

Abstract
Immunotherapy for brain cancer has evolved dramatically over the past decade, owed in part to our improved understanding of how the immune system interacts with tumors residing within the central nervous system (CNS). Glioblastoma (GBM), the most common brain tumor in adults, carries a poor prognosis (<15 months) and only few advances have been made since the FDA’s approval of temozolomide (TMZ) in 2005. Importantly, several immunotherapies have now entered patient trials based on promising preclinical data, and recent studies have shed light on how GBM employs a slew of immunosuppressive mechanisms which may be targeted for therapeutic gain. Altogether, accumulating evidence suggests immunotherapy may soon earn its keep as a mainstay of clinical management for GBM.
Areas Covered
Here, we review cancer vaccines, checkpoint inhibitors, T-cell immunotherapy, and oncolytic virotherapy.
Expert Opinion
Checkpoint blockade induces antitumor activity by preventing negative regulation of T-cell activation. This platform, however, depends on an existing frequency of tumor-reactive T cells, and GBM is weakly immunogenic and GBM patients are typically immunocompromised. Therefore, checkpoint blockade may be most effective when used in combination with a DC vaccine or adoptively transferred tumor-specific T cells generated ex vivo. Both approaches have been shown to induce endogenous immune responses against tumor antigens, providing a rationale for use with checkpoint blockade where both primary and secondary responses may be potentiated.
Keywords: CDX-110, Checkpoint inhibitor, Chimeric antigen receptor T cells, Dendritic cell vaccine, EGFRvIII, GBM, Glioblastoma multiforme, Glioblastoma, Immune system, Immunotherapy, Ipilimumab, Monoclonal antibody, Nivolumab, Oncolytic virus, Peptide vaccine, Rindopepimut, Tumor lysate vaccine
1. Background
Gliomas are the most common primary malignant brain tumors in adults and include ependymomas, oligodendrogliomas, and astrocytomas. The highest grade astrocytoma, glioblastoma (GBM), is the most prevalent in adults and affects 3.19 in 100,000 individuals in the United States.1, 2 GBM typically presents as a heterogeneous lesion in the white matter with a hypodense necrotic core, surrounded by a ring of enhancement that is usually the target of most conventional therapies. Diagnosing GBM and measuring tumor responsiveness to therapy can be difficult through standard imaging alone; contrast or post-contrast findings can result from vascular leakage, iatrogenic impact, or physiological changes within the brain parenchyma, rather than reflecting strict changes in tumor size. For these reasons, radiological (and clinical) criteria defining tumor progression are incomplete, and overall survival (OS) is considered the most clinically useful metric in evaluating outcomes. Despite an aggressive clinical standard of care, patients with GBM carry a median survival of less than 15 months and a 5 year survival rate of just 2–4%, drawing significant interest in novel therapies to improve patient outcomes.3–6
2. Medical need
GBM is clinically debilitating for afflicted patients, manifesting with seizures, nausea, vomiting, headaches, and progressive deficits in memory, personality, and neurologic function.7–9 Corticosteroids are commonly used to counteract neurological symptoms caused by peritumoral edema, but steroid use can result in substantial side effects that can sometimes be intolerable. Surgery and chemotherapy are requisite to relieve mass effect and provide palliation, but tumor recurrence is inevitable and patients will uniformly succumb to their disease. With the exception of temozolomide (TMZ), relatively few advances have translated into standard therapies for GBM patients despite promising preclinical therapeutics that have recently come of age. This may be explained, in part, by small patient population size, which can complicate patient recruitment for well-powered clinical studies and discourage investment from the pharmaceutical industry for developing novel therapies. Moreover, GBM places a significant financial burden on the healthcare system, due in part to frequent inpatient visits which can catapult expenditure for the first year of standard of care treatment to more than $184,000 in the United States.8, 9 In fact, brain tumor treatments are among the costliest with the least return, raising ethical considerations in this new climate of healthcare reform. These insurmountable costs in combination with poor quality of life and prognosis have created a desperate need for more effective and safe therapies.
3. Existing treatment
The current standard of care was defined in 2005 after the European Organization for Research and Treatment of Cancer (EORTC) reported results from a randomized phase III clinical trial showing that the addition of TMZ to postoperative fractionated radiotherapy significantly improved median survival to 14.1 months compared to 12.1 months with radiotherapy alone in patients with newly-diagnosed GBM.7 A long-term follow confirmed that this survival advantage lasts for at least 5 years,10 but tumors ultimately recurred and caused death in most patients. Although TMZ represents a major clinical advance for GBM (the first in several decades), there remains a challenge to further improve outcomes, as overall survival is <10% 5 years after follow-up. Moreover, radiation and TMZ are non-specific therapies by nature and can cause collateral damage to normal tissues, leading to systemic and sometimes intolerable side effects, including myelosuppression and increased risk of opportunistic infections.11, 12 The limitations of the current approach are underscored by the fact that complete tumor resection is usually impossible as tumor boundaries can be indistinguishable from normal brain, and even in cases where they are, the extent of resection may be limited by the need to preserve eloquent cortex. Novel techniques, however, are beginning to safely enhance the extent of resection through the use of fluorescence guided surgery (e.g. using 5-aminolevulinic acid), which has resulted in an increase in the number of macroscopically complete resections. Furthermore, GBM cells are highly infiltrative,13 and single cells have been shown to migrate into regions distant from initial tumor mass and well beyond radiographically defined boundaries of tumor burden. In sum, these factors may explain why GBM recurrence is nearly inevitable.14
4. Current research goals
Patients with GBM require novel therapies which target residual tumor tissue within resection cavities as well as migratory micro-metastases elsewhere in the brain, and in a manner that is highly-specific so as to avoid off-target damage to normal tissues. Immunotherapy has emerged as an ideal strategy to meet this need. Immunotherapy aims to establish anticancer immune responses through a number of distinct mechanisms, and is particularly attractive for malignant brain tumors given that specialized immune cells have migratory capacity, a mechanism to discriminate between normal and neoplastic tissue, and development of immunological memory. Importantly, the ability of T cells to extravasate from vasculature and migrate within the brain parenchyma in response to chemotactic cues may be a critical feature that prevents highly invasive tumor cells from escaping therapy.15 This migratory behavior overcomes limitations of drug delivery imposed by variable intratumoral microvasculature, which can disproportionately concentrate drug when relying on diffusion gradients alone. The pharmacokinetics and limitations of TMZ diffusion, for example, have been well described, and other chemotherapeutic agents that rely on similar delivery methods carry the same constraint.16, 17 An impressive number of immunotherapies have entered the clinical arena for formal evaluation in phase I-III trials following decades of preclinical development. Among the most notable include cancer vaccines using peptides or autologous antigen-presenting dendritic cells (DCs), monoclonal antibodies (mAbs) designed to overcome immune checkpoints, and adoptive T-cell immunotherapy wherein tumor-specific T-cells are generated ex vivo and transferred back into hosts to mount antitumor responses. Select immunotherapies for GBM under current investigation in phase I-III clinical trials are summarized in Table 1.
Table 1.
Select immunotherapies under clinical evaluation for GBM.
