

Abstract
We recently demonstrated that WRAP53β acts as a key regulator of ubiquitin-dependent repair of DNA double-strand breaks. Here, we applied the proximity ligation assay (PLA) to show that at such breaks WRAP53β accumulates in close proximity to γH2AX and, furthermore as demonstrated by their co-immunoprecipitation (IP) binds to γH2AX, in a manner dependent on the ATM and ATR kinases. Moreover, formation of complexes between MDC1 and both its partners RNF8 and phosphorylated ATM was visualized. The interaction of MDC1 with RNF8, but not with ATM requires WRAP53β, suggesting that WRAP53β facilitates the former interaction without altering phosphorylation of MDC1 by ATM. Furthermore, our findings highlight PLA as a more sensitive method for the analysis of recruitment of repair factors and complex formation at DNA breaks that are difficult to detect using conventional immunofluorescence.
Keywords: ATM, Double-strand breaks, DNA damage response, MDC1, PLA, RNF8, TCAB1, WRAP53, WDR79, γH2AX
Abbreviations
- 53BP1
p53-binding protein 1
- ATM
Ataxia Telangiectasia Mutated
- ATR
Ataxia Telangiectasia and Rad3-related protein
- BRCA1
Breast Cancer 1
- BrdU
5-Bromo-2´-deoxyuridine
- BSA
Bovine serum albumin
- ChIP
Chromatin immunoprecipitation
- DAPI
4′,6-diamidino-2-phenylindole
- DNA-PKcs
DNA-dependent protein kinase, catalytic subunit
- Gy
Gray
- IP
Immunoprecipitation
- IR
Ionizing radiation
- MDC1
Mediator of DNA damage checkpoint 1
- NP40
Nonidet P-40
- PARP
Poly ADP ribose polymerase
- PCR
polymerase chain reaction
- PLA
Proximity ligation assay
- RAD51
RAD51 recombinase
- RNF8
Ring finger protein 8
- RNF168
Ring Finger protein 168
- U2OS
U-2 osteosarcoma
- UV-A
Ultraviolet-A
- WRAP53
WD40-encoding RNA antisense to p53.
Introduction
Ubiquitin-mediated repair of DNA double-strand breaks
The human genome is under constant threat from various endogenous and exogenous sources. If double-strand breaks, one of the most severe types of DNA damage, are not repaired properly and in a timely manner, they may contribute to the development of degenerative diseases and cancer.1 To counteract such potentially life-threatening events, cells have evolved several sophisticated mechanisms of DNA repair, of which homologous recombination and non-homologous end joining are used to repair double-strand breaks.
Both these pathways require numerous factors that accumulate at the site of damage under regulation primarily by post-translational modifications, including phosphorylation and ubiquitylation, either of the repair factors themselves or of other proteins associated with the damaged DNA, such as histones. These modifications enable repair proteins to interact in an appropriate manner. For example, upon double-strand break formation, the histone variant H2AX is rapidly phosphorylated at Serine 139 (termed γH2AX) by the ATM/ATR/DNA-PKcs kinases,2 which enables it to bind MDC1 and recruit this factor to the site of DNA damage.3 Subsequent phosphorylation of MDC1 (on its TQXF motifs) by ATM promotes binding of the E3 ubiquitin ligase RNF8.4,5 Together with RNF168, RNF8 catalyzes ubiquitylation of histones in the chromatin flanking the double-strand break, a process critical for assembly of the downstream repair factors BRCA1, RAD51 and 53BP1.4,5
The WD40 protein WRAP53β orchestrates ubiquitin-mediated repair
Proteins containing WD40 domains consisting of several repeats approximately 40 amino acids long, with a C-terminal tryptophan (W) - aspartic acid (D), often act as scaffolds for large protein complexes. The repeats form a circular β-propeller structure, allowing several proteins to interact with the domain simultaneously.6,7
The gene encoding one such protein, WRAP53β (WD40-encoding RNA antisense to p53), is located on chromosome 17p13 and overlaps the p53 gene in a head-to-head fashion.8 This gene was shown in our laboratory to also encode WRAP53α, which regulates the expression of p53 RNA.8 WRAP53α is produced when transcription is initiated from exon 1α, one of the 3 alternative starting exons (1α, 1β, 1γ) in the gene, whereas transcription starting from exon 1β gives rise to WRAP53β, which acts independently of WRAP53α and does not regulate p53. Instead this latter protein facilitates interactions between and localization of factors involved in splicing, telomere elongation and DNA repair,9-12 as well as playing a critical role in the structural maintenance of the nuclear organelles known as Cajal bodies.9
