

Abstract
Ursodeoxycholic acid (UDCA), a natural, hydrophilic nontoxic bile acid, is clinically effective for treating cholestatic and chronic liver diseases. We investigated the chronic effects of UDCA on age-related lipid homeostasis and underlying molecular mechanisms. Twenty-week-old C57BL/6 male and female mice were fed a diet with or without 0.3% UDCA supplementation for 25 weeks. UDCA significantly reduced weight gain, adiposity, hepatic triglyceride, and hepatic cholesterol without incidental hepatic injury. UDCA-mediated hepatic triglyceride reduction was associated with downregulated hepatic expression of peroxisome proliferator-activated receptor-γ, and of other genes involved in lipogenesis (Chrebp, Acaca, Fasn, Scd1, and Me1) and fatty acid uptake (Ldlr, Cd36). The inflammatory cytokines Tnfa, Ccl2, and Il6 were significantly decreased in liver and/or white adipose tissues of UDCA-fed mice. These data suggest that UDCA exerts beneficial effects on age-related metabolic disorders by lowering the hepatic lipid accumulation, while concurrently reducing hepatocyte and adipocyte susceptibility to inflammatory stimuli. [BMB Reports 2016; 49(2): 105-110]
Keywords: Anti-inflammatory, Bile acid, Lipogenesis, Peroxisome proliferator-activated receptor-γ, Ursodeoxycholic acid
INTRODUCTION
Bile acids are cholesterol-derived bioactive amphipathic molecules that play essential roles in many processes required for the maintenance of a healthy lifespan (1). In addition to their classical role as endogenous detergents that regulate bile flow and facilitate intestinal absorption of lipids, nutrients, and lipid-soluble vitamins (2), bile acids are also metabolically active signaling molecules that modulate their own levels, and also regulate triglyceride, cholesterol, glucose, and energy metabolism. Bile acid composition changes as mice age, and several bile acids are increased in the serum of long-lived mouse strains that exhibit attenuated pro-aging pathway signaling (3, 4).
Ursodeoxycholic acid (UDCA) is a natural, hydrophilic bile acid that is used to treat various hepatic and digestive diseases, including cholestasis and primary biliary cirrhosis (5, 6). Oral UDCA administration increases the UDCA levels in serum and bile by competitively inhibiting the ileal absorption of natural, toxic endogenous bile salts (7, 8). The main mode of action of UDCA is likely via displacement of endogenous hepatotoxic bile salts to increase the contribution of non-toxic hydrophilic bile acids in the total bile acid pool. In liver disease, UDCA provides a clinical benefit by mediating the hepatocyte cytoprotection, choleresis, and immunomodulatory functions. The cytoprotective effect of UDCA partly results from the inhibition of cell apoptosis (9). UDCA also reduces the mitochondrial membrane permeability and the release of hydrolytic enzymes from damaged hepatocytes, and improves cell resistance to reactive oxygen species (10, 11). Recently, UDCA was reported to improve hepatic steatosis and insulin sensitivity by inducing the excretion of hepatic lipids, inhibiting hepatic long-chain free fatty acid uptake, and suppressing the miR-34a/SIRT1/p53 pathway in obese mice (12-14). All these effects support the hypothesis that UDCA could be a useful molecule for preventing age-related obesity, steatosis, and inflammation.
We investigated the influence of UDCA on age-related accumulation of body fat, and the molecular mechanisms involved in the anti-obesity effects of UDCA in the liver and white adipose tissue. The results of the present study provide a basis for considering UDCA administration as a therapeutic strategy for treating age-related obesity.
