

Abstract
Domestication and breeding have influenced the genetic structure of plant populations due to selection for adaptation from natural habitats to agro-ecosystems. Here, we investigate the effects of selection on the contents of 51 primary kernel metabolites and their relationships in three Triticum turgidum L. subspecies (i.e., wild emmer, emmer, durum wheat) that represent the major steps of tetraploid wheat domestication. We present a methodological pipeline to identify the signature of selection for molecular phenotypic traits (e.g., metabolites and transcripts). Following the approach, we show that a reduction in unsaturated fatty acids was associated with selection during domestication of emmer (primary domestication). We also show that changes in the amino acid content due to selection mark the domestication of durum wheat (secondary domestication). These effects were found to be partially independent of the associations that unsaturated fatty acids and amino acids have with other domestication-related kernel traits. Changes in contents of metabolites were also highlighted by alterations in the metabolic correlation networks, indicating wide metabolic restructuring due to domestication. Finally, evidence is provided that wild and exotic germplasm can have a relevant role for improvement of wheat quality and nutritional traits.
Keywords: population genomics, domestication, wheat, QST, metabolomics.
Introduction
Agriculture has had an evolutionary effect on crop species by modifying the wild progenitors to adapt them to new environments and human needs. During crop domestication, human and agro-ecosystem demands led to the selection of similar traits—known as the domestication syndrome—in a range of plant species, thereby supporting the occurrence of convergent phenotypic evolution (Gaut 2015). In seed-propagated crops, important domestication-associated traits include the increase in seed size, the loss of dormancy, and seed dispersal mechanisms as well as a reduced, or loss of, photoperiod sensitivity (Gepts and Papa 2002). For most of these traits, quantitative trait loci (QTL) have been identified, and, in some cases, the underlying genes have been cloned (for reviews, see Lenser and Theißen 2013; Olsen and Wendel 2013a, 2013b). The idea that only a few traits, controlled by major genes, describe the essence of the domestication process has been partly abandoned. This idea was known as the “rapid transition” model, whereby domestication was considered a process that includes only a short predomestication cultivation of wild species, together with a relatively rapid rise (over a few hundreds of years) of the domestication syndrome (Wang et al. 1999; Gepts 2014). Recent archaeological and genetic data suggest the occurrence of a long and complex temporal period of transition from gathering to cultivation of wild plants, followed by a lengthy subsequent process of selection for adaptation to both the agro-ecosystem and the human needs (Meyer and Purugganan 2013; Gepts 2014).
The seminal work of Wright et al. (2005) and subsequent studies in maize (Yamasaki et al. 2005; Zheng et al. 2008; Hufford et al. 2012; Swanson-Wagner et al. 2012), together with the evidence available for other crops such as common bean (Sotelo et al. 1995), finger millet (Barbeau and Hilu 1993), and sunflower (Chapman and Burke 2012), indicate that many traits have been the target of selection, including those associated with nutritional value and amino acid metabolism. Recently, Bellucci et al. (2014) reported that, in addition to selection at target loci, domestication had a deep impact on the architecture of expression of the whole transcriptome in common bean. These findings suggest a similar or even deeper impact of domestication on the phenotypic expression at the metabolite level.
Seed metabolites are associated not only with nutritional value, but also with physiological properties such as seed maturation, desiccation, and germination (Rao et al. 2014). Metabolite profiling appears to be relevant for the description of the geographical structure of natural populations (Kleessen et al. 2012), as well as for QTL identification by Genome-Wide Association Studies in maize (Riedelsheimer et al. 2012; Wen et al. 2014) and in rice (Chen et al. 2014). Skogerson et al. (2010) identified 119 metabolites in maize kernels and found that their variation was associated with genotypic variation. Similar results were observed in a limited set of modern varieties of durum wheat (Beleggia et al. 2013), which support the conclusion that metabolite profiling can be exploited as a molecular phenotyping approach to study crop domestication.
Tetraploid wheats, Triticum turgidum L. (2n = 4x = 28; AABB genome), were domesticated in the Fertile Crescent alongside with einkorn and barley. They offer an interesting model to study the effects of selection associated to domestication. About 12,000 years ago, emmer (T. turgidum ssp. dicoccum) was domesticated from wild emmer (T. turgidum ssp. dicoccoides) (Nesbitt and Samuel 1998; Tanno and Willcox 2006). Emmer spread following human migrations throughout Europe and Asia, and became the most important crop in the Fertile Crescent until the early Bronze Age, 10,000 years BC (Bar-Yosef 1998). Free-threshing tetraploid wheats (T. turgidum ssp. turgidum) subsequently originated from emmer. This event was followed by the selection of durum wheat (T. turgidum ssp. turgidum convar. durum), as a crop specialized for the production of pasta, couscous, traditional/typical bread and bulgur, and its spread in the Mediterranean region. It can be useful to consider the evolution of tetraploid wheats as consisting of a least two steps: primary domestication, from wild emmer to emmer, and secondary domestication, from emmer to durum wheat (Gioia et al. 2015).
The aim of this study was to assess the phenotypic variation of primary metabolites in the kernels of three T. turgidum populations that represent both the primary and secondary domestication processes. This corresponds to determining whether the primary and secondary domestication events were associated with changes in the content of specific metabolites. To this end, we tested whether selection, other than neutral processes, might explain the changes observed in primary metabolites using the QST versus FST comparisons (Leinonen et al. 2013). Moreover, we also sought to determine whether the signal of selection observed at the metabolite level can be explained by processes of indirect selection, that is, by the correlation between metabolite content and other “classical” kernel traits associated with the domestication syndrome. Additionally, we investigated whether metabolite coabundance profiles changed over the evolutionary trajectory from primary to secondary domestication, by using network analysis on the metabolites of the three tetraploid wheat populations considered.
Results
Molecular Divergence
We estimated the neutral FST by surveying 26 microsatellite loci across a panel of tetraploid wheat consisting in 12 accession of wild emmer (T. turgidum ssp. dicoccoides), 10 accession of emmer wheat (T. turgidum ssp. dicoccum), and 15 accession of durum wheat (T. turgidum ssp. turgidum convar. durum) (table 1). The neutral FST was estimated by excluding loci that carried signature of divergent selection (P < 0.05). FST estimates were obtained by considering the three taxa simultaneously (FST = 0.149, P < 10−5, excluding two loci), between wild emmer and emmer (FST = 0.062, P < 10−5, no loci were excluded) and between emmer and durum wheat (FST = 0.185, P < 10−5, excluding one locus).
Table 1.
List of Wild and Domesticated Accessions of Triticum turgidum Considered in This Study.
| Taxonomic Classification | Accession | Country |
|---|---|---|
| Wild emmer (Triticum turgidum ssp. dicoccoides) | PI 346783 | n.a. |
| PI 343446 | Israel | |
| PI 481539 | Israel | |
| PI 352323 | Asia Minor | |
| PI 352324 | Lebanon | |
| PI 355459 | Armenia | |
| PI 470944 | Syria, Al Qunaytirah | |
| PI 470945 | Syria, Al Qunaytirah | |
| MG 4343 | n.a. | |
| MG 4328/61 | n.a. | |
| MG 5444/235 | n.a. | |
| MG 4330/66 | n.a. | |
| Emmer (T. turgidum ssp. dicoccum) | Farvento | Italy |
| Lucanica | Italy | |
| Molise selection Colli | Italy | |
| MG 5350 | Ethiopia | |
| MG 4387 | United Kingdom | |
| MG 5473 | Spain | |
| MG 5344/1 | Ethiopia | |
| MG 5293/1 | Italy | |
| MG 5323 | n.a. | |
| MG 3521 | n.a. | |
| Durum wheat (T. turgidum ssp. turgidum convar. durum) | Cappelli | Italy |
| Timilia | Italy | |
| Capeiti-8 | Italy | |
| Trinakria | Italy | |
| Appulo | Italy | |
| Creso | Italy | |
| Neodur | France | |
| Simeto | Italy | |
| Ofanto | Italy | |
| Cirillo | Italy | |
| PR22D89 | Italy | |
| Pedroso | Spain | |
| CER16 | Italy | |
| CER58 | Italy | |
| CER132 | Italy |
Note.—n.a. = not available; PI = accession numbers for USDA National Small Grains Collection, Aberdeen, ID;
MG = accession numbers for CNR Institute of Plant Genetics, Bari, Italy;
CER = accession numbers for CREA-CER Cereal Research Centre, Foggia, Italy.
Metabolite Profiling
In the whole set of tetraploid wheats, in total were detected 51 polar and nonpolar metabolites from 9 compound classes: amino acids, organic acids, polyols, sugars, saturated fatty acids (SFAs), unsaturated fatty acids (UFAs), fatty alcohols, tocopherols, and phytosterols and the metabolite data are available on Dryad Digital Repository DOI:10.5061/dryad.12s8s. Metabolite levels within each taxon, their heritabilities, and the estimates of metabolic divergence among the taxa are reported in table 2. Twenty-nine metabolites were common to all studied lines and 13 were shared among taxa, but not among all of the lines of the same taxon. In total, 42 metabolites out of 51 (82.4%) were shared by the 3 taxa. Absence of 17.6% of the metabolites from specific lines can be due either to a state of silencing of some metabolic steps or to the detection limit of the profiling methods. Based on the Tukey’s test for each of the nine compound classes, wild emmer and emmer were significantly different in the mean level of SFAs and UFAs. No significant differences were detected between emmer and durum wheat. For individual metabolites, wild emmer was different from emmer in eight cases corresponding to seven fatty acids and to fructose. Emmer differed from durum wheat in the mean level of 12 metabolites (i.e., 7 amino acids, 3 sugars and montanyl alcohol, β-tocopherol), all present with smaller contents in the second. In contrast, the content of maltose was significantly higher in durum wheat (table 2).
