

Abstract
Mounting an immune response sufficient to eradicate a tumor is the goal of modern immunotherapy. Single agent therapies with checkpoint inhibitors or costimulatory molecule agonists are effective only for a small portion of all treated patients. Combined therapy, e.g., CTLA-4 and PD-1 checkpoint blockade, is a more effective treatment modality, but in preclinical studies OX40 agonism with CTLA-4 blockade using monoclonal antibodies (aOX40/aCTLA-4) failed to induce tumor regression of larger, more established tumors. We hypothesized that administration of a vaccine with a tumor-associated antigen targeted to the appropriate antigen presenting cell could make combined aOX40/aCTLA-4 therapy more effective. We administered an antibody-based vaccine targeting HER2 to the DEC-205 endocytic receptor on cross-presenting dendritic cells (anti-DEC-205/HER2; aDEC-205/HER2) and a potent adjuvant (poly (I:C)) to assist with maturation, along with aOX40/aCTLA-4 therapy. This therapy induced complete regression of established tumors and a pronounced infiltration of effector CD8 and CD4 T cells, with no effect on regulatory T cell infiltration compared to aOX40/aCTLA-4 alone. To be maximally effective, this therapy required expression of both OX40 and CTLA-4 on CD8 T cells. These data indicate that vaccination targeting cross-presenting dendritic cells with a tumor-associated antigen is a highly effective immunization strategy that can overcome some of the limitations of current systemic immunotherapeutic approaches that lack defined tumor-directed antigenic targets.
Keywords: Immunotherapy, Cytotoxic CD8 T cell, OX40, CTLA-4, Checkpoint blockade, Co-stimulation, Dendritic cell, Vaccine, Anergy, Tolerance
Background
Immunotherapy is quickly garnering attention and enthusiasm as some patients with metastatic disease have achieved long-term remission. However, combinations of immunotherapies and/or targeted therapies will be needed to achieve complete tumor regression for a larger portion of patients. Our lab has been investigating the efficacy of OX40 agonism in combination with CTLA-4 blockade. OX40 is costimulatory molecule expressed by both CD4 and CD8 T cells following T cell receptor (TCR) ligation [1]. Preclinical data demonstrate that treatment with agonist anti-OX40 monoclonal antibodies (aOX40) induced tumor regression by boosting effector CD8 and CD4 T cell expansion and function [2–6]. Another successful approach is the blockade of a co-inhibitory molecule, CTLA-4, which limits an active immune response. Our previous research has demonstrated that combination aOX40/aCTLA-4 therapy significantly improved survival in preclinical models [7]. Surprisingly, this therapy also induced a profound Th2 bias in CD4 T cells. It is known that TCR-mediated recognition of low-affinity antigens can promote a Th2 bias, which limits an effective antitumor immune response, and that promoting a Th1 bias results in more favorable outcomes for patients [8–13]. In order to circumvent a Th2 bias and promote a more robust Th1 response, we opted to augment a CD8 T cell response directly via DEC205 expressing cross-presenting dendritic cells (DCs) [14]. It was previously demonstrated that mice defective in cross-presentation have impaired tumor rejection and that in cancer, DC function is frequently impaired [15, 16]. We hypothesized that vaccination targeting a tumor-associated antigen toward cross-presenting dendritic cells (aDEC-205/HER2 with poly (I: C)) combined with aOX40/aCTLA-4 immunotherapy would promote a robust effector CD8 T cell response capable of clearing established tumors.
