

Abstract
A nucleosomal protein, high mobility group box 1 (HMGB1) is known to be a late mediator of sepsis. Dabrafenib is a B-Raf inhibitor and initially used for the treatment of metastatic melanoma therapy. Inhibition of HMGB1 and renewal of vascular integrity is appearing as an engaging therapeutic strategy in the administration of severe sepsis or septic shock. Here, we examined the effects of dabrafenib (DAB) on the modulation of HMGB1-mediated septic responses. DAB inhibited the release of HMGB1 and downregulated HMGB1-dependent inflammatory responses by enhancing the expressions of cell adhesion molecules (CAMs) in human endothelial cells. In addition, treatment with DAB inhibited the HMGB1 secretion by CLP and sepsis-related mortality and pulmonary injury. This study demonstrated that DAB could be alternative therapeutic options for sepsis or septic shock via the inhibition of the HMGB1 signaling pathway. [BMB Reports 2016; 49(4): 214-219]
Keywords: Barrier integrity, Dabrafenib, HMGB1, Sepsis
INTRODUCTION
Sepsis is systematic inflammatory response syndrome caused by infection and can lead to shock, multiple organ failure, and death if not treated promptly (1). Sepsis is the major causes of death in intensive care units (ICUs) and the tenth leading cause of death overall in high-income countries (1), despite advances in antibiotic therapy and intensive care (2, 3). Recombinant activated protein C (APC, Xigris, Eli Lilly) was licensed in 2001 by the Food and Drug Administration (FDA) and in 2002 by the European Medicine Agency for the treatment of severe sepsis and septic shock (4). In October 2011, however, the only FDA-approved drug for severe sepsis, APC was withdrawn from the market because of side effects and PROWESS and septic shock trials showed its inability to enhance survival rate on 28-day mortality (5). Therefore, we have no drug for sepsis or septic shock, currently. Therefore, developing more effective therapeutic options for sepsis are needed.
The pathogenesis of sepsis is rather complex, tumor necrosis factor (TNF)-α, interleukin (IL)-1, interferon-γ, and macrophage migration inhibitory factor were upregulated early by endotoxin (6). For instance, TNF-α neutralizing antibodies decreased lethality of mouse endotoxemic shock model (7). In contrast, the rapid kinetics of the TNF-α responses in clinical trial made its targeting difficult (7), which drive us to find another late septic mediator such as high mobility group box 1 protein (HMGB1) as potential therapeutic targets for sepsis. In mouse models of sepsis, HMGB1 is first detectable in the circulating blood 8 h after the onset of the disease, and finally increasing to plateau levels from 16 to 32 h (8). In contrast to TNF-α and other early proinflammatory cytokines, the circulating concentrations of HMGB1, as a late acting mediator of sepsis, proceeds and is similar to the severity of sepsis (8). HMGB-1, an important chromatin protein present in almost all eukaryotic cells, is known to interact with nucleosomes, transcription factors, and histones in the nucleus (9). HMGB1 can be secreted into the extracellular space through a passive process initiated by necrotic cells, thereby allowing the innate immune system to recognize damaged cells (10). The secretion of HMGB1 in the endothelium upregulates the expression of cell adhesion molecules (CAMs), such as vascular cell-adhesion molecule (VCAM), intercellular adhesion molecule (ICAM), and E-selectin, which promote inflammation via the recruitment of leukocytes (11). Enhanced blood concentrations of HMGB1 are quantitative in septic patients upto7 days after a diagnosis of sepsis, and these concentrations are associated with the severity of organ damage (12, 13). Large therapeutic options for clinical intervention for sepsis treatment were provided by HMGB1, as a late mediator of sepsis (14).
In our search for repositioning FDA-approved drugs (total 1,163), 327 drugs were selected which are related to vascular inflammation and infection. Among selected drugs, high contents screening system (PerkinElmer Operetta, Waltham, MA) was used to select the compounds which control HMGB1-mediated vascular barrier disruption, and we found that dabrafenib (DAB) inhibited HMGB1- or CLP-induced septic responses. Dabrafenib (brand name; TafinlarTM) is a kinase inhibitor indicated as a single agent for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test (15). Here, we showed the anti-septic effects of DAB on HMGB1-mediated vascular barrier disruption in vitro and in vivo.