| Class | Company | Treatment | Target | Phase | Identifier |
|---|---|---|---|---|---|
|
| |||||
| Peptide vaccines | Stemline Therapeutics |
SL-701 | IL-13Rα, EphA2, survivin | I/II | NCT02078648 |
| Agenus | HSPPC-96 | heat shock protein | II | NCT01814813 | |
| Epitopoietic | ERC-1671 | Tumor lysate and cells from 3 donor patients | II | NCT01903330 | |
|
| |||||
| DC vaccines | – | CMV-specific DCs | CMV antigen pp65 | II | NCT02366728 |
| Northwest Biotherapeutics | DC-VaxL | Antigens derived from patient tumor | III | NCT00045968 | |
| – | DCs pulsed with tumor lysate | Antigens derived from patient tumor | II | NCT01204684 | |
| ImmunoCellular Therapeutics/Targepeutics | ICT107 | HER2, TRP-2, gp100, MAGE-1, IL-13Rα, AIM-2 | II | NCT01280552 | |
|
| |||||
| Checkpoint inhibitors | Tracon Pharmaceuticals/Santen | TRC105 | endoglin | I/II | NCT01648348 |
|
| |||||
| Gilead Sciences | Ipilimumab | CTLA-4 PD-1 VEGF |
III | NCT02017717 | |
|
| |||||
| Ono Pharmaceutical/Bristol-Myers Squibb | Nivolumab | ||||
|
| |||||
| – | Bevacizumab | ||||
|
| |||||
| CureTech/Medivation/Teva | Pidilizumab | PD-1 | I/II | NCT01952769 | |
|
| |||||
| Ipilimumab Nivolumab |
CTLA-4 PD-1 |
I | NCT02311920 | ||
|
| |||||
| Adoptive transfer | – | CAR T Cells | EGFRvIII | I/II | NCT01454596 |
| – | CAR T Cells | IL-13Rα2 | I | NCT02208362 | |
| – | CAR T Cells | Her2 | I | NCT01109095 | |
|
| |||||
| Oncolytic virotherapy | VectorLogics (now DNAtrix) | DNX2401 | Tumor selective adenovirus | I | NCT01956734 |
| Virttu | HSV1716 | Tumor selective herpes simplex virus | I | NCT02031965 | |
| DNX2401 | Tumor selective adenovirus | I | NCT02197169 | ||
| – | PVSRIPO | Tumor selective poliovirus | I | NCT01491893 | |
5. Scientific Rationale
The immune system’s role in cancer development and rejection was first established in the 19th century, when scientists noted remission of established solid tumors in febrile patients.18 This correlation was documented in 1893, when Coley and colleagues published their seminal observations of sarcoma regression following repeated Streptococcus inoculations, hypothesizing that an underlying infection nonspecifically activated the immune system to control tumor progression.19 Since this instrumental finding, the clinical interest in utilizing the immune system for cancer therapy has grown in parallel with our understanding of the mechanisms that govern humoral and cell-mediated immunity. The advent and evolution of DNA recombinant technology, molecular biology, and genetic sequencing have revealed the interplay between the immune system and recognition of aberrant neoplastic cells.20, 21 Carcinomas, for example, are estimated to contain up to 11,000 de novo genomic alterations, many of which create novel antigens that serve as potential sites for immune recognition that may be leveraged to generate antitumor immune responses.22 GBM is a tumor exemplified by genomic alterations with defined targets that have been exploited for this purpose. The type III mutation of the epidermal growth factor receptor (EGFRvIII), for example, confers tumorigenic and invasive properties to tumor cells, is not expressed by normal cells, and is found in greater than 40% of patients with high-grade gliomas.23–28 Other GBM targets of note include mutations in the isocitrate dehydrogenase (IDH) gene, viral antigens resulting from the expression of human β-herpesvirus Cytomegalovirus within GBM (e.g. pp65 and IE1), and tumor-associated antigens such as Her2/neu, Trp-2, and gp100.29–34 The recent identification of these targets has made it possible to evaluate novel immunotherapies in experimental animal models and early trials in humans, and importantly, it has become clear that such therapies can readily traverse the blood-brain barrier to mount meaningful responses against malignant tumors within the CNS.35, 36
6. Competitive environment
Cancer cells can express aberrant proteins as cell-surface targets or as peptide antigens through class I and II major histocompatibility complex (MHC) molecules, which are recognized by CD8+ cytotoxic T-cell lymphocytes (CTLs) and CD4+ helper T cells, respectively. Recently, high-throughput technologies and whole-exomic sequencing have allowed us to identify cancer mutations that can be recognized by T cells.37 Not all mutations can be recognized, however, and new MHC prediction algorithms are attempting to narrow the pool of targetable epitopes by determining the relative binding strength of their respective peptides to individual MHC molecules.38 By selecting a single candidate or pool of immunogenic peptides, for example, it is possible to inoculate patients with peptides to stimulate endogenous immune responses against tumor-specific (TSA) or tumor-associated antigens (TAAs). Similarly, DCs can also be pulsed with peptides, total tumor lysate, or RNA encoding the target antigen(s) and administered as immunotherapy. These cancer vaccines are designed in principle as an active immunization, and have drawn tremendous enthusiasm based on the success of this approach in prophylaxis against viral infection.
6.1 Peptide vaccines
The EGFRvIII vaccine for GBM is an example of the peptide-based approach and represents one of the very few cancer vaccines to have entered phase III clinical trials for GBM. This vaccine consists of a 14-amino acid peptide derived from the EGFRvIII neoepitope (PEPvIII) conjugated to the highly immunogenic carrier protein keyhole limpet hemocyanin (KLH), and is admixed with the potent adjuvant granulocyte macrophage colony-stimulating factor (GM-CSF).39 Celldex Therapeutics (Phillipsburg, NJ), who is developing this vaccine (CDX-110 or rindopepimut), recently completed several clinical trials in patients with newly-diagnosed and recurrent GBM. In two single-arm phase II studies, rindopepimut was well-tolerated and generated impressive anti-EGFRvIII immune responses that translated into significant improvements in overall survival (OS) and progression-free-survival (PFS) against newly-diagnosed disease compared to a control cohort matched for study eligibility and standard of care.40 These findings were confirmed in a single-arm, multicenter phase II trial in a cohort of 65 patients receiving rindopepimut with standard of care (NCT00458601). All three phase II trials demonstrated a median OS of approximately 24 months and a median PFS of 9.2–15.3 months, both from diagnosis.41–43 These results also demonstrated the tolerability and potential synergy of novel immunotherapies when used in combination with standard of care or dose-intensified TMZ.44
Based on these data, rindopepimut entered clinical evaluation in an international two-arm, randomized, phase III clinical trial (NCT01480479) for EGFRvIII-positive newly-diagnosed GBM. In March 2016, Celldex Therapeutics announced discontinuation of this phase III study after the independent Data Safety and Monitoring Board reported that the study would not reach statistical significance for OS. Importantly, rindopepimut performed consistently with data obtained from prior studies (median OS: 20.4 months), but the control arm significantly outperformed historical controls (21.1 months). The unexpected performance of the control arm may be one explanation as to why rindopepimut failed to show a survival benefit. Despite these results, rindopepimut is still being evaluated in recurrent GBM, based on interim results released in 2014 from a phase II clinical trial which demonstrated dramatic improvements in OS and PFS in patients receiving rindopepimut combined with bevacizumab compared to patients receiving bevacizumab alone.45 Formal results from this trial and sub-group analysis of the phase III clinical study mentioned above are highly anticipated.
One important and consistent observation arising from studies targeting EGFRvIII has been the loss of EGFRvIII expression and eventual outgrowth of antigen-negative tumors. This ‘immunologic escape’ is highlighted by the fact that 82% of recurrent tumors do not express EGFRvIII, and may indicate a need to target a greater repertoire of tumor antigens to prevent tumor-escape cell variants.42 This is supported by preliminary reports of immunologic escape with similar peptide-based vaccines, such as a phase I/II trial targeting the glioma-associated antigen (GAA) SL-701 (NCT02078648), a phase I trial evaluating a vaccine against CMV-associated antigens (NCT00639639), and a recently completed phase II trial targeting heat shock protein HSPP-96 (NCT00905060).
6.2 Dendritic Cell Vaccines
DCs, originally described by Steinman and Cohn, are professional antigen presenting cells (APCs) that serve as a crucial link between innate and adaptive immunity, constantly surveilling peripheral tissues for incoming pathogens and danger signals.46 Upon encountering antigen in the periphery, DCs engulf antigenic proteins, process antigen-derived peptides, assemble these peptides onto their MHC-I and MHC-II molecules, and upregulate key lymphoid homing receptors for efficient trafficking to draining lymph nodes (LNs), where they present antigen to naïve T cells and serve as co-stimulatory vehicles for the induction of T-cell effectors.47 Preclinical studies demonstrate that DCs are potent activators of de novo and recall humoral and cellular immune responses.48 As such, DC-based platforms have been pursued as an alternative to peptide vaccination by using ex vivo generated DCs derived from peripheral blood monocyte precursors.49 DCs pulsed with peptide, tumor antigen RNA, or whole tumor lysate can be administered to patients and have been shown to prime CD8+ T cell responses in vivo.50, 51
In 2001, Yu and colleagues reported the feasibility, safety, and bioactivity of a DC vaccine in 4/7 glioma patients who received DCs pulsed with peptides eluted from the surface of glioma cells.52 Systemic cytotoxic responses were detectable, and importantly, 2 of 4 patients who underwent reoperation demonstrated robust CD8+ and memory (CD45RO+) tumor-infiltrating lymphocytes (TILs) within the tumor microenvironment. In a similar study conducted by Liau and colleagues,53 12 GBM patients received DCs pulsed with acid-eluted peptides in three biweekly vaccinations. Six of these patients developed systemic antitumor CTL responses, 4 out of 8 patients who underwent operation demonstrated increased numbers of TILs, and one patient demonstrated an objective clinical response. This study also reported 4 long-term survivors (>2 years from diagnosis); per the trial’s protocol, these patients received DC therapy initially and TMZ after recurrence.