Main text
To elaborate on our previous studies, we tested the effect of combination aOX40/aCTLA-4 therapy on antigen-specific T cell expansion and the kinetics of this response. Combination aOX40/aCTLA-4 therapy significantly increased the frequency, function, and persistence of antigen-specific CD8 T cells in the periphery over time. To determine whether this was a direct or indirect effect on CD8 T cells, we used OX40-deficient and human CTLA-4 knock-in transgenic mice. OX40-/- OT-I cells had a significantly reduced ability to proliferate, differentiate into effector cells, and produce inflammatory cytokines following combination therapy, indicating the requirement for OX40. To determine whether CTLA-4 expression on CD8 T cells was required for the efficacy of combination therapy, we used transgenic mice in which the extracellular portion of the mouse CTLA-4 receptor is swapped with the human version (huCTLA-4 mice), rendering them unresponsive to anti-mouse CTLA-4 antagonism [17]. Surprisingly, CTLA-4 expression on CD8 T cells was required to induce maximal expansion and function of this population following combined aOX40/aCTLA-4 treatment. Furthermore, CD4 T cells were required to induce a potent CD8 T cell response. A key observation we made in our previous study was that aOX40/aCTLA-4 therapy was not sufficient to improve survival of mice with larger, more established tumors. Notably, when aDEC-205/HER2 vaccination was combined with aOX40/aCTLA-4, we observed regression of established tumors (100-150 mm2). This corresponded with a significant increase in inflammatory cytokine and chemokine production by CD4 and CD8 T cells, and a notable decrease in Th2 cytokines from CD4 T cells, which we had observed previously. The triple combination induced profound CD8 and CD4 effector T cell infiltration in the tumor. It is known that T cell anergy is a major obstacle to effective antitumor immunity. To investigate whether this triple combination could overcome T cell anergy, we combined a mouse model of anergy using POET-1 (probasin ovalbumin expressing transgenic-1) combined with a spontaneous prostate cancer model – TRAMP (transgenic adenocarcinoma of the mouse prostate) transgenic mice [18, 19]. POET-1 mice express membrane-bound ovalbumin (mOVA) in the prostate driven by the rat probasin promoter. Thus, POET-1 x TRAMP (TRAMP-OVA) mice express mOVA as a self/tumor-associated antigen that renders ovalbumin-specific CD8 T cells anergic. Combined aOX40/aCTLA-4 therapy with aDEC-205/OVA vaccination rescued anergic tumor-specific CD8 T cells and significantly improved their activation, proliferation, and cytokine production (Fig. 1).
Fig. 1.
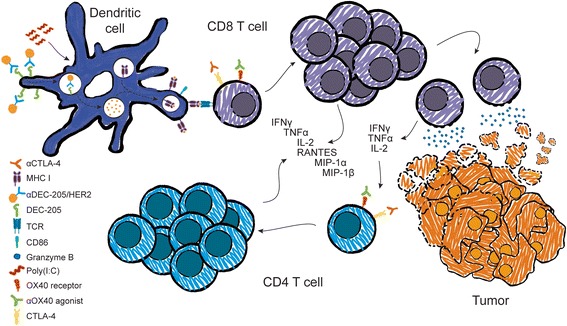
Vaccination using aDEC-205/HER2 combined with adjuvant poly (I:C) induces dendritic cell maturation and costimulatory molecule expression, thereby resulting in more efficient antigen presentation to CD8 T cells. Activation of CD8 T cells via the T cell receptor (TCR) and OX40 using an OX40 agonist induces robust CTL activation, while CTLA-4 blockade releases the brakes on the activated CTL. Effector CD8 T cells can now traffic into the tumor, where they accumulate and induce cancer cell death using cytolytic granule proteins. T cell activation and cancer cell death leads to increased cytokine (IFN-γ, TNF-α, IL-2) and chemokine (CCL3/MIP-1α; CCL4/MIP-1β; CCL5/RANTES) production leading to additional recruitment of effector T cells. OX40 agonism and CTLA-4 blockade also lead to CD4 T cell activation and expansion. Together, this robust T cell response results in tumor eradication and improved long-term survival
Conclusions
