

Abstract
Rapamycin is a well-known inhibitor of the Target of Rapamycin (TOR) signaling cascade; however, the impact of this drug on global genome function and organization in normal primary cells is poorly understood. To explore this impact, we treated primary human foreskin fibroblasts with rapamycin and observed a decrease in cell proliferation without causing cell death. Upon rapamycin treatment chromosomes 18 and 10 were repositioned to a location similar to that of fibroblasts induced into quiescence by serum reduction. Although similar changes in positioning occurred, comparative transcriptome analyses demonstrated significant divergence in gene expression patterns between rapamycin-treated and quiescence-induced fibroblasts. Rapamycin treatment induced the upregulation of cytokine genes, including those from the Interleukin (IL)-6 signaling network, such as IL-8 and the Leukemia Inhibitory Factor (LIF), while quiescent fibroblasts demonstrated up-regulation of genes involved in the complement and coagulation cascade. In addition, genes significantly up-regulated by rapamycin treatment demonstrated increased promoter occupancy of the transcription factor Signal Transducer and Activator of Transcription 5A/B (STAT5A/B). In summary, we demonstrated that the treatment of fibroblasts with rapamycin decreased proliferation, caused chromosome territory repositioning and induced STAT5A/B-mediated changes in gene expression enriched for cytokines.
Keywords: primary fibroblast, quiescence, rapamycin, RNA-seq, STAT5A/B
Abbreviations
- AMPK
adenosine monophosphate protein kinase
- CLOVER
cis-element overrepresentation
- CXCL-6
chemokine (C-X-C motif)
- ELISA
enzyme linked immuno-sorbent reaction
- GO
gene ontology
- H33342
Hoechst 33342
- IL-6
Interleukin 6
- IL-8
Interleukin 8
- KB
kilobase
- KDa
kilodalton
- LC3
Light Chain 3
- LIF
Leukemia inhibitory factor
- MB
megabase
- mTORC
mammalian target of Rapamycin complex
- NFAT
Nuclear factor
- PI3K
phosphoinositide 3-kinase
- PRO
Proliferative
- qRT-PCR
Quantitative reverse transcriptase PCR
- QUI
quiescent
- RAP
Rapamycin
- RNA-seq
RNA sequencing
- SOCS
suppressor of cytokine synthesis
- STAT
signal transducer and activator of transcription
- TSS
transcriptional start site.
Introduction
The evolutionarily conserved target of rapamycin (TOR) complexes function as major signaling hubs downstream of several essential pathways necessary for growth and development. These pathways include the phosphoinositide 3 kinase (PI3K)/adenosine monophosphate protein kinase (AMPK) and insulin signaling networks with TOR complexes providing the essential link to processes such as translational control and autophagy. Mammalian TOR (mTOR) signaling is centered on the TOR protein kinase, which forms the backbone of 2 complexes in higher eukaryotes; mTOR complex (TORC) 1 and mTORC2.1-3 TORC1 is the most studied of the 2 complexes and has been identified as the major complex mediating nutrient sensing at the molecular level.4,5 Under conditions of caloric restriction, a state of reduced nutrient availability without causing malnutrition, TORC1 function is down-regulated causing decreased protein translation and increased autophagy.6-8 At the cellular level, decreased availability of critical molecules, such as essential amino acids,9,10 also leads to inhibition of TORC1 function, decreased rates of protein translation and increased autophagy. The inhibition of TORC1 function has been documented to significantly increase lifespan in a number of model organisms and non-human primates, as well as significantly reducing the development of age-related pathologies, such as cardiovascular disease, diabetes and cancer.6,11-13 Based on these observations, disruption of TOR signaling and function could be a viable strategy for increasing health and promoting healthier aging.
Rapamycin is a macrocyclic lactone-based drug originally isolated from the bacterium Streptomyces hygroscopicus. Rapamycin is bound between the mTOR kinase and FKBP12 subunits of the TORC1 complex resulting in functional inhibition. Although it primarily inhibits TORC1, high concentrations or chronic exposure of rapamycin have also been reported to inhibit TORC2.14-17 This compound has attracted a great deal of attention due to its proposed ability to mimic caloric restriction, resulting in decreased protein translation, increased autophagy and increased health and lifespan.18-22 These benefits were observed in both mice fed high-fat diets20 and older mice23 when fed rapamycin. Rapamycin has been suggested to elicit its anti-aging properties by preventing senescence under conditions that would normally cause cells to irreversibly exit the cell cycle.24,25 In addition, rapamycin stimulates the degradation of the mutant Progerin protein in cells from patients suffering from the premature aging disease Hutchinson Gilford Progeria Syndrome (HGPS).26
Genome organization is tightly linked to normal and disrupted cellular function. Numerous studies have highlighted how folding of the genome in 3 dimensional space, such as the interaction between the locus control region and the β-globin gene cluster,27-29 can facilitate gene expression. These changes in genome organization can also be observed at a much larger scale through the monitoring of chromosome territories. Chromosome territories can reorganize within the nuclear volume upon stimulation of normal dermal fibroblasts into quiescence and senescence.30,31 This reorganization occurred rapidly, taking only minutes in quiescent stimulated cells, demonstrating the dynamic nature of genome organization and its responsiveness to stimuli. Given this rapid and dynamic response and the importance of TOR in regulating cellular function, significant changes in both genome function and organization could result from TOR repression.
Our primary aim was to determine the impact of rapamycin-induced inhibition of TORC1 signaling on genome function and organization. To investigate this, we treated early passage (<20) primary human foreskin fibroblasts with rapamycin in culture, resulting in decreased proliferative rates as well as a decrease in the proportion of cells exhibiting the proliferative marker Ki67 or actively replicating DNA. Rapamycin also induced chromosome territory re-positioning within the nuclei of treated cells, representing a shift in genome organization. Decreased proliferative rates and re-localization of chromosome territories were comparable to those changes observed in fibroblasts induced into quiescence by decreased levels of serum, indicating that both treatments may result in similar cellular responses. Comparative transcriptome analyses of RNA sequencing (RNA-seq) datasets; however, demonstrated that rapamycin promoted significant divergence in transcript profiles. Serum reduction upregulated genes involved with the complement and coagulation cascade, while rapamycin treatment resulted in the upregulation of cytokine genes, including many of those involved with the Interleukin 6 (IL-6) cascade. Transcription factor motif analysis and subsequent chromatin immuno-precipitation (ChIP) assays revealed an increase in Signal Transducer and Activator of Transcription (STAT) 5 A/B promoter occupancy, implicating this factor in regulating rapamycin responsive cytokine-related genes.
Results
Rapamycin reduces cell proliferative rates in normal human fibroblasts
To determine the impact of rapamycin on proliferating cells, we treated proliferating primary human dermal fibroblasts isolated from foreskin (designated 2DD)32 with 500 nM rapamycin for 48h, 120h, 168h and 216h, and measured the doubling times as well as the total number of population doublings within culture (Fig. 1A and 1B). We observed a significant increase in the doubling time from an average of 31h under proliferating conditions to a maximum of 224h following rapamycin exposure. In concordance with this, we also observed a significant decrease in the total number of cell population doublings, with cells under proliferative conditions doubling 7.0 times while rapamycin treated cells only doubled a total of 2.1 times within a 216h time frame. To further determine the impact of rapamycin on proliferation, we measured the percentage of 2DD cells exhibiting Ki67, a nucleolar protein that is a marker of actively proliferating cells.33,34 Only 31% of rapamycin-treated fibroblasts were positive for Ki67 while 70.0% of fibroblasts were positive in proliferating cultures (Fig. 1C) indicating that fewer cells were actively in the cell cycle following rapamycin treatment. To confirm that rapamycin treatment resulted in the expected inhibition of mTOR function, western blot analyses were performed and a decrease in activated phosphorylated mTOR was observed (Fig. S1A). In addition, we observed diminished levels of phosphorylated p70-S6 kinase, a downstream target of TORC1, which indicated decreased TOR activity. Inhibition of TORC1 function induces autophagy which increases the number of light chain (LC) 3 containing autophagosomes.35 Immuno-fluorescence for LC3 identified that 72% of rapamycin-treated cells contained LC3 positive foci in comparison to only 18% in proliferative cultures (Fig. S1B). Together these observations indicate that rapamycin inhibited mTOR function in conjunction with decreased proliferative status of normal fibroblasts.
Figure 1.
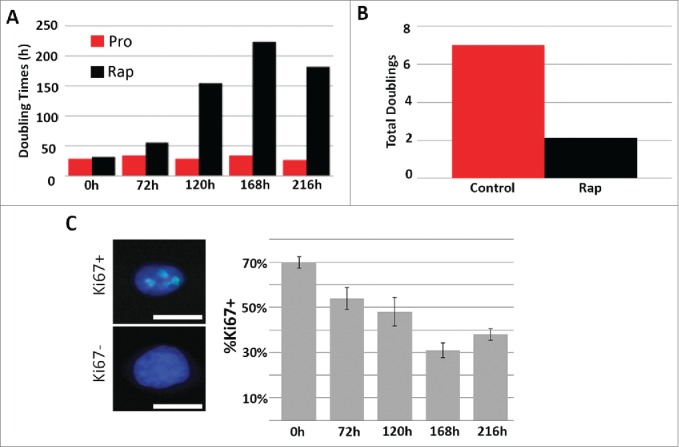
Rapamycin decreases the rate of fibroblast proliferation. 2DD fibroblasts were grown under normal culture conditions or in the presence of 500 nM rapamycin. Cell numbers were monitored and the population doubling times (Y axis) calculated over 0h, 72h, 120h, 168h, and 216h (X axis) (red bars represent proliferative fibroblasts and black bars represent rapamycin-treated fibroblasts for both panels). The total number of population doublings (Y axis) were plotted for control (red) and rapamycin-treated cultures (black). Data represent single biological replicates and repeated 2 additional times (data not shown) demonstrating parallel trends. Ki67 was immuno-labeled in cells cultured in rapamycin for 0h, 72h, 120h, 168h and 216h (C). Ki67 positive (Ki67+) and Ki67 negative (Ki67-) cells are shown on the left side of the panel. Ki67 was false colored green. Chromatin was counterstained with H33342 (blue). The percentage of Ki67 positive (%Ki67+, Y axis) cells calculated for each time point are shown (X axis). Error bars represent the SEM of the percent cells from individual counts. Scale bar = 10 μm.
Previous studies have demonstrated that decreased levels of serum in culture media (from 10% to 0.5%) induced quiescence and caused 2DD fibroblasts to increase cell population doubling times and decrease the number of Ki67 positive cells.30 In this study, quiescence induction of 2DD fibroblasts using 0.5% serum for 120h resulted in 4.3% Ki67 positive cells (Fig. S2A) with doubling times consistently ≥500 hours in several replicates, indicating that significantly fewer cells were actively proliferating. In addition, both quiescent induction with reduced serum and rapamycin treatment caused similar morphological changes, with cells becoming more flattened while still appearing healthy (Fig. S2B). Neither treatment exhibited deleterious effects on cell cultures; quiescent and rapamycin-treated cultures exhibited 96.4% and 97.3% viability via Trypan blue staining within the respective populations, while proliferative cultures had 97.3% viability (Fig. S2C). We further determined the number of cells actively replicating DNA by incubation with the thymidine analog, 5'-ethynyl-2'deoxyuridine (EdU) under each condition. In agreement with our Ki67-based observations, fewer cells incorporated EdU following rapamycin treatment and quiescence induction, indicative of a decrease in the number of cells in S-phase and actively replicating DNA (Fig. S2D). Furthermore, cells cycle analysis of propidium iodide stained proliferating, quiescent and rapamycin-treated 2DD fibroblasts demonstrate that 64.4%, 91.0% and 72.0% of the cells were in G1/G0 respectively (Fig. S2E). Although quiescence induction had a greater impact on reducing the number of actively proliferating 2DD fibroblasts (as measured by doubling times, Ki67 levels, EdU incorporation and flow cytometry), both reduced serum levels and rapamycin treatment exhibited similar phenotypic consequences.