Table 2.
Mean Metabolite Level Detected in Different Tetraploid Wheat Taxa, with Hereditability and QST Estimates.
| No. | Metabolite | Level (μg/g dry weight) |
Range | Heritability2 (h) | QST | ||
|---|---|---|---|---|---|---|---|
| Wild emmera | Emmera | Durum wheata | |||||
| Amino acids (×103) | 2.154 a | 2.038 a | 1.560 a | 0.498–4.779 | 0.99 | 0.15 | |
| Organic acids (×103) | 2.227 a | 2.288 a | 2.073 a | 1.144—3.904 | 0.92 | 0.06 | |
| Polyols | 155.685 a | 136.703 a | 128.470 a | 68.951–411.151 | 0.97 | 0.09 | |
| Sugars (×103) | 86.825 a | 96.495 a | 87.261 a | 66.540–106.600 | 0.66 | 0.00 | |
| Saturated fatty acids (×103) | 1.686 a | 0.991 b | 1.029 b | 0.651–3.499 | 0.75 | 0.41 | |
| Unsaturated fatty acids (×103) | 6.401 a | 3.422 b | 2.541 b | 1.791–15.031 | 0.79* | 0.51* | |
| Fatty alcohols | 4.841 a | 3.153 a | 4.995 a | 0.149–12.861 | 0.91 | 0.00 | |
| Tocopherols | 18.830 a | 18.015 a | 15.501 a | 11.124–27.399 | 0.41 | 0.22 | |
| Phytosterols | 506.740 a | 452.748 a | 442.054 a | 356.352–683.84 | 0.70 | 0.25 | |
| 1 | Alanine | 93.53 a | 89.56 a | 7.32 b | 0.0126–233.907 | 1.00* | 0.55* |
| 2 | Valine | 41.64 a | 55.42 a | 9.07 b | 0.047–91.834 | 0.99** | 0.66** |
| 3 | Leucine | 23.61 a | 30.07 a | 10.40 b | 0.099–51.538 | 0.98 | 0.46 |
| 4 | Proline | 76.53 a | 69.02 a | 30.20 a | 0.036–362.162 | 1.00 | 0.15 |
| 5 | Isoleucine | 2.124 ab | 8.640 a | 0.612 b | 0.002–38.413 | 0.99 | 0.26 |
| 6 | Glycine | 45.594 a | 40.911 a | 34.365 a | 15.887–110.764 | 0.98 | 0.12 |
| 7 | Serine | 30.563 ab | 41.764 a | 22.743 b | 11.093–80.087 | 0.97 | 0.35 |
| 8 | Threonine | 25.834 ab | 29.272 a | 17.876 b | 9.338–50.665 | 0.98 | 0.31 |
| 9 | β-Alanine | 0.157 a | 1.211a | 0.134 a | 0–8.288 | 1.00 | 0.15 |
| 10 | Asparagine | 682.269 a | 665.774 a | 605.207 a | 123.393–1,626.143 | 0.98 | 0.04 |
| 11 | Arginine | 0.042 a | 0.290 a | 0.036 a | 0–2.477 | 0.99 | 0.14 |
| 12 | Aspartic acid | 329.798 a | 372.715 a | 405.380 a | 123.024–803.541 | 0.98 | 0.00 |
| 13 | Glutamic acid | 661.924 a | 570.462 a | 468.735 a | 130.246–1,792.413 | 0.99 | 0.13 |
| 14 | γ-Aminobutiric acid | 32.905 ab | 71.490 a | 28.601 b | 0.081–199.878 | 0.99 | 0.27 |
| 15 | Malic acid (×103) | 1.402 a | 1.373 a | 1.410 a | 0.688–2.439 | 0.91 | 0.00 |
| 16 | Citric acid | 478.164 a | 484.869 a | 486.390 a | 224.592–1,091.296 | 0.89 | 0.00 |
| 17 | Quinic acid | 4.214 a | 6.423 a | 0.941 a | 0–29.071 | 1.00 | 0.17 |
| 18 | Gluconic acid | 342.400 a | 373.813 a | 209.886 a | 1.003–1,074.836 | 0.99 | 0.16 |
| 19 | Sorbitol/galactitol | 61.050 a | 52.426 a | 46.313 a | 10.227–338.772 | 0.99 | 0.04 |
| 20 | Myo-Iinositol | 87.362 a | 86.367 a | 86.451 a | 52.678–131.536 | 0.89 | 0.01 |
| 21 | Arabinose | 8.488 a | 8.470 a | 9.508 a | 0.035–29.289 | 0.99 | 0.02 |
| 22 | Fructose | 978.644 b | 1,527.779 a | 805.612 b | 460.547–2,198.266 | 0.96* | 0.57* |
| 23 | Glucose | 797.125 ab | 1,060.501 a | 625.652 b | 424.344–1,628.938 | 0.64 | 0.50 |
| 24 | Sucrose (×103) | 30.388 a | 30.226 a | 30.263 a | 24.189–44.493 | 0.80 | 0.01 |
| 25 | Maltose (×103) | 4.189 b | 4.108 b | 5.450 a | 2.654–7.759 | 0.96 | 0.31 |
| 26 | Raffinose (×103) | 53.260 a | 57.363 a | 49.307 a | 26.022–68.533 | 0.80 | 0.00 |
| 27 | Myristic acid | 18.511 a | 23.512 a | 14.159 a | 11.794–79.954 | 0.38 | 0.00 |
| 28 | Pentadecanoic acid | 6.659 a | 7.124 a | 6.519 a | 4.533–27.352 | 0.50 | 0.00 |
| 29 | Palmitic acid (×103) | 1.509 a | 0.861 b | 0.897 b | 0.546–3.179 | 0.76 | 0.42 |
| 30 | Margaric acid | 5.836 a | 4.059 a | 4.209 a | 2.843–11.532 | 0.72 | 0.29 |
| 31 | Stearic acid | 88.572 a | 69.011 a | 68.277 a | 50.068–167.587 | 0.67 | 0.27 |
| 32 | Arachidic acid | 12.164 a | 6.605 b | 4.338 b | 0.637–26.032 | 0.81* | 0.53* |
| 33 | Behenic acid | 19.393 a | 12.438 b | 10.150 b | 2.046–33.803 | 0.73 | 0.50 |
| 34 | Lignoceric acid | 24.745 a | 18.853 b | 17.753 b | 14.047–39.677 | 0.69 | 0.38 |
| 35 | Palmitoleic acid | 10.324 a | 6.723 ab | 3.999 b | 0.657–23.2 | 0.72 | 0.35 |
| 36 | Linoleic acid (×103) | 4.857 a | 2.649 b | 1.989 b | 1.223–11.002 | 0.80* | 0.52* |
| 37 | Oleic acid (×103) | 1.432 a | 0.728 b | 0.526b | 0.341–3.779 | 0.76 | 0.46 |
| 38 | Gondoic acid | 97.377 a | 40.371 b | 23.037 b | 3.973–238.384 | 0.78* | 0.56* |
| 39 | Stearyl alcohol | 1.071 a | 0.269 a | 0.754 a | 0.012–3.915 | 0.88 | 0.00 |
| 40 | Palmityl alcohol | 0.135 a | 0.146 a | 0.489 a | 0.005–5.728 | 0.90 | 0.07 |
| 41 | Heneicosyl alcohol | 0.276 a | 0.346 a | 0.418 a | 0.004–9.381 | 0.96 | 0.00 |
| 42 | Lignoceryl alcohol | 2.453 a | 1.725 ab | 1.502 b | 0.158–4.595 | 0.81 | 0.33 |
| 43 | Ceryl alcohol | 0.390 a | 1.771 a | 0.993 a | 0.005–9.014 | 0.97 | 0.13 |
| 44 | 1-Heptacosanol | 0.139 a | 0.134 a | 0.074 a | 0.001–4.031 | 0.94 | 0.00 |
| 45 | Montanyl alcohol | 0.022 ab | 0.322 a | 0.018 b | 0–1.338 | 0.95 | 0.28 |
| 46 | α-Tocopherol | 11.234 a | 11.276 a | 11.343 a | 7.34–16.204 | 0.29 | 0.00 |
| 47 | β-Tocopherol | 7.216 a | 6.753 a | 4.423 b | 2.765–12.176 | 0.66 | 0.37 |
| 48 | Campsterol | 167.606 a | 151.943 a | 152.854 a | 107.308–238.318 | 0.70 | 0.13 |
| 49 | Stigmasterol | 21.188 a | 20.645 a | 20.326 a | 12.664–66.48 | 0.85 | 0.02 |
| 50 | β-Sitosterol | 299.080 a | 267.341 ab | 255.826 b | 220.422–373.998 | 0.71 | 0.32 |
| 51 | Stigmastanol | 15.784 a | 14.346 a | 14.381 a | 10.279–22.676 | 0.70 | 0.13 |
Note.—Different letters in the same row indicate significant differences (Tukey tests; P < 0.05).
aWild emmer = Triticum turgidum L. ssp. dicoccoides; emmer = T. turgidum ssp. dicoccum; durum wheat = T. turgidum ssp. turgidum convar. durum.
2*P ≤ 0.05; **P ≤ 0.01.
