

Abstract
Prenatal exposure to tobacco smoke has lifelong health consequences. Epigenetic signatures such as differences in DNA methylation (DNAm) may be a biomarker of exposure and, further, might have functional significance for how in utero tobacco exposure may influence disease risk. Differences in infant DNAm associated with maternal smoking during pregnancy have been identified. Here we assessed whether these infant DNAm patterns are detectible in early childhood, whether they are specific to smoking, and whether childhood DNAm can classify prenatal smoke exposure status. Using the Infinium 450 K array, we measured methylation at 26 CpG loci that were previously associated with prenatal smoking in infant cord blood from 572 children, aged 3–5, with differing prenatal exposure to cigarette smoke in the Study to Explore Early Development (SEED). Striking concordance was found between the pattern of prenatal smoking associated DNAm among preschool aged children in SEED and those observed at birth in other studies. These DNAm changes appear to be tobacco-specific. Support vector machine classification models and 10-fold cross-validation were applied to show classification accuracy for childhood DNAm at these 26 sites as a biomarker of prenatal smoking exposure. Classification models showed prenatal exposure to smoking can be assigned with 81% accuracy using childhood DNAm patterns at these 26 loci. These findings support the potential for blood-derived DNAm measurements to serve as biomarkers for prenatal exposure.
Keywords: Epigenetics, Prenatal smoking exposure, Childhood, DNA methylation, Biomarker
1. Introduction
A considerable proportion (11%) of women in the United States actively smoke during pregnancy, a major risk factor for pregnancy complications(Castles et al., 1999; Shah and Bracken, 2000) and adverse health outcomes during infancy, childhood and later life (Salmasi et al., 2010; Shah and Bracken, 2000). Understanding the impact of early life exposure to tobacco smoke on future health has important public health implications.
DNA methylation (DNAm) is a type of epigenetic modification central to development and gene regulation. It is of interest as a mediating mechanism in exposure-disease associations, and may also have utility as a biological marker of exposure, even if not mechanistically implicated (Ladd-Acosta, 2015). Several groups have investigated associations between DNAm levels and in utero exposure to tobacco smoke. Using global (Guerrero-Preston et al., 2010), candidate gene-based (Murphy et al., 2012; Suter et al., 2010), and genome-scale (Joubert et al., 2014, 2012; Richmond et al., 2014; Suter et al., 2011) approaches, they identified associations between maternal smoking during pregnancy and DNAm levels in placental tissue and in DNA from cord blood (Lee and Pausova, 2013; Richmond et al., 2014). A recent study, using a low-density DNAm array showed detectible prenatal smoking associations in childhood, but could not assess the reported birth sample associations now confirmed by several groups, given the incompatible array content (Breton et al., 2014). A candidate gene-based study (Novakovic et al., 2014) of 11 individuals showed that comparable differences in DNAm at AHRR, a tobacco-related gene, were detectable at birth as well as at 18 months. Finally, a recent longitudinal investigation revealed some smoking-related DNAm alterations, initially detected in their sample at birth, persist within the same individuals over time (Richmond et al., 2014). While a few of the loci identified by that paper overlap with previous studies, the study did not specifically examine the set of 26 loci (Joubert et al., 2012) that have now been well replicated in other birth samples.
Here we attempted to replicate prenatal smoking-associated DNAm differences observed in infant cord blood, reported by Jou-bert et al. (2012)), in an independent set of 572 early childhood blood samples to determine if the DNAm pattern in childhood is consistent with DNAm “signatures” of prenatal smoking detected at birth. This study, focused on prenatal smoking, assesses the potential utility of a DNAm signature measured later in life as an epigenetic biomarker of prenatal exposure. This study also examines other issues relevant to DNAm’s potential as a biomarker for prenatal smoke exposure. First, since it is possible that the DNAm changes previously reported in cord blood could be related to downstream responses to a range of prenatal exposures and, thus, are not tobacco smoke-specific, we explored associations of DNAm changes with other prenatal exposures, including maternal alcohol and medication use. Second, we evaluated associations between the previously reported DNAm changes and trimester-specific and sustained prenatal smoke exposure. Finally, we used machine learning and 10-fold cross-validation to assess whether childhood DNAm levels at these 26 sites can predict prenatal exposure to smoking.