RESULTS
UDCA decreases weight gain and fat accumulation
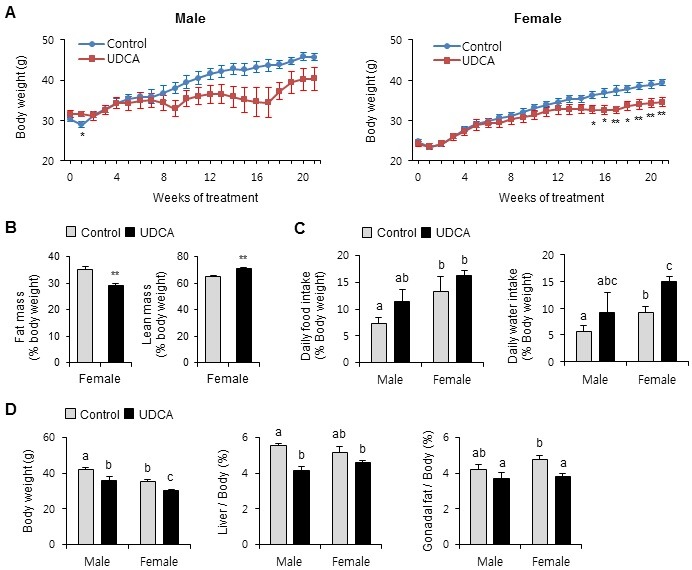
To determine whether UDCA affects age-related body fat accumulation, adult male and female mice were fed a diet with or without 0.3% UDCA for 25 weeks, with body weight monitored weekly (Fig. 1A). Body weight gain was similar in Control and UDCA-fed mice until 8 weeks, and thereafter tended to decrease in the UDCA-fed mice. The difference in body weight between Control and UDCA-fed mice was significant after 15 weeks in females (P < 0.05). Although UDCA also slowed weight gain in male mice after 13 weeks, this difference did not achieve statistical significance (p=0.06-0.07). The decreased body weight in female mice by UDCA was mainly due to decreased total body fat (↓17%) and not lean body mass (↑9%) (Fig. 1B). To rule out reduced food intake causing declining body weight gain, we monitored food and water intake in metabolism cages. Food intake did not decrease in UDCA-fed mice, but was slightly increased in male UDCA-fed mice (Fig. 1C). Water intake also showed a similar trend of increased drinking by UDCA mice vs Controls (Fig. 1C). Next, we removed and weighed the livers and white adipose tissue (gonadal fat). Liver and gonadal adipose weights were decreased by UDCA treatment in both genders, but to differing extents. Male mice showed significantly reduced liver mass (↓26%), whereas female mice showed significantly decreased white adipose tissue mass (↓20%) by UDCA feeding. These results indicate that UDCA ameliorates age-related body weight gain and adiposity.
Fig. 1. Ursodeoxycholic acid decreases age-related weight gain and fat accumulation. Male and female mice were fed diets with or without 0.3% UDCA for 25 weeks, starting at 20-week-old. (A) Body weight was monitored weekly. (B) At the end of the experiment, lean mass and fat mass were measured. *P < 0.05 vs Controls; **P < 0.01 vs Controls. (C) After Control or UDCA feeding for 21 weeks, food and water intake were measured in metabolic cages. (D) At the end of the experiment, body, liver, and gonadal fat weights were measured. n = 3 mice/group for males, and 6 mice/group for females. Results are expressed as the mean ± SEM. Statistical analysis was performed by the two-way analysis of variance, using gender and drug as factors. A significant interaction was observed between gender and drug, demonstrating that the difference of gender results in altered response to UDCA. Bars with different letters are significantly different, P < 0.05.
UDCA changes plasma or liver parameters and is not hepatotoxic
To characterize the impact of UDCA on metabolic parameters, we examined the liver and plasma TG and cholesterol levels (Fig. 2). Plasma TG and total cholesterol levels were both significantly 1.9-fold higher in male vs female Control mice (Fig. 2A). However, neither parameter was altered by UDCA treatment, except for the increased plasma TG in male UDCA vs Control mice.
Fig. 2. Ursodeoxycholic acid changes plasma and liver parameters and is not hepatotoxic. (A) Plasma ALT, AST, and lipid profiles (B) Liver triglyceride (TG) and cholesterol. Results are expressed as the mean±SEM. Statistical analysis was performed by the two-way analysis of variance, using gender and drug as factors. No significant differences among groups were observed for ALT and AST. A statistical difference due to gender (but not drug) was evident for total cholesterol and LDL cholesterol. For the remaining parameters, a significant interaction was observed between gender and drug, demonstrating that the difference of gender results in altered response to UDCA. Bars with different letters are significantly different, P < 0.05. (C) Hematoxylin and eosin staining of liver sections show reduced hepatic adiposity after UDCA treatment in both male and female mice. n = 3 mice/group for males, and 6 mice/group for females.