RESULTS AND DISCUSSION
Drug repositioning for development of new drug by using existing FDA-approved drugs has gained significant attention because of the time and cost benefits (16-18). Here we report the results of DAB on the basis of the inhibition of HMGB1-mediated septic activity in vitro and in vivo.
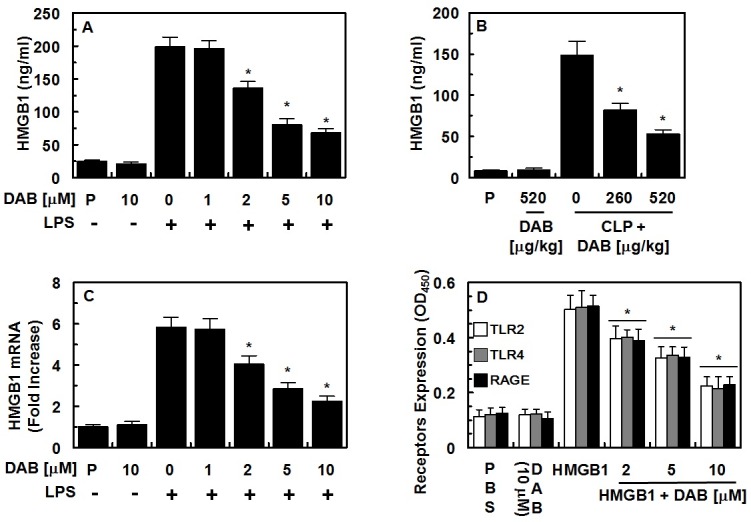
Effects of DAB on LPS and CLP-mediated release of HMGB1
HMGB1 is actively released by immune cells such as monocytes and macrophages in response to LPS, as well as by necrotic cells (11, 19-21). HMGB1 levels rise slowly after 8 h and are corresponded to the severity of sepsis (22). Thus, we tested whether DAB could reduce HMGB1 secretion by LPS in HUVECs. As shown in Fig. 1A, DAB reduced LPS-mediated HMGB1 release by dose-dependent manner, with a maximal effective concentration of 2 μM. In order to confirm the inhibitory effects of DAB on HMGB1 secretion in vivo, DAB was evaluated for its ability to inhibit HMGB1 release using a mouse model of CLP-induced sepsis. Data showed that DAB also inhibited CLP-induced HMGB1 secretion (Fig. 1B). Because the total volume of blood in a mouse is 72 ml/kg (23) and the average weight of used mouse in this study is 27 g, the amount of DAB (260 or 520 μg/kg) injected yielded a maximum concentration of 5, or 10 μM in the peripheral blood. We next determined underlying molecular mechanism of the inhibitory effects of DAB on the secretion of HMGB1 on LPStreated HUVECs. To do this, the effects of DAB on the transcriptional regulation of HMGB1 by LPS in HUVECs were tested. Thus, we determined the effect of DAB on LPS-induced HMGB1 mRNA levels using real time qRT-PCR. Data showed that LPS increased DAB treatment suppressed increased expressions of HMGB1 mRNA by LPS (Fig. 1C). We next assessed the effects of DAB and HMGB1 on the levels of the HMGB1 receptors (TLR2, TLR4, and RAGE) were evaluated. HMGB1 induced the expression of HMGB1 receptors such as TLR2, TLR4, and RAGE in HUVECs, and DAB inhibited the expressions of HMGB1 receptors (Fig. 1D). However, DAB alone did not affect the expressions of HMGB1 receptors. We next tested the effects of DAB on the cell viability. To do this, cell viability assays were applied to probe the toxicity of DAB in HUVECs after 48 h. At the concentrations used (up to 20 μM), DAB did not affect cell viability (data not shown). Collectively, prevention of LPS- or CLP-induced release of HMGB1 by DAB indicates the possible use of DAB in the management of sepsis.