Mutations within the WD40 domain of WRAP53β cause a rare progressive congenital disorder referred to as dyskeratosis congenita, the symptoms of which include bone marrow failure, premature aging and predisposition for cancer.13 In addition, loss of WRAP53β function has been associated with the neurodegenerative disease spinal muscular atrophy,9 as well as with reduced survival and radioresistance in patients with head and neck cancer.14 Moreover, single nucleotide polymorphisms in WRAP53 are correlated with an elevated risk for and poorer survival from various sporadic tumors, including ovarian and breast cancer.15-17
We recently demonstrated that WRAP53β acts as a scaffold for MDC1 and RNF8 during DNA double-strand break repair, binding these proteins simultaneously via its highly conserved WD40 domain, and thereby facilitating their interaction and the accumulation of RNF8 at double-strand breaks.12 RNF8 is the first E3 ligase to be recruited to DNA breaks and WRAP53β is thus required for ubiquitylation at sites of DNA damage and assembly of downstream repair proteins, including 53BP1, BRCA1 and RAD51. Consequently, loss of WRAP53β disrupts repair by homologous recombination and non-homologous end joining and enhances the frequency of spontaneous DNA breaks, highlighting its major role in the repair of double-strand breaks.12
The proximity ligation assay as a tool to visualize factors at DNA double-strand breaks
Recruitment of repair proteins to DNA lesions caused by ionizing radiation (IR) can be assessed from the formation of immunofluorescent foci representing their local accumulation, referred to as IR-induced foci. However, not all repair proteins form accumulations that are detectable in this manner.18
The in situ proximity ligation assay (PLA) allows direct visualization, as well as quantification and precise subcellular localization of protein-protein interactions/associations in fixed cells. The proteins of interest are targeted by specific antibodies conjugated with oligonucleotides and if in close proximity, ligation of the oligonucleotide moieties creates a DNA sequence that can be amplified exponentially by PCR to obtain powerful signal amplification. In this manner, each protein-protein association generates a fluorescent signal detectable under the fluorescence microscope.19
The present investigation was designed to evaluate whether PLA can be applied to monitor repair proteins at sites of DNA damage that do not form detectable IR-induced foci. Employing this procedure, we confirmed our previous findings and achieved deeper insight into the involvement of WRAP53β in the DNA damage response cascade.
Results
PLA visualizes the localization and interactions of DNA repair proteins
To assess whether PLA can detect repair proteins at sites of damage, we initially applied this method to MDC1 and γH2AX (a marker of DNA damage), which are known to interact only at sites of DNA damage. Localization of MDC1 to DNA lesions was first confirmed by immunofluorescent staining that, only after irradiation, revealed foci that exactly overlapped γH2AX foci (Fig. 1A).
Figure 1.
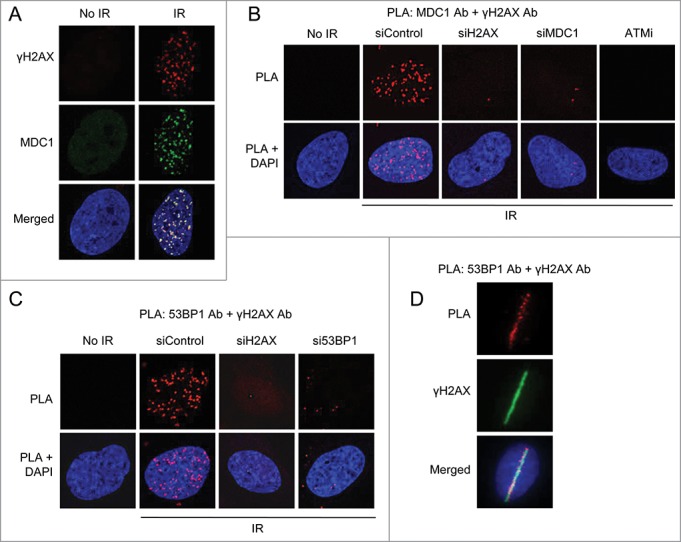
PLA visualizes complex formation and localization of repair factors at DNA breaks. (A) Immunofluorescent staining of γH2AX, a marker for DNA double-strand breaks, and MDC1 in U2OS cells not treated or whole-cell irradiated (6 Gy, 1 hour recovery). Nuclei were stained with DAPI (in blue) in all immunofluorescence and PLA experiments. (B) PLA detection of MDC1-γH2AX interactions visible as distinct fluorescent dots in U2OS cells. Approximately 90% of non-irradiated cells showed no signals and the remainder 2 dots/cell. In irradiated (6 Gy) cells, 100% of cells displayed >10 dots/cell. U2OS cells were transfected with the indicated siRNAs for 48 hours or treated with ATMi for 16 hours, irradiated and 15 minutes later subjected to PLA using MDC1 and γH2AX antibodies. (C) PLA detection of 53BP1-γH2AX interactions. U2OS cells were transfected with the indicated siRNAs for 48 hours, irradiated (6 Gy) and 15 minutes later subjected to PLA using 53BP1 and γH2AX antibodies. (D) U2OS cells were micro-irradiated and fixed after 5 minutes. PLA was performed to detect 53BP1-γH2AX interactions and cells were counterstained for γH2AX to visualize DNA double-strand breaks.