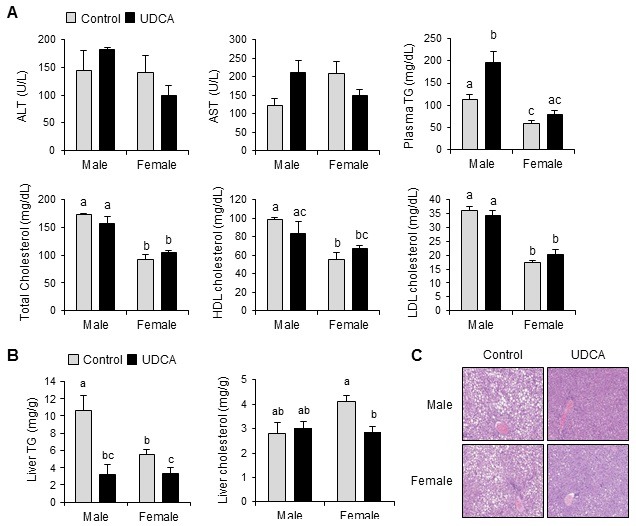
Liver TG levels were also significantly 1.9-fold higher in male vs female Control mice (Fig. 2B). UDCA feeding significantly decreased the liver TG by ≈31% and ≈61% in male and female mice, respectively, resulting in similar final absolute levels. UDCA also decreased the hepatic total cholesterol levels by ≈30% in female mice, although no decreased hepatic cholesterol levels were observed in male mice (Fig. 2B). In accordance with these results, H&E staining revealed decreased lipid droplets in liver sections from UDCA vs Control mice of both genders (Fig. 2C). We examined the plasma ALT and AST levels as indicators of liver-injury status (Fig. 2A). Neither ALT nor AST levels were significantly changed by UDCA, in either gender in mice. Taken together, these results indicate that chronic UDCA administration significantly lowers liver lipid accumulation and is not hepatotoxic.
UDCA affects transcription of hepatic genes associated with lipid homeostasis
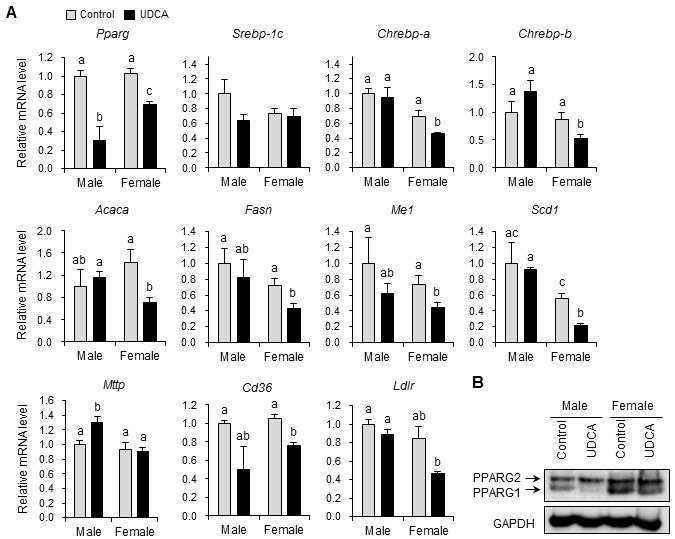
To understand the molecular mechanisms underlying the lipid phenotypes observed in UDCA-fed mice, we analyzed the mRNA expression levels of several genes associated with lipid homeostasis in the liver (Fig. 3). Hepatic lipid homeostasis is tightly regulated by balanced lipid synthesis (lipogenesis), catabolism (fatty acid oxidation), and secretion. The transcription factors responsible for lipid synthesis include PPARG, sterol regulatory element binding protein 1c (SREBP-1c), and carbohydrate response element binding protein (ChREBP). UDCA significantly reduced Pparg mRNA expression in both male and female mouse livers (Fig. 3A). This was reflected at the protein level, whereby UDCA significantly reduced the liver expression of PPARG-1, but not -2, in both male and female mice (Fig. 3B). UDCA showed gender-specific effects on two transcription factors, Srebp-1c and Chrebp, which are responsible for insulin- and glucose-induced lipogenic gene expression, respectively (15). UDCA tended to repress Srebp1c mRNA expression in male mice, and significantly reduced expression of the ChREBP isoforms Chrebp-a and Chrebp-b in female mice (Fig. 3A). Expression of primary lipogenic enzyme targets of these transcription factors, including acetyl-CoA carboxylase alpha (Acaca), fatty acid synthase (Fasn), malic enzyme 1, NADP(+)-dependent, cytosolic (Me1), and stearoyl-Coenzye A desaturase 1 (Scd1), was significantly decreased by UDCA-feeding in female, but not male, mice (Fig. 3A). The mRNA expression of genes involved in fatty acid oxidation, including PPAR alpha (Ppara) and PPAR beta/delta (Pparb/d), and their target genes such as carnitine palmitoyltransferase 1a (Cpt1a), Cpt1b, and Cpt2, were not significantly altered by UDCA in mice of either gender (not shown). Next, we determined the expression of genes involved in lipid secretion and uptake. Gene expression of the microsomal triglyceride transfer protein (Mttp), a key regulator in the assembly and secretion of chylomicrons and very low-density lipoprotein in the intestine