Fig. 1. Effects of DAB on HMGB1 release and expression of HMGB1 receptors. (A) HUVECs were treated with the indicated concentrations of DAB for 6 h, after stimulation with LPS (100 ng/ml, 16 h), and HMGB1 release was measured by ELISA. (B) Male C57BL/6 mice that underwent CLP were administered indicated DAB (intravenously 12 h after CLP, n = 5). Mice were euthanized 24 h after CLP. Serum HMGB1 levels were measured by ELISA. (C) The same as (A) except that real time qRT-PCR analysis was performed using specific primers for HMFB1 and GAPDH, as described in the materials and methods section. (D) Confluent HUVECs were activated with or without HMGB1 (1 μg/ml, 16 h), followed by incubation with DAB for 6 h. Expression of TLR2 (white bar), TLR4 (gray bar) or RAGE (black bar) was determined by cell-based ELISA. The results shown are mean ± SEM from three separate experiments in different days with triplicate wells. P = PBS is the vehicle control. *P < 0.05 versus LPS alone (A, C), CLP alone (B) or HMGB1 alone (D).
Effect of DAB on HMGB1-mediated vascular integrity
It is well known that LPS and HMGB1 could cause the cleavage and disruption of the endothelial barrier (24). Therefore, a permeability assay was applied to test whether DAB could restore LPS- or HMGB1-induced vascular disruptive responses in HUVECS. HUVECs were treated with various concentrations of DAB for 6 h following the addition of LPS (100 ng/ml) or HMGB1 (1 μg/ml). Data showed that DAB dose-dependently inhibited LPS- and HMGB1-mediated membrane disruption (Fig. 2A and 2B). The in vivo effects of DAB on vascular permeability were assessed to corroborate the in vitro results. Fig. 2C shows that DAB induced a marked inhibition of the peritoneal dye leakage, induced by HMGB1. HMGB1 elicits an inflammatory response through diverse pathways, including MyD88/IRAK/TRAF, which leads to NF-κB activation; the Rac1/PI3K pathway; via the activation of ERK 1/2 and p38 MAPK downstream of TLR2/4; and the Ras/p38 pathway downstream of RAGE (25-27). Therefore, DAB could affect the p38 pathway, HUVECs were stimulated by HMGB1 and treated with DAB. HMGB1 upregulated the expression of phosphorylated p38, which was clearly reduced by treatment with DAB (Fig. 2D). In addition, treatment with DAB (5 or 10 μM) inhibited the formation of HMGB1-induced paracellular gaps with the formation of dense F-actin rings (Fig. 2E). However, DAB alone did not affect the formation of dense F-actin rings. The reduction in HMGB1-induced permeability and p38 activation by DAB points towards a promising role as ant-sepsis drugs.
Fig. 2. Effects of DAB on HMGB1-mediated permeability in vitro and in vivo. Effects of treatment with different concentrations of DAB for 6 h on barrier disruption caused by LPS (A, 100 ng/ml, 4 h) or HMGB1 (B, 1 μg/ml, 16 h) were monitored by measuring the flux of Evans blue-bound albumin across HUVECs. (C) The effects of DAB on HMGB1-induced (2 μg/mouse, i.v.) vascular permeability in mice were examined by measuring the amount of Evans blue in peritoneal washings (expressed μg/mouse, n = 5). (D) HUVECs were activated with HMGB1 (1 μg/ml, 16 h), followed by treated with different concentrations of DAB for 6 h. The effects of DAB on HMGB1-mediated expression of phospho-p38 were determined by ELISA. (E) Staining for F-actin. HUVEC monolayers grown on glass coverslips were stimulated with or without HMGB1 for 1 h, and then treated with DAB (5 or 10 μM) for 6 h, and stained for F-actin. Arrows indicate intercellular gaps. The representative images were from three separate experiments in different days with similar results. Results are expressed as the mean ± SEM of three separate experiments in different days. *P < 0.05 versus LPS (A) or HMGB1 (B-D).