2. Materials and methods
2.1. Sample inclusion
Study participants included in this analysis are a subset of children enrolled in the Study to Explore Early Development (SEED) (Schendel et al., 2012). SEED is a US national case-control study that has enrolled over 2800 children, and their parents, with approximately equal numbers of children with an autism spectrum disorder (ASD), children from the general population (POP controls), and children with a non-ASD developmental delay (DD controls) (Schendel et al., 2012) that were all born during the same time frame and same geographic areas. SEED has collected extensive information on prenatal exposures and biospecimens suitable for DNAm analysis from the same individuals. We measured DNAm in whole blood collected from 608 SEED children (mean (SD) age at collection was 59.7 (6.1) months), for whom we had genome-wide genotyping data, a complete caregiver interview, and a sufficient amount of DNA. Both ASD cases (N=289) and typically developing control (N=319) children were included to increase power to detect exposure-associated DNAm changes. To ensure exposure-based differences in DNAm were not driven by ASD status, we performed conditional analyses by case control status and in a control-only subset. After filtering by DNAm quality and cigarette smoke and covariate missing data (Supplementary Fig. 1; available as supplementary data online), 572 samples were included in the final analyses.
2.2. Prenatal exposure variables
The prenatal exposure data were collected in the SEED caregiver interview (CGI) with the mother, as described previously (Schendel et al., 2012). Retrospective self-report of prenatal smoking, while certainly not invulnerable to reporting error, has been shown to be comparable to medical records (Rice et al., 2007). Additionally, recall of smoking during pregnancy six years later had 90% agreement with smoking assessed during the pregnancy (Hensley Alford et al., 2009).
Prenatal exposures were assessed during four exposure windows: the overall pregnancy period, and the first (T1), second (T2), and third trimesters (T3). For direct comparison with previous birth sample findings (Joubert et al., 2012), we focused our main analyses on exposure during T2.
Cigarette smoking
In utero exposure to either passive household smoking or active maternal smoking was determined from CGI responses to questions concerning timing and amount of cigarette smoke exposure. Smoking exposures were dichotomized as ‘exposed’ or ‘unexposed’ during each exposure window. Active smoking during an exposure window was defined as either any exposure for > 2 months during the exposure window, or average consumption of ≥ 1 cigarette/day for ≥ 1 month during the exposure window. Passive smoking was determined from report of another household member smoking ≥ 1 cigarette/day for ≥ 1 month during the exposure window. Individuals were classified as “unexposed” if they had no active or passive smoking exposure throughout the duration of the pregnancy to provide the same comparison group for all exposure categories. In addition, for active maternal smoking in each exposure window, a three level dose-response variable was created: unexposed, 1 to < 10 cigarettes/day, and ≥ 10 cigarettes per day. Lastly, we used 3 additional categories of prenatal exposure: (1) sustained active, defined as the mother smoking ≥ 1 cigarette per day, on average, for the duration of pregnancy; (2) sustained passive, household member smoking ≥ 1 cigarette per day, on average, for the duration of pregnancy and no active maternal smoking at any time during pregnancy; and (3) trimester 1 (T1) quitters, mother smoking ≥ 1 cigarette per day, on average, only during the first trimester. The 32 individuals that did not have trimester 2-specific smoking data and 1 individual missing maternal age and education variables (used as covariates) were excluded from all analyses, leaving 572 subjects (Supplementary Fig. 1). We removed 41 individuals with no active smoking exposure during T2 but some active or passive exposure at some point during pregnancy, leaving 531 total individuals for our T2 smoking analyses. All self-reported exposure data passed through extensive logic checks to identify responses that did not fit expected skip patterns or had inaccurate or nonsensible values. Each discrepancy was resolved or coded as missing.
Other prenatal exposures
To evaluate specificity of the DNAm signature to smoking exposure, we examined three other prenatal exposures, maternal use of alcohol and two types of medications, based on their availability in SEED (see Supplementary data, available online, for details).