and liver, was increased in the livers of UDCA-fed male mice, but not in the livers of female mice (Fig. 3A). This result correlated with increased plasma TG levels observed in UCDA-fed male, but not female mouse livers (Fig. 2A). The mRNA levels of the free fatty acid uptake transporter CD36 molecule (Cd36), known to be associated with hepatic steatosis, and low-density lipoprotein receptor (Ldlr), were decreased in the livers of UDCA-fed female, but not male, mice (Fig. 3A).
Fig. 3. Ursodeoxycholic acid modulates lipid homeostasis gene expression in the liver. (A) Hepatic mRNA levels of genes involved in lipogenesis and lipid uptake were measured by quantitative real-time PCR. All mRNA levels were normalized to large ribosomal protein Rplp0 expression. n = 3 mice/group for males, and 6 mice/group for females. Results are expressed as the mean ± SEM. Statistical analysis was performed by the two-way analysis of variance, using gender and drug as factors. No significant differences among groups were observed for Srebp-1c. For the remaining genes, a significant interaction was observed between gender and drug, demonstrating that the difference of gender results in altered response to UDCA. Bars with different letters are significantly different, P < 0.05. (B) Protein levels of hepatic peroxisome proliferator-activated receptor gamma (PPARG) were measured by western blotting.
UDCA downregulates inflammatory gene expression in liver and adipose tissues
We examined whether UDCA positively influences age-related inflammatory status, because bile acids reportedly have profound anti-inflammatory properties against liver injury induced by a high-fat diet (16). We examined inflammatory and anti-inflammatory cytokine expression in liver and adipose tissues. Hepatic expression of the key inflammatory cytokine, tumor necrosis factor-alpha (Tnfa) was significantly decreased in male mice by UDCA (Fig. 4A). Although not statistically significant, UDCA decreased Tnfa mRNA levels in female mouse livers to ≈48% of the Control values. Chemokine (C-C motif) ligand 2 (Ccl2) was significantly decreased in both genders by UDCA. However, anti-inflammatory cytokine Cd163 expression was significantly increased by UDCA in female, but not male, mouse livers.
Fig. 4. Ursodeoxycholic acid downregulates inflammatory gene expression in liver and adipose tissues. mRNA levels of inflammatory genes from (A) liver and (B) white adipose tissues were measured by quantitative real-time PCR. All mRNA levels were normalized to large ribosomal protein Rplp0 expression. n = 3 mice/group for males, and 6 mice/group for females. Results are expressed as the mean ± SEM. Statistical analysis was performed by two-way analysis of variance using gender and drug as factors. A statistical difference due to drug (but not gender) was evident for Ccl2 in liver. For the remaining genes, a significant interaction was observed between gender and drug, demonstrating that the difference of gender results in altered response to UDCA. Bars with different letters are significantly different, P < 0.05.