No PLA signals indicative of interaction between γH2AX and MDC1 were detected in non-irradiated cells. In contrast, several such signals were detected following irradiation and these γH2AX-MDC1 PLA signals yielded a very similar pattern as foci formation of these proteins (Fig. 1B). Knockdown of H2AX or MDC1 or inhibition of H2AX phosphorylation with an inhibitor of ATM reduced the number of these signals, indicating that the method is both specific and sensitive (Fig. 1B). Similar results were obtained with the repair protein 53BP1 and γH2AX (Fig. 1C) and moreover, following laser-microirradiation these proteins associated specifically at the laser stripes (Fig. 1D). Clearly, PLA can be used to visualize repair proteins at sites of DNA damage in fixed cells.
PLA reveals an association between WRAP53β and γH2AX at DNA double-strand breaks
Recently, we identified WRAP53β as a novel player in the DNA damage response that orchestrates ubiquitin-dependent assembly of repair factors at double-strand breaks.12 Although utilizing both immunofluorescence and chromatin immunoprecipitation (ChIP) we could demonstrate that WRAP53β accumulates at sites of damage,12 most WRAP53β antibodies do not provide visualization of this protein in repair foci. On the other hand, PLA utilizing one such antibody revealed an association between WRAP53β and γH2AX in response to irradiation, i.e. accumulation of WRAP53β at sites of DNA damage (Fig. 2A). This association was specific, since no PLA signals were obtained when the WRAP53β and γH2AX antibodies were combined with mouse or rabbit IgG antibodies, respectively (Fig. 2A). In laser micro-irradiated cells this association was observed only at laser stripes (Fig. 2B), a localization also confirmed by immunofluorescent staining (Fig. 2C). Together, these findings show that PLA can be used to visualize recruitment of WRAP53β to sites of DNA damage, also in cases when this protein is undetectable in IR-induced foci.
Figure 2.
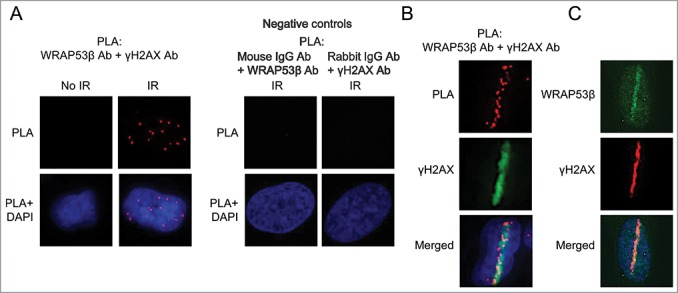
WRAP53β associates with γH2AX at sites of DNA damage. (A) PLA detection of WRAP53β-γH2AX interactions in U2OS cells. Approximately 70% of non-irradiated cells showed no signals and the remainder 1–2 dots/cell. All irradiated cells (6 Gy, 15 minutes recovery) contained >10 dots each. Negative controls for the PLA, showing the detection of WRAP53β and γH2AX combined with the indicated normal IgG antibody in irradiated (6 Gy, 15 min recovery) U2OS cells. (B) U2OS cells were micro-irradiated and fixed after 5 minutes. PLA was performed to detect WRAP53β-γH2AX interactions and cells were counterstained for γH2AX to visualize DNA double-strand breaks. (C) U2OS cells were micro-irradiated, fixed 5 minutes later and immunostained for WRAP53β and γH2AX.
WRAP53β binds γH2AX in an ATM and ATR-dependent manner
To confirm the specificity of the WRAP53β-γH2AX interaction, immunoprecipitation (IP) was performed in non-irradiated and irradiated U2OS and H1299 cells. This showed that WRAP53β co-precipitates γH2AX in both these cell lines. Reciprocal IP of γH2AX verified the interaction with WRAP53β. Moreover, these interactions were enhanced in response to DNA damage (Fig. 3A and B).
Figure 3.
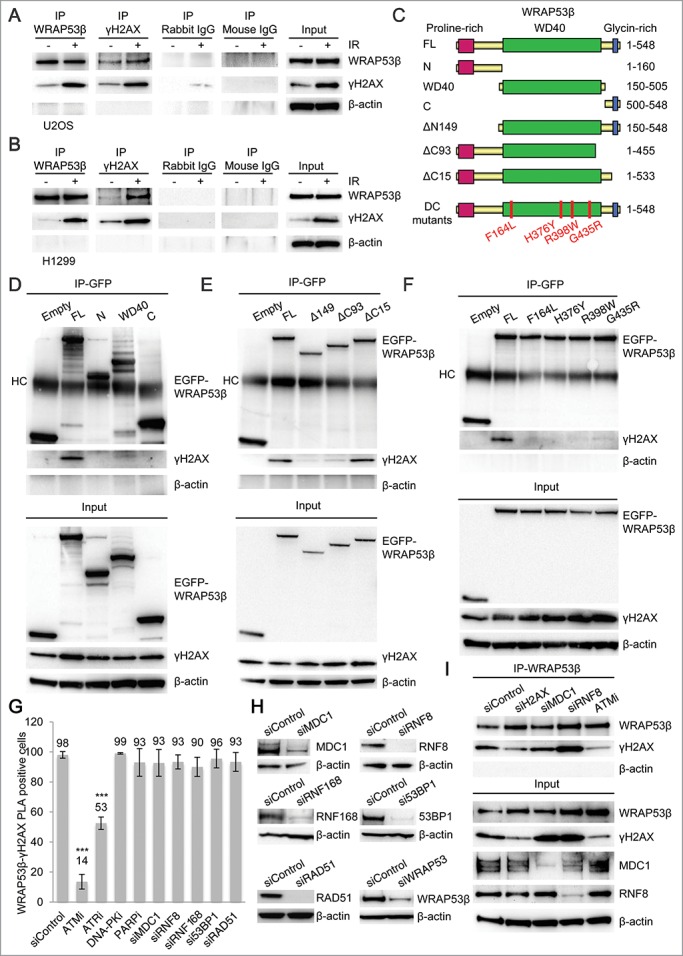