Our recent studies suggest that finding appropriate vaccination methods to combine with checkpoint inhibitors (e.g., aCTLA-4) and costimulatory molecule agonism (e.g., aOX40) will be more effective at reducing tumor burden and improving survival than any single agent. In particular, the use of aOX40/aCTLA-4 alone was insufficient to eliminate larger, more established tumors, which may be due to increased Th2-associated cytokines or tumor-induced anergy [7]. One possible explanation for the reduced efficacy of combination therapy in the absence of vaccination is because it relies on TCR-mediated recognition of endogenous antigens. Due to mechanisms of central and peripheral tolerance the majority of these T cells are likely to be of low affinity for their respective tumor-associated antigens. In the absence of competition from T cells with higher affinity or an abundance of antigen, a Th2 response predominates [20, 21]. By administering both an adjuvant to promote DC maturation and a tumor-associated antigen targeted to an endocytic receptor present on DCs, we were able to very effectively prime an antitumor cytotoxic T lymphocyte (CTL) response. CTL activation through the TCR is known to induce expression of both OX40 and CTLA-4 receptors, thus providing targets for aOX40/aCTLA-4 therapy. This triple combination—using OX40 agonism to step on the gas, CTLA-4 blockade to release the brakes, and vaccination using aDEC-205/HER2 to steer the immune response in the right direction—was able to generate profound CTL infiltration into the tumor leading to tumor regression (Fig. 1). One possible explanation for the observed increase in Th1 polarization and concomitant reduction in Th2 cytokine production following triple combination therapy is that CTL-mediated cancer cell death will release an abundance of antigens, including those derived from over-expressed and/or mutated self-proteins. CD4 T cell recognition of these epitopes on mature antigen presenting cells expressing the appropriate costimulatory molecules would favor a Th1-polarized response. These and previous data also suggest an effect on CD8 T cell differentiation as a possible mechanism for the increased efficacy of the therapy. Our lab is currently investigating the molecular mechanisms underlying this process. Currently, there are multiple clinical trials testing various combinations of immune-based therapeutic modalities, including checkpoint inhibitors, targeted therapy with small molecule inhibitors, adoptive cell therapy, and standard of care chemotherapy or radiation. Dual treatment with CTLA-4 and PD-1 blockade (ipilimumab and nivolumab, respectively) was recently approved, and while it improves the overall response rate, the majority of patients succumb to their disease. The incidence of Grade 3-4 toxicities also increases with dual therapy, which is not surprising given the importance of these two molecules in preventing rampant autoimmunity. Perhaps combining a method of vaccination with a checkpoint inhibitor and a costimulatory molecule agonist, such as monoclonal antibodies activating OX40, 4-1BB, or GITR, will provide greater efficacy for patients, as it may more easily direct the immune response in the direction desired—away from normal self-antigens and toward a tumor-associated antigen. In the growing age of bioinformatics and personalized medicine, it seems that personalized vaccination is becoming a more feasible possibility for patients. Combining vaccination using a patient’s own tumor-associated neoepitopes with checkpoint inhibition and/or costimulatory molecule agonism will likely promote a more directed T cell response and may benefit a majority of patients, even with a minimal baseline presence of T cells. In fact, it is in this scenario where the efficacy of OX40 agonists may truly shine.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of supporting data
Not applicable.
Acknowledgements
The authors would like to thank Diego Barragan-Echenique for his assistance in designing the figure.
Funding
This work was supported by grants from the NIH 1R21CA190790 (W.L.R.), Susan G. Komen CCR15329664 (W.L.R.), and the American Cancer Society 2014 Roaring Fork Valley Postdoctoral Fellowship PF1424901LIB (S.N.L.).
Abbreviations
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- TCR
T cell receptor
- TIL
tumor infiltrating lymphocyte
Footnotes
Competing interests
W.L.R has received commercial research grants, consulting fees, and/or royalties from Bristol-Myers Squibb, Merck, Galectin Therapeutics, and Nektar Therapeutics. S.N.L has received royalties from Galectin Therapeutics. No non-financial competing interests exist for any of the authors.