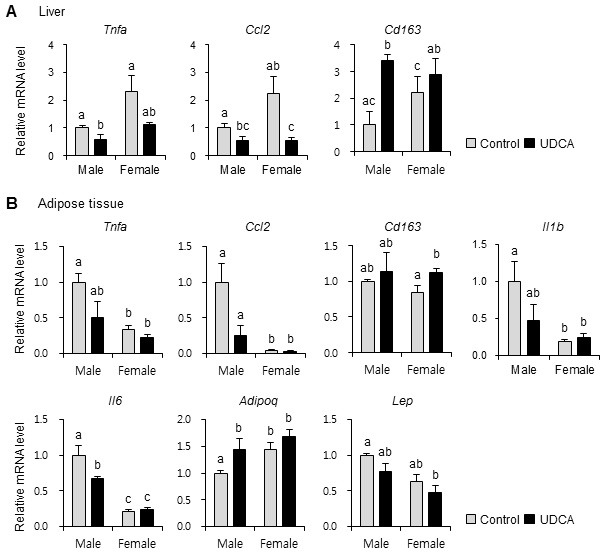
In adipose tissue, UDCA administration tended to decrease mRNA levels of Tnfa and Ccl2 in both genders. Cd163 expression was significantly increased by UDCA administration in the adipose tissues of female, but not male, mice (Fig. 4B). Inflammatory cytokine levels, such as interleukin-1-beta (Il1b) and interleukin 6 (Il6), were not changed by UDCA in female mouse adipose tissues. In male mouse adipose tissues, however, Il1b tended to decrease, and Il6 was significantly decreased by UDCA. Adiponectin is an adipokine with well-established anti-atherogenic, anti-inflammatory, and insulin-sensitizing properties (17). Adiponectin (Adipoq) gene expression was increased in UDCA-fed male, but not female, mouse adi pose tissue (Fig. 4B). Leptin (Lep) mRNA tended to be decreased by UDCA in adipose tissue of both genders, although this change did not achieve statistical significance.
DISCUSSION
UDCA is effective in treating cholestasis and chronic liver diseases. High-dose UDCA effectively improves ALT, serum fibrosis markers, and other relevant metabolic parameters (18, 19). Animal models of nonalcoholic fatty liver disease have shown that UDCA ameliorates insulin sensitivity, and liver steatosis and inflammation (16, 20). Most previous studies on the potential beneficial effects of UDCA on glucose and lipid metabolism were performed in metabolic disease models of obesity and fatty liver (12, 21, 22). Therefore, we undertook to evaluate the chronic effects of UDCA on normal physiology in both male and female mice.
UDCA was fed to adult (20-week-old) mice until 45 weeks of age (old adult), to model human adults who self-administer oral UDCA as a health supplement. The present study demonstrates that chronic UDCA administration protects against agerelated weight gain, adiposity, and inflammation. Protection against hepatic fat accumulation in UDCA-fed mice was associated with significant downregulation of genes involved in hepatic lipid synthesis (PPARG, Chrebp-a/-b, Acaca, Fasn, Me1, and Scd1) and lipid uptake (Cd36 and Ldlr). Moreover chronic UDCA administration decreased inflammatory cytokines such as Tnfa and Ccl2, while increasing anti-inflammatory cytokine Cd163 expression in the liver and white adipose tissue. Interesting findings from the present study show gender-specific regulation of lipogenesis and inflammation by UDCA in old-adult mice.
As previously shown (23), plasma TG and cholesterol levels were higher in male as compared to female mice. Although a UDCA-phospholipid conjugate reportedly reduces elevated serum TG and cholesterol in the nonalcoholic fatty liver and steatohepatitis disease models, unmodified UDCA had no effect on serum TG and cholesterol (16). In accordance with those results, we failed to detect an inhibitory effect of UDCA on plasma TG and cholesterol in both genders; rather, male mice conversely showed slightly increased plasma TG levels after UDCA administration. Therefore, different UDCA formulations should be evaluated for better therapeutic efficacy in improving plasma lipid profiles.