In a 2011 phase I/II study, 58% of GBM patients treated with a peptide-pulsed DC vaccine directed against the GAAs EphA2, IL13Rα2, YKL-40 and gp100 epitopes demonstrated multiple CD8+ T-cell responses to 3/4 targeted GAAs, providing the first evidence of generating a directed response against multiple tumor antigens.54 The scalability of this approach to additional or alternative GAAs, however, has been somewhat limited by the cost and lengthy time required for the appropriate selection and preparation of multiple immunogenic peptides. Additionally, the identification of antigens on autologous cultured tumor cells can be difficult or impossible in patients with non-resectable or recurrent tumors. These results have, however, demonstrated that vaccines designed to elicit responses against multiple antigens may be a viable approach to treating heterogeneous tumors. Several follow-up trials, including two phase II trials (NCT01280552 and NCT02366728), are underway to explore this approach in combination with potent adjuvants designed to improve the immunogenicity of tumor vaccines.
DCs pulsed or co-cultured with whole tumor lysate have also been used to generate T-cell responses against previously undefined tumor antigens. Despite concerns over potential for autoimmunity (due to inclusion of self-antigens) or inflammatory toxicity, such vaccines have been tested in humans with promising results. A phase II trial in patients with newly-diagnosed GBM demonstrated an improvement in median OS to 31.9 months, compared to just 15.0 months with standard chemoradiation therapy alone.55 Although the sample size of this initial trial was small (less than 20 patients in each treatment arm), there are multiple phase II/III trials in progress examining this approach, including DCVax-L (NCT00045968 and NCT02146066), proteome-based DC vaccines (NCT01759810), and trials examining the optimal adjuvant for lysate vaccines (NCT01204684). Importantly, one current study is also assessing the safety and efficacy of DC vaccines loaded with tumor lysate for the treatment of both adult and pediatric patients (≥ 13 years old) with refractory malignant glioma (NCT01808820). This study is expected to reach completion in July 2018.
Although results from these trials are encouraging and have proven valuable, vaccine trials have rarely produced convincing clinical data in advanced cancers.50 The reasons for this discrepancy are thought to be multifactorial and remain under active investigation. The migratory profile and kinetics of injected DCs have recently been identified as critical factors to the threshold of required antitumor responses in experimental studies in animals and humans.
One area of active investigation is the efficiency at which injected DCs reach vaccine site-draining lymph nodes (LNs). This has been investigated in the context of advanced melanoma patients receiving DC vaccines. In these clinical trials, only a maximum of 4% of intradermally-administered DCs reached these draining LNs.56, 57 A seminal preclinical study revealed that preconditioning the vaccine site with inflammatory cytokines or mature, unpulsed DCs significantly increases the migration of subsequently injected antigen-specific DCs to draining LNs, which then proportionally increases the magnitude and quality of induced T cell responses.58 The concept of vaccine site preconditioning entails administering an inflammatory agent locally in the skin prior to administering a cellular vaccine, with the goal of activating draining lymphatics and inducing key migratory receptors needed for effective lymph node homing. Recently, a blinded, randomized study demonstrated that preconditioning the DC vaccine site with Tetanus-diphtheria (Td) toxoid resulted in significantly increased DC migration compared to the cohort receiving a dose of unpulsed DCs. Furthermore, patients randomized to Td showed significantly improved PFS (range 15.4 – 47.3 months) and OS (range 20.6 – 47.3 months) compared to the unpulsed DC cohort. This study was recently published with corroborating preclinical evidence that Td, given locally at a single DC vaccine site, stimulated a systemic response enabling the migration of bilateral DC vaccine sites to reach their respective draining LNs.59 These observations are being evaluated in a higher-powered phase II clinical trial (NCT02366728).
Studies elsewhere are also attempting to improve the DC vaccine strategy through combinatorial therapy with checkpoint inhibitors that may improve the priming of naïve CD8+ T cells or prevent the functional inhibition of CTLs within tumor microenvironments. More recently, DC vaccines have also been utilized as cellular adjuvants in patients with GBM to provide in vivo antigenic stimulation to adoptively transferred T cells, where they may function to enhance T-cell polyfunctionality and expansion (NCT00693095).
6.3 Checkpoint blockade
An extensive amount of clinical trial data has been now accumulated from patients with both solid and blood-borne cancers, providing insight into how the nature of individual cancers may dictate patient responsiveness to immune-based therapies. Melanoma, for example, is a highly immunogenic cancer that responds remarkably well to immunotherapy, and clinical trials in patients with metastatic disease have achieved objective responses rates of up to 72%.60 With the exception of melanoma, this success has not yet been realized in many other solid cancers, which have remained somewhat resistant to immunotherapy. The relative bulk of disease and anatomical tumor site have been proposed as two important factors in determining therapeutic outcomes, although concerns over the immune-privileged status of the CNS have been put to rest in the context of diseased brain, where local inflammation can increase the permeability of the blood-brain barrier to enhance T-cell trafficking for active immunosurveillance.61, 62 The immunosuppressive nature of brain tumors, however, remains of critical importance, as the local milieu of tumor microenvironments can diminish or completely abrogate the antitumor activity of effector cells altogether. The brain parenchyma is immunosuppressive by nature, as normal CNS immune responses are skewed towards type 2 CD4+ helper T cell responses characterized by strong humoral responses and suppressed cell mediated immunity.63 GBM further contributes to this immunosuppression through several well-defined mechanisms, including the recruitment and accumulation of myeloid-derived suppressor cells (MSDCs),64 CD4+ CD25+ FOXP3+ regulatory T cells (TREG),65, 66 and alternatively activated M2-type microglia/macrophages.67 Other mechanisms include altered human leukocyte antigen (HLA) expression, and increased levels of immunosuppressive factors including TGF-β, IDO, IL-10, COX2, and PGE2, among others.68–70 The dysregulation of these mechanisms, and others not mentioned here, collectively contribute to a state of immune evasion that is typified by systemic lymphopenia, low numbers of effector T cell tumor infiltrates, a high fraction of TREG within a dwindled CD4+ compartment, extensive immunosuppressive environments within draining cervical CLNs, and perhaps most notably, low to non-existent T-cell responsiveness.66, 71
The possibility that T cells remain unresponsive despite their capacity to recognize tumor antigens has propelled us to better understand the biology of intratumoral immune synapses, which has culminated into strategies that promote effector T cell function by overcoming tumor-induced dysregulation of immune checkpoints. The B7/CTLA-4 and PD-1/PD-L1 axes are archetypal examples of immune regulators and have been extensively reviewed elsewhere.72 CTLA-4 is an inhibitory cell-surface receptor expressed by all T cells and competes with the co-stimulatory receptor, CD28, for the ligands B7-1 (CD80) and B7-2 (CD86). CTLA-4 engagement therefore not only delivers self-inhibitory signals to T cells but can also prevent necessary costimulation by CD28 to amplify TCR signals by acting as a ligand sink, thereby preventing sufficient T-cell activation.72–75 Importantly, CTLA-4 is also constitutively expressed by TREG, and CTLA-4 engagement dramatically enhances TREG suppression of effector T-cells. In contrast, the PD-1/PD-L1 axis operates through a distinct and non-redundant checkpoint mechanism. Like CTLA-4, PD-1 is an inhibitory cell-surface receptor that is expressed on activated T-cells and other immune cells, and a wide range of tumors as well as APCs have been shown to upregulate one of its two major ligands, PD-L1.76, 77 Following promising preclinical data, antibodies against CTLA-4, PD-1, and other immune checkpoints have entered clinical trials and have produced highly encouraging results against several cancers.
CTLA-4 blockade was approved by the U.S. Food and Drug Administration (FDA) in 2010 for metastatic melanoma,78 and is under active investigation for several other advanced cancers, including GBM, renal cell carcinoma, and cancers of the lung, pancreas, and liver. Of note, anti-CTLA-4 has been associated with significant rates of dose-dependent inflammatory toxicity in up to 21% of patients,79 drawing some concern over the potential for systemic damage by this class of drugs.80–82 Nivolumab is a fully human IgG4 antibody that blocks the interaction of the PD-1 receptor to its ligands. Early clinical trials in patients with non-small cell lung cancer, melanoma, and renal cell carcinoma have demonstrated response rates ranging from 18% to 28%; responses are even higher (36%) for patients with histologically-confirmed PD-L1-expressing tumors.83 In fact, a 2015 report from a phase III clinical trial demonstrated an objective response rate of 40% in patients with previously untreated, advanced melanoma.84 Clinical trials evaluating checkpoint blockade in combination with other drugs and standard of care in patients with GBM are ongoing. These studies include a phase I trial evaluating combination therapy with anti-PD1 and anti-CTLA-4 in the context of TMZ (NCT02311920), and a phase III trial comparing the safety and efficacy between anti-PD1 alone, combination therapy with anti-PD1 and anti-CTLA-4, and bevacizumab alone (NCT02017717).