WRAP53β binds γH2AX in a manner dependent on DNA damage, ATM and ATR. (A) IP of WRAP53β and γH2AX from untreated or irradiated (6 Gy, 30–60 minutes recovery) U2OS cells followed by immunoblotting with the indicated antibodies. Rabbit and mouse IgG were used as negative controls. (B) IP of WRAP53β and γH2AX from untreated or irradiated (6 Gy, 60 minutes recovery) H1299 cells followed by immunoblotting with the indicated antibodies. (C) Schematic illustration of the EGFP-tagged WRAP53β deletion and mutation constructs. (D-F) U2OS cells were transiently transfected with the indicated EGFP-WRAP53β plasmids for 24 h; irradiated with 6 Gy and 1 hour later subjected to IP of GFP; followed by immunoblotting for γH2AX, GFP-WRAP53β and β-actin. HC indicates heavy chain of the antibody. (G) U2OS cells were treated with the inhibitors and siRNAs indicated for 24 hours and 48 hours, respectively, irradiated with 6 Gy and fixed 15 minutes later. PLA signals of WRAP53β-γH2AX interactions were quantified in 100 cells for each experiment (n=3) and nuclei containing ≥4 signals were counted as positive cells. No significant change in the number of PLA signals per cell was observed after treatment with the drugs/siRNAs that did not influence the WRAP53β-γH2AX interactions. Instead, these cells displayed the same number of PLA signal per cell as the untreated ones (>10 dots/cell). In the case of ATM and ATR inhibitors, the PLA signal almost disappeared in the negative cells (the majority had less than 2 dots/cell) or remained unchanged compared to control cells (>10 dots/cell). Error bars, s.e.m.; n=3, * p<0.05, *** p<0.001, Student's t-test. (H) Western blot analysis of MDC1, RNF8, RNF168, 53BP1, RAD51 and WRAP53β levels in U2OS cells treated with the indicated siRNAs for 48 hours. (I) U2OS cells were transfected with the indicated siRNAs for 48 hours or treated with ATMi for 16 hours, irradiated (6 Gy) and 15 minutes later subjected to IP with WRAP53β antibody followed by immunoblotting for WRAP53β, γH2AX, MDC1, RNF8 and β-actin.
To determine the region(s) of WRAP53β that interacts with γH2AX, a series of EGFP-WRAP53β deletion and mutation constructs was used (Fig. 3C). Deletion mutants of WRAP53β containing only the N-, WD40- or C-region all failed to bind γH2AX, indicating that several regions of WRAP53β are involved in binding γH2AX (Fig. 3D). Alternatively, the binding site(s) could be located in the borderlines of the WRAP53β deletions. To test this idea, another set of WRAP53β deletion constructs was used, in which the borderline sequences were intact. Deletion of either the N- (ΔN149) or C-terminal (ΔC93) region flanking the WD40 domain of WRAP53β prevented the interaction with γH2AX, while a construct lacking only the glycin-rich sequence (ΔC15) was fully capable of binding to γH2AX (Fig. 3E). Thus, the amino acids 1–533 of WRAP53β are critical for efficient binding to γH2AX.
Missense mutations in WRAP53β cause the cancer predisposition-syndrome dyskeratosis congenita.13 These mutations disrupt the folding of WRAP53β by the chaperonin TRiC/CCT (TCP-1 Ring Complex, also called CCT for chaperonin containing TCP-1), which has been suggested to cause the defective function of WRAP53β in dyskeratosis congenita.20 Interestingly, when analyzing these mutants for binding to γH2AX, none of them could interact with γH2AX (Fig. 3F). This indicates that TRiC-dependent folding of WRAP53β is required for binding to γH2AX and that impaired WRAP53β-γH2AX interaction may contribute to dyskeratosis congenita.
To further characterize the interaction between WRAP53β and γH2AX, we explored what factors that regulate this interaction. Applying both PLA and IP, the interaction between WRAP53β and γH2AX was shown to be markedly reduced by inhibition of ATM or ATR, whereas inhibition of DNA-PK or PARP or, alternatively, siRNA depletion of the DNA repair factors MDC1, RNF8, RNF168, 53BP1 or RAD51 had no influence (Fig. 3G-I). Thus, in response to DNA damage, WRAP53β associates with γH2AX in a manner dependent on TRiC, ATM and ATR, but no downstream repair factors.