Heritability (h2) was high for all classes of compounds, ranging from 0.41 for tocopherols to 0.99 for amino acids (table 2). In addition, the heritability for single metabolite ranged from 0.29 for α-tocopherol to ∼1 for alanine, proline, and quinic acid. Based on the coefficient of additive genetic variation (Houle 1992; Hansen et al. 2011), the measured metabolites were highly correlated between wild emmer and emmer (r = 0.539, P < 0.0001). This was not the case for emmer versus durum wheat (r = −0.140, P = 0.326), nor for wild emmer versus durum wheat (r = −0.081, P = 0.572). More specifically, with regard to the coefficient of additive genetic variation in single metabolites, durum wheat showed larger differences in comparison with wild emmer and emmer (supplementary table S1, Supplementary Material online).
For each metabolite, the level of divergence (QST) was calculated (see Materials and Methods; fig. 1) to determine whether selection (not only neutral processes) has been at least partially responsible for the observed differences among taxa. For 25 metabolites the observed QST was higher than expected under neutrality. Specifically, the strongest evidence for selection was found for 16 and 6 metabolites in the steps from wild to domesticated emmer and from emmer to durum wheat, respectively (P < 0.05). Moreover, the evidence for selection was found in both steps for 3 metabolites. With the exception of γ-aminobutyric acid (GABA), the metabolites that significantly varied when comparing wild emmer versus emmer were mainly those of the nonpolar fraction, and in particular all of the SFAs, most of the UFAs, three fatty alcohols, and β-sitosterol. Thirteen of these metabolites showed a decrease from wild emmer to emmer, with the exception of GABA as well as ceryl and montanyl alcohols. On the other hand, the metabolites for which the QST was significant in the emmer versus durum wheat comparison were principally the amino acids and β-tocopherol, which showed a significant decrease (table 2). Finally, the metabolites for which QST was significant in both comparisons were glucose, fructose, and oleic acid. Although for oleic acid a decrease was noted during both evolutionary steps, the two sugars increased from wild emmer to emmer and then decreased to durum wheat (table 2). Following the application of the sequential Bonferroni correction, seven metabolites showed a signature of selection: five during the transition from wild emmer to emmer (i.e., fructose, gondoic acid, behenic acid, palmitic acid, lignoceryl alcohol) and two during the step from emmer to durum wheat (i.e., alanine and valine; fig. 1). With the exception of lignoceryl alcohol, all these metabolites showed significant differences between taxa (Tukey–Kramer tests; table 2).
Fig. 1.

QST distribution associated with the evolutionary steps wild emmer versus emmer (primary domestication) and emmer versus durum wheat (secondary domestication).
It is possible that variations in the metabolite levels between taxa could be driven by correlations with other classical kernel phenotypic traits that were under selection during domestication (Golan et al. 2015). To distinguish such clearly indirect effects, seven kernel traits have been considered: 1,000 kernel weight (KW); protein content (pc); embryo weight (EW); seed weight without embryo (i.e., endosperm weight, ENW); embryo weight/total seed weight ratio (EW/TW); endosperm weight/total seed weight ratio (ENW/TW); and endosperm weight/embryo weight (ENW/EW). The seven traits were strongly correlated among each other (data not shown) and a principal component (PC) analysis indicates that the three PCs can effectively summarize the information carried by the seven variables explaining the 98.99% of the total variance (supplementary table S2, Supplementary Material online). The first, PC1, is highly correlated with three original variables that mainly describe the size of the embryo relative to that of the seed; the second, PC2, is correlated with embryo and seed weights, while the third, PC3, mainly describes the seed pc (supplementary table S2, Supplementary Material online).
We next treated PC1–3 as new (latent) covariates (LC1–3) and determined their correlation to the content of each metabolite. We found that the metabolite contents were often strongly correlated with the LCs, particularly with embryo size and pc (supplementary table S3, Supplementary Material online). Based on analysis of covariance (ANCOVA), the metabolites were categorized into two groups: those with a nonsignificant and those with a significant interaction between LCs and taxa, denoted by nsINT and sINT. We then tested if the two groups of metabolites show difference with respect to the number of metabolites with significant or nonsignificant QST. For the comparison of wild emmer versus emmer, the number of metabolites with significant QST was statistically higher in the sINT than the nsINT group (Fisher exact test: two-tailed, P = 0.0053; supplementary fig. S1, Supplementary Material online). Although this was not true for the comparison of emmer versus durum wheat (P = 0.4619), it held for the comparison of wild emmer versus durum wheat (P = 0.0097). The differences for the mean QST followed the same pattern, with P values of 0.0053, 0.6040, and 0.0008 for three contrasts, respectively. The estimates for QST upon correcting for indirect effects of LCs (henceforth QST1) were obtained by repeating the ANCOVA for the metabolites for which the interactions with the three LCs were not significant, thus allowing to obtain the “corrected” between and within taxa variance components to determine the QST1 values (table 3). When the three taxa were considered simultaneously, the QST1[][] values were significant only for alanine, fructose, and glucose; moreover, for the two sugars the correction with the covariates produced a larger value compared with the respective QST. Based on QST1, only seven metabolites were significantly affected by selection in the step wild emmer versus emmer, three of which were previously identified as significantly changed based on QST (i.e., GABA, myristic acid, β-sitosterol). Moreover, upon correcting for indirect effects of LCs, we found evidence for selection (based on the corrected QST) for ten metabolites in the step emmer versus durum wheat; for seven of these (i.e., alanine, leucine, serine, threonine, fructose, glucose, β-tocopherol) we also found evidence for selection based on the QST. All the fatty alcohols and the UFAs (i.e., oleic, linoleic) were no longer significant once covariates were considered. For fructose, glucose, and β-sitosterol, we found evidence for selection based on both QST and QST1 measures.
Table 3.
QST1 Values Calculated for all Three Taxa, for Wild Emmer Versus Emmer, and for Emmer Versus Durum Wheat Comparisons.a
| Metabolite |
QST1 |
||
|---|---|---|---|
| All Three Taxa | Wild Emmer vs. Emmer | Emmer vs. Durum Wheat | |
| Organic acids | 0.12 | 0.00 | 0.57* |
| Polyols | 0.08 | 0.04 | 0.00 |
| Sugars | 0.00 | 0.00 | 0.00 |
| Tocopherols | 0.00 | 0.00 | 0.68** |
| Phytosterols | 0.12 | 0.20 | 0.32 |
| Alanine | 0.55* | 0.00 | 0.91*** |
| Leucine | 0.42 | 0.11 | 0.66** |
| Proline | 0.18 | 0.00 | 0.37 |
| Isoleucine | 0.26 | 0.13 | 0.38 |
| Glycine | 0.17 | 0.00 | 0.53* |
| Serine | 0.36 | 0.13 | 0.68** |
| Threonine | 0.34 | 0.00 | 0.61** |
| β-Alanine | 0.14 | 0.12 | 0.10 |
| Aspartic acid | 0.00 | 0.00 | 0.04 |
| Glutamic acid | 0.15 | 0.00 | 0.18 |
| γ-Aminobutiric acid | 0.26 | 0.25* | 0.41 |
| Citric acid | 0.00 | 0.06 | 0.00 |
| Gluconic acid | 0.19 | 0.00 | 0.31 |
| Sorbitol/galactitol | 0.00 | 0.00 | 0.00 |
| Myo-inositol | 0.11 | 0.38** | 0.00 |
| Arabinose | 0.00 | 0.00 | 0.00 |
| Fructose | 0.62** | 0.53*** | 0.74*** |
| Glucose | 0.57* | 0.31** | 0.82*** |
| Sucrose | 0.25 | 0.46*** | 0.00 |
| Maltose | 0.16 | 0.00 | 0.32 |
| Raffinose | 0.00 | 0.00 | 0.00 |
| Myristic acid | 0.00 | 0.37** | 0.00 |
| Pentadecanoic acid | 0.00 | 0.19 | 0.00 |
| Stearyl alcohol | 0.01 | 0.00 | 0.00 |
| Palmityl alcohol | 0.00 | 0.00 | 0.00 |
| 1-Heptacosanol | 0.00 | n.d. | 0.00 |
| Montanyl alcohol | 0.26 | 0.16 | 0.29 |
| α-Tocopherol | 0.00 | 0.00 | 0.93*** |
| β-Tocopherol | 0.11 | 0.00 | 0.54* |
| Campsterol | 0.11 | 0.17 | 0.00 |
| Stigmasterol | 0.00 | 0.00 | 0.00 |
| β-Sitosterol | 0.16 | 0.24* | 0.49* |
| Stigmastanol | 0.08 | 0.13 | 0.00 |
aWild emmer = Triticum turgidum L. ssp. dicoccoides; emmer = T. turgidum ssp. dicoccum; durum wheat = T. turgidum ssp. turgidum convar. durum. n.d., not detected.
*P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Comparative Analysis of Partial Correlations between Metabolic Profiles across Taxa
Having analyzed the difference in metabolite contents between taxa upon correction for the LCs, we next investigated the extent of difference between partial correlations controlling for the LCs (referred to correlations, see Materials and Methods) in the three wheat populations. We found that the number of significant positive correlations is at least 4-fold greater than the number of significant negative correlations in each taxon (table 4). Durum wheat had the largest number of significant positive correlations, followed by wild emmer and emmer; this ordering of taxa was reversed when inspecting the number of negative correlations (table 4).
Table 4.