Previous work has demonstrated that one of the hallmarks of quiescence induction is the re-positioning of chromosome 18 within the nuclear volume of fibroblasts.30 We re-capitulated this observation; reduced serum levels resulted in re-location of chromosome 18 from the nuclear periphery to a more central position within the nucleus (Fig. 2). We performed 2D-DNA fluorescence in situ hybridization (FISH) to 'paint' specific chromosomes and determined the location of these chromosomes within the nucleus by performing erosion analysis. Briefly, erosion analysis breaks the nucleus into 5 concentric rings or shells of area, with the outer most shell at the nuclear periphery considered shell 1 and the inner most ring, shell 5. The percent of each chromosome that falls into each ring was then measured and divided by the counter stain signal for chromatin to normalize for DNA content in each shell. Treatment of rapamycin induced a similar repositioning of chromosome 18 within the nuclear volume, exhibiting a more interior nuclear location. Rapamycin treatment also caused significant re-localization of chromosome 10, moving from an intermediate position (shells 3 and 4) to a more peripheral position, further indicating a shift in chromosome territory positioning and genome reorganization (Fig. 2). Repositioning of both chromosomes 18 and 10 was significant with regards to proliferative samples (p values ≤0.01). Under all conditions identified for 2DD cells, the X chromosome remains at the periphery of the nucleus.30 Although the statistical analyses (Student's T-tests) did show that there was a difference between quiescent and rapamycin-treated samples in the ratio of chromosome signal to chromatin, correlation calculations demonstrate that the X chromosome is found toward the periphery of the nucleus. Negative correlation trends were demonstrated for chromosome 18 in proliferative vs. quiescent (R2 value = −0.83) and proliferative vs. rapamycin-treated fibroblasts (R2 = −0.99), indicating a significant shift of chromosome position in response to quiescence induction or rapamycin treatment. Previous reports also demonstrate that chromosome 10 moves from an intermediate position toward the periphery of the nucleus upon quiescence induction.30 Although T-test's of our data did not demonstrate significance when comparing the ratios of signal intensity, correlation calculations exhibit more similarities in chromosome 10 positioning between quiescent and rapamycin treated fibroblasts (R2 = 0.87) than between proliferative and quiescent (R2 = 0.43). These correlations indicate that there is a stronger relationship in the positioning of chromosome 10 following the removal of serum and rapamycin treatment than with actively dividing proliferative cells.
Figure 2.
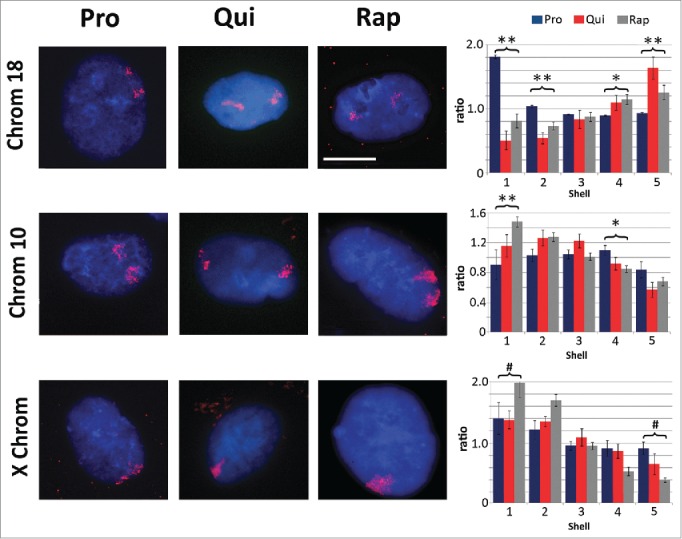
Chromosome re-localization following rapamycin treatment and quiescence induction. Chromosomes 18 (top row), 10 (middle row) and the X chromosome (bottom row) were identified in proliferating (Pro, first column), quiescent (Qui, second column) and rapamycin-treated (Rap, third column) 2DD fibroblasts by chromosome painting. Red signal represents the identified chromosomes; chromatin was counter stained with H33342 (blue). Scale bar = 5 μm for all images. Following painting, erosion analysis was performed to designate the location of the chromosomes within the nuclear volume. Nuclei were broken into 5 concentric shells of area, shell 1 being most exterior and shell 5 being most interior. Graphs for each specific chromosome represents the measured ratio of % chromosome signal divided by the % H33342 signal in each shell (Y axis) to normalize for DNA content in each shell (X axis). Proliferative positioning is represented by blue bars, quiescent by red and rapamycin-treated in gray. Error bars = SEM. Two tailed Student's test for unequal variance were used to demonstrate significant difference in chromosome positioning. ** indicates that both proliferative and quiescent measurements had p values ≤0 .01. * indicates that only rapamycin had significant changes in chromosome positioning. # indicates a significant difference (p values ≤0 .01) between quiescent and rapamycin-treated samples. Correlation calculations were also performed to demonstrate if the trend in chromosome positioning was similar or divergent between the samples. Chromosome 18 Pro vs. Qui R2 = −0.83, Pro vs. Rap R2 = −0.99 and Qui vs. Rap R2 = 0.87. Chromosome 10 Pro vs. Qui R2 = 0.43, Pro vs. Rap R2 = 0.64 and Qui vs. Rap R2 = 0.95. X chromosome: Pro vs. Qui R2 = 0.84, Pro vs. Rap R2 = 0.98 and Qui vs. Rap R2 = 0.91.
Rapamycin treatment and serum restriction induced divergent transcript profiles
Both rapamycin and quiescence induction reduced proliferative rates, decreased the number of cells exhibiting Ki67 or actively replicating DNA, and caused the repositioning of chromosomes 18 and 10. These similarities demonstrate that both treatments may have common or overlapping consequences on cellular function. The mTOR pathway is downstream of many signaling and growth factor receptors. Decreased serum levels (which include signaling and growth factors required for cell growth in culture) induce phenotypic changes and decreased growth responses similar to those of direct inhibition via rapamycin. To determine if there were significant similarities between these conditions, we performed comparative transcriptome analyses of RNA-seq data generated from mRNAs isolated from proliferative, quiescent and rapamycin-treated fibroblasts. Raw sequence reads from Illumina-based sequencing were mapped to a reference genome (GRh37/hg19), normalized using the RPKM (reads per kilobase of gene per million reads) method, and then analyzed using the tool, SeqMonk. For initial comparisons, scatter plots were generated, indicating the log number of reads from quiescent fibroblasts (Fig. 3A) or the log number of reads from rapamycin-treated fibroblasts (Fig. 3B), against the log number of reads from proliferative fibroblasts. We highlighted a subset of genes that changed transcript profiles ≥5-fold in quiescent vs. proliferative or rapamycin-treated vs. proliferative, and genes that changed ≥5-fold in both data sets. In quiescent 2DD fibroblasts, we identified 751 genes that exhibited a ≥5-fold change in expression (428 genes up-regulated and 323 genes downregulated) (Table S3). Rapamycin treatment resulted in 537 genes exhibiting a ≥5-fold change (421 genes upregulated and 116 down-regulated) (Table S4). Although the majority of genes from quiescent and rapamycin-treated 2DD fibroblasts had similar read count values to those of proliferating cells (correlations of 0.971 and 0.975 respectively), the scatter plots indicated that there was little overlap in the genes that changed expression between these 2 treatments. Examination of both gene lists revealed that only 123 genes (76 up-regulated and 47 downregulated) were common between quiescent and rapamycin-treated fibroblasts (Fig. 3C; Table S5), indicating that the responses to each condition were predominantly divergent and unique.
Figure 3.
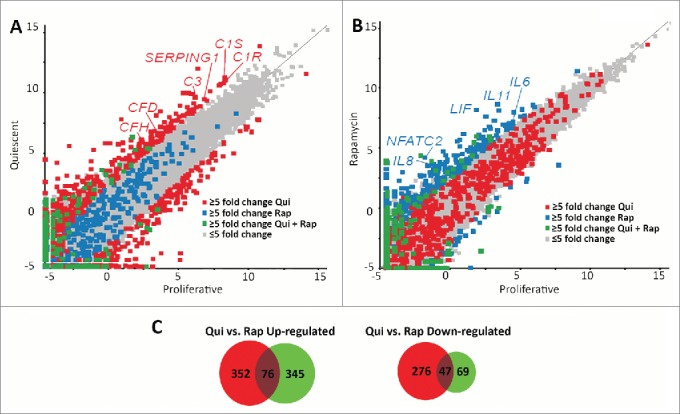
Scatter plots demonstrate different transcript profiles following quiescence induction or rapamycin treatment. Scatter plot comparing transcript abundance in quiescent and proliferating fibroblasts. The number of counts for each transcript identified by RNA-seq for proliferative (X axis) and quiescent (Y axis) fibroblasts was log-base-2 transformed with each square representing a single transcript. Transcripts exhibiting ≥5-fold increase and decrease in quiescent fibroblasts when compared to proliferative samples are marked in red (A). Gray squares represent transcripts that did not change abundance ≥5-fold. The number of counts for each transcript identified by RNA-seq for proliferative (X axis) and rapamycin-treated (Y axis) fibroblasts was log-base-2 transformed with each square representing a single transcript. Gray squares represent transcripts that did not change abundance ≥5-fold. Transcripts exhibiting ≥5-fold increase and decrease in rapamycin treated fibroblasts when compared to proliferative samples are marked in blue (B). Blue squares in panel A represent those transcripts that had a ≥5-fold increase and decrease in rapamycin treated fibroblasts and the red squares in panel B represent those transcripts that had a ≥5-fold increase and decrease in quiescent fibroblasts. Green squares in both panels represent transcripts that had a ≥5-fold increase or decrease under both quiescence induction and rapamycin treatment when compared to proliferative fibroblasts. Red or blue text highlights specific genes within each scatterplot. Venn diagrams demonstrating the number of genes up-regulated (left) or down regulated (right) from quiescent (red circles) or rapamycin-treated (green circles) fibroblasts in comparison to proliferating fibroblasts (C). Numbers in the overlapped areas indicate the number of genes changing expression in both quiescent and rapamycin treated fibroblasts.
We validated the change in expression of genes from both quiescent and rapamycin-treated fibroblasts by reverse transcriptase-quantitative PCR (RT-qPCR) to ensure that our observations from RNA-seq were accurate (Fig. S6A). Of the 22 genes analyzed to have increased ≥5-fold by RNA-seq in the quiescence dataset, 19 (86%) were also increased ≥5-fold by qRT-PCR. Of the 11 genes upregulated ≥5-fold by RNA-seq in rapamycin-treated data sets, 7 (64%) were ≥5-fold up-regulated by qRT-PCR (Fig. S6B). In addition, we tested 17 genes identified from the rapamycin RNA-seq dataset that were below our ≥5-fold cut-off and observed that all of these genes (100%) did not change ≥5-fold when measured by RT-qPCR (Fig. S6B). BEDgraphs of the raw sequence reads demonstrate that the ActC1 gene had the highest fold increase as a function of rapamycin treatment (Fig. S6C). Western blots confirmed that this equated to an increase in ACTC1 protein (Fig. S6D). These results demonstrate that the observed changes in transcript abundance likely result in biologically relevant changes in protein levels for other identified genes.