UDCA significantly reduced hepatic TG and cholesterol content in a gender-dependent manner. Hepatic TG levels were decreased by UDCA in both genders, but hepatic cholesterol levels were only decreased in female mice. Of note, the basal hepatic TG levels were higher in male vs female mice, whereas basal hepatic cholesterol levels were higher in females. Interestingly, plasma insulin and glucose levels were decreased by UDCA, suggesting improved insulin sensitivity (data not shown). An inverse relationship between hepatic TG and insulin sensitivity has been reported (24); therefore, it is reasonable to suggest that UDCA improved hepatic insulin resistance by decreasing levels of hepatic lipids. However, further experiments to specifically define UDCA effects on age-related insulin resistance are needed.
Consistent with decreased hepatic TG and cholesterol, a major effect of UDCA on lipid metabolism was the decreasing hepatic lipid synthesis and lipid uptake in a gender-specific manner. In female, but not male, mice, decreased hepatic TG was associated with reduced expression of important lipogenesis genes, such as Chrebp, Acaca, Fasn, and Scd1. In male mice, lipogenesis-related gene expression was unchanged, but genes involved in hepatic lipid uptake and secretion were changed by UDCA. Therefore, the effects of UDCA on lipid metabolism are gender-dependent, and associated with reduced hepatic TG. The effects of UDCA on fatty acid oxidation remain controversial; fatty acid oxidation was increased by UDCA in obese Zucker rats, but remained unchanged in lean rats and high-fat diet-fed mice (20, 25). In our study, the expression of genes involved in fatty acid oxidation remained unchanged by UDCA (data not shown). Therefore, we speculate that UDCA decreases the hepatic TG by inhibiting two pathways: ChREBP-medicated lipogenesis and PPARG-mediated lipid uptake/secretion.
Chronic inflammation is a pathological characteristic of aging, and increased inflammatory cytokines, such as IL-6, IL-1β, and TNFα, are linked to many age-related disorders (26). Chronic inflammation is also associated with insulin resistance (27). Production of inflammatory cytokines is stimulated by activated NFᴋB and JNK pathways. Phosphorylated NFᴋB and IᴋBα levels were not changed in livers of mice of either gender by UDCA, but hepatic phospho-JNK was decreased in UDCAfed mice of both genders (data not shown). This observation suggests that chronic UDCA administration suppresses age-related inflammatory responses.
In summary, this study provides evidence that chronic UDCA administration can improve age-related obesity and insulin resistance. The underlying molecular mechanisms for these effects may include reduced hepatic lipogenesis and decreased inflammatory responses. These findings support the beneficial effects of UDCA in preventing and treating age-related chronic metabolic diseases, such as insulin resistance and type-2 diabetes.
MATERIALS AND METHODS
Animals
Animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Lee Gil Ya Cancer and Diabetes Institute, Gachon University. Male and female C57BL/6 mice (12-weeks-old) were purchased from Orientbio (Gyeonggi Province, Korea). Mice were maintained on a normal diet (PicoLabⓇ Rodent Diet 20, Orientbio) under a 12-h light/dark cycle until age 20 weeks, and then were fed either a control formulation (D12450B; Research Diets, Inc., New Brunswick, NJ), or one supplemented with 0.3% (w/w) UDCA (Sigma-Aldrich, St. Louis, MO) for 25 weeks, until sacrifice. Body composition was assessed by 1H-magnetic resonance spectroscopy (BioSpin, Bruker, Billerica, MA). After control or UDCA feeding for 21 weeks, mice were housed individually in metabolic cages for a 4-day period, and food and water intake was measured during the last 2-day period. At the 25-week experimental endpoint, mice were euthanized at the beginning of the light cycle and weighed. Blood samples were collected via the hepatic portal vein, and plasma was obtained by centrifugation at 300 × g for 15 min at 4℃. Livers and gonadal fats were removed, weighed, and either snap-frozen in liquid nitrogen, or were formalin-fixed. Liver and plasma samples were stored at −80℃ until RNA isolation or biochemical analysis.
Biochemical analysis
Plasma ALT, AST, TG, and total cholesterol levels were determined by automated analysis (Model AU-480; Olympus, Tokyo, Japan). Liver TG and cholesterol profiles were determined using a Cleantech TG-S kit (Asan Pharmacy, Inc., Seoul, Korea) and Cholesterol/Cholesteryl Ester Quantitation Kit (BioVision, Inc., Milpitas, CA), respectively.