6.4 Adoptive T-cell immunotherapy
The clinical significance of immunotherapy is nowhere better exemplified than in the context of adoptive T cell transfer, where nearly three decades of discovery and innovation have culminated into producing consistent and durable responses against highly aggressive and advanced cancers. This strategy is exemplified in the context of stage IV metastatic melanoma, where the isolation, ex vivo expansion, and adoptive transfer of tumor-infiltrating T-cells (TILs) with high-dose IL-2 has produced objective clinical responses in up to 72% of patients recovering from chemotherapy.60 Importantly, even large metastases in the brain have responded to systemic therapy.36 Based on this proof-of-concept, significant efforts have been directed towards the ex vivo generation of tumor-specific T cells for similar adoptive transfer therapies, as the direct isolation of T cells from tumor specimens is laborious, technically difficult, or near impossible in most cancers. This has been accomplished by genetically engineered patient T-cells with transgenic TCRs, and more recently, with chimeric antigen receptors (CARs), which have already demonstrated their potential in eliciting long-term and complete responses in patients with hematological cancers.85, 86 CARs are cell-surface receptors, created by adjoining the antigen-binding variable regions of monoclonal antibodies (mAbs) with cytoplasmic TCR signaling molecules through short linker and transmembrane residues. CARs can therefore be derived from high-affinity mAb clones reactive against antigens of interest, and effectively redirect CAR-expressing T cells to destroy tumor cells.
The majority of available clinical trial data is derived from studies targeting B-cell malignancies with CD19- or CD20- directed CARs, which have routinely resulted in partial or complete remissions.85–88 In fact, Kochenderfer and colleagues reported study results this year in which 15 patients with advanced B-cell malignancies were treated with autologous anti-CD19 CAR T cells after receiving cyclophosphamide and fludarabine chemotherapy, where they achieved eight complete remissions, four partial remissions, and one stable lymphoma.89 Based on such success, CAR therapy has been under active investigation for the treatment of solid tumors, and preclinical studies have shown that systemic administration of EGFRvIII-specific CARs can eradicate intracranial GBM in syngeneic animal models with the potential to confer immunologic protection against tumor rechallenge (Figure 2). The therapeutic potential of CAR T cells against GBM has been corroborated by several groups.90–93
Figure 2. Proposed mechanism of induced immunological protection.
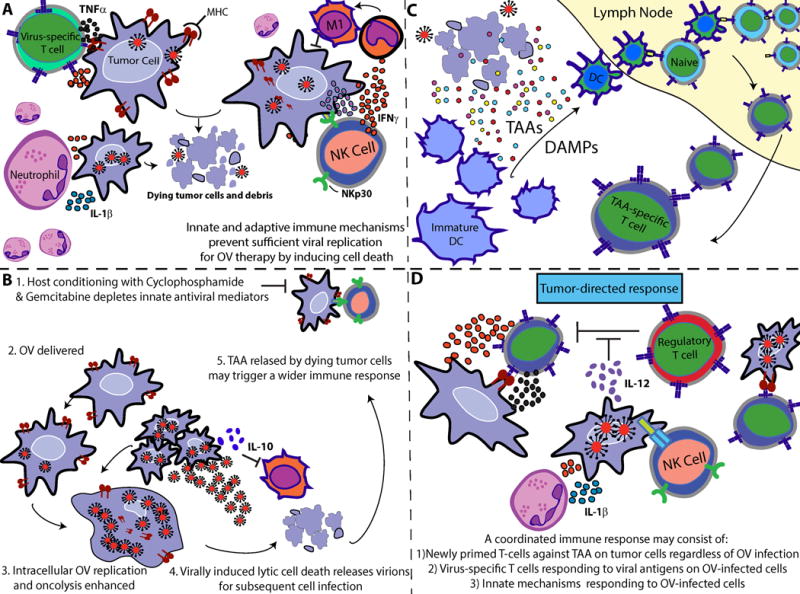
(A) CARs migrate to, engage, and induce cytotoxicity against tumor cells in an antigen-specific manner. Immunogenic debris from dying tumor cells (B) drain or are carried by professional APCs into local lymph nodes, where naïve T-cells become primed against tumor antigens. (C) Newly-primed T cells exit the lymph nodes and migrate to distant tumor sites to engage malignant cells.
One key benefit of this platform is the ability of CAR T cells to recognize targets without the need for TCR:MHC complex formation, a necessary component of normal T cell recognition of antigen. This is of particular importance for GBM, where MHC class I expression is frequently downregulated in migratory tumor cells that invade surrounding brain tissue.94 CAR constructs have also been designed to include additional signaling molecules that enhance the survival and persistence of engineered T cells, and importantly, T cells can also be further modified to influence responsiveness or confer resistance to local immunosuppression. As mentioned previously, CTLs are susceptible to and can be rendered dysfunctional through local intratumoral immunosuppression through a variety of mechanisms, including GBM-mediated secretion of TGF-β. To counteract this suppression, one novel approach has been the manipulation of intracellular micro-RNAs, which are key regulators of gene expression. In CTLs, for example, micro RNA-23a (mIR-23a) has been identified as a negative regulator of BLIMP-1, whose expression is required for effector T-cell immunocompetence.95 TGF-β has been shown to control mIR-23a levels to reduce CTL functionality, and mIR-23a blockade has been shown to confer resistance to TGFβ-mediated suppression.95 TGF-β inhibitors have been shown to be well-tolerated in humans, but this approach warrants caution as deleterious consequences can arise with systemic and non-specific antagonism of a pleiotropic cytokine. This serves to highlight an important safety benefit of restricting these highly sensitive interactions exclusively to cancer sites by using engineered T cells specific for tumor antigens. For example, T cells can be engineered to express micro RNAs or decoy micro RNAs to favorably regulate gene expression, or overexpress pro-inflammatory cytokines to alter local immune milieu. These T-cell modifications can theoretically be regulated in a way that is dependent on CAR activation, which would be an advantage over the systemic administration of clinical reagents aimed for similar purposes. Therefore, as our understanding of cancer immunobiology continues to evolve, so too will our ability to modulate key intracellular mechanisms to engineer the perfect CAR T cell.
EGFRvIII-CARs are currently being explored for recurrent GBM in a phase I/II clinical trial at the National Cancer Institute (NCT01454596) and the University of Pennsylvania (NCT02209376); and IL13Rα2-CARs and HER2-CARs are also under clinical investigation for recurrent or refractory GBM at City of Hope Medical Center (NCT02208362) and Baylor College of Medicine (NCT02442297), respectively.
6.5 Oncolytic Viruses
Viruses have been studied extensively in cancer, for both their role as causative agents and as potential tools for therapy. These origins date back over a century, when in 1912, N.G. De Pace found that vaccinating a woman (who was bitten by a rabid dog) with an attenuated rabies virus vaccine induced regression of her cervical carcinoma. Virotherapy has been commonly used to genetically modify cancer cells, where it has been shown to increase tumor sensitivity to exogenous agents, prevent angiogenesis, and correct oncogenetic defects.96–99 More recently, virotherapy has expanded beyond purposes of gene delivery to more directly leverage the ability of viruses to selectively target cancer cells, induce cell death, amplify, and spread within tumor. Viruses can exert antitumor benefits by 1) inducing tumor cell lysis (oncolysis), 2) recruiting immune cells indirectly through the release of pro-inflammatory cytokines and chemoattractants by dying tumor cells, and 3) inducing immune responses against themselves, infected tumor cells, and uninfected tumor cells through bystander effects.100 Importantly, these secondary immune responses can sometimes be unproductive, as effective virotherapy seems to require a delicate balance between antitumor and antiviral activity such that sufficient viral infection of tumor occurs before viral particles are cleared through innate immune mechanisms. Neutrophils and myeloid-derived cells (i.e. macrophages and monocytes) are components of innate immunity and are recruited to tumor beds following oncolytic virus (OV) infection. In preclinical models, antibody-mediated depletion of neutrophils is coincident with enhanced viral replication,101 and macrophages – which assume an M1 pro-inflammatory phenotype in response to viral infection – have been associated with a dramatic clearance in viral titers that can be reversed upon macrophage depletion.102, 103 Chiocca and colleagues have also eloquently shown that depletion of NK cells facilitates OV replication and improves survival in preclinical models of GBM.104 These studies have supported the notion that at least during the initial period of OV infection, suppression of innate immune mechanisms may be required for robust OV antitumor activity, although this may depend on the tumor model and viral vector used. It is clear, however, that when a productive OV infection does occur, it can engender immunogenic tumor cell death and induce long-term and robust CD8+ T cell antitumor responses (Figure 1) both in preclinical models and in patients with solid tumors receiving combinatorial treatment with OV, TMZ, and cyclophosphamide.105 More than 7 different OVs have been evaluated in clinical trials for GBM, including adenovirus (AdV), herpes simplex virus (HSV), Newcastle disease virus, reovirus, measles virus, H1 parovirus, and poliovirus, all of which have been extensively reviewed elsewhere.106
Figure 1.