Network analyses reveal enrichment in cytokine-related pathways following rapamycin treatment
To determine which biological pathways were enriched for by quiescence induction and rapamycin treatment, we performed network analyses using identified genes that had significantly changed expression as a result of each treatment. Cytoscape36 with ReactomeFI37 was used to classify highly interacting groups of genes (modules) from our data sets, and identify the associated pathway annotation terms (Kyoto Encyclopedia of Genes and Genome; KEGG terms). Twenty-four individual modules were identified in the network dataset of genes that were over-expressed in quiescent 2DD fibroblasts (Table S7) (p-value <0.05, FDR <0.05). Examination of the KEGG terms associated with these modules showed significant enrichment in the complement and coagulation cascade as well as for infection response (Table 1). Examination of the network associated with this term shows several genes that have either increased or decreased in transcript abundance (Fig. S8). For example, transcripts from the Serping1, C2 and CFD genes increased while Serpine1, F2 and F12 show significantly decreased transcript levels in response to quiescence induction. These data demonstrated a previously unreported enrichment in KEGG-associated terms in upregulated genes for the complement and coagulation pathways in response to reduced serum levels in normal human fibroblasts. Analysis of quiescence up-regulated genes demonstrated enrichment for Gene Ontology (GO) terms involved with the negative regulation of cell proliferation and negative regulation of mRNA synthesis/transcription (p-value <0.05) (Fig. S9A).
Table 1.
Network pathway annotation terms enriched in quiescence upregulated genes. Genes ≥5 fold up-regulated in response to quiescence induction were analyzed for enrichment of KEGG terms. The identified enriched pathway KEGG annotation terms (GeneSet) from network analyses are listed. The total number of genes from the network identified to be upregulated in response to quiescence and belonging to a specific pathway was identified. p-value (with 0 values equating to<0.0001) and false discovery rates (FDR) are presented. Nodes identify specific genes/proteins from our data sets present in the networks.
| GeneSet | Genes From Network | p-value | FDR | Nodes |
|---|---|---|---|---|
| Complement and coagulation cascades | 9 | 0 | <1.000e-03 | CFH,CFD,SERPING1,C3,C1R,BDKRB2,C1S,C2,CFB |
| Extracellular matrix organization | 13 | 0 | <5.000e-04 | LTBP4,NCAM1,CTSK,VCAN,MFAP4,FMOD,LUM,DCN,BMP4,BMP2,TNXB,FBLN1,COL14A1 |
| Staphylococcus aureus infection | 7 | 0 | <3.333e-04 | CFH,CFD,C3,C1R,C1S,C2,CFB |
| Metabolism of xenobiotics by cytochrome P450 | 5 | 0.0003 | 2.73E-02 | ADH1B,ADH1A,AKR1C2,AKR1C1,ALDH3A1 |
| Pertussis | 5 | 0.0003 | 2.32E-02 | SERPING1,C3,C1R,C1S,C2 |
| BMP receptor signaling | 4 | 0.0003 | 1.97E-02 | CHRDL1,BMP4,BMP2,CHRD |
| Complement cascade | 7 | 0.0007 | 4.16E-02 | CFH,CFD,C3,C1R,C1S,C2,CFB |
| Cadherin signaling pathway | 5 | 0.0011 | 5.34E-02 | CDH23,PCDHB5,PCDHB4,PCDHB14,PCDHB12 |
| Wnt signaling pathway | 8 | 0.0011 | 4.79E-02 | CDH23,PCDHB5,PCDHB4,SMARCA5,PCDHB14,PCDHB12,SFRP2,SFRP4 |
Ten individual modules were identified in the under-expressed gene data set as a function of quiescence induction (Table S10). Enriched KEGG terms included cell cycle, mitosis, Aurora B signaling and p53 signaling (Table 2) in addition to GO term enrichment for biological processes in cell cycle progression and mitosis (p-value <0.05) (Fig. S9B). These observations indicated that gene expression patterns for cellular replication are inhibited in the absence of growth factors.
Table 2.
Network pathway annotation terms enriched in genes downregulated in quiescent fibroblasts. Genes ≥5 fold down-regulated in response to quiescence induction were analyzed for enrichment of KEGG terms. The identified enriched pathway annotation terms (GeneSet) from network analyses are listed. The total number of genes from the network identified to be downregulated in response to quiescence and belonging to a specific pathway were identified. p-value (with 0 values equating to<0.0001) and false discovery rates (FDR) are presented. Nodes identify the genes/proteins from our datasets present in the networks. For brevity only the top 20 most significant annotations are presented.
| GeneSet | Genes From Network | p-value | FDR | Nodes |
|---|---|---|---|---|
| Mitotic Prometaphase | 15 | 0 | <1.000e-03 | AURKB,KIF2C,CDCA8,CDK1,MAD2L1,ZWINT,NCAPH,BUB1,CENPM,CENPF,BIRC5,CDC20,CCNB1,CCNB2,PLK1 |
| Cell cycle | 14 | 0 | <5.000e-04 | E2F2,PKMYT1,PTTG1,CDC45,CCNA2,CDK1,MCM5,MAD2L1,SFN,BUB1,CDC20,CCNB1,CCNB2,PLK1 |
| Aurora B signaling | 9 | 0 | <3.333e-04 | KIF23,AURKA,AURKB,KIF2C,CDCA8,NCAPH,BUB1,BIRC5,KIF20A |
| FOXM1 transcription factor network | 9 | 0 | <2.500e-04 | AURKB,CCNA2,CDK1,FOXM1,CENPF,BIRC5,CCNB1,CCNB2,PLK1 |
| Mitotic Metaphase and Anaphase | 13 | 0 | <2.000e-04 | PTTG1,AURKB,KIF2C,CDCA8,UBE2C,MAD2L1,ZWINT,BUB1,CENPM,CENPF,BIRC5,CDC20,PLK1 |
| APC/C-mediated degradation of cell cycle proteins | 10 | 0 | <1.667e-04 | AURKA,PTTG1,AURKB,CCNA2,CDK1,UBE2C,MAD2L1,CDC20,CCNB1,PLK1 |
| Oocyte meiosis | 11 | 0 | <1.429e-04 | PKMYT1,AURKA,PTTG1,CDK1,MAD2L1,BUB1,CDC20,CCNB1,CCNB2,RPS6KA1,PLK1 |
| PLK1 signaling events | 8 | 0 | <1.250e-04 | PRC1,AURKA,CDK1,BUB1,CDC20,CCNB1,PLK1,KIF20A |
| Mitotic G2-G2/M phases | 10 | 0 | <1.111e-04 | PKMYT1,AURKA,CCNA2,CDK1,FOXM1,MYBL2,CENPF,CCNB1,CCNB2,PLK1 |
| Progesterone-mediated oocyte maturation | 9 | 0 | <1.000e-04 | PKMYT1,CCNA2,CDK1,MAD2L1,BUB1,CCNB1,CCNB2,RPS6KA1,PLK1 |
| p73 transcription factor network | 8 | 0 | <9.091e-05 | SERPINE1,CCNA2,CDK1,RAD51,SFN,BUB1,CCNB1,PLK1 |
| Senescence-Associated Secretory Phenotype (SASP) | 8 | 0 | <8.333e-05 | HIST1H2BL,CCNA2,UBE2C,HIST1H4A,IL6,HIST1H2BH,RPS6KA1,HIST1H2AJ |
| Cell Cycle Checkpoints | 9 | 0 | <7.692e-05 | PKMYT1,CDC45,CDK1,UBE2C,MCM5,MAD2L1,CDC20,CCNB1,CCNB2 |
| Mitotic G1-G1/S phases | 9 | 0 | <7.143e-05 | E2F2,PKMYT1,CDC45,CCNA2,CDK1,MCM5,RRM2,MYBL2,CCNB1 |
| p53 signaling pathway | 7 | 0 | <6.667e-05 | GTSE1,SERPINE1,CDK1,RRM2,SFN,CCNB1,CCNB2 |
| Mitotic Prophase | 8 | 0 | <6.250e-05 | HIST1H2BL,CDK1,HIST1H4A,HIST1H2BH,CCNB1,CCNB2,PLK1,HIST1H2AJ |
| Nucleosome assembly | 6 | 0 | 5.88E-05 | HIST1H2BL,MIS18A,HIST1H4A,HJURP,HIST1H2BH,HIST1H2AJ |
| Gastrin-CREB signaling pathway via PKC and MAPK | 10 | 0 | 5.56E-05 | MCHR1,MMP3,KISS1,CDK1,F2,GNG13,OXTR,P2RY6,RPS6KA1,ADRA1D |
| E2F transcription factor network | 6 | 0 | 4.21E-04 | E2F2,SERPINE1,CCNA2,CDK1,RRM2,MYBL2 |
| Meiotic Recombination | 5 | 0.0001 | 1.55E-03 | HIST1H2BL,RAD51,HIST1H4A,HIST1H2BH,HIST1H2AJ |
| DNA Damage/Telomere Stress Induced Senescence | 5 | 0.0001 | 2.24E-03 | HIST1H2BL,CCNA2,HIST1H4A,HIST1H2BH,HIST1H2AJ |
| Aurora A signaling | 4 | 0.0001 | 2.41E-03 | AURKA,AURKB,TACC3,BIRC5 |
| Mitotic Telophase/Cytokinesis | 3 | 0.0002 | 3.96E-03 | KIF23,PLK1,KIF20A |
| Oxidative Stress Induced Senescence | 5 | 0.0007 | 1.26E-02 | E2F2,HIST1H2BL,HIST1H4A,HIST1H2BH,HIST1H2AJ |
| Extracellular matrix organization | 8 | 0.0011 | 1.87E-02 | MMP3,PLOD2,SERPINE1,TNC,COL9A2,COL7A1,COMP,ITGA3 |
| Meiotic Synapsis(R) | 4 | 0.0011 | 1.81E-02 | HIST1H2BL,HIST1H4A,HIST1H2BH,HIST1H2AJ |
| Telomere Maintenance | 4 | 0.0013 | 2.03E-02 | HIST1H2BL,HIST1H4A,HIST1H2BH,HIST1H2AJ |
| Chromatin modifying enzymes | 5 | 0.0015 | 2.30E-02 | HIST1H2BL,HIST1H4A,ACTB,HIST1H2BH,HIST1H2AJ |
| GPCR ligand binding | 10 | 0.0021 | 3.10E-02 | MCHR1,KISS1,GAL,F2,CXCL3,GNG13,OXTR,P2RY6,PENK,ADRA1D |
| ATR signaling pathway | 3 | 0.0032 | 4.37E-02 | CCNA2,RAD51,PLK1 |
In contrast to quiescence, the rapamycin-induced network contained 22 distinct modules (p-value ≤0.05, FDR <0.05), 19 from the upregulated gene dataset and 3 from the down- regulated gene data set (Table S11). In the rapamycin up-regulated genes, we observed a significant enrichment for KEGG pathway annotation terms associated with PI3K-AKT signaling (Table 3), linking our observations to known mTOR-associated pathways. However, the most significant pathway annotation term identified in the up-regulated gene dataset was related to cytokine-cytokine receptor interactions. Examination of the network associated with this term demonstrates the up regulation of several cytokines (Fig. S12) and provides a functional relationship between IL-6ST, IL-11, IL-1B, INHBA, CXCL-1, CXCL-3, CXCL-2, PF4V1, LIF, IFNA1, BMP2, IL-6, IL-8 and TGFBR1 genes. Four of these genes (LIF, IL-8, IL-11, INHBA) ranked within the top 20 of all genes from the up-regulated data set that exhibited a ≥5-fold change in expression. We further observed that many of these genes belong to the IL-6 signaling pathway. Enzyme linked immuno-sorbent assays (ELISA) for IL-6 and IL-8 confirmed that these cytokines were produced in response to rapamycin and secreted into the cell media (Fig. 4A). Western blot analysis also confirmed elevated levels of the Leukemia Inhibitory Factor (LIF), also linked to IL-6 signaling, within cell lysates of rapamycin-treated fibroblasts (Fig. 4B). In addition, other cytokine/chemokine genes not associated with IL-6 signaling were upregulated, indicating that rapamycin elicits a response in fibroblasts broadly enriched for in cytokines.