WRAP53β regulates binding between RNF8 and MDC1 but not the association between phosphorylated ATM and MDC1
PLA demonstrated that in irradiated cells WRAP53β associates with its known partners MDC1 and RNF8 in a specific manner (Fig. 4A), as also confirmed by IP (Fig. 4B). Since the extent of these interactions was the same with and without irradiation, as well as in ATMi-treated cells (Fig. 4B), we conclude that they are independent of both DNA damage and ATM.
Figure 4.
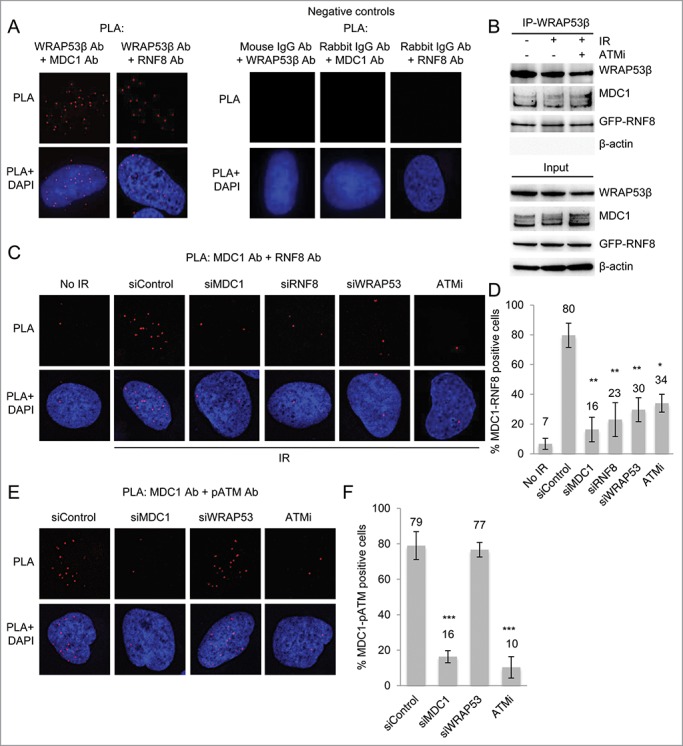
WRAP53β facilitates MDC1-RNF8 interaction. (A) PLA signals of WRAP53β-MDC1 and WRAP53β-RNF8 interactions in irradiated (6 Gy, 15 minutes recovery) U2OS cells. Negative controls for PLA, showing the detection of MDC1, RNF8 or WRAP53β combined with the indicated normal IgG antibody in irradiated (6 Gy, 15 minutes recovery) U2OS cells. The images show representative numbers of interactions. (B) U2OS cells were either left untreated, irradiated with 6 Gy or treated with ATMi for 16 hours prior to irradiation with 6 Gy. Fifteen minutes later, IP of WRAP53β was performed followed by immunoblotting of WRAP53β, MDC1, GFP-RNF8 and β-actin. (C) PLA signals of MDC1-RNF8 interactions in U2OS cells treated with siControl, siMDC1, siRNF8 or siWRAP53#2 for 48 hours or ATMi for 24 hours, irradiated with 6 Gy and fixed after 15 minutes. (D) Quantification of the results in (C). PLA signals were quantified in 100 cells for each experiment and nuclei containing ≥4 signals were counted as positive cells. The majority of the positive cells showed the same amount of PLA signals per cell as the corresponding positive control (>10 dots/cell), whereas the negative cells mostly had less than 2 dots/cell. (E) PLA signals of MDC1-pATM interactions in U2OS cells treated with siControl, siMDC1, siWRAP53#2 for 48 hours or ATMi for 24 hours, irradiated with 6 Gy and fixed after 15 minutes. (F) Quantification of the results in (E). PLA signals were quantified in 100 cells for each experiment and nuclei containing ≥4 signals were counted as positive cells. Error bars, s.e.m.; n=3, *** p<0.001, Student's t-test.
WRAP53β acts as a scaffold for formation of a complex between RNF8 and MDC1,12 an interaction that is also dependent on phosphorylation of MDC1 by ATM,4,5,21 Accordingly, in cells lacking WRAP53β the extent of interaction between MDC1 and RNF8 was clearly reduced (Fig. 4C). 80% of the control cells exhibited PLA signals reflecting MDC1-RNF8 interactions, whereas the corresponding value following WRAP53β depletion was only 30% (Fig. 4D). The specificity of the PLA signals was confirmed by knockdown of MDC1, RNF8 or inhibition of MDC1 phosphorylation with an inhibitor of ATM (Fig. 4C).
The interaction between phosphorylated ATM and MDC1 was, however, unchanged in WRAP53β depleted cells (Fig. 4E and F), both the amount of positive cells as well as the number of PLA signals per cell. We conclude that WRAP53β mediates the interaction between RNF8 and phosphorylated MDC1, but is not involved in formation of a complex between ATM and MDC1.
Discussion
Here, we introduce the novel PLA procedure for visualization of proteins and their association at sites of DNA damage. This method accurately detects complexes between the established repair proteins MDC1 and 53BP1 and γH2AX at such lesions and is more powerful than traditional immunofluorescent staining in cases where IR-induced foci are difficult to detect, as demonstrated for the repair factor WRAP53β. In addition, application of PLA to laser micro-irradiated cells confirmed that these protein interactions occur at sites of DNA damage (laser stripes).