Comparisons among the Three Taxa of Partial Correlations (FDR = 5%) among Metabolite Levels.
| Taxon | Partial Correlation |
Comparison | Shared Partial Correlation |
Nonshared Partial Correlation | Ratio | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Positive (n) | Negative (n) | Positive (n) | Negative (n) | Correlation (r) among Positive | Positive to Negative | Negative to Positive | Positive and Negative | Shared/Nonshared Partial Correlation | ||
| Wild emmer | 333 | 47 | Wild emmer vs. emmer | 165 | 6 | 0.47* | 25 | 19 | 153 | 1.12 |
| Emmer | 317 | 74 | Emmer vs. durum wheat | 155 | 2 | 0.29 | 41 | 17 | 136 | 1.15 |
| Durum wheat | 439 | 45 | Wild emmer vs. durum wheat | 191 | 2 | 0.31* | 27 | 18 | 247 | 0.78 |
*P = 0.05.
A correlation found in one taxon may not have the same magnitude and sign in the others. Therefore, we inspected the number of shared positive partial correlations and the correspondence of their values (see Materials and Methods section for definition). We observed that the number of metabolite pairs that were positively correlated in emmer and durum wheat was smallest in comparison with the other two contrasts (table 4). However, although wild emmer and durum wheat shared the largest number of positive correlations, the correspondence of their values was significantly (P < 0.05) smaller than in the case of wild emmer and emmer. The largest number of shared partial correlations that switched from positive to negative value was observed for the comparison emmer versus durum wheat, and minor differences among taxa were observed when considering the number of correlations that switched from negative to positive value. Taking into account the number of nonshared partial correlations between pairs of taxa, durum wheat was closer to emmer than to wild emmer. Based on the ratio between the shared and nonshared partial correlations, wild emmer and durum wheat appeared as the two most divergent taxa.
This correlation-based analysis indicates that a large fraction of correlations are taxa specific, despite the finding of shared partial correlations; moreover, a fraction of changes in sign of significant partial correlations supports the claim for large rewiring of the underlying regulatory and metabolic networks. The data also indicate that the step leading from domesticated emmer to durum wheat might have had a greater impact on metabolism compared with the process responsible for the evolution from wild to domesticated emmer. Moreover, the observed difference in metabolic restructuring between the two domestication events is in line with our findings based on the coefficient of additive genetic variation, above. This is supported by the smallest number of shared correlations, lack of correspondence between the values of the shared correlations, and the largest number of correlations which change sign between emmer and durum wheat.
Network Analysis
The correlation structure among the content of metabolites in each of the three taxa can be represented by a network. The nodes in this network denote metabolites and the edges stand for the presence of significant partial correlations between pairs of metabolites (fig. 2). A change in a metabolite content is then expected to propagate across the network edges and cause alterations in the contents in the rest of the metabolites. The effect on such changes can be summarized by the centrality of a metabolite (i.e., a node) in the network. Therefore, in the following, we determine the centralities of metabolites in each taxon-specific network, and compare them between the taxa.
Fig. 2.

Metabolite partial correlation networks for wild emmer (A), emmer (B), and durum wheat (C). Squares denote isolated nodes (i.e., metabolites which are not involved in any significant correlation). The compound classes are denoted in colors indicated in the legend. The nodes are numbered, and the corresponding names can be found in table 2.
We investigated five classical measures of node centrality, namely, the degree, eigenvalue centrality, node betweeness, node closeness, and node subgraph centrality (Toubiana et al. 2013) at false discovery rate of 5% for the creation of the network edges. The degree of a node is determined by the number of edges incident on it. The eigenvalue centrality is based on the idea that a node is more central if its neighbors (i.e., nodes directly accessible via an edge) are central. A node is considered more central according to the betweenness centrality if a larger fraction of shortest paths for any pairs of nodes pass through it. By the closeness centrality, a node is more central if it has smaller total distance (i.e. , length of shortest paths) to all other nodes in the network. Finally, a node is deemed central based on the subgraph centrality if it participates in a larger number of closed loops (Estrada and Rodriguez-Velazquez 2005).
We found that the centralities were more conserved between wild emmer and emmer than between emmer and durum wheat (table 5). This is consistent across four of the five centrality measures (i.e., degree, closeness, subgraph centrality, and, to a lesser extent, eigenvalue centrality). Therefore, we conclude that the restructuring of the networks appears more profound for the step leading to durum wheat from emmer. This is in line with the conclusions drawn by the correlation-based analysis and the coefficient of additive genetic variation. However, in contrast to some of the findings from the previous analyses, the network centrality measures of durum wheat tended to be more concordant to those of wild emmer than of emmer.
Table 5.
Comparisons among Metabolite Correlation Networks (FDR = %).
| Comparison | Node Degree | Node Eigenvalue | Betweeness | Closeness | Subgraph Centrality |
|---|---|---|---|---|---|
| Wild emmer vs. emmer | 0.61* | 0.49 | −0.13 | 0.80* | 0.60* |
| Emmer vs. durum wheat | 0.49* | 0.37 | 0.03 | 0.67* | 0.32 |
| Wild emmer vs. durum wheat | 0.59* | 0.61* | 0.37 | 0.85* | 0.46* |
Note.—For each of the five centrality measures, the Pearson correlation (r) between the three possible pairs of the Triticum taxa are reported.
*P = 0.05.
We also wanted to know whether the ranking of metabolites based on the absolute values of the differences between their centralities corresponded to the rank based on their QST values (table 6). We found stronger concordance for wild emmer versus emmer; this was particularly the case when all isolated nodes (i.e., nodes of degree zero) were excluded from the analysis. The notable differences in the centralities reflect changes in metabolic regulation or differences in metabolic pathways. The same analysis was conducted by excluding or considering only metabolites with significant LCs × species interactions (supplementary table S4, Supplementary Material online). Upon excluding metabolites with significant interactions, a significant correlation was observed between subgraph centralities and QST for emmer versus durum wheat, and for wild emmer versus emmer in the second case.
Table 6.
Correlations between QST and Differences of Node Centrality Measures of Degree, Eigenvalue, Betweenness, Closeness, and Subgraph.
| QST Concordance | Domestication (Pearson’s Correlation Coefficient) |
||
|---|---|---|---|
| Primary | Secondary | Both | |
| Wild emmer/emmer s.s. = 49 (37) | Emmer/durum wheat s.s. = 51 (40) | Wild emmer/durum wheat s.s. = 48 (38) | |
| Degree | 0.329* (0.410*) | 0.031 (−0.092) | 0.217 (0.222) |
| Eigenvalue | 0.336* (0.412*) | 0.075 (−0.081) | 0.178 (0.174) |
| Betweenness | 0.238 (0.276) | 0.031 (−0.117) | 0.422** (0.456**) |
| Closeness | 0.259 (0.389*) | 0.156 (−0.106) | 0.049 (0.005) |
| Subgraph | −0.274 (−0.428**) | 0.356* (0.270) | 0.035 (−0.34) |
Note.—Concordance between variables is supported by significant Pearson correlation coefficient. Statistics are calculated both considering all detectable metabolites and excluding those metabolites that were isolated in the pair of networks being compared (reported in brackets). s.s. = sample size.
*P < 0.05; **P < .01.
Finally, we ranked the metabolites in each network based on their participation in taxon-specific correlations. These taxon-specific correlations and respective metabolites can be regarded as the metabolomics “barcode” of each taxon. The lowest number of taxon-specific significant correlations was observed for wild emmer (63), followed by emmer (80) and durum wheat (161). The number of metabolites involved in taxon-specific correlations differed between the taxa: 40 in durum and 31 and 34 respectively in emmer and wild emmer. We observed that palmitoleic acid, lignoceric acid, and maltose were involved in 37.3% of the correlations specific to wild emmer. Moreover, neither palmitoleic acid nor lignoceric acid was involved in taxon-specific correlations in emmer (fig. 3). For this taxon, stearic and arachidic acids were involved 22.5% of taxon-specific correlations. In durum wheat, behenic and gondoic acids and palmityl and stearyl alcohols accounted for 34.5% of the taxon-specific correlations. None of these metabolites were involved in the taxon-specific correlations of wild emmer and emmer. When considering the two evolutionary steps separately, in wild emmer versus emmer the compounds showing the strongest changes in terms of taxon-specific correlations were stearic, palmitoleic, and lignoceric acids (all three with significant QST; fig. 1). Considering emmer versus durum wheat, the metabolites with the strongest change were stearyl alcohol and stearic, gondoic, and behenic acids. None of these had significant QST for emmer versus durum wheat during secondary domestication, but the three acids showed significant QST for wild emmer versus emmer.
Fig. 3.
