Down syndrome was comparable with previous Eisenmenger studies,12 the true prevalence of Eisenmenger patients with learning difficulties in the community remains unknown. While the association between use of DTT and outcome presented here and in a previous study12 are impressive, the cost of DTT is considerable and further studies focusing on cost utility and cost benefit analyses are desirable. Due to the limited sample size, the possibility of type II errors (i.e. the failure to detect an actual effect) should be considered especially as hazard ratios for some parameters are in the range of clinical relevance.
Conclusions
The current study illustrates the alarmingly poor survival prospects of Eisenmenger patients even in the current era with the option of DTT and in a wealthy health system. Treatment naive ES patients had especially high mortality rates approaching 60–70% at 10 years of follow-up. Consistent with a previous report, treatment with DTT was associated with better survival in the current study. In addition, a strategy of escalation of therapy was associated with similar survival benefits of dual therapy compared with DTT monotherapy. The results of this study imply that ES patients should be linked to tertiary specialist centres and a proactive approach of treatment with DTT should be considered for all eligible patients, based on the dismal natural prognosis and the potentially substantial prognostic benefit of therapy.
Supplementary material
Supplementary material is available at European Heart Journal online.
Authors' contributions
G.-P.D., M.-A.K., O.T. performed statistical analysis; H.B., U.M.M.B., F.B., H.K. handled funding and supervision; G.-P.D., M.-A.K., O.T. acquired the data; G.-P.D., H.B., U.M.M.B. conceived and designed the research; G.-P.D., H.B. drafted the manuscript; O.M., U.M.M.B., H.K., F.B. made critical revision of the manuscript for key intellectual content.
Funding
This study was supported by a research grants from the EMAH Stiftung Karla Voellm, Krefeld, Germany, the DZHK (German Centre for Cardiovascular Research), the BMBF (German Ministry of Education and Research), and Actelion Pharmaceuticals Deutschland GmbH.
Conflict of interest: G.D., O.T., F.B., H.B., and H.K. have received educational/travel grants from Actelion, Pfizer, and/or have served on the advisory boards of Actelion, Germany (G.D., H.K.).
Acknowledgements
We thank the following colleagues for recruiting patients and for providing medical information at baseline and during follow-up.
Prof. Dr. med. Gernot Buheitel. Klinikum Augsburg; II. Klinik für Kinder und Jugendliche. Augsburg. Prof. Dr. med. Deniz Kececioglu. Herz- und Diabeteszentrum NRW; Zentrum für Angeborene Herzfehler; Klinik für Kinderkardiologie und angeborene Herzfehler. Bad Oeynhausen. Prof. Dr. med. Felix Berger. Deutsches Herzzentrum Berlin; Abteilung für Angeborene Herzfehler/Kinderkardiologie. DHZ Berlin. Prof. Dr. med. Johannes Breuer. Universitätsklinikum Bonn; Zentrum für Kinderheilkunde; Abteilung für Kinderkardiologie. Bonn. Dr. med. Trong Phi Lê. Klinikum Links der Weser gGmbH; Klinik für strukturelle und angeborene Herzfehler/Kinderkardiologie. Bremen. PD Dr. med. Frank Pillekamp. Universitätsklinikum Düsseldorf; Klinik für Allgemeine Pädiatrie, Neonatologie und Kinderkardiologie. Düsseldorf. Prof. Dr. med. Sven Dittrich. Universitätsklinikum Erlangen; Kinder- und Jugendklinik; Pädiatrische Kardiologie. Erlangen. Dr. med. Ulrich Neudorf. Universitätsklinikum Essen; Klinik für Kinderheilkunde III; Abteilung für Pädiatrische Kardiologie. Essen. Prof. Dr. med. Brigitte Stiller. Universitäts-Herzzentrum Freiburg Bad Krozingen; Zentrum für Kinder- und Jugendmedizin; Klinik für Angeborene Herzfehler/Pädiatrische Kardiologie. Freiburg. Prof. Dr. med. Dietmar Schranz. Universitätsklinikum Gießen und Marburg GmbH; Klinik für Kinderkardiologie und angeborene Herzfehler. Giessen. Prof. Dr. med. Thomas Paul. Kinderherzklinik Göttingen; Abteilung Pädiatrische Kardiologie und Intensivmedizin. Göttingen. Prof. Dr. med. Ralph Grabitz. Universitätsklinikum Halle (Saale); Universitätsklinik und Poliklinik für Pädiatrische Kardiologie. Halle. Prof. Dr. med. Rainer Kozlik-Feldmann. Universitäres Herzzentrum Hamburg GmbH; Klinik und Poliklinik für Kinderkardiologie. Hamburg. Prof. Dr. med. Philipp Beerbaum. Medizinische Hochschule Hannover (MHH); Zentrum für Kinderheilkunde und Jugendmedizin; Kinderkardiologie und pädiatrische Intensivmedizin. Hannover. Prof. Dr. med. Matthias Gorenflo. Universitätsklinikum Heidelberg AöR; Zentrum für Kinder- und Jugendmedizin; Kinderheilkunde II: Pädiatrische Kardiologie/Angeborene Herzfehler. Heidelberg. Prof. Dr. med. Hashim Abdul-Khaliq. Universitätsklinikum des Saarlandes; Klinik für Pädiatrische Kardiologie. Homburg. PD Dr. med. Thomas Kriebel. Westpfalz-Klinikum GmbH; Klinik für Kinder- und Jugendmedizin; Kinderkardiologie. Kaiserslautern. Prof. Dr. med. Ingo Dähnert. Herzzentrum Leipzig GmbH; Klinik für Kinderkardiologie. Leipzig. Prof. Dr. med. Peter Ewert. Deutsches Herzzentrum München; Klinik für Kinderkardiologie und angeborene Herzfehler. DHZ München. Prof. Dr. med. Christian Jux. Universitätsklinikum Münster (UKM); Klinik für Kinder- und Jugendmedizin; Pädiatrische Kardiologie. Münster. Dr. med. Matthias W. Freund. Klinikum Oldenburg gGmbH, Medizinischer Campus Universität Oldenburg; Zentrum für Kinder- und Jugendmedizin—Elisabeth-Kinderkrankenhaus. Oldenburg.