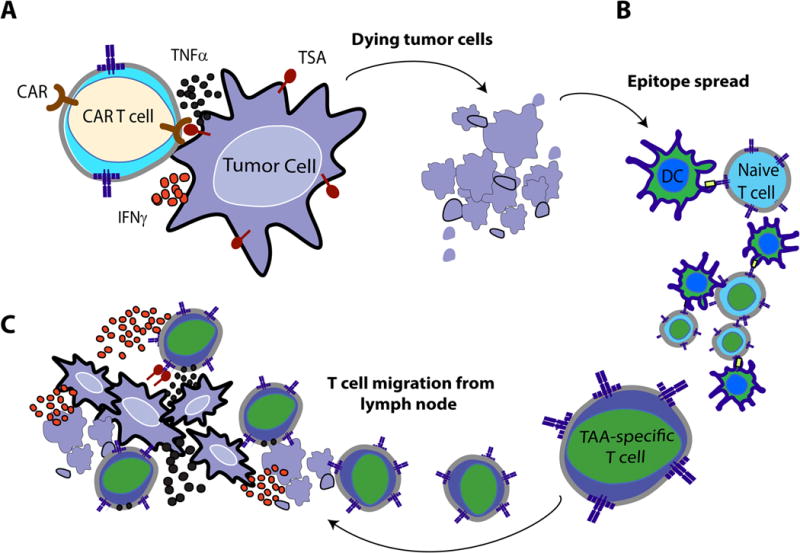
(A) Upon administration of OV therapy, infected tumor cells are recognized and quickly eradicated by antiviral innate mediators, including NK cells, M1 macrophages, neutrophils, and virus-specific T-cells of the cellular compartment. (B) Host-conditioning with cyclophosphamide and gemcitabine chemotherapy can blunt antiviral immune mechanisms to provide a window of sufficient OV replication. OV-infected tumor cells eventually die through lytic mechanisms or by immune-cell recognition during the rebound phase after chemotherapy. Dying tumor cells may lead to an efflux of tumor-associated antigens (TAA) or damage-associated molecular pattern molecules (DAMPs), (C) which can be engulfed by immature dendritic cells (DCs) and later presented to naïve T cells in the local tumor-draining lymph node. Together, these mechanisms may act in concert to promote (D) a coordinated tumor-directed immune response comprising of OV-mediated cell lysis, anti-viral immunity against OV-infected cells, and adaptive immunity responding to TAA. Reproduced with permission from reference.103
Despite the promise of OV therapy, it has not yet achieved convincing clinical success in advanced trials, and no OV has been approved by the U.S. FDA to date, although AdV ONYX-015 has been approved in China for patients with head and neck cancer. A promising candidate may be on the horizon for GBM, however, based on early phase I clinical data reported in 2014–2015. This OV is a genetically modified poliovirus which binds the CD155 cell-surface polio receptor, which is expressed in a high proportion of GBM tumor cells.107 To reduce the potential for neurotoxicity that is typically observed in cases of human poliomyelitis, poliovirus was genetically altered at its internal ribosomal entry site to swap its cognate sequence with a non-pathogenic sequence derived from human rhinovirus type II,108 effectively eliminating neurovirulence in this poliovirus/rhinovirus recombinant chimera (PVSRIPO). Intratumoral administration (via catheter enhanced delivery or CED) of PSVRIPO is currently being evaluated in a phase I dose escalation trial in patients with recurrent GBM, and preliminary results have demonstrated complete and near-complete radiographic responses following viral infusion in the first two patients recruited on study, who remain alive >31 months and >33 months post-PVSRIPO infusion.109, 110
6.6 Gene Transfer Therapy
Gene therapy, or the use of nucleic acids to treat disease, provides a distinct novel approach to treatment of GBM, especially when combined with immunotherapy. One such modality relies on a combination of adenoviral vectors to deliver two genes: thymidine kinase (TK) and fms-like tyrosine kinase-3 ligand (Flt3L). TK works by phosphorylating the prodrug Ganciclovir to induce DNA-crosslinking and subsequent tumor cell death, while Flt3L is a strong DC growth factor and chemoattractant to the microenvironment. TK-mediated tumor cytotoxicity leads to the release of inflammatory molecules and new antigens that can then be taken up by DCs and presented to T cells to prime them for an antigen-specific response against the tumor.111 In multiple animal models of GBM, this combination has led to tumor regression, long-term survival, and development of immunological memory.112–115 A Phase I dose escalation safety study is currently underway to evaluate this combined cytotoxic and immune-stimulatory strategy in patients with resectable GBM (NCT01811992). Other gene therapies have also shown promise in pre-clinical models of brain tumors including adenovirus-mediated delivery of interferon alpha and IL-12.113
7. Potential development issues
The primary objective of immunotherapy for patients with GBM is to enhance OS and improve quality of life through the use of targeted therapies. Over the past two decades, the clinical management of GBM has remained largely the same, although there have been major technological advances in imaging and surgery that have improved tumor resection procedures and reduced complication rates. Despite this, patient outcomes have not significantly changed, and there is a growing consensus that new and finely-tuned approaches are required. Although several immunotherapies have translated from mouse to human, many have failed to proceed beyond phase II studies, due in part to both lack of available funding or unanticipated side effects. One recurring problem has been the difficult task of recruiting sufficient patients for clinical trials, where reduced statistical power can make it difficult to interpret trial outcomes and therefore frustrate opportunities to lure financial investment for next phase development.
The occurrence of adverse events or severe adverse events in patients treated with experimental therapy can also debilitate development, which is why we have repeatedly stated the need for thorough safety and toxicity studies in preclinical models of disease prior to the initiation of human studies. Immunotherapies are commonly evaluated in either immunocompetent (IC) or immunodeficient (ID) animal models, and both carry pros and cons. ID models allow investigators to study novel therapies in the context of human brain tumor xenografts that can be surgically implanted into the brain. ID mice can also be engrafted with human immune cells, and therefore, specific interactions between human immune cells, human tumor tissue, and drug can be studied within this context in vivo. In IC mice, where they are equipped with fully functional immune systems, spontaneously-generated or chemically-induced syngeneic tumor cells lines are required for brain tumor models. Importantly, IC models afford the opportunity to evaluate the potential for novel therapies to induce secondary immune responses and cross-reactivity against other unexplored antigens. When possible, novel immunotherapies should be evaluated in the context of both IC and ID model systems to ensure safety before they enter testing in human subjects.
8. Conclusion
Given the immediate danger imposed by bulky tumors on neurologic function, future treatment of GBM will likely incorporate immunotherapies as an adjunct strategy following tumor resection and chemoradiation. Importantly, immunotherapy has been shown to synergize with the current standard of care, and in most cases, without an increase in the risk for toxicity. Ionizing radiation induces pro-inflammatory signaling, non-specifically activates the immune system, and improves antigen presentation, and the immunologic space created by TMZ-induced lymphopenia has been shown to significantly enhance CD8+ T cell responses after adoptive T cell transfer and vaccination.116 Here, we have reviewed several candidate immunotherapies, including their successes and pitfalls, and made every attempt to highlight the obstacles which remain to achieving long-term and durable responses against this devastating disease. Despite the work ahead, it is becoming clear that immunotherapy for GBM holds tremendous promise and the landscape of clinical management for malignant brain tumors is poised for change.