Table 3.
Network pathway annotation terms enriched in rapamycin up-regulated genes. Genes ≥5 fold upregulated in response to rapamycin treatment were analyzed for enrichment of KEGG terms. The identified enriched pathway annotation terms (GeneSet) from network analyses are listed. The total number of genes from the network identified to be up-regulated in response to rapamycin and belonging to a specific pathway were identified. p-value (with 0 values equating to<0.0001) and false discovery rates (FDR) are presented. Nodes identify the genes/proteins from our data sets present in the networks. Only the top 20 most significant annotations are shown.
| GeneSet | Genes From Network | p-value | FDR | Nodes |
|---|---|---|---|---|
| Cytokine-cytokine receptor interaction | 13 | 0 | <1.000e-03 | IL6ST,IL11,IL1B,INHBA,CXCL1,CXCL3,CXCL2,LIF,IFNA1,IL6,BMP2,IL8,TGFBR1 |
| Salmonella infection | 8 | 0 | <5.000e-04 | IL1B,ROCK2,PKN2,CXCL1,CXCL3,CXCL2,IL6,IL8 |
| Pathways in cancer | 12 | 0 | 3.33E-04 | FGF7,PTGS2,TPR,FGF2,MDM2,LAMC2,ITGAV,IL6,BMP2,IL8,TGFBR1,ITGA2 |
| Legionellosis | 6 | 0 | 2.50E-04 | IL1B,CXCL1,CXCL3,CXCL2,IL6,IL8 |
| Signaling by SCF-KIT | 8 | 0 | 2.00E-04 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF,JAK2 |
| IL23-mediated signaling events | 5 | 0 | 1.67E-04 | IL1B,CXCL1,CD4,IL6,JAK2 |
| PIP3 activates AKT signaling | 7 | 0 | 1.43E-04 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Validated transcriptional targets of AP1 family members Fra1 and Fra2 | 5 | 0 | 2.50E-04 | LIF,NFATC2,IVL,IL6,IL8 |
| Signaling by ERBB4 | 8 | 0 | 2.22E-04 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF,JAK2 |
| TNF signaling pathway | 7 | 0 | 3.00E-04 | PTGS2,IL1B,CXCL1,CXCL3,CXCL2,LIF,IL6 |
| PI3K-Akt signaling pathway | 11 | 0 | 3.64E-04 | FGF7,FGF2,PKN2,MDM2,LAMC2,IFNA1,ITGAV,IL6,ITGA2,NR4A1,JAK2 |
| IL27-mediated signaling events | 4 | 0 | 8.33E-04 | IL6ST,IL1B,IL6,JAK2 |
| ErbB4 signaling events | 4 | 0 | 8.46E-04 | NRG1,MDM2,HBEGF,JAK2 |
| NOD-like receptor signaling pathway | 5 | 0 | 8.57E-04 | IL1B,CXCL1,CXCL2,IL6,IL8 |
| Interleukin-6 signaling | 3 | 0.0001 | 1.27E-03 | IL6ST,IL6,JAK2 |
| Signaling by FGFR | 7 | 0.0001 | 1.94E-03 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Signaling by ERBB2 | 7 | 0.0001 | 1.88E-03 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| ErbB receptor signaling network | 3 | 0.0002 | 3.00E-03 | NRG1,EREG,HBEGF |
| DAP12 interactions | 7 | 0.0002 | 2.75E-03 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Signaling by EGFR | 7 | 0.0002 | 2.75E-03 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Signaling by PDGF | 7 | 0.0002 | 3.29E-03 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Cell junction organization | 5 | 0.0002 | 3.91E-03 | CLDN4,F11R,LAMC2,LIMS1,CLDN14 |
| Haematopoietic cell lineage | 5 | 0.0003 | 5.04E-03 | IL11,IL1B,CD4,IL6,ITGA2 |
| Rheumatoid arthritis | 5 | 0.0004 | 5.46E-03 | IL11,IL1B,CXCL1,IL6,IL8 |
| NF-kappa B signaling pathway | 5 | 0.0004 | 5.60E-03 | PTGS2,IL1B,CXCL2,IL8,ATM |
| Cell adhesion molecules (CAMs) | 6 | 0.0004 | 6.08E-03 | CLDN4,F11R,CLDN14,ITGAV,CD4,CD274 |
| p53 pathway feedback loops 2 | 3 | 0.0006 | 9.15E-03 | MDM2,ATR,ATM |
| Jak-STAT signaling pathway | 6 | 0.0007 | 9.57E-03 | IL6ST,IL11,LIF,IFNA1,IL6,JAK2 |
| Glypican 1 network | 3 | 0.0007 | 9.41E-03 | NRG1,FGF2,TGFBR1 |
| Proteoglycans in cancer | 7 | 0.0008 | 1.09E-02 | FGF7,FGF2,ROCK2,MDM2,ITGAV,ITGA2,HBEGF |
| Amoebiasis | 5 | 0.0009 | 1.13E-02 | IL1B,LAMC2,CXCL1,IL6,IL8 |
| BARD1 signaling events | 3 | 0.001 | 1.33E-02 | TOPBP1,ATR,ATM |
| Epithelial cell signaling in Helicobacter pylori infection | 4 | 0.0012 | 1.46E-02 | F11R,CXCL1,IL8,HBEGF |
| Transcriptional misregulation in cancer | 6 | 0.0013 | 1.56E-02 | BMI1,MDM2,JMJD1C,IL6,IL8,ATM |
| Fc epsilon receptor (FCERI) signaling | 8 | 0.0015 | 1.66E-02 | FGF7,NRG1,FGF2,EREG,MDM2,NFATC2,NR4A1,HBEGF |
| Chemokine signaling pathway | 6 | 0.0018 | 1.96E-02 | ROCK2,CXCL1,CXCL3,CXCL2,IL8,JAK2 |
| signal transduction through il1r | 3 | 0.0018 | 1.92E-02 | IL1B,IFNA1,IL6 |
| Glucocorticoid receptor regulatory network | 4 | 0.0019 | 1.99E-02 | MDM2,IL6,IL8,NR4A1 |
| ATR signaling pathway | 3 | 0.0021 | 2.14E-02 | TOPBP1,MDM2,ATR |
| TGF-β signaling pathway | 4 | 0.0022 | 2.11E-02 | GDF6,INHBA,BMP2,TGFBR1 |
| TGF-β signaling pathway | 4 | 0.0022 | 2.11E-02 | GDF6,INHBA,BMP2,TGFBR1 |
| Signaling by NGF | 7 | 0.0028 | 2.59E-02 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Small cell lung cancer | 4 | 0.0028 | 2.59E-02 | PTGS2,LAMC2,ITGAV,ITGA2 |
| il 6 signaling pathway | 2 | 0.0031 | 2.74E-02 | IL6,JAK2 |
| Hepatitis B | 5 | 0.0031 | 2.78E-02 | IFNA1,NFATC2,IL6,IL8,TGFBR1 |
| Validated transcriptional targets of deltaNp63 isoforms | 3 | 0.0034 | 2.86E-02 | NRG1,MDM2,ATM |
| p53 pathway | 3 | 0.0034 | 2.86E-02 | MDM2,ATR,ATM |
| Fanconi anemia pathway | 3 | 0.0036 | 2.97E-02 | TOPBP1,ATR,ATM |
| Calcineurin-regulated NFAT-dependent transcription in lymphocytes | 3 | 0.0038 | 3.06E-02 | PTGS2,NFATC2,IL8 |
| IL6-mediated signaling events | 3 | 0.0041 | 3.11E-02 | IL6ST,IL6,JAK2 |
| Integrins in angiogenesis | 3 | 0.0041 | 3.11E-02 | FGF2,F11R,ITGAV |
| Malaria | 3 | 0.0046 | 3.39E-02 | IL1B,IL6,IL8 |
| atm signaling pathway | 2 | 0.0047 | 3.40E-02 | MDM2,ATR |
| Sumoylation by RanBP2 regulates transcriptional repression | 2 | 0.0047 | 3.40E-02 | RANBP2,MDM2 |
| SHP2 signaling | 3 | 0.0051 | 3.64E-02 | IL6ST,IL6,JAK2 |
| Chagas disease (American trypanosomiasis) | 4 | 0.0055 | 3.85E-02 | IL1B,IL6,IL8,TGFBR1 |
| Toll-like receptor signaling pathway | 4 | 0.0059 | 4.11E-02 | IL1B,IFNA1,IL6,IL8 |
| Beta5 beta6 beta7 and beta8 integrin cell surface interactions | 2 | 0.006 | 4.08E-02 | ITGAV,TGFBR1 |
| cell cycle: g2/m checkpoint | 2 | 0.006 | 4.08E-02 | MDM2,ATR |
| Signaling by the B Cell Receptor (BCR) | 7 | 0.0064 | 4.33E-02 | FGF7,NRG1,FGF2,EREG,MDM2,NR4A1,HBEGF |
| Influenza A | 5 | 0.0068 | 4.51E-02 | IL1B,IFNA1,IL6,IL8,JAK2 |
| Meiotic Synapsis | 3 | 0.0069 | 4.45E-02 | H2AFB1,ATR,SYNE1 |
| p53 pathway | 3 | 0.0069 | 4.45E-02 | MDM2,ATR,ATM |
Figure 4.
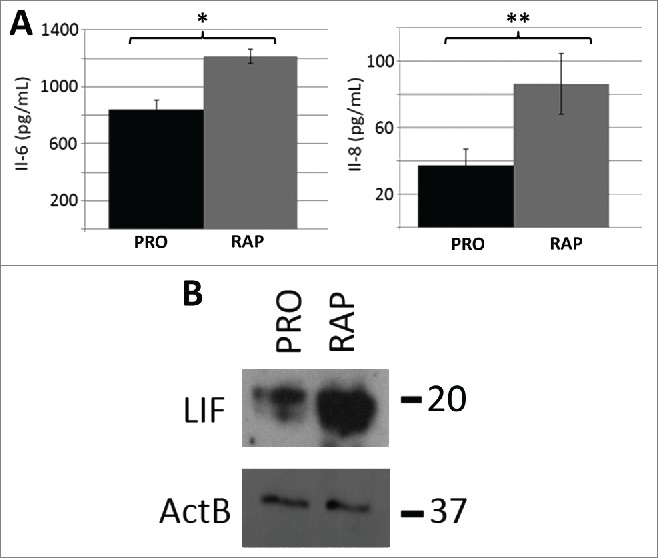
Protein levels of IL-6, IL-8 and LIF increased as a function of rapamycin treatment. ELISA assays were used to confirm that the rapamycin-induced increases in transcript profiles from the IL-6 and IL-8 genes also led to increased levels of secreted cytokines (A). Three biological replicates of culture media from proliferative (PRO) and 5 day rapamycin-treated cells (RAP) were subjected to sandwich ELISA and the concentration of both IL-6 and IL-8 determined. p-values for one tailed Student's t-tests are shown for IL-6 (* p=0.006) and IL-8 (** p=0.038). Error bars demonstrate the SEM. Equivalent amounts of whole-cell protein extracts from proliferative (PRO) and rapamycin-treated (RAP) fibroblasts were subjected to protein gel blotting for the LIF protein (˜20KDa) (B). A blot for β-actin was also used as a load control. Molecular weights (in KDa) are shown to the right of the blots.