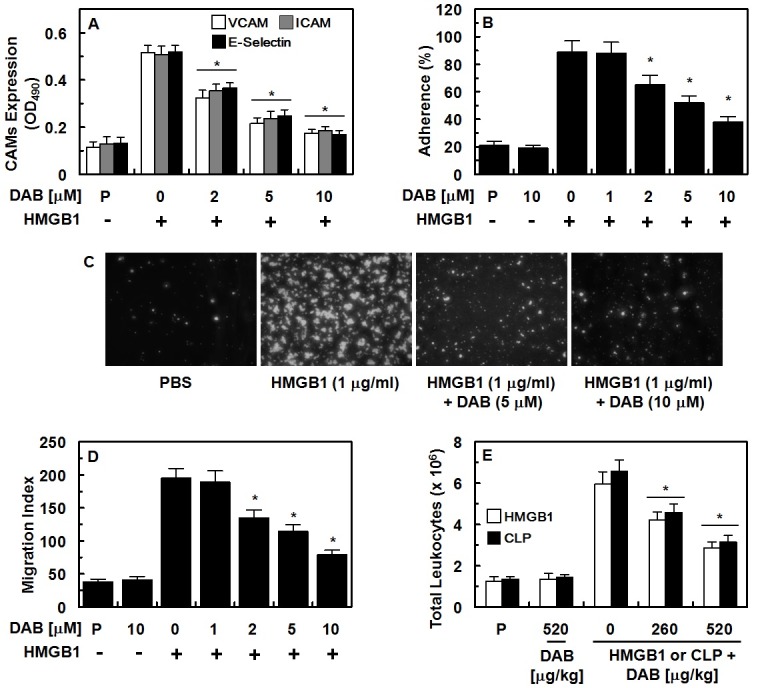
Effects of DAB on HMGB1-mediated expression of CAMs, and adhesion/migration of human neutrophils
Previous report indicated that the expressions of CAMs (ICAM-1, VCAM-1, and E-selectin) were increased by HMGB1, which is correlated with vascular inflammatory diseases (28). CAMs also support the behaviors of leukocytes (adhesion and transmigration toward endothelium to the site of inflammation (28). As can be seen in Fig. 3A, DAB reduced the expression of CAMs by HMGB1 signaling pathway. In addition to reduced CAM expression, DAB reduced the adherence of the human neutrophils to HUVECs and their subsequent migration (Fig. 3B-3D). These results were consistent with in vivo data by the suppression of HMGB1- or CLP-induced migration of leukocytes in the peritoneal space (Fig. 3E).
Fig. 3. Effects of DAB on HMGB1-mediated pro-inflammatory responses. (A-D) HUVECs were stimulated with HMGB1 (1 μg/ml) for 16 h, followed by treatment with DAB for 6 h. HMGB1-mediated (A) expression of VCAM-1 (white bar), ICAM-1 (gray bar), and E-selectin (black bar) in HUVECs, (B, C) adherence of human neutrphils to HUVEC monolayers, and (D) migration of human neutrphils through HUVEC monolayers were analyzed. (E) The effects of treatment with DAB on HMGB1-induced (2 μg/mouse, i.v., white box) or CLP-induced (black box) leukocyte migration into the peritoneal cavities of mice were analyzed. The representative images were from three separate experiments in different days with similar results. All results indicate the mean ± SEM of three separate experiments in different days. *P < 0.05 vs. HMGB1.
Effects of DAB on HMGB1-stimulated activation of NF-κB/ERK and production of IL-6/TNF-α
Vascular inflammatory mediators, such as TNF-α and IL-6, commit to progressions of the systemic severe vascular inflammatory responses and secreted by HMGB1 (29). As outlined above, HMGB1 stimulates the release of pro-inflammatory cytokines through various pathways (14, 27, 30), including ERK 1/2, ultimately leading to the activation of NK-κB and upregulation of inflammatory cytokine release. Therefore, we determined the effects of DAB on the activation of inflammatory signaling molecules and the secretion of TNF-α and IL-6, HUVECs were activated with HMGB1 for 16 h, followed by incubation with DAB for 6 h. Our results indicate that DAB significantly reduced TNF-α and IL-6 levels (Supplementary Fig. 1A and 1B). As shown in Supplementary Fig. 1C and 1D, DAB treatment (6 h) inhibited the the phosphorylation of NF-κB and ERK 1/2 by HMGB1. And, we measured HMGB1-induced translocation of NF-κB from cytosol to nucleus by using p65 NF-κB and fluorescein isothiocyanate (FITC)-conjugated antibody. As a result, data (Supplementary Fig. 1E) indicated that treatment with DAB inhibited the translocation of NF-κB p65 from cytoplasm into nucleus by HMGB1.