Further characterization revealed that WRAP53β and γH2AX are co-precipitated by IP and that WRAP53β binds γH2AX in a manner dependent on ATM and ATR. These findings are in agreement with our previous observations that accumulation of WRAP53β in repair foci depends on the ATM and ATR kinases.12 We previously showed that the WD40 domain of WRAP53β is responsible for binding MDC1 and RNF8.12 Our current findings demonstrate that expression of a larger region of WRAP53β (amino acids 1–533 containing the proline-rich region and the WD40 domain) is required for interaction with γH2AX.
Moreover, we show that single amino acid mutations in WRAP53β found in patients with dyskeratosis congenita13 completely disrupt the capacity of this protein to bind γH2AX. Interestingly, it was recently shown that the chaperonin TRiC, which is involved in the folding of difficult-to-fold proteins, controls the folding and function of WRAP53β.20 By binding its WD40 domain, TRiC folds WRAP53β, which enables binding between WRAP53β and the telomerase RNA component TERC and allows the subsequent function of WRAP53β in telomere elongation.20 Our finding that dyskeratosis congenita-mutants of WRAP53β are unable to bind γH2AX indicates that TRiC-mediated folding of WRAP53β is required for γH2AX binding and opens the possibility that loss of WRAP53β-γH2AX interaction and disturbed DNA repair could contribute to the pathogenesis of dyskeratosis congenita.
Since accumulation of WRAP53β in repair foci requires the MDC1 protein, it was surprising that knockdown of MDC1 did not attenuate WRAP53β-γH2AX PLA signals. Possibly, WRAP53β can interact with γH2AX in the absence of MDC1, but that extensive accumulation of WRAP53β in repair foci requires MDC1. Moreover, recruitment of different pools of WRAP53β to double-strand breaks may involve different factors. In cells containing a site-specific DNA double-strand break introduced by the I-PpoI endonuclease, ChIP revealed that a portion of WRAP53β accumulated in the vicinity of the breakpoint and overlapping γH2AX-positive sites, whereas another portion of WRAP53β accumulated at the breakpoint site itself, a region normally negative for γH2AX.12 Moreover, these different pools of WRAP53β remained for different lengths of time at these two sites, indicating that the recruitment of WRAP53β to DNA breaks is regulated in different ways.
Furthermore, we tested our earlier proposal that by facilitating binding between RNF8 and MDC1, WRAP53β is required for recruitment of RNF8 to double-strand breaks and the subsequent ubiquitylation of the flanking chromatin.12 Both PLA and IP revealed that WRAP53β interacts with RNF8 and MDC1 in a manner independent of DNA damage and ATM. Knockdown of WRAP53β abrogated the interaction between MDC1 and RNF8 without affecting binding between MDC1 and phosphorylated ATM. Thus, this loss of binding between RNF8 and MDC1 in the absence of WRAP53β is not due to attenuated phosphorylation of MDC1, but rather, WRAP53β appears to facilitate re-localization of RNF8 to sites of DNA damage and its subsequent interaction with phosphorylated MDC1. Possibly, such re-localization to DNA breaks involves binding between WRAP53β and γH2AX.
In summary, we demonstrate here that PLA allows sensitive monitoring of the localization of and association between proteins at DNA breaks. Moreover, WRAP53β interacts with γH2AX at sites of DNA damage, and also enables direct binding between MDC1 and RNF8 without altering the association between MDC1 and ATM.
Material and Methods
Cells and culture conditions
U2OS cells were maintained in McCoy's 5A medium (HyClone, Thermo Scientific), supplemented with 10% fetal bovine serum (HyClone) and 2,5 µg/mL Plasmocin (InvivoGen) at 37°C in 5% CO2 humidified incubators.
Ionizing radiation
γ-irradiation was performed with a 137Cs source (Scanditronix, Uppsala, Sweden) at the Karolinska Institutet, Stockholm, at a photon dose rate of 0.5 Gy·minutes−1. Dosimetry was done with an ionization chamber as well as with ferro sulfate.
Laser micro-irradiation
Localized DNA damage was generated by exposure of cells to a UV-A laser. U2OS cells were pre-sensitized with 10 mM 5-Bromo-2′-deoxyuridine (BrdU) for 24 hours at 37°C. Prior to microscopy the medium was replaced for a phenol red-free medium. Micro-irradiation was performed with a confocal microscope equipped with a 365-nm UV-A laser.
Immunofluorescence microscopy
Cells were grown on sterilized cover slips and fixed with 4% paraformaldehyde for 15 minutes at room temperature. They were then permeabilized with 0.1% Triton X-100 for 5 minutes at room temperature, followed by 30 minutes of blocking in blocking buffer (2% BSA, 5% glycerol, 0,2% Tween20, 0,1% NaN3). Coverslips were subsequently incubated for 1 hour in primary antibody and 40 minutes in secondary antibody diluted in blocking buffer. The cover slips were mounted with Vectashield mounting medium with DAPI (Vector laboratories). Images were acquired with a LSM700 confocal microscope (Carl Zeiss Microimaging Inc.), mounted on Zeiss Axio observer.Z1 equipped with Plan-Apochromat 63x/1.4 oil immersion lenses, and processed using Zen 2012 Black or with a Zeiss Axioplan 2 microscope, equipped with an AxioCam HRm Camera using 40 or 63 oil immersion lenses, and processed using Axiovision Release 4.7.