Enrichment for GO terms from rapamycin up-regulated genes further supports the negative regulation of cell proliferation, while demonstrating enrichment in terms associated with positive regulation of mRNA synthesis/transcription (p-value <0.05) (Fig. S9C). Both of these terms were common to quiescence induction and rapamycin treatment, and included genes such as IDI-1, MDM-2, AREG and AREGB. This correspondence of GO terms indicated that although there were only 76 upregulated genes in common between the datasets, many of these are involved with signal transduction as well as negative regulation of the cell cycle and transcription. Other than this, there was no commonality between the KEGG pathway terms and little overlap with GO terms. Network pathway analyses demonstrated only 4 KEGG annotation terms enriched within rapamycin down-regulated modules (Table S13), all of which contained ≤4 genes in each module (Table 4). Analysis of GO terms enrichment indicates that genes promoting general transcription are downregulated as a function of rapamycin treatment (Fig. S9D), further illustrating the divergence in the gene response to quiescence and rapamycin.
Table 4.
Network pathway annotation terms enriched in genes down-regulated in rapamycin-treated fibroblasts. Genes ≥5 fold downregulated in response to rapamycin treatment were analyzed for enrichment of KEGG terms. The identified enriched pathway annotation terms (GeneSet) from network analyses are listed. The total number of genes from the network identified to be down-regulated in response to rapamycin and belonging to a specific pathway were identified. p-value (with 0 values equating to<0.0001) and false discovery rates (FDR) are presented. Nodes identify to the genes/proteins from our datasets present in the networks.
| GeneSet | Genes From Network | p-value | FDR | Nodes |
|---|---|---|---|---|
| GPCR ligand binding | 4 | 0.0001 | 3.30E-02 | MCHR1,F2,GNG13,ADM2 |
| Formation of Fibrin Clot (Clotting Cascade) | 2 | 0.0002 | 2.03E-02 | F2,PF4V1 |
| Thrombin signaling through proteinase activated receptors (PARs) | 2 | 0.0002 | 2.03E-02 | F2,GNG13 |
| Gastrin-CREB signaling pathway via PKC and MAPK | 3 | 0.0003 | 2.05E-02 | MCHR1,F2,GNG13 |
STAT5A/B binding sites are enriched in rapamycin-induced gene promoters
To identify mechanisms driving rapamycin-mediated changes in gene expression, we performed transcription factor motif searches using the cis-element over-representation (CLOVER) algorithm.38 We focused on promoter regions from 1000bp upstream to 50bp downstream of the transcription start site of genes that exhibited ≥5-fold change. Several binding motifs for the Signal Transducer and Activator of Transcription (STAT) family were enriched in the promoters of genes up-regulated by rapamycin. We observed an increase in the expression of several genes, including as IL-6ST and LIF,39 which are known to be regulated by the STAT5A/B transcription factor. Unlike other members of the STAT family, STAT5A/B also had a significant number of reads in our RNA-seq data sets, indicating that it was expressed. We, therefore, focused our analysis on this transcription factor to determine if STAT5A/B played a role in mediating changes in gene expression in response to rapamycin. Of the 421 genes identified to be upregulated, 144 (34.2%) (Table S14) had a STAT5A/B consensus binding element (Fig. 5A), a significant enrichment when compared against the genomic background (p-value <0.05). Immuno-fluorescence microscopy and subsequent line scans (Fig. S15A) demonstrated cells treated with rapamycin have larger, brighter foci of STAT5A/B within the nucleus. Furthermore, immuno-fluorescence for activated phosphor-tyrosine 694 STAT5A/B (STAT5A/B-Y694P) demonstrated brighter nuclear localization (Fig. 5B). However, it did appear that not all cells responded identically to the treatment with only a sub-population (∼40%) of labeled cells exhibiting brighter foci. Examination of our RNA-seq datasets did not show an increase in STAT5A/B transcripts; however, western blot analysis of protein extracts from rapamycin-treated 2DD cells did show an increase in the amount of STAT5A/B (Fig. S15B). Our observations indicate that rapamycin may lead to the stabilization of STAT5A/B protein levels and drive the transcription of STAT5A/B-mediated genes.
Figure 5.
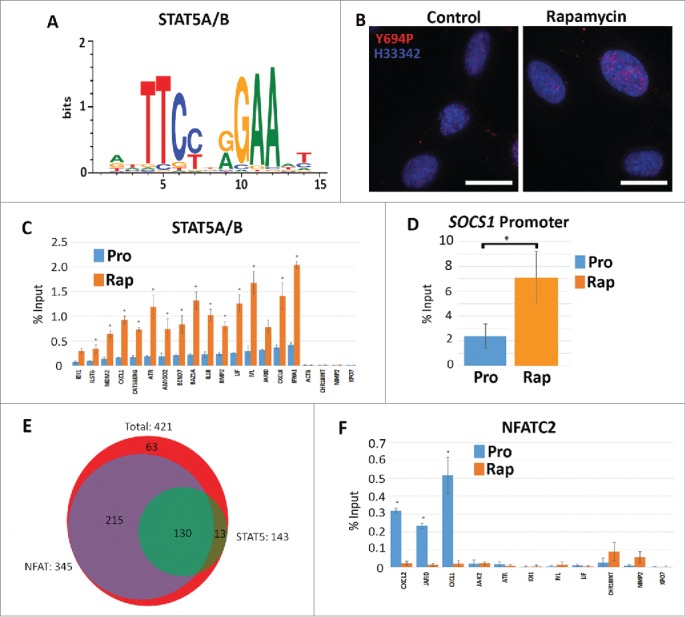
Increased STAT5A/(B)promoter occupancy but not NFACT2 in genes upregulated in response to rapamycin. Using CLOVER, the promoters of genes that increased expression following rapamycin treatment were enriched in STAT5A/B transcription factor binding sites. Position weight matrix/sequence logo of this binding site is shown (A). Scale to the left represents the log-base-2 of the information content of each nucleotide and the bottom scale represents the position of those nucleotides within the binding site. Immuno-fluorescence for phosphorylated Y694-STAT5A/B (Y694P; red) in proliferating (Control) and rapamycin-treated (Rapamycin) fibroblasts (B). Chromatin is counterstained with H33342 (blue). Scale bar = 10 μm. ChIP assays were used to compare promoter occupancy of STAT5A/B in proliferative (Pro/blue) and rapamycin treated (Rap/orange) samples (C). Promoters analyzed are given (X-axis) and the percent enrichment over input reported (Y-axis). For all ChIP assays, error bars represent the SEM and * indicate significant enrichment with p-values from one tailed t-tests ≤0 .05. ChIP assays for STAT5A/B promoter occupancy in the promoter of the SOCS1 gene under proliferative (Pro) and rapamycin treatment (Rap) (D). Venn diagram indicating the number of genes up-regulated by rapamycin (red; 421 genes) that shared STAT5A/B (green; 143 genes) and NFAT (purple; 345 genes) binding sites (E). ChIP assays were also performed for NFATC2 promoter occupancy. Proliferative (Pro/blue) and rapamycin treated (Rap/orange) samples were compared. Promoters analyzed are given at the bottom and the percent enrichment over input reported (F).
We conducted ChIP assays to determine if the increased presence of STAT5A/B within the nucleus resulted in increased chromatin binding. We confirmed increased STAT5A/B occupancy within the promoters of 15 rapamycin up-regulated genes, 4 of which encoded cytokines (Fig. 5C). We also tested MMP2, XPO7, Actin β, which do not contain STAT5A/B elements, as well as an intergenic region of chromosome 18, and did not see significant enrichment above input. ChIP for RNA polymerase II was used to validate enrichment of our assays (Fig. S16). These observations demonstrate that STAT5A/B promoter occupancy increased in fibroblasts treated with rapamycin and may identify this factor as key to mediating the observed cytokine response.
The Suppressor of Cytokine Synthesis (SOCS) family of proteins are negative regulators of cytokine signaling which have been documented to control STAT5 localization; binding and sequestering these molecules in the cytoplasm. In a number of systems, SOCS1 has been shown to control the localization and transcriptional activation function of STAT5.40,41 Examination of our RNA-seq data sets demonstrated that rapamycin treatment resulted in a 5.05 fold decrease in SOCS1 transcripts (Table S4). In biological replicates, we demonstrated a 2.7+/−0.06 fold decrease SOCS1 transcript abundance by qRT-PCR validation (data not shown). Furthermore, ChIP assays for STAT5A/B demonstrated increased occupancy of the SOCS1 promoter in response to rapamycin (Fig. 5D). Although the levels of transcript were reduced, our data indicates that other repressors are likely bound to this region inhibiting transcription, with increased levels of STAT5A/B bound at the promoter to quickly drive expression once the rapamycin stimulus is removed.
In addition to STAT5A/B consensus sites, CLOVER screens of rapamycin up-regulated genes showed a significant enrichment in binding elements for the Nuclear Factor of Activated T Cells (NFAT) transcription factor family (346/422; 82.0%) (Table S17). We also observed a significant increase in the level of NFATC2 transcript (also known as NFAT1) as a function of rapamycin treatment (Table S4). NFAT family members have putative roles in cell proliferation, apoptosis and tumorigenesis, and have been hypothesized to drive the expression of several cytokines.42 In addition, it has been reported that NFATC2 acts to repress STAT5A/B function.43 In our analyses of promoters, we found that 130/421 (30.9%) promoters contained both STAT5A/B and NFAT binding elements (Fig. 5E). Furthermore, we found that 130/143 (90.9%) promoters that had STAT5A/B sites also had an NFAT consensus element. We tested the promoters of the CXCL-2, JARID and CXCL-1 genes which had increased STAT5A/B promoter occupancy, and found that they exhibited decreased NFATC2 binding in rapamycin-treated cells when compared to proliferative cells (Fig. 5F). Other genes queried (JAK2, ATR, IDI-1, IVL and LIF) showed little or no binding of NFATC2 under both proliferative or rapamycin treatments.
Discussion
We observed that rapamycin treatment induced phenotypic changes of normal human foreskin fibroblasts similar to that of serum reduction, as well as causing the re-localization of chromosomes 18 and 10 within the nuclear volume. In addition, rapamycin induced a significantly divergent transcriptional profile than that of quiescence, up-regulating cytokine/chemokine-related genes. These observations confirmed that although rapamycin decreased cell proliferation without causing cell death, it did not result in a cellular state similar to quiescence. Increased occupancy of STAT5A/B in the promoters of these genes implicate this factor as part of the mechanism mediating rapamycin-induced changes in gene expression.
Network analyses of datasets from both quiescent and rapamycin-treated fibroblasts demonstrated enrichment of several distinct biological pathways. Interestingly, genes from the complement and coagulation cascade (Table 1) were enriched in quiescent fibroblasts. As the protein products of these genes are secreted into the extracellular space and are linked with serine-type peptidase/endopeptidase activity, they might have roles in modulating the extracellular matrix. This alteration of the matrix by these proteins could in turn be recognized by neighboring cells to gauge environmental conditions. A previous study examined the changes in gene expression from normal fetal lung fibroblasts under conditions of serum reduction.44 While this previous study did not identify significant enrichment in GO terms associated with the complement and coagulation pathway, genes such as SERPING1 and CFHL1 were up-regulated by serum/mitogen removal. A similar serine-type peptidase/endopeptidase activity could be common between both lung and foreskin fibroblasts. Down-regulated genes, such as those related to transcription (FOXM1) and regulation of the cell cycle (CCNB1), were also present in our RNA-seq data set as well as the study by Coller and colleagues.44 These observations demonstrate commonalities between these different, although similar, cell types in response to quiescence induced by serum reduction.