9. Expert Opinion
As our understanding of immune regulation and the mechanisms governing effective tumor immunity has evolved exponentially, so too has our ability to approach clinical trials with rationale and intelligent design. Recent evidence indicates GBM can be subdivided into distinct subtypes (neural, proneural, classical, or mesenchymal) based on genomic abnormalities and specific expression patterns in the p53, IDH1, EGFR, and NF1 genes.117 These genetic subtypes have been shown to differ in both their clinical features and response to standard of care therapy.118 Importantly, results from a recent phase I trial in patients with GBM also suggest that tumors defined by the mesenchymal gene expression profile may be more immunogenic and responsive to immune-based therapies, as patients with tumors in this subtype experienced increased survival following tumor-lysate pulsed DC vaccination with TLR agonists compared to historical controls with tumors of the same subtype.119 Interestingly, clinical samples from this trial have also demonstrated increased numbers of TILs in tumors defined by the mesenchymal subtype compared to tumors expressing other gene signatures. These observations warrant investigation into determining how these four gene expression signatures dictate tumor immunogenicity, as these studies will be critical to understanding the mechanisms for why and how the population in which immunotherapy for GBM may succeed or fail.
Combination therapy with CTLA-4 and PD-1 blockade is specifically geared to counteract the separate and non-redundant immunosuppressive mechanisms of inadequate T-cell activation and intratumoral cellular inhibition. This approach is currently under investigation in a phase III study in patients with recurrent GBM (NCT02017717). The excitement concerning the clinical success of checkpoint blockade against peripheral tumors is well-deserved, as this line of drugs may offer new hope for patients with advanced cancers. One criticism of this platform, however, has been the absolute requirement of immunogenic tumor material to observe therapeutic responses. We now appreciate the seemingly paradoxical roles of the immune system in cancer development; Schreiber and colleagues have eloquently described in the ‘immunoediting’ hypothesis that cancer results, at least in part, from the selection of ‘fit’ tumor cells that have evolved to evade immune detection.120 Whereas some cancers may have evolved mechanisms to usurp local immune regulation, others may lack the antigens to provoke immune responses altogether. Human TCR repertoires are generated to identify a virtually infinite number of foreign- and self-epitopes. Although the majority of T cells that recognize self-antigens are eliminated during thymic development, a small frequency of autoreactive T-cells with degenerate TCRs can exist through adulthood, requiring constant TCR stimulation or exposure to a high density of antigen for complete activation.121, 122 The implication here is that certain cancers may have too few (or no) immunogenic antigens, and checkpoint blockade in this context would have little consequence on a CD3+ T cell compartment that either lacks the TCRs required to recognize tumor antigens or contain TCRs that have too low an avidity to be sufficiently activated. In these cases, checkpoint inhibitors would only be effective as adjuvants to a primary therapy that either endows a cancer patient with tumor-directed T-cells or sufficiently primes a nascent precursor frequency of these cells. This conundrum is further complicated by the fact that many aggressive cancers, including GBM, frequently downregulate MHC expression and thus bypass T cell recognition of tumor cells altogether, even when effector cells are poised to kill. As such, checkpoint inhibitors will likely achieve their greatest potential against GBM when used in combination with a primary immunotherapy where these limitations can be appropriately addressed through ex vivo manipulation of autologous immune cells.
Whether this primary immunotherapy is comprised of a cancer vaccine or engineered T-cells remains equivocal, and several arguments can be made in favor of either approach. Clinical studies evaluating the EGFRvIII vaccine, for example, have demonstrated the limitations of immunotherapies targeting single antigens. From this perspective, the ‘ideal’ approach would be to target as large a repertoire of tumor antigens as possible while avoiding damage to normal tissue. Although DCs can be manipulated to express as many of these antigens as desired (by electroporation of RNA, constructed mini-genes, or tumor lysate), DC vaccines are limited by inefficient migration to LNs and priming of T cells. These areas are being actively explored to improve the DC platform. CAR T cells, on the other hand, overcome key limitations of cancer vaccines, as these T cells are already tumor-specific and previously activated. It is possible to infuse diverse populations of CAR T cells specific for different tumor antigens, but this may be an unrealistic approach for GBM due to a paucity of known cell-surface, tumor-specific targets expressed in a high proportion of patients. Furthermore, the expense of producing CAR T cells specific for multiple antigens through retroviral engineering will likely be cost-prohibitive, although significant efforts are underway to produce allogeneic ‘off-the-shelf’ CAR T cells by the pharmaceutical industry.
Preclinical studies of CAR T cells targeting EGFRvIII-positive tumors have also indicated the possible induction of endogenous immune responses against other tumor antigens, in a process known as epitope spreading. This is exemplified by studies which show that mice previously cured of EGFRvIII-positive tumors by EGFRvIII-CARs are protected against re-challenge with EGFRvIII-negative tumors. This lends support to future studies that combine checkpoint blockade with CAR T cell therapy; if EGFRvIII-CARs can sufficiently produce immune responses against non EGFRvIII-expressing tumor cells, checkpoint inhibitors may serve to prevent the dysfunction of newly-primed T cells, enhance their proliferation and the likelihood of tumor eradication.
In summary, we believe the future of this field will require significant investment in understanding which patients stand to benefit from therapy, why certain patients do not, and overcoming mechanisms of evolved resistance. As such, emphasis must be placed on proper patient selection to discern the relevance of age, minimal residual disease status, and immunological profile of patients. Moreover, therapies which have demonstrated promising efficacy in preclinical glioma models and favorable safety profiles in phase I clinical trials have seldom reached higher-powered studies, as they are commonly conducted in patients with advanced stage cancer or recurrent disease. Assessing therapeutic efficacy in this refractory patient population may contribute to the low success rate observed in higher-powered trials. In order to adequately judge efficacy, clinical trials should be conducted on patients with earlier stage disease to fairly assess efficacy against newly-established tumors. With that said, we remain highly optimistic surrounding the promise of immunotherapy for the treatment of malignant brain tumors, and maintain that a complete cure will one day be within our reach.
Acknowledgments
This work was supported by funding from the National Institutes of Health: 1R01CA177476, (J.H. Sampson), P50-CA190991 (J.H. Sampson), 5R01-CA135272 (J.H. Sampson), T32-CA009111 (C.M. Suryadevara). Additional support was provided by the Pediatric Brain Tumor Foundation (D.D. Bigner and J.H. Sampson), and the Ben and Catherine Ivy Foundation (J.H. Sampson).
Footnotes
This work was completed in Durham, NC, USA.
Declaration of Interests
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ostrom QT, Gittleman H, Liao P, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-oncology. 2014 Oct;16(Suppl 4):iv1–iv63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Farah P, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-oncology. 2013 Nov;15(Suppl 2):ii1–56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeAngelis LM. Brain tumors. The New England journal of medicine. 2001 Jan 11;344(2):114–23. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- 4.Rulseh AM, Keller J, Klener J, et al. Long-term survival of patients suffering from glioblastoma multiforme treated with tumor-treating fields. World J Surg Oncol. 2012 Oct 24;10 doi: 10.1186/1477-7819-10-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLendon RE, Halperin EC. Is the long-term survival of patients with intracranial glioblastoma multiforme overstated? Cancer. 2003 Oct 15;98(8):1745–48. doi: 10.1002/cncr.11666. [DOI] [PubMed] [Google Scholar]
- 6.Buckner JC. Factors influencing survival in high-grade gliomas. Seminars in oncology. 2003 Dec;30(6 Suppl 19):10–4. doi: 10.1053/j.seminoncol.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 7.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005 Mar 10;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 8.Ray S, Bonafede MM, Mohile NA. Treatment Patterns, Survival, and Healthcare Costs of Patients with Malignant Gliomas in a Large US Commercially Insured Population. American health & drug benefits. 2014 May;7(3):140–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Kutikova L, Bowman L, Chang S, Long SR, Thornton DE, Crown WH. Utilization and cost of health care services associated with primary malignant brain tumors in the United States. J Neuro-Oncol. 2007 Jan;81(1):61–65. doi: 10.1007/s11060-006-9197-y. [DOI] [PubMed] [Google Scholar]
- 10.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology. 2009 May;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 11.Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant astrocytomas. Annals of neurology. 1990 Dec;28(6):818–22. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 12.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clinical Cancer Research. 2000;6(7):2585–97. [PubMed] [Google Scholar]