In situ PLA
Cells were cultured on coverslips, fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 for 5 minutes, followed by 1 hour blocking in blocking buffer. For the visualization of protein interactions, samples were incubated with the primary antibodies for 1 hour at room temperature. Duolink in situ PLA was performed according to the manufacturer's protocol (OLINK Bioscience, Sweden) using PLA probe anti-mouse minus and PLA probe anti-rabbit plus. Goat α-mouse Alexa Fluor 488 secondary antibody was added in order to counterstain for γH2AX.
Antibodies
Primary antibodies: Rabbit α-WRAP53-C2 (cat# PA-2020–100, Innovagen AB, Sweden), mouse α-γH2AX (cat# 05–636, Millipore), rabbit α-γH2AX (cat# 2577, Cell Signaling), rabbit α-MDC1 (cat# ab11169, abcam), mouse α-MDC1 (cat# ab50003, abcam), mouse α-pATM (cat# sc-47739, Santa Cruz Biotechnology), mouse α-RNF8 (cat# sc-271462, Santa Cruz Biotechnology), rabbit α-RNF168 (cat# ABE367, Millipore), rabbit α-53BP1 (cat# NB100–904, Novus Biologicals), rabbit α-RAD51 (cat# sc-8349, Santa Cruz Biotechnology), mouse α-β-actin (cat# A5441, Sigma), rabbit α-GFP (cat# ab290, abcam), normal rabbit IgG (cat# sc-2027, Santa Cruz Biotechnology) and normal mouse IgG (cat# sc-2025, Santa Cruz Biotechnology).
Secondary antibodies: sheep α-mouse HRP (cat# NA931V, GE Healthcare), donkey α-rabbit HRP (cat# NA934V, GE Healthcare), goat α-rabbit HRP (cat# 7074, Cell Signal), horse α-mouse HRP (cat# 7076, Cell Signal), goat α-rabbit Alexa Fluor 488 (cat# A11008, Life technologies), goat α-mouse Alexa Fluor 488 (cat# A11029, Life technologies) and donkey α-mouse Alexa Fluor 594 (cat# A21203, Life technologies).
siRNA transfections
siRNA oligonucleotides used: siWRAP53#2 (cat# SI00388948, Qiagen), siH2AX (cat# SI00032844, Qiagen), siMDC1 (cat# L-003506–00–0005, Dharmacon), siRNF8 (cat# L-006900–00–0005, Dharmacon), siRNF168 (cat# SI04143251, Qiagen), si53BP1 (cat# SI02663731, Qiagen), siRAD51 (cat# SI02663682, Qiagen) and siControl (cat# 1027280, Qiagen). Ten–20 nM of siRNA was transfected into cells using HiPerfect (Qiagen) transfection reagent in accordance with the supplier's recommendations.
Treatment with small-molecule inhibitors
ATM (KU55933) and DNA-PK (NU7441) inhibitors were obtained from TOCRIS bioscience. The ATR inhibitor (VE-821) was obtained from Axon MedChem (cat# Axon 1893). The PARP inhibitor Olaparib was provided by Thomas Helleday. Where appropriate, 10 μM ATMi, 2 μM DNA-PKi, 2.5 μM ATRi and 10 μM PARPi were added to the culture medium 16–24 hours prior to IR.
Immunoprecipitation
Cells were lysed in NP40 buffer (150mM NaCl, 50mM Tris-HCL pH 8,0, 1% NP40, 1% protease inhibitor cocktail) for 15 minutes on ice, followed by 3×5 seconds sonication. Protein lysates were spun down at 6000 rpm for 5 minutes and protein concentrations were quantified by Bradford assay (Biorad). Proteins were immunoprecipitated with 1 µg antibody per 1 mg protein and 10 µl Dynabeads Protein G (Life technologies) overnight at 4°C. The beads were washed 4×15 minutes in 1 ml NP40 buffer and prepared for western blotting.
Western blotting
Cell extracts for protein gel blot analysis: cells were harvested, washed and lysed in ice cold lysis buffer (100mM Tris-HCL pH 8, 150mM NaCl, 1% NP-40, 1% PMSF, 1% protease inhibitor cocktail) for 30 minutes on ice followed by sonication. Lysates were centrifuged at 14000 rpm for 15 minutes at 4°C and protein concentrations were determined using Bradford assay (Biorad). Western blotting was performed according to standard procedures.
Statistical analysis
The analyses were performed using Microsoft Office Excel 2011. Two-tailed Student's t test was used to determine statistical significance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the Swedish Cancer Society (Cancerfonden), the Swedish Research Foundation (VR), the Strategic Research Program in Cancer (StratCan), The Association for International Cancer Research (AICR), the Swedish Childhood Cancer Society (Barncancerfonden), the Cancer Society of Stockholm (Cancerföreningen) and the Karolinska Institutet.