2.3. DNA methylation measurements and quality control
DNAm was isolated from whole blood and measured on the Infinium 450 K Array (Illumina, San Diego, CA) according to manufacturer protocols (see Supplementary data, available online). All quality assurance analyses were performed using Bioconductor and R-3.0.x. Illumina idat files were obtained and processed using the minfi package (version 1.8.9) (Aryee et al., 2014). We generated a sample quality control report using the qcReport function. We then assessed the correlation of replicate samples across plates to identify problems with particular plates/batches and to assess the accuracy of the DNAm values. Next, for each sample and each CpG probe, we computed three methylation metrics: (a) the total probe intensity (methylated+unmethylated channel values); (b) beta (ratio of methylated versus total probe intensity) and (c) M-value (logit (beta)). We removed samples with low overall intensities based on total probe intensity (n=2). Sex of each child was empirically determined using total probe intensities on the X and Y chromosomes and compared to parent-report for discrepancy. Finally, quantile normalization was performed (separately for type I, type II, autosome, and sex chromosome probes) and M-values computed using the preprocessQuantile minfi function. For plotting, beta values (β), obtained via the getBeta function in minfi were used to allow more intuitive interpretation, since these range from 0% to 100% methylated. Beta values were also used to estimate cell type proportions per sample (see Supplementary data, available online).
2.4. Statistical analysis
We determined the association between prenatal exposure to smoking and DNAm for the 26 CpG sites previously implicated in cord blood (Joubert et al., 2012) via general linear regression. For each of the 26 loci we fit DNAm M-values as a function of exposure status and potential confounders: E(M)= α+ βE+γ1A1+ γ2C2+ γ3M3+ γ4S4+ γ5Y5+ΓX+ɛ, where E represents prenatal cigarette smoke exposure status, A1 represents genetically estimated racial ancestry (European, African, Admixed), C2 represents ASD case versus POP control, M3 represents male child, S4 represents maternal education (less than high school, high school of equivalent, college but less than Bachelor’s, Bachelor’s degree, Master’s degree or higher), Y5 represents maternal age, and X represents a vector of cell type proportions for granulocytes, monocytes, B cells, NK cells, CD4+ T cells, and CD8 + T cells, α, β, γ1−γ5, Γ represent linear regression coefficients and ɛ represents error. Covariates were chosen given reports that they may be associated with DNAm. Maternal age and education were included as measures of socioeconomic status (SES), which has been associated with changes in DNAm (McGuinness et al., 2012; Tehranifar et al., 2013). The proportion of individuals with African, European, and Admixed ancestries, which are correlated with SNP alleles and DNAm, varied across exposure groups (Barfield et al., 2014; Drong et al., 2013; Kerkel et al., 2008; Liu et al., 2013, 2014; Schmitz et al., 2013; Smith et al., 2014). We used P values based on F statistics from linear regression and a significance threshold of 1.92E-03 to account for multiple testing of 26 loci. Similar models were run for the other exposure variables. P-values for dose-response trends were computed using a Cochran-Armitage test for trend via the Bioconductor coin package.
Classification analyses were performed using the Bioconductor caret package (Kuhn, 2008). We built a support vector machine (SVM) classification model using a radial basis function kernel and SEED childhood DNAm measurements at the 26 sites under investigation in this study as attributes. We implemented 10-fold cross-validation to assess the predictive accuracy of the SVM classifier, i.e. ability to discriminate between prenatally exposed versus unexposed individuals. Using the same SVM-10 fold cross validation procedure, we assessed the specificity of the SVM classifier in two ways. First, using the 26 DNAm attributes associated with prenatal smoking exposure, we built a SVM classifier to predict prenatal exposure to each of three other exposures including maternal use of alcohol, B2AR and SSRIs. Second, to determine how well other DNAm signatures based on 26 sites can predict prenatal exposure to smoking, we generated 1000 sets of 26 randomly chosen DNAm sites, also measured on the 450 K, to use as input attributes for the SVM classifier. We report measures of sensitivity (proportion of individuals with reported prenatal exposure that are predicted to be prenatally exposed using a classifier built with childhood DNAm patterns), specificity (the proportion of individuals with no reported prenatal exposure that are predicted to be unexposed using childhood DNAm patterns) as well as a measure of overall balanced accuracy ([sensitivity+specificity]/2) that has been shown to be more suitable for datasets, like ours, with an imbalanced number of samples in the outcome classes (Velez et al., 2007).