Protective effect of DAB in the CLP-induced septic lethality
It is well known that upregulated expressions of HMGB1 contribute the progression and severity of sepsis (14, 31). In this study, we showed the inhibitory and cytoprotective effects of DAM on HMGB1 and underlying signaling effects. This prompted us to validate the effects of DAB on the survival study of mice with sepsis following CLP surgery. To determine whether DAB protects mice from CLP-induced sepsis lethality DAB was administered to mice after CLP. A single administration of DAB (260 or 520 μg/kg, 12 h after CLP) did not prevent CLP-induced death (data not shown). Thus, we next administered same amounts of DAB twice (once 12 h after CLP and once 50 h after CLP). This increased the survival rate from 30 to 50%, according to the Kaplan-Meier survival analysis (P < 0.00001, Fig. 4A). The marked improvement in survival rate achieved by the administration of DAB indicated that the inhibitory effects of DAB on not only HMGB1 secretion but inflammatory responses were therapeutic strategy for management of sepsis and septic shock.
Fig. 4. Effects of DAB on lethality or pulmonary injury after CLP. (A) Male C57BL/6 mice (n = 20) were administered DAB at 260 μg/kg (i.v. □) or at 520 μg/kg (i.v. ■) at 12 h and 50 h after CLP. Animal survival was monitored every 12 hours after CLP for 15 days. Control CLP mice (●) and sham-operated mice (○) were administered sterile saline (n = 20). Kaplan-Meier survival analysis was used to determine the overall survival rates versus CLP treated mice. (B) Male C57BL/6 mice underwent CLP and were administered DAB intravenously at 12 h and 50 h after CLP (n = 5). Mice were euthanized 96 h after CLP. Histopathological scores for the lung tissue were recorded as described in methods. (C) Photomicrographs of lung tissues (H&E staining, ×200). Sham group (grade 1); CLP group (grade 3); Right, CLP + DAB group, (grade 2). Illustrations indicate representative images from three independent experiments in different days with similar results. All results indicate the mean ± SEM of three separate experiments in different days with similar results. (D-G) The same as (B, C) except that mice were bled and euthanized. AST, ALT, BUN, and creatinine level in plasma was measured. All results indicate the mean ± SEM of three separate experiments. *indicates P < 0.05 vs. CLP.

Protective effect of DAB in the CLP-induced tissues injury
We determined the effects of DAB on CLP-induced pulmonary injury to confirm the protective effects of DAB on CLP-induced death. We did not find the significant differences between the lungs of sham mice and sham mice treated with DAB (data not shown). In CLP-operating mice group, we found that the numbers of infiltrated inflammatory cells into the interstitium and alveolar spaces were increased and that the areas of damaged pulmonary architecture were increased (Fig. 4B and 4C). However, DAB treatment decreased damaged pulmonary architecture and the lung injury score was decreased in CLP-operating mice (Fig. 4B and 4C). Multiple organ failure, such as liver and kidney, was found in severe vascular inflammatory conditions, sepsis (32). As shown in Fig. 4, treatment of DAB reduced CLP-induced plasma level of ALT and AST (markers of hepatic injury, Fig. 4D), and creatinine and BUN (markers of renal injury, Fig. 4E and F). DAB also reduced the general tissue damage marker, LDH, in CLP-operating mice (Fig. 4G).