Histological staining
Freshly-isolated liver and gonadal fat samples were fixed in neutral-buffered formalin and embedded in paraffin blocks; sections were stained with hematoxylin and eosin (H&E).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from mouse liver and gonadal fat using RNAiso Plus (Takara, Shiga, Japan). Purified total RNA was treated with RNase-free DNase (Roche, Penzberg, Germany), and reverse-transcribed using a QuantiTectⓇ Reverse Transcription Kit (Qiagen, Hilden, Germany). Gene-specific primers (Supplementary Table S1) were designed using Primer Express Software (PerkinElmer Life Sciences, Waltham, MA), and validated by analysis of template titration and dissociation curves. Quantitative gene expression analyses were performed on a 7900HT Fast Real-Time PCR System (Life Technologies, Carlsbad, CA) using SYBRⓇ Premix Ex TaqTM II, ROX Plus (Takara). Expression levels were calculatd by the comparative Cτ method using ribosomal protein, large, P0 (Rplp0) as the invariant control.
Immunoblot analysis
Total protein was isolated from mouse liver using PRO-PREP Protein Extraction Solution (Intron Biotechnology, Daejeon, Korea) supplemented with phosphatase inhibitors. Immunoblot analysis used the following antibodies: anti-peroxisome proliferator-activated receptor gamma (PPARG) was from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was from Millipore Inc., (Billerica, MA).
Statistical analysis
Data were expressed as the mean ± standard error of the mean (SEM). Statistical analysis was performed using SPSS (v.17.0; SPSS/IBM Inc., Chicago, IL). When we compared treatment group to control, we used the Mann-Whitney U test. In that case, we used * or ** to show statistical difference (*, P < 0.05; **, P < 0.01). However, when we compared two factors, gender and drug, we used the Kruskal-Wallis test. If a significant interaction was evident, group comparisons with the Mann-Whitney U test were performed. Different letters denote groups that are significantly different (P < 0.05).
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2012R1A1A3018738), and by Gachon University (GCU-2012-5167), both awarded to J-Y Cha.
References
- 1.Arlia-Ciommo A, Piano A, Svistkova V, Mohtashami S, Titorenko VI. Mechanisms underlying the anti-aging and anti-tumor effects of lithocholic bile acid. Int J Mol Sci. (2014);15:16522–16543. doi: 10.3390/ijms150916522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. (2009);50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amador-Noguez D, Yagi K, Venable S, Darlington G. Gene expression profile of long-lived Ames dwarf mice and Little mice. Aging Cell. (2004);3:423–441. doi: 10.1111/j.1474-9728.2004.00125.x. [DOI] [PubMed] [Google Scholar]
- 4.Amador-Noguez D, Dean A, Huang W, Setchell K, Moore D, Darlington G. Alterations in xenobiotic metabolism in the long-lived Little mice. Aging Cell. (2007);6:453–470. doi: 10.1111/j.1474-9726.2007.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poupon R, Chretien Y, Poupon RE, Ballet F, Calmus Y, Darnis F. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. (1987);1:834–836. doi: 10.1016/S0140-6736(87)91610-2. [DOI] [PubMed] [Google Scholar]
- 6.Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid 'mechanisms of action and clinical use in hepatobiliary disorders'. J Hepatol. (2001);35:134–146. doi: 10.1016/S0168-8278(01)00092-7. [DOI] [PubMed] [Google Scholar]
- 7.Zollner G, Fickert P, Fuchsbichler A, et al. Role of nuclear bile acid receptor, FXR, in adaptive ABC transporter regulation by cholic and ursodeoxycholic acid in mouse liver, kidney and intestine. J Hepatol. (2003);39:480–488. doi: 10.1016/S0168-8278(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 8.Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. (1998);339:1217–1227. doi: 10.1056/NEJM199810223391707. [DOI] [PubMed] [Google Scholar]