Previous computationally-based studies predicted rapamycin treatment may lead to increased cytokine production, in particular those related to IL-6.45-47 While we confirmed this prediction, we were surprised that such a large number of genes associated with cytokines/chemokines were upregulated in fibroblasts. IL-6 production has previously been linked to stress-induced premature senescence (SIPS)/replicative senescence and the senescence-associated secretory phenotype (SASP).48,49 A recent study by Lagerge and colleagues indicated that mTOR inhibition with rapamycin or knockdown of mTOR components causes decreased IL-6/cytokine production in senescent fibroblasts.50 Although we observed the opposite effect, with increased IL-6 and IL-8 production, our system used proliferating fibroblasts exposed to higher concentrations of rapamycin. 2DD fibroblasts treated with rapamycin were still able to proliferate, albeit at a much slower rate, indicating that our treatments were likely not causing SIPS/SASP. It is a possibility that rapamycin is inducing cytokine production in proliferating cells while repressing SASP in senescent cells, mediating a balance between the two states. Rapamycin enhances tumor necrosis factor (TNF)-α stimulated production of IL-6 and IL-8 in proliferating human orbital fibroblasts45 supporting our observed link between rapamycin and cytokine production. Although rapamycin has been partially linked with cytokine production in other cell types,45-47 we demonstrate, at least in part, the mechanisms involved with this stimulation, identifying STAT5A/B and decreased levels of SOCS1 transcription as being involved with this rapamycin-mediated response.
ELISAs provided further evidence of IL-6 and IL-8 production, displaying an increase in the amount of both proteins secreted from fibroblasts treated with rapamycin. Increased levels of these cytokines were detected within the culture media after 120h, but the fold change values were not as dramatic as those observed at the transcript level. This finding raises the possibility that IL-6 and IL-8 could have effects on adjacent cells within the environment. Furthermore, western blotting confirmed increased levels of LIF, which is also an IL-6-related secreted protein. Future investigations of these proteins should reveal if these signals are part of the program caused by disrupted nutrient sensing which promotes increased cellular health and longevity.
Transcription factor motif screens and subsequent ChIP analysis identified STAT5A/B as one of the factors binding up-regulated gene promoters in response to rapamycin in 2DD fibroblasts. One of the genes identified to have a STAT5A/B binding site in the promoter encodes the Suppressor of Cytokine Signaling (SOCS) family member, SOCS1. The SOCS1 protein, which is a negative regulator of Janus kinases (JAK) and STAT proteins, has previously been reported to sequester STAT5A/B in the cytoplasm.51 In rapamycin-treated fibroblasts, SOCS1 transcript abundance was decreased 5.05 fold in our RNA-seq dataset. This increase in STAT5A/B-mediated transcription of several cytokine genes agrees with the model in which SOCS1 forms part of the negative feedback loop; decreased expression of the SOCS1 gene possibly leads to decreased protein levels in the cytoplasm.52 This loss of SOCS1 protein would, therefore, allow STAT5A/B to translocate into the nucleus, where it binds and drives the expression of a large number of genes.41 In addition, several of the cytokines we observed to be up-regulated, such as IL-6 and LIF, are reported to up-regulate the expression of SOCS1.53 We did not observe an up-regulation of SOCS1 expression; however, this could be due to the ongoing treatment with rapamycin. Future studies will need to examine how STAT5A/B and decreased expression of the SOCS1 gene are linked to inhibition of mTOR by rapamycin. An indirect interaction between STAT5A/B and mTOR has been proposed in T cells and in response to cytokine signaling54; however, the mechanism involved with this is unclear.
Other SOCS family members have a range of functions in different cell types, often acting to form a negative feedback loop for cytokine signaling.52 Immune responses implicated in signaling from SOCS family members have been linked to obesity and type-2 diabetes.51 While decreased expression of other SOCS family members was observed, these genes did not reach our criteria for significance. However, these observations may implicate SOCS family members as potential global regulators controlling rapamycin-mediated transcription programs in other cell types.
In addition to STAT5A/B involvement with rapamycin-mediated gene induction, we also identified NFATC2 binding sites to be significantly enriched in these promoters. Previous studies have identified a physical interaction between STAT5A/B and NFATC2, with NFATC2 acting as a negative regulator.43 The increased transcription of the NFATC2 gene may be a response to increased activity of STAT5A/B; the resulting NFATC2 protein functioning to sequester and inhibit continued STAT5A/B-mediated transcription. Our ChIP analysis showed, at least in a subset of promoters analyzed, that NFATC2 is not interfering with STAT5A/B promoter occupancy.
In conclusion, we have demonstrated that rapamycin induced a significant change in the transcript profiles of normal human foreskin fibroblasts, distinguishing rapamycin treatment from quiescence induction by serum reduction. Furthermore, we observed increased cytokine/chemokine expression and production, including IL-6, IL-8 and LIF, which coincided with increased STAT5A/B promoter occupancy at many of these genes. This STAT5A/B-mediated regulation of gene expression provides novel insight into the mechanisms through which rapamycin treatment impacts cell proliferation, gene expression and cytokine production. Our observations link TOR inhibition with changes in genome function and organization independent of quiescence; changes which have the potential to regulate cellular health and longevity which, in turn may influence the aging process at the tissue/organ/organismal level.
Materials and Methods
Cell culture, cell counts and treatments
The normal human foreskin fibroblast cell line, designated 2DD (previously described in30), was cultured in high glucose (5mg/ml) DMEM (GE Health Care) no later than passage 20. For proliferative cultures, fibroblasts were grown in media containing 10% fetal bovine serum (FBS) (GE Health Care) and 1% penicillin/streptomycin (Sigma). Cultures were never allowed to proceed past 70% confluence to prevent contact inhibition. For passaging and counting, cells were dissociated from the tissue culture flasks using TrypLE Xpress (Invitrogen) and counted using a haemocytometer. For determination of cell viability counts, cells in suspension were mixed 1:1 with 0.4% Trypan blue dye (VWR international), and the total number of stained and unstained documented. Cells were induced into quiescence by the reduction of serum to 0.5% for 5 d Rapamycin treatments were identical to proliferative cultures but included 500 nM rapamycin (LC Laboratories, cat #; R-5000) dissolved in dimethyl sulfoxide (DMSO) in the media (stock solutions of 1.09 mM). Final concentrations of DMSO in media were at 0.046%. All parallel proliferative cultures were treated with DMSO to avoid artifacts. Media was changed every 2-3 d to ensure the presence of the drug over time.
Immuno-labeling
Proliferative, quiescent and rapamycin-treated cells grown on coverslips were fixed with 4% formaldehyde (FA) in phosphate buffered saline (PBS) for 10 min at RT. Cells were dehydrated using an ethanol series up to 100% followed by further extraction with methanol/acetone (1:1 mixture; 5 min RT). Cells were rehydrated and further extracted with 0.5% Triton X-100/PBS before blocking in 1% bovine serum albumin (BSA)/PBS for 20 min. Following this, immuno-labeling with rabbit anti-Ki67 (Novacastra Cat #: NCL-Ki67p_CE) (1:5,000 dilution in 1% BSA/PBS) and secondary antibody incubation with goat anti-rabbit A488 (Stratech/Jackson Scientific, Cat #: 111–545–003-JIR) (1:200 dilution in 1% BSA/PBS) was conducted. Nuclei were counterstained with Hoechst 33342 (H33342) dye (1:10,000 dilution in mounting media). Images were collected at 40X magnification with a constant exposure time. Gray-scale images were imported into Image J software and an arbitrary threshold selected for all images. Any nuclei containing label above the threshold was scored as positive for Ki67.
For immuno-labeling, cells grown on coverslips were fixed in 4% FA/PBS. Cells were permeablized with 0.5% Triton X-100/PBS. Rabbit anti-LC3 antibody (Abcam, Cat #: ab48394) (1:2000 dilution) was used to label autophagosome following rapamycin treatment. Rabbit anti-STAT5A/B (Abcam, Cat #: ab7969) (1:200) or rabbit anti-STAT5 phospho-tyrosine 694 (Thermo Fisher, Cat #: J.536.2) (1:100) was used for the localization of STAT5A/B protein. Secondary labeling was achieved using the above mentioned antibody. All images were collected in gray-scale, false-colored in Adobe Photoshop (CS5/6) and merged. Line scans were performed using the McMaster Biophotonics ImageJ software developed by McMaster University (https://www.macbiophotonics.ca/software.htm).
EdU incorporation/labeling and flow cytometry
Following 2 and 5 day rapamycin treatment or quiescence induction, live cells were incubated for 2h with 5μM 5-ethynyl-2-deoxyuridine (EdU) and fixed using the methods previously described for immuno-fluorescence. Following permeabilization, cells were labeled using the Click-It dye reagents (Life Technologies Cat #: C10338) following the manufacturer's instructions. For flow cytometry studies, proliferative, quiescent, and rapamycin treated cells were grown for 5 d before being fixed in a final concentration of 70% ethanol. Fixed cells were pelleted and washed in 1 X PBS. Fibroblasts were then re-suspended in 1ml dH2O and 20 μl of propidium iodide solution (VWR Cat #: CA421301-BL). 100 μg of RNase was added and samples were incubated at 37°C for 20 minutes. Analysis was performed using the BD Accuri C6 flow cytometer, flow rate was set to slow and 10,000 events recorded. Events were plotted using forward scatter (FSC, X-axis) and side scatter (SSC, Y-axis) parameters in order to identify single cells, and gates were used to remove cellular debris and doublets from the analysis. A histogram with linear axes plotting fluorescence intensity units (X-axis) against events counted (Y-axis), and gates were set to determine the percentage of cells in G1/G0, S and G2 phase based on DNA content.
Western blotting
Western blot analyses were performed on protein extracts generated from proliferative, quiescent and rapamycin-treated cultures. Following disruption of cells from dishes using TrypLE Express (Invitrogen), cells were pelleted followed by disruption in RIPA (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) buffer. Equivalent protein amounts were loaded (60 ug) per lane, transferred to nitrocellulose membrane, and blocked with 5% skim milk powder in Tris-buffered saline containing 0.5% Triton X-100 (SMP/TBST). Primary antibodies used were rabbit anti-LIF (1:500 dilution, Abcam Cat #: ab113262), rabbit anti-p70-S6 kinase-phospho421/424 (1:500 dilution, Cell Signaling, Cat #: 9204S), rabbit anti-Actin B (1:2000 dilution, Abcam Cat #: ab8227), rabbit anti-ActC1 (1:500 dilution, Abcam Cat #: ab151787), rabbit anti-STAT5A/B (Abcam, Cat #: ab7969) (1:2000) and rabbit anti-phosphor-mTOR (ser2448) (1:500 dilution, Cell Signaling, Cat #: 5536). Secondary antibodies used were goat anti-rabbit horse radish peroxidase (HRP) (1:5000 dilution) or donkey anti-mouse HRP (1:5000 dilution, Jackson Scientific/Cedarlane Cat #: 715–035–150).