The present work was undertaken to evaluate the protective effect of DAB on vascular barrier integrity. Management of vascular integrity is appearing as pivotal therapeutic strategies for the control of sepsis. HMGB1 is involved in the advancement of sepsis and associated with severe vascular inflammatory diseases (14). Here, our data indicated that the anti-septic responses of DAB occur via the suppression of HMGB1 release and HMGB1-induced vascular inflammatory responses. The molecular mechanism of anti-septic responses by DAB is attributed by the inhibition of HMGB1 secretion (Fig. 1A, 1B), the expressions of HMGB1 receptors (Fig. 1C) and HMGB1-induced vascular disruption (Fig. 3B, 3C, 3E) via suppression of the activation of p38 (Fig. 2D). In addition, DAB inhibited the adhesion and migration of leukocytes toward HUVECs by suppression of the expressions of CAMs (Fig. 3). The inhibitory mechanism of DAB on septic condition is the downregulation of the production of inflammatory cytokines (TNF-α, IL-6, Supplementary Fig. 1A, 1B) and the activation of inflammatory transcriptional factor (NF-κB, ERK1/2, Supplementary Fig. 1C, 1D). DAB also inhibited the translocation of NF-κb from cytosol to nucleus (Supplementary Fig. 1E).
Taken together, the results of this study suggested that DAB inhibited both LPS- and CLP-mediated secretion of HMGB1, expressions of HMGB1 receptors, and HMGB1-mediated vascular disruption by increasing barrier integrity and suppression of the expression of CAMs. Further, DAB reduced the adhesion and migration of leukocytes toward HUVECs. The barrier protective effects of DAB were confirmed in a mouse model, in which treatment with DAB reduced CLP-induced mortality and pulmonary injury. Our findings indicate that DAB could be one of potential drug candidates for treatment of sepsis and septic shock.
MATERIALS AND METHODS
See supplementary information for this section.
Acknowledgments
This study was supported by the Gachon University research fund of 2014 (GCU 2014-5099), the National Research Foundation of Korea (NRF) funded by the Korea government [MSIP] (Grant No. 2014R1A2A1A11049526) and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C0001).
References
- 1.Russell JA. Management of sepsis. N Engl J Med. (2006);355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 2.Andreu Ballester JC, Ballester F, Gonzalez Sanchez A, Almela Quilis A, Colomer Rubio E, Penarroja Otero C. Epidemiology of sepsis in the Valencian Community (Spain), 1995-2004. Infect Control Hosp Epidemiol. (2008);29:630–634. doi: 10.1086/589583. [DOI] [PubMed] [Google Scholar]
- 3.Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. (2001);29:S109–116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- 4.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. (2001);344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 5.Thachil J, Toh CH, Levi M, Watson HG. The withdrawal of Activated Protein C from the use in patients with severe sepsis and DIC [Amendment to the BCSH guideline on disseminated intravascular coagulation]. Br J Haematol. (2012);157:493–494. doi: 10.1111/j.1365-2141.2011.09019.x. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia M, He M, Zhang H, Moochhala S. Sepsis as a model of SIRS. Front Biosci. (2009);14:4703–4711. doi: 10.2741/3561. [DOI] [PubMed] [Google Scholar]
- 7.Tracey KJ, Fong Y, Hesse DG, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. (1987);330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Yang H, Czura CJ, Sama AE, Tracey KJ. HMGB1 as a late mediator of lethal systemic inflammation. Am J Respir Crit Care Med. (2001);164:1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 9.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. (2005);15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Ulloa L, Tracey KJ. The "cytokine profile": a code for sepsis. Trends Mol Med. (2005);11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 11.Bae JS, Rezaie AR. Activated protein C inhibits high mobility group box 1 signaling in endothelial cells. Blood. (2011);118:3952–3959. doi: 10.1182/blood-2011-06-360701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. (2005);33:564–573. doi: 10.1097/01.CCM.0000155991.88802.4D. [DOI] [PubMed] [Google Scholar]