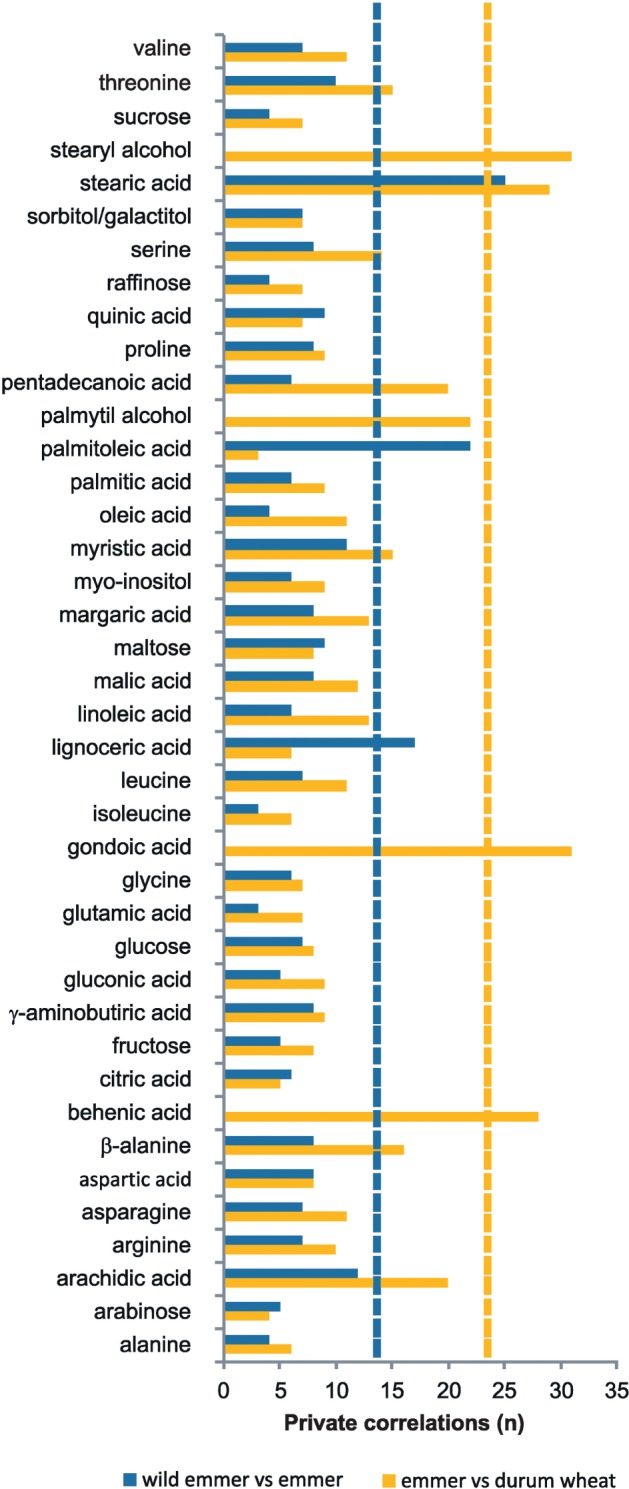
Changes in the numbers of taxon-specific correlations during primary domestication and secondary domestication. The changes are shown only for metabolites which are involved in taxon-specific network from at least one taxon. The bars indicate the sums of the taxon-specific correlations in the compared networks (blue for wild emmer vs. emmer, yellow for emmer vs. durum wheat). Dashed vertical lines denote twice the value of the mean number of taxon-specific correlations for the respective domestication event.
Discussion
The phenotypic variation for kernel metabolite composition has been characterized by comparing significant steps of tetraploid wheat taxa evolution, during and after domestication. We identified significant changes among compound classes and single metabolites and associated the observed changes with selection during domestication by explicitly testing hypotheses using QST versus FST comparisons. To make claims about a role of selection in choosing diversity, appropriate tests of neutrality or direct measures of fitness are needed. Moreover, changes in kernel metabolite composition may also be attributed to correlations with plant and organ traits associated to domestication. In this respect, our results show that selection signatures concerning metabolic changes were, at least in some cases, independent from the variations at morpho-agronomic traits related to the domestication syndrome. This suggests that domestication and breeding have an unexpectedly large genetic basis. Moreover, our metabolomics data support the genome-wide effects of domestication identified using genome scans for signatures of selection in maize and common bean (Wright et al. 2005; Yamasaki et al. 2005; Hufford et al. 2012; Bellucci et al. 2014). Furthermore, although genomic scans cannot distinguish between selection and hitchhiking, testing directly the neutral distribution of phenotypic variation is less likely to be influenced by the reduced recombination at linked loci.
Even if we cannot estimate the number of loci involved in the observed metabolic changes, the relatively high number of metabolic features associated with the domestication of tetraploid wheats provides stronger support for models of domestications based on a relatively long transition from the cultivation of wild plants to the complete transition to domesticated crop (Gepts 2014). In addition, the correlation network analysis indicated that domestication and breeding have reshaped the metabolite coabundance patterns, as previously observed for transcriptomic data in common bean (Bellucci et al. 2014) and maize (Swanson-Wagner et al. 2012). Taken together, these results could explain the complex segregation observed in populations derived from hybridization between modern varieties and exotic germplasm, along with the difficulty to use in modern breeding wild relatives and landraces.
Here we report strong divergence among the three groups of tetraploid wheats, particularly for the amino acids alanine and valine and for the UFAs as a whole, with the largest effects for gondoic, linoleic, and arachidic acids. Altogether, it appears that the emergence of emmer mainly impacted the metabolism of the SFAs and UFAs, while the changes leading to durum wheat largely concerned amino acid metabolism. The possibility that the processes of domestication of tetraploid wheats have induced significant changes at the metabolomic level, and in particular that they might have affected amino acid and fatty acid biosyntheses, has been suggested for several crop species. Screens for signatures of selection conducted at genomic levels have identified genes that underlie subtle phenotypic changes, such as those supporting metabolic shifts. Such methods applied to the domestication process do not require any a priori assumption concerning the traits that were subject to selection.
In a seminal study, Wright et al. (2005) compared the sequence diversity between inbred maize and teosinte at 774 genes to determine the consequences of the domestication processes. The top 4% of genes that were identified as candidates under selection during domestication were enriched in functions related to amino acid biosynthesis. This was further confirmed by Yamasaki et al. (2005) with very high statistical support based on 1,200 maize genes. More recently, genome-wide resequencing of 75 wild, landrace, and improved maize lines (Hufford et al. 2012) identified genes with signals of selection stronger than those shown to control major morphological changes; in addition, several of these candidates are involved in nitrogen metabolism, like those encoding glutamine synthetase and nitrate reductase. In Heliantuhus annuus, a survey with 492 microsatellite loci derived from Expressed Sequence Tags of a large panel of wild accessions, landraces, and improved lines allowed the identification of genes under selection involved in amino acid synthesis and protein catabolism (Chapman et al. 2008).
Bellucci et al. (2014) considered the consequences of the domestication process of Phaseolus vulgaris, using RNAseq analysis; three genes were identified as having key roles in carbon/nitrogen interactions. In maize, the reshaping of the transcriptome during domestication includes a significant overrepresentation of genes related to amino acid salvage and the biosynthesis of the sulfur-containing amino acids (Swanson-Wagner et al. 2012).
The data provided here together with those from genomic scans for selection signatures suggest that, in several crops, selection acting on components of amino acid metabolism can lead to convergent phenotypic changes participating to the domestication syndrome. This is also coherent with the observation that improved growth due to heterosis may be due to changes in protein metabolism (Goff 2011).
We speculate that the selection for nutritional quality together with the selection for adaptation to a new agro-ecosystem characterized by a greater provision of nitrogen have acted as major factors dictating the need of change. In durum wheat, an important trait is gluten quality, which is mostly determined by the protein composition of the kernel. As shown by Laidò et al. (2013, 2014), the evolution of durum wheat has been associated with selection for protein composition, in particular with a strong reduction in glutenin diversity, the most important fraction responsible for gluten quality.
UFAs have exhibited a strong reduction across the step from wild emmer to emmer, and a further reduction in durum wheat. It can be hypothesized that during domestication these changes were associated with a genetic variation at a key step in UFA metabolism, as also suggested by the comparisons of the corresponding networks. In an analysis of several legume species, an undirected change in the content of fatty acids was found, showing an increase in some species and a decrease in others following domestication (Fernández-Marín et al. 2014). These authors argued that part of the detected changes might have made membranes less prone to oxidation and speculate that this modification was driven by selection for seed storability. Consistent with this hypothesis, in our experiment the reduction of UFAs was stronger than that for SFAs, which resulted in a decreasing UFA/SFA ratio. The step from wild emmer to emmer reduced the UFA/SFA ration by ∼9%, a third of the effect for the step emmer versus durum wheat (∼28%). It should be noted that a high UFA/SFA ratio in the human diet helps in the prevention of cardiovascular diseases (Mozaffarian and Wu 2011). Thus, our results indicating a reduction of the UFA/SFA ratio may suggest that not necessarily all of the metabolic variations that occurred during domestication have proceeded toward an amelioration of the nutritional quality. This is likely due to the observation that yield-related traits were given priority during the domestication process. Indeed, the reduction in fatty acids might have favored increased grain yield, as noted in an oat selection experiment (Holland et al. 2001), or higher kernel and flour conservation (Kopfler et al. 2012). Moreover, fatty acid composition affects seed odor and flavor because specific fatty acids have different susceptibilities to peroxidation leading to rancidity. Interestingly, the decrease in poly-UFAs, those most susceptible to oxidation, accompanied domestication of grain legumes (Fernández-Marín et al. 2014). In this regard, it is interesting to note that linoleic acid, the only poly-UFA detected, was under selection based on QST.
The correlation-based network analyses indicate that selection might have modified the metabolic structure in different pathways, but also during different evolutionary steps of tetraploid wheats. The present analysis supports a restructuring of the associations between measured metabolites more evident for the step emmer versus durum wheat of the domestication process.
We pointed out that there was concordance between the change in the node centrality between wild emmer and emmer and the values of QST for this contrast. We also noted that the network analysis suggested that wild emmer is closer to durum wheat than to emmer with respect to their network centrality measures. The reason for this finding is that the centrality measures, as determined in our analysis, take into consideration only the network structure, but not the values for the correlations based on which the networks are established. This opens the possibility for future research in the direction of developing theoretical models for changes in correlation network structure due to domestication.