References
- 1.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481:287-94; PMID:22258607; http://dx.doi.org/ 10.1038/nature10760 [DOI] [PubMed] [Google Scholar]
- 2.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434:605-11; PMID:15758953; http://dx.doi.org/ 10.1038/nature03442 [DOI] [PubMed] [Google Scholar]
- 3.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213-26; PMID:16377563; http://dx.doi.org/ 10.1016/j.cell.2005.09.038 [DOI] [PubMed] [Google Scholar]
- 4.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007; 131:901-14; PMID:18001825; http://dx.doi.org/ 10.1016/j.cell.2007.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007; 131:887-900; PMID:18001824; http://dx.doi.org/ 10.1016/j.cell.2007.09.040 [DOI] [PubMed] [Google Scholar]
- 6.Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci 2010; 35:565-74; PMID:20451393; http://dx.doi.org/ 10.1016/j.tibs.2010.04.003 [DOI] [PubMed] [Google Scholar]
- 7.Xu C, Min J. Structure and function of WD40 domain proteins. Protein Cell 2011; 2:202-14; PMID:21468892; http://dx.doi.org/ 10.1007/s13238-011-1018-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahmoudi S, Henriksson S, Corcoran M, Mendez-Vidal C, Wiman KG, Farnebo M. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol Cell 2009; 33:462-71; PMID:19250907; http://dx.doi.org/ 10.1016/j.molcel.2009.01.028 [DOI] [PubMed] [Google Scholar]
- 9.Mahmoudi S, Henriksson S, Weibrecht I, Smith S, Soderberg O, Stromblad S, Wiman KG, Farnebo M. WRAP53 is essential for Cajal body formation and for targeting the survival of motor neuron complex to Cajal bodies. PLoS Biol 2010; 8:e1000521; PMID:21072240; http://dx.doi.org/ 10.1371/journal.pbio.1000521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tycowski KT, Shu MD, Kukoyi A, Steitz JA. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell 2009; 34:47-57; PMID:19285445; http://dx.doi.org/ 10.1016/j.molcel.2009.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 2009; 323:644-8; PMID:19179534; http://dx.doi.org/ 10.1126/science.1165357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henriksson S, Rassoolzadeh H, Hedstrom E, Coucoravas C, Julner A, Goldstein M, Imreh G, Zhivotovsky B, Kastan MB, Helleday T, et al.. The scaffold protein WRAP53beta orchestrates the ubiquitin response critical for DNA double-strand break repair. Genes Dev 2014; 28:2726-38; PMID:25512560; http://dx.doi.org/ 10.1101/gad.246546.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, Artandi SE. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 2011; 25:11-6; PMID:21205863; http://dx.doi.org/ 10.1101/gad.2006411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garvin S, Tiefenbock K, Farnebo L, Thunell LK, Farnebo M, Roberg K. Nuclear expression of WRAP53beta is associated with a positive response to radiotherapy and improved overall survival in patients with head and neck squamous cell carcinoma. Oral Oncol 2015; 51:24-30; PMID:25456005; http://dx.doi.org/ 10.1016/j.oraloncology.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Closas M, Kristensen V, Langerod A, Qi Y, Yeager M, Burdett L, Welch R, Lissowska J, Peplonska B, Brinton L, et al.. Common genetic variation in TP53 and its flanking genes, WDR79 and ATP1B2, and susceptibility to breast cancer. Int J Cancer 2007; 121:2532-8; PMID:17683073; http://dx.doi.org/ 10.1002/ijc.22985 [DOI] [PubMed] [Google Scholar]
- 16.Schildkraut JM, Goode EL, Clyde MA, Iversen ES, Moorman PG, Berchuck A, Marks JR, Lissowska J, Brinton L, Peplonska B, et al.. Single nucleotide polymorphisms in the TP53 region and susceptibility to invasive epithelial ovarian cancer. Cancer Res 2009; 69:2349-57; PMID:19276375; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medrek K, Magnowski P, Masojc B, Chudecka-Glaz A, Torbe B, Menkiszak J, Spaczynski M, Gronwald J, Lubinski J, Gorski B. Association of common WRAP 53 variant with ovarian cancer risk in the Polish population. Mol Biol Rep 2013; 40:2145-7; PMID:23192612; http://dx.doi.org/ 10.1007/s11033-012-2273-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409-33; PMID:21363960; http://dx.doi.org/ 10.1101/gad.2021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al.. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3:995-1000; PMID:17072308; http://dx.doi.org/ 10.1038/nmeth947 [DOI] [PubMed] [Google Scholar]
- 20.Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, Artandi SE. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 2014; 159:1389-403; PMID:25467444; http://dx.doi.org/ 10.1016/j.cell.2014.10.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al.. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007; 318:1637-40; PMID:18006705; http://dx.doi.org/ 10.1126/science.1150034 [DOI] [PMC free article] [PubMed] [Google Scholar]