- 13.Gibot S, Massin F, Cravoisy A, et al. High-mobility group box 1 protein plasma concentrations during septic shock. Intensive Care Med. (2007);33:1347–1353. doi: 10.1007/s00134-007-0691-2. [DOI] [PubMed] [Google Scholar]
- 14.Bae JS. Role of high mobility group box 1 in inflammatory disease: Focus on sepsis. Arch Pharm Res. (2012);35:1511–1523. doi: 10.1007/s12272-012-0901-5. [DOI] [PubMed] [Google Scholar]
- 15.Li JX, Feng JM, Wang Y, et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. (2014);5:e1278. doi: 10.1038/cddis.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padhy BM, Gupta YK. Drug repositioning: re-investigating existing drugs for new therapeutic indications. J Postgrad Med. (2011);57:153–160. doi: 10.4103/0022-3859.81870. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Zhu F, Ma XH, et al. Predicting targeted polypharmacology for drug repositioning and multi- target drug discovery. Curr Med Chem. (2013);20:1646–1661. doi: 10.2174/0929867311320130005. [DOI] [PubMed] [Google Scholar]
- 18.O'Connor KA, Roth BL. Finding new tricks for old drugs: an efficient route for public-sector drug discovery. Nat Rev Drug Discov. (2005);4:1005–1014. doi: 10.1038/nrd1900. [DOI] [PubMed] [Google Scholar]
- 19.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. (2008);11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 20.El Gazzar M. HMGB1 modulates inflammatory responses in LPS-activated macrophages. Inflamm Res. (2007);56:162–167. doi: 10.1007/s00011-006-6112-0. [DOI] [PubMed] [Google Scholar]
- 21.Mullins GE, Sunden-Cullberg J, Johansson AS, et al. Activation of human umbilical vein endothelial cells leads to relocation and release of high-mobility group box chromosomal protein 1. Scand J Immunol. (2004);60:566–573. doi: 10.1111/j.0300-9475.2004.01518.x. [DOI] [PubMed] [Google Scholar]
- 22.Czura CJ, Yang H, Tracey KJ. High mobility group box-1 as a therapeutic target downstream of tumor necrosis factor. J Infect Dis. (2003);187(Suppl 2):S391–396. doi: 10.1086/374753. [DOI] [PubMed] [Google Scholar]
- 23.Diehl KH, Hull R, Morton D, et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. (2001);21:15–23. doi: 10.1002/jat.727. [DOI] [PubMed] [Google Scholar]
- 24.Lee W, Ku SK, Lee YM, Bae JS. Anti-septic effects of glyceollins in HMGB1-induced inflammatory responses in vitro and in vivo. Food Chem Toxicol. (2014);63:1–8. doi: 10.1016/j.fct.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 25.Qin YH, Dai SM, Tang GS, et al. HMGB1 enhances the proinflammatory activity of lipopolysaccharide by promoting the phosphorylation of MAPK p38 through receptor for advanced glycation end products. J Immunol. (2009);183:6244–6250. doi: 10.4049/jimmunol.0900390. [DOI] [PubMed] [Google Scholar]
- 26.Sun C, Liang C, Ren Y, et al. Advanced glycation end products depress function of endothelial progenitor cells via p38 and ERK 1/2 mitogen-activated protein kinase pathways. Basic Res Cardiol. (2009);104:42–49. doi: 10.1007/s00395-008-0738-8. [DOI] [PubMed] [Google Scholar]
- 27.Palumbo R, Galvez BG, Pusterla T, et al. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J Cell Biol. (2007);179:33–40. doi: 10.1083/jcb.200704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo Y, Li SJ, Yang J, Qiu YZ, Chen FP. HMGB1 induces an inflammatory response in endothelial cells via the RAGE-dependent endoplasmic reticulum stress pathway. Biochem Biophys Res Commun. (2013);438:732–738. doi: 10.1016/j.bbrc.2013.07.098. [DOI] [PubMed] [Google Scholar]
- 29.Erlandsson Harris H, Andersson U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. (2004);34:1503–1512. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 30.Park JS, Gamboni-Robertson F, He Q, et al. High mobility group box 1 protein interacts with multiple Tolllike receptors. Am J Physiol Cell Physiol. (2006);290:C917–924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 31.Yang EJ, Ku SK, Lee W, et al. Barrier protective effects of rosmarinic acid on HMGB1-induced inflammatory responses in vitro and in vivo. J Cell Physiol. (2013);228:975–982. doi: 10.1002/jcp.24243. [DOI] [PubMed] [Google Scholar]
- 32.Astiz ME, Rackow EC. Septic shock. Lancet. (1998);351:1501–1505. doi: 10.1016/S0140-6736(98)01134-9. [DOI] [PubMed] [Google Scholar]