In conclusion, this study indicates that selection appears to have operated to modify the metabolomic profile of durum wheat during its evolution. Overall, this work provides additional support for domestication models that assume a relatively slow transition from the cultivation of wild plants to the development of fully domesticated crops. Indeed, the larger the number of independent traits that have been modified by selection, the longer the transition to fully domesticated crop. Our findings illustrate that selection supporting a change in metabolites content might have operated partly independently of variations at well-known morpho-agronomic traits associated with the domestication syndrome, and that the observed metabolomic changes might be a result of modifications both in the relationships between the metabolites (i.e., regulation and participation in metabolic pathways) and between metabolites and phenotypic traits. This study also illustrates that selection might have operated in different directions. Indeed, primary domestication mainly involved changes in fatty acid metabolism, while during secondary domestication changes were more evident for the metabolism of amino acids. It is speculated that such changes might be consequences of crop adaptation to the agro-ecosystem, probably in relation to agricultural practices. Finally, as selection seems to have supported, at least in the case of UFAs, the reduction of some nutritional and quality traits, most likely in the wild and exotic germplasm useful traits can be identified and further incorporated into modern elite varieties to improve specific traits.
Materials and Methods
Plant Material
The tetraploid wheat (T. turgidum L., 2n = 4x = 28; AABB genome) collection classified according to van Slageren (1994) consisted of 37 accessions of wild emmer, emmer, and durum wheat as shown in table 1. The wild emmer and emmer wheat accessions were kindly provided by the National Small Grain Collection (Aberdeen, USA), John Innes Centre (Norwich, UK), the Institute of Crop Production, GeneBank Department (Czech Republic), and the CNR Institute of Plants Genetics (Bari, Italy). The durum wheat varieties represent a selection of Italian accessions (except “Neodur” and “Pedroso”) that were collected by the CREA Cereal Research Centre (Foggia, Italy) and the Department of Environmental and Agro-Forestry Biology and Chemistry, University Aldo Moro (Bari, Italy). Twenty plants of each accession were sown at Valenzano (Bari, Italy), and a single plant that represented the prevalent biotype of the accession was selected and grown to maturity to produce self-seed. The collection was grown in the year 2009/2010 under a conventional farming system in Foggia (southern Italy). The kernels were obtained from a randomized field experiment with three replicates, with the exception of T. dicoccum MG4387 and T. dicoccoides MG4343, MG4328/61, PI343446, PI355459, and PI470945, for which only two replicates were available. The samples were collected at the 11.4 stage (ripe for cutting, straw dead) of the Feekes scale (Feekes 1941).
Genotyping:DNA Extraction and SSR Analysis
To obtain a neutral benchmark for the genetic divergence among the three taxa and for each of the three possible pairs of contrasts, we conducted molecular marker assays with 26 microsatellite (SSR) loci. These loci were the same as those used to study the genetic diversity and population structure of a collection of tetraploid wheats (T. turgidum L.) (Laidò et al. 2013). The leaf tissue of the plants that represented the prevalent biotypes of the accessions was used for DNA extraction, according to the protocol described by Sharp et al. (1988). The wheat collection was genotyped with 26 SSR markers selected from the durum wheat maps developed by the Cereal Research Centre of Foggia (Italy) and the Department of Environmental and Agro-Forestry Biology and Chemistry of Aldo Moro University (Bari, Italy). The selection of the SSR markers was carried out considering the following criteria: locus-specific amplification, low complexity, robust amplification, and good genome coverage (one maker for each chromosome arm).The wheat DNA samples were divided into three plates, in which two control DNA samples of known molecular weight were included, to correct possible electrophoretic migration differences between groups according to Somers et al. (2007). Polymerase chain reaction (PCR) amplification was carried out in 15 μL volumes containing 2 μL DNA (≈80 ng), 10× PCR buffer (EuroClone), 0.4 μM of each microsatellite primer (the forward primers were fluorescently labeled), 1.5 mM MgCl2 (EuroClone), 0.2 mM of deoxynucleotide triphosphate (dNTP) mixture (Fermentas), and 1 U Taq DNA-polymerase (EuroClone). The PCR was carried out using a thermal cycler (BIO-RAD) as follows: 95 °C for 3 min, followed by 35 cycles of PCR (94 °C for 30 s, Ta specific for each primer for 30 s, 72 °C for 1 min), and a final extension at 72 °C for 10 min. The PCR products were detected by capillary electrophoresis using an ABI PRISM 3130 Analyser and analyzed using GeneMapper version 4.0 genotyping software. The internal molecular weight standard was 500-ROX.
Metabolite Profiling
After collection, the samples were freeze dried, milled using a laboratory mill (Udy-Cyclone 1093 Foss Tecator), passed through a 0.5-mm sieve, and stored at −25 °C until analysis. The samples were analyzed within 3 months of the freeze drying. The extraction, derivatization, and analysis of these samples by gas chromatography–mass spectrometry (GC-MS) for the profiling of the polar and nonpolar metabolites were performed following protocols described previously (Beleggia et al. 2013) as part of the metabolomics platform of CRA-CER (Foggia). Briefly, 100-mg dry weight of each sample was extracted using a mixture of methanol, ultrapure water, and CHCl3 (1:1:3 v/v/v). The samples were stored at 4 °C for 30 min, and then centrifuged at 4,000 × g for 10 min. Aliquots (50 μl) of the polar and nonpolar phases (500 μl) were dried in a Speed-vac for further analysis. The polar residues were redissolved and derivatized for 90 min at 37 °C in methoxyamine hydrochloride in pyridine (70 μl; 20 μg/ml), followed by incubation with N-methyl-N-(trimethylsilyl)trifluoro-acetamide (MSTFA, 120 μl) at 37 °C for 30 min. The nonpolar fraction was redissolved and derivatized for 30 min at 37 °C in MSTFA (70 μl). The polar and nonpolar metabolites were analyzed using GC (Agilent 6890N; Agilent Technologies, USA) coupled with quadrupole MS (Agilent 5973; Agilent Technologies) (Beleggia et al. 2013). The chromatograms and mass spectra were evaluated using the AMDIS program, while the GC-MS quantification was performed using a Chemstation program. The metabolites were identified by comparing the mass spectroscopy data with those of the NIST 2008 database and with a custom library obtained with reference compounds, while the absolute contents of the polar and nonpolar metabolites were determined by comparisons with standard calibration curves obtained in the ranges of 0.04–2.00 ng and 0.05–3.00 ng, respectively. For both polar and nonpolar profiling, the batches for analysis included 17 runs with 2 calibration standard mixes at the beginning and end of each batch. Before the GC-MS injection, the samples were randomized, with the instrumental performance monitored by internal standards added after the extractions. The standards and all of the chemicals used (HPLC grade) were from Sigma-Aldrich Chemical Co. (Deisenhofen, Germany), while MSTFA was from Fluka.
Statistics
Genotyping
To determine the levels of genetic divergence among the T. turgidum taxa, we calculated the FST statistics (Wright 1931) adopting analysis of molecular variance (Excoffier et al. 1992) and considering the individuals belonging to each taxon as a population. In this framework, the FST was calculated as , where is the (co)variance of the SSR allele frequencies between populations (taxa) and is the total variance (the sum of the between and within population [taxa][co]variance components, ). The significance of FST was determined by permuting individuals across populations (105 randomizations) with the Arlequin version 3.5 software (Excoffier and Lischer 2010).
We first calculated FST considering all of the 26 SSR loci. However, such estimates can be seen as the results of selective and nonselective (neutral) processes. To disentangle these effects, as far as possible, and to obtain estimates of the level of (putatively) neutral population divergence, we used a two-step approach: 1) We applied an FST-based outlier test of selection to identify loci that were putatively under selection, that is, with “too high” or “too low” observed FST values compared with the simulated neutral expectations; and (2) we discarded the outlier loci and we retained the putatively neutral SSR loci only. These two steps were reiterated until no SSR loci with signature of selection were detected. Estimates of “neutral” FST were then obtained considering the final “neutral data set.” To detect signatures of selection, we adopted the FDIST approach (Beaumont and Nichols 1996; Beaumont and Balding 2004), implemented in Arlequin version 3.5, with 106 simulations. In particular, in this method, the distribution of FST across loci as a function of heterozygosity between populations is obtained by performing simulations under a finite-island model. The outlier loci were those present in the tails of the generated distribution.
Metabolite Profiling
For the statistical analyses here we considered as quantitative traits the total content of each of the 9 classes of metabolites: Amino acids, organic acids, polyols, sugars, SFAs, UFAs, fatty alcohols, tocopherols, and phytosterols, as given by the levels of each of the single 51 metabolites determined for these Triticum genotypes. Three biological and three analytical replicates of each genotype were considered. We processed the data using two different statistical models. First, we adopted the nested analysis of variance (NANOVA) using the REML procedure. For each quantitative trait (metabolite level), this analysis allowed partitioning of the total variance () into genetic variance components due to differences between taxa (), between genotypes within taxa (), and to error/environment (arising among individuals of the same genotype; that is, among the three replicates) (). Error/environmental variance () was taken away from the total phenotypic variance () to determine the total genetic variance (). We then calculated the heritability of each quantitative (metabolite level) trait as . However, it is possible that variations in the metabolite levels among these taxa were due to correlation with other phenotypic traits that were associated to selection during primary domestication and secondary domestication (e.g., to pleiotropic effects). For this reason, we studied the correlations between the metabolite levels and seven other seed traits that were putatively under selection during wheat evolution: EW, seed weight without embryo (indicated as ENW), EW/TW, 1,000 KW, ENW/TW, ENW/EW, and pc. To obtain QST estimates corrected as equalizing taxa for seed traits (henceforth: QST1), we applied ANCOVA. If a covariate is highly related to another covariate (at a correlation of 0.5 or more), then it will not adjust the dependent variable over and above the other covariates. Thus, we reduced dimensionality of data by applying PC analysis. The seven original covariates can be summarized by three PCs (see Results) that we treated as LCs. First, for each metabolite, we fit the model including two main terms (taxa, genotypes within taxa), three covariates (LC1, LC2, LC3), and three interactions (LC1 × taxa, LC2 × taxa, LC3 × taxa). For each metabolite, we decided that when at least one of the covariate × taxa interactions was significant, ANCOVA was not applicable; that is, the homogeneity of slopes cannot be assumed if interactions are significant, and ANCOVA cannot be used to “equalize” taxa for seed traits. For the metabolites for which all three interactions were not significant, we reran ANCOVA without entering interaction terms. In this case, we set taxa and genotypes within taxa as random factors and we calculated the corrected between and within taxa variance components using the REML procedure. These variances were then used to determine the QST1 values. Thus, this analysis allowed the differences among taxa for the metabolite levels to be tested while factoring out differences for covariate variables; that is, as if the phenotypic traits have the same value for all three of the taxa. As we treated the three botanical taxa of T. turgidum as populations, for each quantitative trait the level of among-taxa divergence was estimated using the QST statistic through equation (1) (Bonnin et al. 1996):
| (1) |
where is the between-taxa genetic variance component, is the within-taxa genetic variance component, and FIS is the inbreeding coefficient that measures the reduction in the heterozygosity observed within individuals relative to expectation assuming the Hardy–Weinberg equilibrium. It should be noted that with our design, includes both additive and nonadditive components. Consideration of nonadditive gene actions appears to contribute to underestimation of QST (Whitlock 1999). However, when the goal was to study diversifying selection, the test will remain conservative (Kronholm et al. 2010).