2D FISH/Chromosome Painting and Erosion Analysis
Chromosome painting was performed on untreated 2DD normal human fibroblasts or fibroblasts treated with 500 nM Rapamycin for 5 d following the protocol detailed by Mehta and colleagues30 for both the painting procedure and DOP-PCR of chromosomes to create probes containing biotinylated uridine residues (biotin-16-UTP, Roche). Digital gray-scale images of random nuclei were captured using a Photometrics cooled charged-coupled device (CCD) camera, false-colored and merged in Adobe Photoshop (CS6). Smart Capture VP V1.4, a Leica fluorescence microscope (Leitz DMRB) with Plan Fluotar 100 × oil-immersion lens were used to collect images. New software called the Cell Nucleus Analyzer was developed (using Python coding language) for nuclear segmentation and analysis and based on the analysis software used previously.55 Briefly, the imaging software divides the DAPI image into 5 concentric shells of equal area, the first shell being most peripheral, and the innermost denoting the interior of the nucleus. The script measures the pixel intensity of DAPI and the chromosome probe in these 5 shells. Normalization of the probe signal was conducted by dividing the percentage of the probe by the percentage of DAPI signal in each shell. ≥ 35 nuclei were measured for each chromosome under proliferating conditions or following Rapamycin treatment. Ratios from each shell were averaged and the standard error of the mean calculated. Data were plotted as bar graphs with the X axis representing the shell and the Y axis the % signal chromosome/% signal H33342. Overlapping cells in the images were segmented using a semi-automatic method based on the watershed algorithm56 and user-specified markers. Both 2 tailed Student's T-tests for unequal variance and correlation calculations were performed in Microsoft Excel to demonstrate significance alterations in positioning.
RNA extractions, cDNA library synthesis and RT-qPCR
RNA was extracted from proliferative, quiescent and rapamycin treated cells using either Trizol (Invitrogen) or the FastRNA Pro Green Kit with FastPrep-24 instrument (MP Biomedicals) according to the manufacturer's instructions. RNA extractions for proliferative, quiescent and rapamycin treated fibroblasts were conducted at the same time for each replicate. Cultures for treatment were expanded from a single flask of fibroblasts to prevent any variation in passage or chronological age of the culture. The integrity of RNA was determined using the Bioanalyzer (Agilent Technologies) with RNA with an RNA integrity number (RIN) above 9.0 used for further analysis. For RT-qPCR, 5 ug of RNA was used in each reaction with 50 ng of random hexamer used as primers in SuperScriptIII reactions (Invitrogen) as per manufacturer's instructions. Following precipitation, cDNAs were resuspended in 100 ul H2O. cDNAs were diluted 1:10 and a 1 ul was used in each 10 ul reaction with 300 nM forward and reverse primer, 1.5 ul H2O and 5 ul 2X IQ RT-qPCR mastermix (Biorad). RT-qPCR reactions were conducted using the RotorGene RT-qPCR machine (Qiagen). Each reaction was run in triplicate with non-template controls to ensure that there was no contamination. In addition, melt curve analyses were performed to confirm the presence of only one product in each reaction. RT-qPCR results were quantified using the ΔΔCt method against 5 normalizing genes: PRDX5, EFEMP2, FAU, SPARC and FKBP10. The fold change for each gene of interest was compared to each of the normalizers and the average fold change between the 5 genes calculated. SEM were calculated for all genes of interest. A complete list of primers used is given in supplemental information (Table S18).
RNA sequencing and mapping
Extracted RNAs were submitted to Center for the Analysis of Genome Evolution and Function, (University of Toronto, Toronto, Canada) and poly-adenylated RNAs purified. cDNA libraries were constructed and subjected to paired-end sequencing using the Illumina GIIx to generate 75bp reads from 2 biological replicates for proliferative, quiescent or rapamycin treated samples. All reads were mapped back to a human reference genome (GRCh37/hg19 assembly from Ensembl) using the TUXEDO software package including TopHat2 with the following parameters: –bowtie1 -p 8 -r 20 –solexa-quals –coverage-search –microexon-search–library-type fr-unstranded. No clipping of reads was performed. The resulting BAM files were imported into the quantification and visualization tool SeqMonk (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk). Using the feature probe generator function, 149,135 probes were produced and the reads quantified within these probes using the reads per kilobase of gene per million reads (RPKM) normalization method as previously defined within.57 A read value of 0.05 was added to each probe to avoid infinite values during quantification. Examination of several of the transcripts demonstrated that genes from rapamycin-treated or quiescence samples had sufficient read depth indicating that the ≥5-fold change in these genes is not an artifact of low data volume. Probes were converted to genes in SeqMonk, and genes that had significant fold change in quiescent and rapamycin-treated fibroblasts were calculated against proliferative reads as baseline. Genes that had transcript abundance changes, either increased or decreased ≥5-fold, were considered significant and used for further analysis.
ELISA assays
Cells were plated as described above. Three mL of media were used for each flask of cells (with or without rapamycin) and maintained for the duration of the experiment. After 5 days, the media was removed, aliquoted and stored at −80°C. ELISA assays for IL-6 and IL-8 were performed as per manufacturer's instructions (R&D Systems) and quantified. One tailed Student's t-tests were performed indicating that the observed increase in protein levels was significant.
Network analysis
Cytoscape36 and ReactomeFI37 were used to identify the enrichment of network annotation terms (KEGG) to categorize specific pathways either up-regulated or downregulated (≥5-fold) as a function of quiescence induction or rapamycin treatment. Lists of Ensembl gene IDs were used as input to the Gene Set/Mutation Analysis function along with the following parameters: 2013 network version and gene set file format. Once the networks were constructed, all of the built-in functions within ReactomeFI Analyze Network Functions were used to determine KEGG and GO term enrichment within the network. The networks were also clustered into a subset of modules using the Cluster FI Network function within ReactomeFI. The resulting modules were analyzed for KEGG and GO term enrichment using the built-in functions within ReactomeFI Analyze Module Functions (Pathway Enrichment). Each module was required to contain a minimum of 2 genes to be included in the analysis for KEGG and GO term enrichment. GeneCodis58,59 was used for the identification of GO terms associated with biological processes for lists of genes that were upregulated or down-regulated as a function of quiescence induction or rapamycin treatment. For visualization of the networks, the most significantly enriched KEGG annotations terms were uploaded to Cytoscape using CytoKEGG. The nodes were colored based on fold change with upregulation represented in green and down-regulation in red with more intensely colored nodes representing those that had the greatest fold change.
Promoter analysis
CLOVER,38 using the TRANSFAC database, was used to identify over-represented transcription factor binding sites in the promoters of genes that exhibited a ≥5-fold change in transcript abundance during quiescence induction or rapamycin treatment. The complete set of human promoter sequences was used as a background baseline. Transcription factor binding sites were ranked based on raw score and only contained those with p-values <0.05.
Chromatin immuno-precipitation
2DD cells were grown to 70% confluence in 150 mm diameter dishes and treated as previously described. The cells were fixed with 1% FA in 10 mL media for 10 min at 37°C and washed 2X with 10 mL PBS containing 1X protease and phosphatase inhibitors (Sigma). Cell pellets were lysed for 10 min on ice in 600 uL SDS Lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8.0) with added inhibitors. The cells were sonicated on ice, for 3 cycles of 60 sec at 30% duty cycle, into chromatin fragments ˜500 bp in length. Five ug of anti-STAT5A/B (Abcam, Cat #: ab7969) was added to 107 cells and incubated overnight at 4°C, followed by binding of complexes of protein A Dynabeads (Invitrogen) overnight at 4°C. For NFATC2 (Abcam, Cat #: ab2722) or RNAPII-S5 (Abcam, Cat #: ab5131) ChIP assays, 5 ug of antibodies were bound to protein G (EDM Millipore Cat #: 16–201) or protein A agarose beads (EDM Millipore Cat #: 16–157), respectively, for 2h at 4°C. For non-specific binding controls, goat anti rabbit antibodies were bound to both protein A agarose and Dynabeads. The samples were washed with Low Salt buffer (0.1% SDS, 1% Triton X, 2 mM EDTA, 20 mM Tris pH 8.0, 150 mM NaCl), High Salt buffer (0.1% SDS, 1% Triton X, 2 mM EDTA, 20 mM Tris pH 8.0, 500 mM NaCl) and TE buffer, and eluted with 500 uL Elution buffer (1% SDS, 0.1 M NaHCO3). Crosslinks were reversed with 20 uL 5M NaCl incubated at 65°C for 5h, followed by phenol chloroform extraction and DNA precipitation. DNA was re-suspended in 30 uL ddH2O and 0.67 uL used in 10 uL RT-qPCR reactions carried out with gene-specific (300 nM) primers. A list of ChIP primers can be found in supplemental information (Table S19). ChIP-qPCR data was normalized by the percent input method and enrichment calculated. Standard error was calculated as a function of the standard deviations between triplicates.
RNA-seq files submitted to the Gene Expression Omnibus; accession number GSE65145).
Research Support
CHE is funded through support by the Royal Society Research Grant (UK), University of Saskatchewan NSERC President's Fund and NSERC Discovery Grant. ZEG is supported by a scholarship from the Saskatchewan Innovation and Opportunity Fund. KM was supported in part by the NSERC USRA program. JAM is funded by the Canadian Institutes of Health Research. JMB and IRK are funded through the Brunel Progeria Research Fund (UK). AK is funded through NSERC Discovery Grant program.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. A. Van Kessel (Animal and Poultry Science, UofS), Dr. N. Hogan (Animal and Poultry Science, UofS), Dr. K. Lessard (Agriculture Canada, UofS Campus) and Dr. D. Korber (Food and Bioproduct Sciences, UofS) for the generous use of laboratory space and equipment. We would also like to thank Dr. N. Low (Food and Bioproduct Sciences, UofS) and Dr. M. Nickerson (Food and Bioproduct Sciences, UofS) for the support as well as Mrs. P. Hind, Dr. H. Foster, Ms. E. Beaton-Brown, Mrs. M. Malo and Dr. T. Harkness for their technical advice.
Author's Contributions
ZEG performed cell growth/Ki67 studies, RNA extractions, RT-qPCR assays and assisted with manuscript construction. KM performed pathway and promoter analyses as well as linear clustering. MS/AW performed Western blot. MS performed ChIP assays and assisted with manuscript construction. BT assisted in bioinformatics screens. WD/JG performed ELISA assays and assisted with analysis. ME designed the erosion software. JMB and IRK assisted with the initial setup of the culture model used. AK assisted with computational analyses and manuscript preparation. JAM assisted with the RNA-seq preparation/analysis manuscript preparation. CHE performed RNA extracts, RT-qPCR and RNA-seq data analysis, as well as constructing the manuscript.