Authors’ contributions
SNL and WLR drafted the manuscript and edited the final version. Both authors reviewed and approved the final manuscript.
Authors’ information
Not applicable.
Contributor Information
Stefanie N. Linch, Email: Stefanie.linch@providence.org
William L. Redmond, Phone: +503-215-3841, FAX: +503-215-6841, Email: William.redmond@providence.org
References
- 1.Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29(3):187–201. doi: 10.1615/CritRevImmunol.v29.i3.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gough MJ, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33(8):798–809. doi: 10.1097/CJI.0b013e3181ee7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gough MJ, et al. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68(13):5206–5215. doi: 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 4.Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol. 2007;179(11):7244–7253. doi: 10.4049/jimmunol.179.11.7244. [DOI] [PubMed] [Google Scholar]
- 5.Redmond WL, Gough MJ, Weinberg AD. Ligation of the OX40 co-stimulatory receptor reverses self-Ag and tumor-induced CD8 T-cell anergy in vivo. Eur J Immunol. 2009;39(8):2184–2194. doi: 10.1002/eji.200939348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti-OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. Eur J Immunol. 2007;37(1):157–166. doi: 10.1002/eji.200636428. [DOI] [PubMed] [Google Scholar]
- 7.Redmond WL, Linch SL, Kasiewicz MJ. Combined Targeting of Costimulatory (OX40) and Coinhibitory (CTLA-4) Pathways Elicits Potent Effector T Cells Capable of Driving Robust Antitumor Immunity. Cancer Immunol Res. 2014;2(2):142–153. doi: 10.1158/2326-6066.CIR-13-0031-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeiffer C, et al. Altered peptide ligands can control CD4 T lymphocyte differentiation in vivo. J Exp Med. 1995;181(4):1569–1574. doi: 10.1084/jem.181.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao X, Grant C, Constant S, Bottomly K. Induction of IL-4-producing CD4+ T cells by antigenic peptides altered for TCR binding. J Immunol. 1997;158(9):4237–4244. [PubMed] [Google Scholar]
- 10.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 11.Tatsumi T, et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4 (+) T cell responses against MAGE-6 in HLA-DRB10401 (+) patients with renal cell carcinoma or melanoma. J Exp Med. 2002;196(5):619–628. doi: 10.1084/jem.20012142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shiao SL, et al. TH2-Polarized CD4+ T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol Res. 2015;3(5):518–25. doi: 10.1158/2326-6066.CIR-14-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ochi A, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209(9):1671–1687. doi: 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuji T, et al. Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol. 2011;186(2):1218–1227. doi: 10.4049/jimmunol.1000808. [DOI] [PubMed] [Google Scholar]
- 15.Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinzon-Charry A, Maxwell T, Lopez JA. Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol Cell Biol. 2005;83(5):451–461. doi: 10.1111/j.1440-1711.2005.01371.x. [DOI] [PubMed] [Google Scholar]
- 17.Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lees JR, et al. Deletion is neither sufficient nor necessary for the induction of peripheral tolerance in mature CD8+ T cells. Immunology. 2006;117(2):248–261. doi: 10.1111/j.1365-2567.2005.02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurwitz AA, Foster BA, Allison JP, Greenberg NM, Kwon ED, et al. The TRAMP mouse as a model for prostate cancer. In: Coligan JE, et al., editors. Current protocols in immunology. 2001. [DOI] [PubMed] [Google Scholar]
- 20.Milner JD, Fazilleau N, McHeyzer-Williams M, Paul W. Cutting edge: lack of high affinity competition for peptide in polyclonal CD4+ responses unmasks IL-4 production. J Immunol. 2010;184(12):6569–6573. doi: 10.4049/jimmunol.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4 (+) T cell polarization In Vivo. Immunity. 2014;41(1):63–74. doi: 10.1016/j.immuni.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]