Given the strict autogamy of T. turgidum (Chabra and Sethi 1991; Nevo 2011) and the results of the analysis of polymorphism at 26 SSR loci where no heterozygotes were found (not shown), we posed FIS = 1, giving equation (2) (Bonnin et al. 1996):
| (2) |
Due to the use of two statistical models, we calculated two sets of QST statistics, one named QST (not using covariates) and the second QST1 (using covariates). We obtained QST and QST1 values to estimate the mean divergence among the three taxa, as also between the three possible pairs of taxa. These comparisons allowed disentangling the effects due to primary domestication (wild emmer vs. emmer) from those due to secondary domestication events (emmer vs. durum wheat). The comparison of wild emmer versus durum wheat represents the combination of these two effects. For metabolites and classes of compounds that were determined as significantly different from the previous comparisons, mean differences for the taxa were tested using Tukey’s tests. The two NANOVA models were investigated using the JMP software (SAS Institute Inc., version 8). To test the significance of the observed QST, we used the method of Whitlock and Guillaume (2009). Instead of directly comparing estimates of QST (the divergence for a quantitative trait) with estimates of FST (the molecular divergence used as a neutral benchmark), this method compares the observed QST with the null distribution of QST expected for neutrally evolving traits. In particular, the method predicts the sample variance that would be expected from QST of a neutral trait by simulating it with information on (neutral) FST and the within-population additive variance of the trait. Specifically, we followed these steps:
Determination of the expected among-population variance component for a neutral trait, which is given by , where in our case FST is the genetic divergence among taxa estimated using neutral SSR markers.
Provide parametric estimation of the sampling distribution of by multiplying by a random number, drawn from a χ2 distribution with two degrees of freedom [d.f. = 3 (i.e., the number of taxa) − 1] (Whitlock 2008).
Calculation of the expected QST of a neutral trait using the measured within-population variation, , and the expected among-population variation , following equation (2).
For each trait and for each comparison among taxa, we obtained the distribution of the test statistic QST − FST from 500,000 repetitions. The resulting P-value was determined by observing the quantile of the simulated distribution that had more extreme values than the observed value. Finally, we divided the metabolites into two groups, those with significant QST (sQST) and those with not significant QST (nsQST). We also divided the metabolites into two groups based on the significance of the interactions species × LCs, considering the three taxa simultaneously (sINT, nsINT). We then tested the associations between the significance of the QST and the significance of the interactions species × LCs, using contingency analysis and Fisher exact tests. We also tested the hypothesis that the sINT and nsINT groups differ for the mean QST level, using one-way ANOVA.
Correlation and Network Analysis
Here, we first determined the correlations based on the metabolite profiles from each of the three wheat populations. Prior to the analysis of correlations, the missing values for the contents of the measured metabolites and compound classes were imputed with a nonparametric approach based on random forest (Stekhoven and Buehlmann 2012), which has been shown to outperform other approaches with respect to the imputation error (Waljee et al. 2013). To this end, we used the R package missForest. Although analysis of the differential behaviors of the levels of the metabolites can indicate quantitative differences between the wheat species, this approach focussed instead on the correlative behavior of individual metabolites across the three wheat populations. To investigate the metabolic profiles with respect to these scenarios, we first used the Pearson correlation coefficient, which we in turn use to derive networks. The correlations were controlled for the effects of the seven phenotypic traits (partial correlations). To arrive at correlations from which the effects of these seven phenotypic traits were partially out, denoted by , we used the approach of Velicer (1976) according to equation (3):
| (3) |
where is the covariance between metabolites, is the covariance between the metabolic and phenotypic traits, is the covariance between the phenotypic traits, and diag denotes the operator for a diagonal matrix. The derived partial correlations were tested with permutation tests (separate permutation of the replicates for each metabolite) with B repetitions, and empirical P-values were given by the ratio of the number of more extreme observations incremented by one and (B + 1). We noted that only partial correlations at prespecified FDR were retained for the analysis (FDR 5%). Moreover, the correlation structure can be represented by a network in which nodes represent metabolites and edges denote the presence of significant correlation over all 1,000 imputations. The network structure can be used to determine the metabolites for which the connections to the rest of the network change the most between two species. In addition, using various centrality measures, it was possible to investigate the changes in the position/role of metabolites in the network of data-established associations. In such a way, this moves away from investigating only the local aspects of the effects of a metabolite, and instead brings about the systems point of view. In addition, network properties can be used to further describe the global differences between the structures of significant correlations between species, and how these arise from local differences. We thus obtained three taxon-specific, correlation-based networks (i.e., for wild emmer, emmer, durum wheat). To characterize each network, we calculated the following seminal properties (Newman 2003) of the derived correlation-based networks at an FDR of 5%: Average path length, diameter, number of components, and isolated nodes. Also, to gain insight into the relative importance of each node (metabolite) within a network, we obtained five different measures of centrality: Node degree, eigenvalue, betweenness, closeness, and subgraph. Thus, to quantify differences in the metabolite correlation structure among taxa, we determined the agreement of the node centrality measures between the taxon-specific correlation-based networks. Moreover, to further determine the effects of selection (as measured by QST) on the rewiring of the metabolic profiling of the wheat seeds during domestication, we determined the concordance between QST and the differences in the node centrality measures between taxon-specific networks. In both cases, the degree of agreement was quantified by Person’s r coefficients of correlation. The correlation and network-based analyses were carried out in R. To calculate the network properties, we used the R package igraph (Csardi and Nepusz 2006).
Supplementary Material
Supplementary tables S1–S4 and figure S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgement
This work was supported by the PON a3 PlASS project.