- 13.Kesari S. Understanding glioblastoma tumor biology: the potential to improve current diagnosis and treatments. Seminars in oncology. 2011 Dec;38(Suppl 4):S2–10. doi: 10.1053/j.seminoncol.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Miao H, Choi BD, Suryadevara CM, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PloS one. 2014;9(4):e94281. doi: 10.1371/journal.pone.0094281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews Immunology. 2012 Apr;12(4):269–81. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostermann S, Csajka C, Buclin T, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004 Jun 1;10(11):3728–36. doi: 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- 17.Portnow J, Badie B, Chen M, Liu A, Blanchard S, Synold TW. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: potential implications for the current approach to chemoradiation. Clin Cancer Res. 2009 Nov 15;15(22):7092–8. doi: 10.1158/1078-0432.CCR-09-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiemann B, Starnes CO. Coley’s toxins, tumor necrosis factor and cancer research: a historical perspective. Pharmacology & therapeutics. 1994;64(3):529–64. doi: 10.1016/0163-7258(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 19.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clinical orthopaedics and related research. 1991 Jan;(262):3–11. [PubMed] [Google Scholar]
- 20.Parish CR. Cancer immunotherapy: the past, the present and the future. Immunology and cell biology. 2003 Apr;81(2):106–13. doi: 10.1046/j.0818-9641.2003.01151.x. [DOI] [PubMed] [Google Scholar]
- 21.Fenton RG, Longo DL. Genetic instability and tumor cell variation: implications for immunotherapy. Journal of the National Cancer Institute. 1995 Feb 15;87(4):241–3. doi: 10.1093/jnci/87.4.241. [DOI] [PubMed] [Google Scholar]
- 22.Stoler DL, Chen N, Basik M, et al. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 1999 Dec 21;96(26):15121–6. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inda MD, Bonavia R, Mukasa A, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Gene Dev. 2010 Aug 15;24(16):1731–45. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boockvar JA, Kapitonov D, Kapoor G, et al. Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci. 2003 Dec;24(4):1116–30. doi: 10.1016/j.mcn.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Moscatello DK, Holgado-Madruga M, Godwin AK, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer research. 1995 Dec 1;55(23):5536–9. [PubMed] [Google Scholar]
- 26.Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer research. 1991 Apr 15;51(8):2164–72. [PubMed] [Google Scholar]
- 27.Libermann TA, Razon N, Bartal AD, Yarden Y, Schlessinger J, Soreq H. Expression of epidermal growth factor receptors in human brain tumors. Cancer research. 1984 Feb;44(2):753–60. [PubMed] [Google Scholar]
- 28.Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proceedings of the National Academy of Sciences of the United States of America. 1992 Apr 1;89(7):2965–9. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato Y, Jin GL, Kuan CT, McLendon RE, Yan H, Bigner DD. A monoclonal antibody IMab-1 specifically recognizes IDH1(R132H), the most common glioma-derived mutation. Biochem Bioph Res Co. 2009 Dec 18;390(3):547–51. doi: 10.1016/j.bbrc.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodges TR, Choi BD, Signer DD, Yan H, Sampson JH. Isocitrate dehydrogenase 1: what it means to the neurosurgeon A review. J Neurosurg. 2013 Jun;118(6):1176–80. doi: 10.3171/2013.3.JNS122282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nair SK, De Leon G, Boczkowski D, et al. Recognition and killing of autologous, primary glioblastoma tumor cells by human cytomegalovirus pp65-specific cytotoxic T cells. Clin Cancer Res. 2014 May 15;20(10):2684–94. doi: 10.1158/1078-0432.CCR-13-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang JG, Eguchi J, Kruse CA, et al. Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics. Clin Cancer Res. 2007 Jan 15;13(2 Pt 1):566–75. doi: 10.1158/1078-0432.CCR-06-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saikali S, Avril T, Collet B, et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J Neurooncol. 2007 Jan;81(2):139–48. doi: 10.1007/s11060-006-9220-3. [DOI] [PubMed] [Google Scholar]
- 34.Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004 Jul 15;64(14):4980–6. doi: 10.1158/0008-5472.CAN-03-3504. [DOI] [PubMed] [Google Scholar]
- 35.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009 Jul 16;114(3):535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong JJ, Rosenberg SA, Dudley ME, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010 Oct 1;16(19):4892–8. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinschenk T, Gouttefangeas C, Schirle M, et al. Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer research. 2002 Oct 15;62(20):5818–27. [PubMed] [Google Scholar]
- 38.Nielsen M, Lund O, Buus S, Lundegaard C. MHC class II epitope predictive algorithms. Immunology. 2010 Jul;130(3):319–28. doi: 10.1111/j.1365-2567.2010.03268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gedeon PC, Choi BD, Sampson JH, Bigner DD. Rindopepimut: anti-EGFRvIII peptide vaccine, oncolytic. Drugs of the future. 2013 Mar;38(3):147–55. doi: 10.1358/dof.2013.038.03.1933992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heimberger AB, Hussain SF, Aldape K, et al. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. J Clin Oncol. 2006 Jun 20;24(18):107s–07s. [Google Scholar]
- 41.Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-oncology. 2015 Jan 13; doi: 10.1093/neuonc/nou348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010 Nov 1;28(31):4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-oncology. 2011 Mar;13(3):324–33. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sampson JH, Archer GE, Bigner DD, et al. Effect of EGFRvIII-targeted vaccine (CDX-110) on immune response and TTP when given with simultaneous standard and continuous temozolomide in patients with GBM. J Clin Oncol. 2008 May 20;26(15) [Google Scholar]
- 45.Reardon D, Schuster J, Tran D, et al. ReACT: A Phase II Study of Rindopepimut Vaccine (CDX-110) plus Bevacizumab in Relapsed Glioblastoma. Neuro-oncology. 2014 Nov;:16. [Google Scholar]
- 46.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. Journal of Experimental Medicine. 1973;137(5):1142–62. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annual Review of Immunology. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 48.Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. Journal of Experimental Medicine. 1997;186(7):1177–82. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair S, Archer GE, Tedder TF. Isolation and generation of human dendritic cells. Current Protocols in Immunology. 2012:1–23. doi: 10.1002/0471142735.im0732s99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nature reviews Cancer. 2012;12(4):265–77. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fecci PE, Mitchell DA, Archer GE, et al. The history, evolution, and clinical use of dendritic cell-based immunization strategies in the therapy of brain tumors. J Neuro-Oncol. 2003 Aug-Sep;64(1):161–76. doi: 10.1007/BF02700031. [DOI] [PubMed] [Google Scholar]
- 52.Yu JS, Wheeler CJ, Zeltzer PM, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001 Feb 1;61(3):842–7. [PubMed] [Google Scholar]
- 53.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clinical Cancer Research. 2005;11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 54.Okada H, Kalinski P, Ueda R, et al. Induction of CD8(+) T-Cell Responses Against Novel Glioma-Associated Antigen Peptides and Clinical Activity by Vaccinations With alpha-Type 1 Polarized Dendritic Cells and Polyinosinic-Polycytidylic Acid Stabilized by Lysine and Carboxymethylcellulose in Patients With Recurrent Malignant Glioma. J Clin Oncol. 2011 Jan 20;29(3):330–36. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho DY, Yang WK, Lee HC, et al. Adjuvant Immunotherapy with Whole-Cell Lysate Dendritic Cells Vaccine for Glioblastoma Multiforme: A Phase II Clinical Trial. World Neurosurg. 2012 May-Jun;77(5–6):736–44. doi: 10.1016/j.wneu.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 56.De Vries I, Krooshoop D, Scharenborg N, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Research. 2003;63(1):7–12. [PubMed] [Google Scholar]
- 57.Verdijk P, Aarntzen EH, Lesterhuis WJ, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15(7):2531–40. doi: 10.1158/1078-0432.CCR-08-2729. [DOI] [PubMed] [Google Scholar]
- 58.Martin-Fontecha A, Sebastiani S, Hopken UE, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. Journal of Experimental Medicine. 2003;198:615–21. doi: 10.1084/jem.20030448. 2003 Aug 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015 Mar 19;519(7543):366–9. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011 Jul 1;17(13):4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, Sontheimer H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nature communications. 2014;5:4196. doi: 10.1038/ncomms5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolburg H, Noell S, Fallier-Becker P, Mack AF, Wolburg-Buchholz K. The disturbed blood-brain barrier in human glioblastoma. Molecular aspects of medicine. 2012 Oct-Dec;33(5–6):579–89. doi: 10.1016/j.mam.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 63.Harling-Berg CJ, Park TJ, Knopf PM. Role of the cervical lymphatics in the Th2-type hierarchy of CNS immune regulation. J Neuroimmunol. 1999 Nov 15;101(2):111–27. doi: 10.1016/s0165-5728(99)00130-7. [DOI] [PubMed] [Google Scholar]
- 64.Raychaudhuri B, Rayman P, Ireland J, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-oncology. 2011 Jun;13(6):591–9. doi: 10.1093/neuonc/nor042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006 Jul;8(3):234–43. doi: 10.1215/15228517-2006-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer research. 2006 Mar 15;66(6):3294–302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 67.Wei J, Gabrusiewicz K, Heimberger A. The controversial role of microglia in malignant gliomas. Clinical & developmental immunology. 2013;2013:285246. doi: 10.1155/2013/285246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lohr J, Ratliff T, Huppertz A, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. 2011 Jul 1;17(13):4296–308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 69.Hishii M, Nitta T, Ishida H, et al. Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery. 1995 Dec;37(6):1160–6. doi: 10.1227/00006123-199512000-00016. discussion 66–7. [DOI] [PubMed] [Google Scholar]