- 9.Bellentani S. Immunomodulating and anti-apoptotic action of ursodeoxycholic acid: where are we and where should we go? Eur J Gastroenterol Hepatol. (2005);17:137–140. doi: 10.1097/00042737-200502000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acidinduced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. (1998);4:165–178. doi: 10.1007/s0089480040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lukivskaya O, Patsenker E, Buko VU. Protective effect of ursodeoxycholic acid on liver mitochondrial function in rats with alloxan-induced diabetes: link with oxidative stress. Life Sci. (2007);80:2397–2402. doi: 10.1016/j.lfs.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 12.Quintero P, Pizarro M, Solis N, et al. Bile acid supplementation improves established liver steatosis in obese mice independently of glucagon-like peptide-1 secretion. J Physiol Biochem. (2014);70:667–674. doi: 10.1007/s13105-014-0336-1. [DOI] [PubMed] [Google Scholar]
- 13.Nie B, Park HM, Kazantzis M, et al. Specific bile acids inhibit hepatic fatty acid uptake in mice. Hepatology. (2012);56:1300–1310. doi: 10.1002/hep.25797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castro RE, Ferreira DM, Afonso MB, et al. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human nonalcoholic fatty liver disease. J Hepatol. (2013);58:119–125. doi: 10.1016/j.jhep.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. (2008);118:829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pathil A, Mueller J, Warth A, Chamulitrat W, Stremmel W. Ursodeoxycholyl lysophosphatidylethanolamide improves steatosis and inflammation in murine models of nonalcoholic fatty liver disease. Hepatology. (2012);55:1369–1378. doi: 10.1002/hep.25531. [DOI] [PubMed] [Google Scholar]
- 17.Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol Metab. (2013);2:133–141. doi: 10.1016/j.molmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van de Meeberg PC, Houwen RH, Sinaasappel M, Heijerman HG, Bijleveld CM, Vanberge-Henegouwen GP. Low-dose versus high-dose ursodeoxycholic acid in cystic fibrosis-related cholestatic liver disease. Results of a randomized study with 1-year follow-up. Scand J Gastroenterol. (1997);32:369–373. doi: 10.3109/00365529709007686. [DOI] [PubMed] [Google Scholar]
- 19.Roda E, Azzaroli F, Nigro G, et al. Improved liver tests and greater biliary enrichment with high dose ursodeoxycholic acid in early stage primary biliary cirrhosis. Dig Liver Dis. (2002);34:523–527. doi: 10.1016/S1590-8658(02)80112-8. [DOI] [PubMed] [Google Scholar]
- 20.Tsuchida T, Shiraishi M, Ohta T, Sakai K, Ishii S. Ursodeoxycholic acid improves insulin sensitivity and hepatic steatosis by inducing the excretion of hepatic lipids in high-fat diet-fed KK-Ay mice. Metabolism. (2012);61:944–953. doi: 10.1016/j.metabol.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 21.Lee JM, Gang GT, Kim DK, et al. Ursodeoxycholic acid inhibits liver X receptor alpha-mediated hepatic lipogenesis via induction of the nuclear corepressor SMILE. J Biol Chem. (2014);289:1079–1091. doi: 10.1074/jbc.M113.491522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mueller M, Thorell A, Claudel T, et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol. (2015);62:1398–1404. doi: 10.1016/j.jhep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Link JC, Chen X, Prien C, et al. Increased High-Density Lipoprotein Cholesterol Levels in Mice With XX Versus XY Sex Chromosomes. Arterioscler Thromb Vasc Biol. (2015);35:1778–1786. doi: 10.1161/ATVBAHA.115.305460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voshol PJ, Haemmerle G, Ouwens DM, et al. Increased hepatic insulin sensitivity together with decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology. (2003);144:3456–3462. doi: 10.1210/en.2002-0036. [DOI] [PubMed] [Google Scholar]
- 25.Nunes PM, Jones JG, Rolo AP, Palmeira CM, Carvalho RA. Ursodeoxycholic acid treatment of hepatic steatosis: a (13)C NMR metabolic study. NMR Biomed. (2011);24:1145–1158. doi: 10.1002/nbm.1672. [DOI] [PubMed] [Google Scholar]
- 26.Chung HY, Cesari M, Anton S, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. (2009);8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. (2006);116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]