Reference
- 1.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP1 2-Rapamycin complex interacting with the binding domain of human FRAP. Science 1996; 273:239-41; PMID:8662507; http://dx.doi.org/ 10.1126/science.273.5272.239 [DOI] [PubMed] [Google Scholar]
- 2.Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. Isolation of a protein taeget of the FKBP12-Rapamycin complex in mammalian cells. J Biol Chem 1995; 270(2):815-22; PMID:7822316; http://dx.doi.org/ 10.1074/jbc.270.2.815 [DOI] [PubMed] [Google Scholar]
- 3.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Science 1994; 369:756-8. [DOI] [PubMed] [Google Scholar]
- 4.Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab 2010; 11(6):453-65; PMID:20519118; http://dx.doi.org/ 10.1016/j.cmet.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andre C, Cota D. Coupling nutrient sensing to metabolic homoeostasis: the role of the mammalian target of rapamycin complex 1 pathway. Proc Nutr Soc 2012; 71(4):502-10; PMID:22877732; http://dx.doi.org/ 10.1017/S0029665112000754 [DOI] [PubMed] [Google Scholar]
- 6.Lee C, Longo VD. Fasting vs dietary restriction in cellular protection and cancer treatment: from model organisms to patients. Oncogene 2011; 30(30):3305-16; PMID:21516129; http://dx.doi.org/ 10.1038/onc.2011.91 [DOI] [PubMed] [Google Scholar]
- 7.Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 2013; 38(5):233-42; PMID:23465396; http://dx.doi.org/ 10.1016/j.tibs.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SG, Buel GR, Blenis J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol Cells 2013; 35(6):463-73; PMID:23694989; http://dx.doi.org/ 10.1007/s10059-013-0138-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 2012; 18(9):524-33; PMID:22749019; http://dx.doi.org/ 10.1016/j.molmed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 2013; 14(3):133-9; PMID:23361334; http://dx.doi.org/ 10.1038/nrm3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson RM, Shanmuganayagam D, Weindruch R. Caloric restriction and aging: studies in mice and monkeys. Toxicol Pathol 2009; 37(1):47-51; PMID:19075044; http://dx.doi.org/ 10.1177/0192623308329476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun 2014; 5:3557; PMID:24691430; http://dx.doi.org/ 10.1038/ncomms4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weindruch R, et al. . Symposium: calorie restriction: effects on body composition, insulin signaling and aging. J Nutr 2001; 131(3):918s-923s; PMID:11238786 [DOI] [PubMed] [Google Scholar]
- 14.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17(6):596-603; PMID:16226444; http://dx.doi.org/ 10.1016/j.ceb.2005.09.009 [DOI] [PubMed] [Google Scholar]
- 15.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 2013; 126(Pt 8):1713-9; PMID:23641065; http://dx.doi.org/ 10.1242/jcs.125773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14(14):1296-302; PMID:15268862; http://dx.doi.org/ 10.1016/j.cub.2004.06.054 [DOI] [PubMed] [Google Scholar]
- 17.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22(2):159-68; PMID:16603397; http://dx.doi.org/ 10.1016/j.molcel.2006.03.029 [DOI] [PubMed] [Google Scholar]
- 18.Bjedov I, Partridge L. A longer and healthier life with TOR downregulation: genetics and drugs. Biochem Soc Trans 2011; 39(2):460-5; PMID:21428920; http://dx.doi.org/ 10.1042/BST0390460 [DOI] [PubMed] [Google Scholar]
- 19.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 2010; 11(1):35-46; PMID:20074526; http://dx.doi.org/ 10.1016/j.cmet.2009.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, Diaz V, Sloane L, Maslin K, Treaster S, et al. . Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci 2014; 69(2):119-30; PMID:23682161; http://dx.doi.org/ 10.1093/gerona/glt056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schröder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, et al. . Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest 2013; 123(8):3272-91; PMID:23863708; http://dx.doi.org/ 10.1172/JCI67674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab 2012; 15(5):713-24; PMID:22560223; http://dx.doi.org/ 10.1016/j.cmet.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. . Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460(7253):392-5; PMID:19587680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8(12):1888-95; PMID:19471117; http://dx.doi.org/ 10.4161/cc.8.12.8606 [DOI] [PubMed] [Google Scholar]
- 25.Xu Y, Li N, Xiang R, Sun P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem Sci 2014; 39(6):268-76; PMID:24818748; http://dx.doi.org/ 10.1016/j.tibs.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med 2011; 3(89):89ra58; http://dx.doi.org/ 10.1126/scitranslmed.3002346 [DOI] [PubMed] [Google Scholar]
- 27.Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nat Genet 2002; 32(4):623-6; PMID:12426570; http://dx.doi.org/ 10.1038/ng1051 [DOI] [PubMed] [Google Scholar]
- 28.Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, van Steensel B. Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet 2011; 7(3):e1001343; PMID:21455484; http://dx.doi.org/ 10.1371/journal.pgen.1001343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. . Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet 2010; 42(1):53-61; PMID:20010836; http://dx.doi.org/ 10.1038/ng.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta IS, Amira M, Harvey AJ, Bridger JM. Rapid chromosome territory relocation by nuclear motor activity in response to serum removal in primary human fibroblasts. Genome Biol 2010; 11(1):R5; PMID:20070886; http://dx.doi.org/ 10.1186/gb-2010-11-1-r5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta IS, Eskiw CH, Arican HD, Kill IR, Bridger JM. Farnesyltransferase inhibitor treatment restores chromosome territory positions and active chromosome dynamics in Hutchinson-Gilford progeria syndrome cells. Genome Biol 2011; 12(8):R74; PMID:21838864; http://dx.doi.org/ 10.1186/gb-2011-12-8-r74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bridger JM, Kill IR, O'Farrell M, Hutchison CJ. Internal lamin structures within G1 nuclei of human dermal fibroblasts. J Cell Sci 1993; 104:297-306; PMID:8505362 [DOI] [PubMed] [Google Scholar]
- 33.Bridger JM, Kill IR, Lichter P. Association of pKi-67 with satellite DNA of the human genome in early G1 cells. Chromosome Res 1998; 6:13-24; PMID:9510506; http://dx.doi.org/ 10.1023/A:1009210206855 [DOI] [PubMed] [Google Scholar]
- 34.Booth DG, Takagi M, Sanchez-Pulido L, Petfalski E, Vargiu G, Samejima K, Imamoto N, Ponting CP, Tollervey D, Earnshaw WC, et al. . Ki-67 is a PP1-interacting protein that organises the mitotic chromosome periphery. Elife 2014; 3:e01641; PMID:24867636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ladoire S, Chaba K, Martins I, Sukkurwala AQ, Adjemian S, Michaud M, Poirier-Colame V, Andreiuolo F, Galluzzi L, White E, et al. . Immunohistochemical detection of cytoplasmic LC3 puncta in human cancer specimens. Autophagy 2012; 8(8):1175-84; PMID:22647537; http://dx.doi.org/ 10.4161/auto.20353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res 2003; 13(1):2498-504; PMID:14597658; http://dx.doi.org/ 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol 2010; 11(5):R53; PMID:20482850; http://dx.doi.org/ 10.1186/gb-2010-11-5-r53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res 2004; 32(4):1372-81; PMID:14988425; http://dx.doi.org/ 10.1093/nar/gkh299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salas EM, García-Barchino MJ, Labiano S, Shugay M, Pérez-Encinas M, Quinteiro C, García-Delgado M, Vizmanos JL, Novo FJ. LIF, a Novel STAT5-Regulated Gene, Is Aberrantly Expressed in Myeloproliferative Neoplasms. Genes Cancer 2011; 2(5):593-6; PMID:21901172; http://dx.doi.org/ 10.1177/1947601911420139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi R, Yoshimura A. SOCS1 and regulation of regulatory T cells plasticity. J Immunol Res 2014; 2014:943149; PMID:25133199; http://dx.doi.org/ 10.1155/2014/943149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peltola KJ, Paukku K, Aho TL, Ruuska M, Silvennoinen O, Koskinen PJ. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood 2004; 103(10); PMID:14764533; http://dx.doi.org/ 10.1182/blood-2003-09-3126 [DOI] [PubMed] [Google Scholar]
- 42.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 1997; 15:707-747; http://dx.doi.org/ 10.1146/annurev.immunol.15.1.707 [DOI] [PubMed] [Google Scholar]
- 43.Zheng J, Fang F, Zeng X, Medler TR, Fiorillo AA, Clevenger CV. Negative cross talk between NFAT1 and Stat5 signaling in breast cancer. Mol Endocrinol 2011; 25(12):2054-64; PMID:21964595; http://dx.doi.org/ 10.1210/me.2011-1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol 2006; 4(3):e83; PMID:16509772; http://dx.doi.org/ 10.1371/journal.pbio.0040083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee WM, Paik JS, Cho WK, Oh EH, Lee SB, Yang SW. Rapamycin enhances TNF-α-induced secretion of IL-6 and IL-8 through suppressing PDCD4 degradation in orbital fibroblasts. Curr Eye Res 2013; 38(6):699-706; PMID:23281820; http://dx.doi.org/ 10.3109/02713683.2012.750368 [DOI] [PubMed] [Google Scholar]
- 46.Zhao J, Benakanakere MR, Hosur KB, Galicia JC, Martin M, Kinane DF. Mammalian target of rapamycin (mTOR) regulates TLR3 induced cytokines in human oral keratinocytes. Mol Immunol 2010; 48(1-3):294-304; PMID:20728939; http://dx.doi.org/ 10.1016/j.molimm.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Domhan S, Schwager C, Wei Q, Muschal S, Sommerer C, Morath C, Wick W, Maercker C, Debus J, Zeier M, et al. . Deciphering the systems biology of mTOR inhibition by integrative transcriptome analysis. Curr Pharm Des 2014; 20(1):88-100; PMID:23530502; http://dx.doi.org/ 10.2174/138161282001140113125549 [DOI] [PubMed] [Google Scholar]
- 48.Kojima H, Inoue T, Kunimoto H, Nakajima K. IL-6-STAT3 signaling and premature senescence. JAKSTAT 2013; 2(4):e25763; PMID:24416650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garbers C, Kuck F, Aparicio-Siegmund S, Konzak K, Kessenbrock M, Sommerfeld A, Häussinger D, Lang PA, Brenner D, Mak TW, et al. . Cellular senescence or EGFR signaling induces Interleukin 6 (IL-6) receptor expression controlled by mammalian target of rapamycin (mTOR). Cell Cycle 2013; 12(21):3421-32; PMID:24047696; http://dx.doi.org/ 10.4161/cc.26431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. . MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015; 17(8):1049-61; PMID:26147250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galic S, Sachithanandan N, Kay TW, Steinberg GR. Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem J 2014; 461(2):177-88; PMID:24966052; http://dx.doi.org/ 10.1042/BJ20140143 [DOI] [PubMed] [Google Scholar]
- 52.Dimitriou ID, Clemenza L, Scotter AJ, Chen G, Guerra FM, Rottapel R. Putting out the fire: coordinated suppression of the innate and adaptive immune systems by SOCS1 and SOCS3 proteins. Immunol Rev 2008; 224(1):265-83; PMID:18759933; http://dx.doi.org/ 10.1111/j.1600-065X.2008.00659.x [DOI] [PubMed] [Google Scholar]
- 53.Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci 2000(113):2813-9; PMID:10910765 [DOI] [PubMed] [Google Scholar]
- 54.Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol 2015; 36(1):21-9; PMID:25592035; http://dx.doi.org/ 10.1016/j.it.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 1999; 145(6):1119-1131; PMID:10366586; http://dx.doi.org/ 10.1083/jcb.145.6.1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vincent L, Soille P. Watersheds in digital spaces: an efficient algorithm based on immersion simulations. IEEE Transactions on Pattern Analysis and Machine Intelligence 1991; 13(6):583-98; http://dx.doi.org/ 10.1109/34.87344 [DOI] [Google Scholar]
- 57.Mitchell JA, Clay I, Umlauf D, Chen CY, Moir CA, Eskiw CH, Schoenfelder S, Chakalova L, Nagano T, Fraser P. Nuclear RNA sequencing of the mouse erythroid cell transcriptome. PLoS One 2012; 7(11):e49274; PMID:23209567; http://dx.doi.org/ 10.1371/journal.pone.0049274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nogales-Cadenas R, Carmona-Saez P, Vazquez M, Vicente C, Yang X, Tirado F, Carazo JM, Pascual-Montano A. GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res 2009; 37(Web Server issue):W317-22; PMID:19465387; http://dx.doi.org/ 10.1093/nar/gkp416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res 2012; 40(Web Server issue):W478-83; PMID:22573175; http://dx.doi.org/ 10.1093/nar/gks402 [DOI] [PMC free article] [PubMed] [Google Scholar]