- 70.Rolle CE, Sengupta S, Lesniak MS. Mechanisms of immune evasion by gliomas. Advances in experimental medicine and biology. 2012;746:53–76. doi: 10.1007/978-1-4614-3146-6_5. [DOI] [PubMed] [Google Scholar]
- 71.Waziri A. Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurgery clinics of North America. 2010 Jan;21(1):31–42. doi: 10.1016/j.nec.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 72.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012 Apr;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freeman GJ, Gribben JG, Boussiotis VA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science. 1993 Nov 5;262(5135):909–11. doi: 10.1126/science.7694363. [DOI] [PubMed] [Google Scholar]
- 74.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (Cd80) and B7-2 (Cd86) Bind with Similar Avidities but Distinct Kinetics to Cd28 and Ctla-4 Receptors. Immunity. 1994 Dec;1(9):793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 75.Schneider H, Downey J, Smith A, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006 Sep 29;313(5795):1972–75. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 76.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nature reviews Immunology. 2008 Jun;8(6):467–77. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 77.Wang C, Thudium KB, Han M, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer immunology research. 2014 Sep;2(9):846–56. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 78.Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proceedings of the National Academy of Sciences of the United States of America. 2003 Jul 8;100(14):8372–7. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006 May 20;24(15):2283–9. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnston RL, Lutzky J, Chodhry A, Barkin JS. Cytotoxic T-lymphocyte-associated antigen 4 antibody-induced colitis and its management with infliximab. Digestive diseases and sciences. 2009 Nov;54(11):2538–40. doi: 10.1007/s10620-008-0641-z. [DOI] [PubMed] [Google Scholar]
- 81.Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009 Sep 1;15(17):5591–8. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 82.Margolin K, Ernstoff MS, Hamid O, et al. Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. The Lancet Oncology. 2012 May;13(5):459–65. doi: 10.1016/S1470-2045(12)70090-6. [DOI] [PubMed] [Google Scholar]
- 83.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012 Jun 28;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Robert C, Long GV, Brady B, et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. New Engl J Med. 2015 Jan 22;372(4):320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 85.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011 Aug 25;365(8):725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014 Oct 16;371(16):1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nature reviews Clinical oncology. 2013 May;10(5):267–76. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Till BG, Jensen MC, Wang J, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012 Apr 26;119(17):3940–50. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015 Feb 20;33(6):540–9. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bullain SS, Sahin A, Szentirmai O, et al. Genetically engineered T cells to target EGFRvIII expressing glioblastoma. J Neuro-Oncol. 2009 Sep;94(3):373–82. doi: 10.1007/s11060-009-9889-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kong S, Sengupta S, Tyler B, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. 2012 Nov 1;18(21):5949–60. doi: 10.1158/1078-0432.CCR-12-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morgan RA, Johnson LA, Davis JL, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human gene therapy. 2012 Oct;23(10):1043–53. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004 Dec 15;64(24):9160–6. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 94.Zagzag D, Salnikow K, Chiriboga L, et al. Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Laboratory investigation; a journal of technical methods and pathology. 2005 Mar;85(3):328–41. doi: 10.1038/labinvest.3700233. [DOI] [PubMed] [Google Scholar]
- 95.Lin R, Chen L, Chen G, et al. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. The Journal of clinical investigation. 2014 Dec;124(12):5352–67. doi: 10.1172/JCI76561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sandmair AM, Loimas S, Puranen P, et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Human gene therapy. 2000 Nov 1;11(16):2197–205. doi: 10.1089/104303400750035726. [DOI] [PubMed] [Google Scholar]
- 97.Chiocca EA, Broaddus WC, Gillies GT, Visted T, Lamfers ML. Neurosurgical delivery of chemotherapeutics, targeted toxins, genetic and viral therapies in neuro-oncology. J Neuro-Oncol. 2004 Aug-Sep;69(1–3):101–17. doi: 10.1023/b:neon.0000041874.02554.b3. [DOI] [PubMed] [Google Scholar]
- 98.Myers R, Harvey M, Kaufmann TJ, et al. Toxicology study of repeat intracerebral administration of a measles virus derivative producing carcinoembryonic antigen in rhesus macaques in support of a phase I/II clinical trial for patients with recurrent gliomas. Human gene therapy. 2008 Jul;19(7):690–8. doi: 10.1089/hum.2008.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wirth T, Samaranayake H, Pikkarainen J, Maatta AM, Yla-Herttuala S. Clinical trials for glioblastoma multiforme using adenoviral vectors. Curr Opin Mol Ther. 2009 Oct;11(5):485–92. [PubMed] [Google Scholar]
- 100.Atherton MJ, Lichty BD. Evolution of oncolytic viruses: novel strategies for cancer treatment. Immunotherapy. 2013 Nov;5(11):1191–206. doi: 10.2217/imt.13.123. [DOI] [PubMed] [Google Scholar]
- 101.Breitbach CJ, Paterson JM, Lemay CG, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Molecular therapy: the journal of the American Society of Gene Therapy. 2007 Sep;15(9):1686–93. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 102.Fulci G, Dmitrieva N, Gianni D, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer research. 2007 Oct 1;67(19):9398–406. doi: 10.1158/0008-5472.CAN-07-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alvarez-Breckenridge CA, Choi BD, Suryadevara CM, Chiocca EA. Potentiating oncolytic viral therapy through an understanding of the initial immune responses to oncolytic viral infection. Current opinion in virology. 2015 Apr 3;13:25–32. doi: 10.1016/j.coviro.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 104.Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nature medicine. 2012 Dec;18(12):1827–34. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liikanen I, Ahtiainen L, Hirvinen ML, et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Molecular therapy: the journal of the American Society of Gene Therapy. 2013 Jun;21(6):1212–23. doi: 10.1038/mt.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer journal. 2012 Jan-Feb;18(1):69–81. doi: 10.1097/PPO.0b013e31824671c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro-oncology. 2004 Jul;6(3):208–17. doi: 10.1215/S1152851703000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proceedings of the National Academy of Sciences of the United States of America. 1996 Mar 19;93(6):2370–5. doi: 10.1073/pnas.93.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gromeier M, Dobrikova E, Dobrikov M, et al. Oncolytic Poliovirus Immunotherapy of Glioblastoma. Neuro-oncology. 2014 Jul;16 [Google Scholar]
- 110.Desjardins AS John, Peters Katherine, Vlahovic Gordana, Randazzo Dina, Threatt Stevie, Herndon James, Boulton Susan, Lally-Goss Denis, McSherry Frances, Lipp Eric, Friedman Allan, Friedman Henry, Bigner Darell, Gromeier Matthias. Oncolytic polio/rhinovirus recombinant (PVSRIPO) against recurrent glioblastoma (GBM): Optimal dose determination. ASCO Annual Meeting; 2015; Chicago, Il. 2015. [Google Scholar]
- 111.Kamran N, Candolfi M, Baker GJ, et al. Gene Therapy for the Treatment of Neurological Disorders: Central Nervous System Neoplasms. Methods Mol Biol. 2016;1382:467–82. doi: 10.1007/978-1-4939-3271-9_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ali S, King GD, C JF, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65(16):7194–204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Curtin JF, King GD, Candolfi M, et al. Combining cytotoxic and immune-mediated gene therapy to treat brain tumors. Curr Top Med Chem. 2005;5(12):1151–70. doi: 10.2174/156802605774370856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ghulam Muhammad AK, Candolfi M, King GD, et al. Antiglioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression. Clin Cancer Res. 2009;15(19):6113–27. doi: 10.1158/1078-0432.CCR-09-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Curtin JF, Liu N, Candolfi M, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6(1):e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanchez-Perez LA, Choi BD, Archer GE, et al. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PloS one. 2013;8(3):e59082. doi: 10.1371/journal.pone.0059082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010 Jan 19;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010 Jan 19;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011 Mar 15;17(6):1603–15. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011 Mar 25;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 121.Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, Sherman LA. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. Journal of immunology. 1998 Jan 15;160(2):643–51. [PubMed] [Google Scholar]
- 122.Gilboa E. The promise of cancer vaccines. Nature reviews Cancer. 2004 May;4(5):401–11. doi: 10.1038/nrc1359. [DOI] [PubMed] [Google Scholar]