References
- Bar-Yosef O. 1998. The Natufian culture in the Levant, threshold to the origins of agriculture. Evol Anthropol. 6(5):159–177. [Google Scholar]
- Barbeau WE, Hilu KW. 1993. Protein, calcium, iron, and amino-acid content of selected wild and domesticated cultivars of finger millet. Plant Foods Hum Nutr. 43(2):97–104. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Balding DJ. 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 13:969–980. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. 1996. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc B. 263:1619–1626. [Google Scholar]
- Beleggia R, Platani C, Nigro F, De Vita P, Cattivelli L, Papa R. 2013. Effect of genotype, environment and genotype-by-environment interaction on metabolite profiling in durum wheat (Triticum durum Desf.) grain. J Cereal Sci. 57:183–192. [Google Scholar]
- Bellucci E, Bitocchi E, Ferrarini A, Benazzo A, Biagetti E, Klie S, Minio A, Rau D, Rodriguez M, Panziera A, et al. 2014. Decreased nucleotide and expression diversity and modified co-expression patterns characterize domestication in the common bean. Plant Cell 26:1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin I, Prosperi JM, Olivieri I. 1996. Genetic markers and quantitative genetic variation in Medicago truncatula (Leguminosae): a comparative analysis of population structure. Genetics 143:1795–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabra AK, Sethi SK. 1991. Inheritance of cleistogamic flowering in durum wheat (Triticum durum). Euphytica 55(2):147–150. [Google Scholar]
- Chapman MA, Burke JM. 2012. Evidence of selection on fatty acid biosynthetic genes during the evolution of cultivated sunflower. Theor Appl Genet. 125:897–907. [DOI] [PubMed] [Google Scholar]
- Chapman MA, Pashley CH, Wenzler J, Hvala J, Tang S, Knapp SJ, Burke JM. 2008. A genomic scan for selection reveals candidates for genes involved in the evolution of cultivated sunflower (Helianthus annus). Plant Cell 20:2931–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Gao Y, Xie W, Gong L, Lu K, Wang W, Li Y, Liu X, Zhang H, Dong H, et al. 2014. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat Genet. 46:714–721. [DOI] [PubMed] [Google Scholar]
- Csardi G, Nepusz T. 2006. The igraph software package for complex network research. InterJ Complex Syst. 1695. [Google Scholar]
- Estrada E, Rodriguez-Velazquez JA. 2005. Subgraph centrality in complex networks. Phys Rev. E71:056103.. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 10:564–567. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse P, Quattro J. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feekes W. 1941. De Tarween haar milieu. Verslogen van de Technische Tarwe Commissie, XVII Editor Gromingen:Hoitsema, 560–561
- Fernández-Marín B, Milla R, Martín-Robles N, Arc E, Kranner I, Becerril JM, García-Plazaola JI. 2014. Side-effects of domestication: cultivated legume seeds contain similar tocopherols and fatty acids but less carotenoids than their wild counterparts. BMC Plant Biol. 14:1599.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS. 2015. Evolution is an experiment: assessing parallelism in crop domestication and experimental evolution: (Nei Lecture, SMBE 2014, Puerto Rico). Mol Biol Evol. 32:1661–1671. [DOI] [PubMed] [Google Scholar]
- Gepts P. 2014. The contribution of genetic and genomic approaches to plant domestication studies. Curr Opin Plant Biol. 18:51–59. [DOI] [PubMed] [Google Scholar]
- Gepts P, Papa R. 2002. Evolution during domestication In: Encyclopedia of life sciences. London: Macmillan Publishers, Nature Publishing Group; p. 1–7. [Google Scholar]
- Gioia T, Nagel KA, Beleggia R, Fragasso M, Ficco DBM, Pieruschka R, De Vita P, Fiorani F, Papa R. 2015. The impact of domestication on the phenotypic architecture of durum wheat under contrasting nitrogen fertilisation. J Exp Bot. 66:5519–5530. [DOI] [PubMed] [Google Scholar]
- Goff SA. 2011. A unifying theory for general multigenic heterosis: energy efficiency, protein metabolism, and implications for molecular breeding. New Phytol. 189:923–937. [DOI] [PubMed] [Google Scholar]
- Golan G, Oksenberg A, Peleg Z. 2015. Genetic evidence for differential selection of grain and embryo weight during wheat evolution under domestication. J Exp Bot. 66:5703–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T, Pélabon C, Houle D. 2011. Heritability is not evolvability. Evol Biol. 38:258–277. [Google Scholar]
- Holland JB, Frey KJ, Hammond EG. 2001. Correlated responses of fatty acid composition, grain quality and agronomic traits to nine cycles of recurrent selection for increased oil content in oat. Euphytica 122:69–79. [Google Scholar]
- Houle D. 1992. Comparing evolvability and variability of quantitative traits. Genetics 130:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufford MB, Xu X, van Heerwaarden J, Pyhäjärvi T, Chia JM, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM, et al. 2012. Comparative population genomics of maize domestication and improvement. Nat Genet. 44:808–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleessen S, Antonio C, Sulpice R, Laitinen R, Fernie AR, Stitt M, Nikoloski Z. 2012. Structured patterns in geographic variability of metabolic phenotypes in Arabidopsis thaliana. Nat Commun. 3:1319.. [DOI] [PubMed] [Google Scholar]
- Kopfler K, Prevo L, Thralls T, Tupper K. 2012. Effect of Flour Age on Sensory Evaluation of Whole Wheat Bread. Bastyr University Food Science, LabTR5115L-D, December 4th, 1–10.
- Kronholm I, Loudet O, De Meaux J. 2010. Influence of mutation rate on estimators of genetic differentiation—lessons from Arabidopsis thaliana. BMC Genet. 11:88.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidò G, Mangini G, Taranto F, Gadaleta A, Blanco A, Cattivelli L, Marone D, Mastrangelo AM, Papa R, De Vita P. 2013. Genetic diversity and population structure of tetraploid wheats (Triticum turgidum L.) estimated by SSR, DArT and pedigree data. PLoS One 8(6):e67280.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laidò G, Marone D, Russo MA, Colecchia S, Mastrangelo AM, De Vita P, Papa R. 2014. Linkage disequilibrium and genome-wide association mapping in tetraploid wheat (Triticum turgidum L.). PLoS One 9(4):e9521.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen T, McCairns RS, O'Hara RB, Merilä J. 2013. QST–FST comparisons: evolutionary and ecological insights from genomic heterogeneity. Nat Rev Genet. 14:179–190. [DOI] [PubMed] [Google Scholar]
- Lenser T, Theißen G. 2013. Molecular mechanisms involved in convergent crop domestication. Trends Plant Sci. 18(12):704–714. [DOI] [PubMed] [Google Scholar]
- Meyer RS, Purugganan MD. 2013. Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet. 14:840–852. [DOI] [PubMed] [Google Scholar]
- Mozaffarian DMD, Wu JHY. 2011. Omega-3 fatty acids and cardiovascular disease effects on risk factors, molecular pathways, and clinical events. J Am Coll Cardiol. 58(20): 2047–2067. [DOI] [PubMed] [Google Scholar]
- Nesbitt M, Samuel D. 1998. Wheat domestication: archaeobotanical evidence. Science 279(5356):1431–1431.9508710 [Google Scholar]
- Nevo E. 2011. Triticum In: Kole C, editor. Wild crop relatives: genomic and breeding resources. Berlin/Heidelberg: Springer; p. 407–456. [Google Scholar]
- Newman MEJ. 2003. The structure and function of complex networks. SIAM Rev. 45(2):167–256. [Google Scholar]
- Olsen KM, Wendel JF. 2013a. Crop plants as models for understanding plant adaptation and diversification. Front Plant Sci. 4:290.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen KM, Wendel JF. 2013b. A bountiful harvest: genomic insights into crop domestication phenotypes. Annu Rev Plant Biol. 64:47–70. [DOI] [PubMed] [Google Scholar]
- Rao J, Cheng F, Hu C, Quan S, Lin H, Wang J, Chen G, Zhao X, Alexander D, Guo L, et al. 2014. Metabolic map of mature maize kernels. Metabolomics 10(5):775–787. [Google Scholar]
- Riedelsheimer C, Lisec J, Czedik-Eysenberg A, Sulpice R, Flis A, Grieder C, Altmann T, Stitt M, Willmitzer L, Melchinger AE. 2012. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc Natl Acad Sci U S A. 109(23):8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PJ, Kreis M, Shewry PR, Gale MD. 1988. Location of β-amylase sequences in wheat and its relatives. Theor Appl Genet. 75:286–290. [Google Scholar]
- Skogerson K, Harrigan GG, Reynolds TL, Halls SC, Ruebelt M, Landolino A, Pandravada A, Glenn KC, Fiehn O. 2010. Impact of genetics and environment on the metabolite composition of maize grain. J Agric Food Chem. 58:3600–3610. [DOI] [PubMed] [Google Scholar]
- Somers DJ, Banks T, DePauw R, Fox S, Clarke J, Pozniak C, McCarteney C. 2007. Genome-wide linkage disequilibrium analysis in bread wheat and durum wheat. Genome 50:557–567. [DOI] [PubMed] [Google Scholar]
- Sotelo A, Sousa H, Sanchez M. 1995. Comparative study of the chemical composition of wild and cultivated beans (Phaseolus vulgaris). Plant Foods Hum Nutr. 47:93–100. [DOI] [PubMed] [Google Scholar]
- Stekhoven DJ, Buehlmann P. 2012. MissForest—non-parameteric missing value imputation for mixed-type data. Bioinformatics 28(1):112–118. [DOI] [PubMed] [Google Scholar]
- Swanson-Wagner R, Briskine R, Schaefer R, Hufford MB, Ross-Ibarra J, Myers CL, Tiffin P, Springer NM. 2012. Reshaping of the maize transcriptome by domestication. Proc Natl Acad Sci U S A. 109:11878–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno KI, Willcox G. 2006. How fast was wild wheat domesticated? Science 311:1886.. [DOI] [PubMed] [Google Scholar]
- Toubiana D, Fernie AR, Nikoloski Z, Fait A. 2013. Network analysis: tackling complex data to study plant metabolism. Trends Biotechnol. 31(1): 29–36. [DOI] [PubMed] [Google Scholar]
- van Slageren MW. 1994. Wild wheats: a monograph of Aegilops L. and Amblyopyrum (Jaub. &Spach) Eig (Poaceae). Wageningen Agriculture University Papers 7:513.
- Velicer WF. 1976. Determining the number of components from the matrix of partial correlations. Psychometrika 41(3):321–327. [Google Scholar]
- Waljee A, Mukherjee AG, Singal AG, Zhang Y, Warren J, Balis U, Marrero J, Zhu J, Higgins PDR. 2013. Comparison of imputation methods for missing laboratory data in medicine. BMJ Open 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RL, Stec A, Hey J, Lukens L, Doebley J. 1999. The limits of selection during maize domestication. Nature 398:236–239. [DOI] [PubMed] [Google Scholar]
- Wen W, Li D, Li X, Gao Y, Li W, Li H, Liu J, Liu H, Chen W, Luo J, et al. 2014. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat Commun. 17(5):3438.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC. 1999. Neutral additive genetic variance in a metapopulation. Genet Res. 74:215–221. [DOI] [PubMed] [Google Scholar]
- Whitlock MC. 2008. Evolutionary inference from QST. Mol Ecol. 17:1885–1896. [DOI] [PubMed] [Google Scholar]
- Whitlock MC, Guillaume F. 2009. Testing for spatially divergent selection: comparing QST to FST. Genetics 183(3):1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. 1931. Evolution in Mendelian populations. Genetics 16(2):97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS. 2005. The effects of artificial selection on the maize genome. Science 308:1310–1314. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Tenaillon MI, Vroh Bi I, Schroeder SG, Sanchez-Villeda H, Doebley JF, Gaut BS, McMullen MD. 2005. A large-scale screen for artificial selection in maize identifies candidate agronomic loci for domestication and crop improvement. Plant Cell 17(11):2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Allen WB, Roesler K, Williams ME, Zhang S, Li J, Glassman K, Ranch J, Nubel D, Solawetz W, et al. 2008. A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat Genet. 40:367–372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.