References
- 1.D'Alto M, Diller GP. Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: current advanced management strategies. Heart 2014;100:1322–1328. [DOI] [PubMed] [Google Scholar]
- 2.Gatzoulis MA, Beghetti M, Landzberg MJ, Galie N. Pulmonary arterial hypertension associated with congenital heart disease: recent advances and future directions. Int J Cardiol 2014;177:340–347. [DOI] [PubMed] [Google Scholar]
- 3.Opotowsky AR, Landzberg MJ, Beghetti M. The exceptional and far-flung manifestations of heart failure in Eisenmenger syndrome. Heart Fail Clin 2014;10:91–104. [DOI] [PubMed] [Google Scholar]
- 4.Dimopoulos K, Wort SJ, Gatzoulis MA. Pulmonary hypertension related to congenital heart disease: a call for action. Eur Heart J 2014;35:691–700. [DOI] [PubMed] [Google Scholar]
- 5.Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation 2007;115:1039–1050. [DOI] [PubMed] [Google Scholar]
- 6.Cantor WJ, Harrison DA, Moussadji JS, Connelly MS, Webb GD, Liu P, McLaughlin PR, Siu SC. Determinants of survival and length of survival in adults with Eisenmenger syndrome. Am J Cardiol 1999;84:677–681. [DOI] [PubMed] [Google Scholar]
- 7.Daliento L, Somerville J, Presbitero P, Menti L, Brach-Prever S, Rizzoli G, Stone S. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J 1998;19:1845–1855. [DOI] [PubMed] [Google Scholar]
- 8.Diller GP, Dimopoulos K, Broberg CS, Kaya MG, Naghotra US, Uebing A, Harries C, Goktekin O, Gibbs JS, Gatzoulis MA. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: a combined retrospective and case-control study. Eur Heart J 2006;27:1737–1742. [DOI] [PubMed] [Google Scholar]
- 9.Diller GP, Kempny A, Inuzuka R, Radke R, Wort SJ, Baumgartner H, Gatzoulis MA, Dimopoulos K. Survival prospects of treatment naive patients with Eisenmenger: a systematic review of the literature and report of own experience. Heart 2014;100:1366–1372. [DOI] [PubMed] [Google Scholar]
- 10.Manes A, Palazzini M, Leci E, Bacchi Reggiani ML, Branzi A, Galie N. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: a comparison between clinical subgroups. Eur Heart J 2014;35:716–724. [DOI] [PubMed] [Google Scholar]
- 11.Bonello B, Renard S, Mancini J, Hubert S, Habib G, Fraisse A. Life span of patients with Eisenmenger syndrome is not superior to that of patients with other causes of pulmonary hypertension. Cardiovasc Diagn Ther 2014;4:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimopoulos K, Inuzuka R, Goletto S, Giannakoulas G, Swan L, Wort SJ, Gatzoulis MA. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation 2010;121:20–25. [DOI] [PubMed] [Google Scholar]
- 13.Sun YJ, Yang T, Zeng WJ, Gu Q, Ni XH, Zhao ZH, Liu ZH, Xiong CM, He JG. Impact of sildenafil on survival of patients with Eisenmenger syndrome. J Clin Pharmacol 2013;53:611–618. [DOI] [PubMed] [Google Scholar]
- 14.Beland MJ, Jacobs JP, Tchervenkov CI, Franklin RC. Report from the Executive of The International Working Group for Mapping and Coding of Nomenclatures for Paediatric and Congenital Heart Disease. Cardiol Young 2002;12:425–430. [DOI] [PubMed] [Google Scholar]
- 15.Mylotte D, Pilote L, Ionescu-Ittu R, Abrahamowicz M, Khairy P, Therrien J, Mackie AS, Marelli A. Specialized adult congenital heart disease care: the impact of policy on mortality. Circulation 2014;129:1804–1812. [DOI] [PubMed] [Google Scholar]
- 16.Freeman JV, Yang J, Sung SH, Hlatky MA, Go AS. Effectiveness and safety of digoxin among contemporary adults with incident systolic heart failure. Circ Cardiovasc Qual Outcomes 2013;6:525–533. [DOI] [PubMed] [Google Scholar]
- 17.Shah M, Avgil Tsadok M, Jackevicius CA, Essebag V, Behlouli H, Pilote L. Relation of digoxin use in atrial fibrillation and the risk of all-cause mortality in patients >/=65 years of age with versus without heart failure. Am J Cardiol 2014;114:401–406. [DOI] [PubMed] [Google Scholar]
- 18.Whitbeck MG, Charnigo RJ, Khairy P, Ziada K, Bailey AL, Zegarra MM, Shah J, Morales G, Macaulay T, Sorrell VL, Campbell CL, Gurley J, Anaya P, Nasr H, Bai R, Di Biase L, Booth DC, Jondeau G, Natale A, Roy D, Smyth S, Moliterno DJ, Elayi CS. Increased mortality among patients taking digoxin – analysis from the AFFIRM study. Eur Heart J 2013;34:1481–1488. [DOI] [PubMed] [Google Scholar]
- 19.Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, Grunig E, Staehler G, Rosenkranz S, Halank M, Held M, Lange TJ, Behr J, Klose H, Claussen M, Ewert R, Opitz CF, Vizza CD, Scelsi L, Vonk-Noordegraaf A, Kaemmerer H, Gibbs JS, Coghlan G, Pepke-Zaba J, Schulz U, Gorenflo M, Pittrow D, Hoeper MM. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014;129:57–65. [DOI] [PubMed] [Google Scholar]
- 20.Baumgartner H, Bonhoeffer P, De Groot NM, de Haan F, Deanfield JE, Galie N, Gatzoulis MA, Gohlke-Baerwolf C, Kaemmerer H, Kilner P, Meijboom F, Mulder BJ, Oechslin E, Oliver JM, Serraf A, Szatmari A, Thaulow E, Vouhe PR, Walma E, Task Force on the Management of Grown-up Congenital Heart Disease of the European Society of Cardiology (ESC), Association for European Paediatric Cardiology (AEPC), ESC Committee for Practice Guidelines (CPG). ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J 2010;31:2915–2957. [DOI] [PubMed] [Google Scholar]
- 21.Broberg C, Ujita M, Babu-Narayan S, Rubens M, Prasad SK, Gibbs JS, Gatzoulis MA. Massive pulmonary artery thrombosis with haemoptysis in adults with Eisenmenger's syndrome: a clinical dilemma. Heart 2004;90:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G, Guidelines ESCCfP. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493–2537. [DOI] [PubMed] [Google Scholar]
- 23.Diller GP, Alonso-Gonzalez R, Dimopoulos K, Alvarez-Barredo M, Koo C, Kempny A, Harries C, Parfitt L, Uebing AS, Swan L, Marino PS, Wort SJ, Gatzoulis MA. Disease targeting therapies in patients with Eisenmenger syndrome: response to treatment and long-term efficiency. Int J Cardiol 2013;167:840–847. [DOI] [PubMed] [Google Scholar]
- 24.Galie N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, Chiossi E, Landzberg M, Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006;114:48–54. [DOI] [PubMed] [Google Scholar]
- 25.Gatzoulis MA, Beghetti M, Galie N, Granton J, Berger RM, Lauer A, Chiossi E, Landzberg M, BREATHE-5 Investigators. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol 2008;127:27–32. [DOI] [PubMed] [Google Scholar]
- 26.Schuuring MJ, Vis JC, Duffels MG, Bouma BJ, Mulder BJ. Adult patients with pulmonary arterial hypertension due to congenital heart disease: a review on advanced medical treatment with Bosentan. Ther Clin Risk Manag 2010;6:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D'Alto M, Romeo E, Argiento P, Sarubbi B, Santoro G, Grimaldi N, Correra A, Scognamiglio G, Russo MG, Calabro R. Bosentan-sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int J Cardiol 2012;155:378–382. [DOI] [PubMed] [Google Scholar]
- 28.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 2010;182:652–660. [DOI] [PubMed] [Google Scholar]
- 29.Drake JI, Gomez-Arroyo J, Dumur CI, Kraskauskas D, Natarajan R, Bogaard HJ, Fawcett P, Voelkel NF. Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol Genomics 2013;45:449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grinnan D, Bogaard HJ, Grizzard J, Van Tassell B, Abbate A, DeWilde C, Priday A, Voelkel NF. Treatment of group I pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med 2014;189:1562–1564. [DOI] [PubMed] [Google Scholar]