3. Results
3.1. Study population
For direct comparison with previous birth samples (Joubert et al., 2012), we focused on second trimester smoking. No substantial differences were identified between exposed and unexposed children with respect to age or sex (Table 1). The proportion of children with African, European, and Admixed ancestries across the exposure groups varied and were adjusted for in methylation analyses. ASD cases had higher levels of prenatal smoking exposure than controls. Stratified and conditional analyses of methylation associations by case control status are reported below.
Table 1.
Descriptive Characteristics of the SEED Samples Examined in this Study, Stratified by In Utero Tobacco Exposure Status.
| Smoke exposure statusa
|
||||||
|---|---|---|---|---|---|---|
| Exposed |
Unexposed |
|||||
| Mean (range) | No. | % | Mean (range) | No. | % | |
| T1 active maternal | 37 | 6.8 | 505 | 93.2 | ||
| T2 active maternal | 26 | 4.9 | 505 | 95.1 | ||
| T3 active maternal | 24 | 4.5 | 505 | 95.5 | ||
| Sustained active maternal | 23 | 4.4 | 505 | 95.6 | ||
| Sustained passive household | 19 | 3.6 | 505 | 96.4 | ||
| Age, months | 59.2 (45.7–68.6) | 59.6(38.7–70.7) | ||||
| Sex | ||||||
| Male | 17 | 5.1 | 319 | 94.9 | ||
| Female | 9 | 4.6 | 186 | 95.4 | ||
| Ancestral populationb | ||||||
| European | 13 | 4.2 | 299 | 95.8 | ||
| African | 6 | 9.8 | 55 | 90.2 | ||
| Asian | 0 | 0 | 20 | 100 | ||
| Admixed | 6 | 4.8 | 119 | 95.2 | ||
| Not available | 1 | 7.7 | 12 | 92.3 | ||
| Phenotype | ||||||
| Control | 7 | 3.0 | 227 | 97.0 | ||
| Autism | 19 | 6.4 | 278 | 93.6 | ||
Abbreviations: T1, trimester 1; T2, trimester 2; T3, trimester 3.
For demographic variables, the exposed group includes individuals with active maternal cigarette smoke exposure during T2 and unexposed individuals are defined as having no active or passive exposure throughout the duration of pregnancy. The 22 individuals with sustained active maternal smoking are also included in the T1, T2, and T3 counts for active maternal smoking.
Assigned using computed ancestry information from genome-wide genotyping data.
3.2. DNA methylation quality assurance
Two samples with low overall 450 K intensities and 1 sample with an outlier blood cell composition were removed, resulting in high quality DNAm data for 605 samples, 572 with T2 prenatal cigarette exposure and covariate data. DNAm was highly reproducible across plates and wells, with correlation coefficients for each pair of replicates (n=8) ranging from 0.989 to 0.994. Our data QC and filtering pipeline is shown in Supplementary Fig. 1. We found no significant differences in blood cell type compositions between prenatal cigarette smoke exposed versus unexposed children (Supplementary Table 1 and Supplementary Fig. 2).
3.3. DNAm changes associated with in utero smoke exposure
Effect sizes for the association between prenatal cigarette smoke exposure and DNAm in SEED children, mean age of 5, show striking similarity to those previously observed in cord blood birth samples for all 26 CpG sites (Joubert et al., 2012) (Fig. 1). After adjusting for potential confounders, statistical significance (P<1.92E-03) was observed for 7 of these 26 CpG sites associated with active T2 smoking (Fig. 1 and Table 2). Effect sizes were similar with and without adjustment for cell type. Analysis of SEED control-only samples revealed a similar pattern of DNAm differences and effect sizes as the complete set of samples (Supplementary Fig. 3). Similarly, stratified analyses showed the overall DNAm differences, effect sizes, and patterns at these 26 loci were similar among males and females (Supplementary Fig. 4; Supplementary Table 2). We observed a sex-specific change at a single CpG site (cg11715943), located near the HLA-DPB2 gene, with increased DNAm among prenatally exposed females and decreased methylation among prenatally exposed males (Supplementary Fig. 4).
Fig. 1.
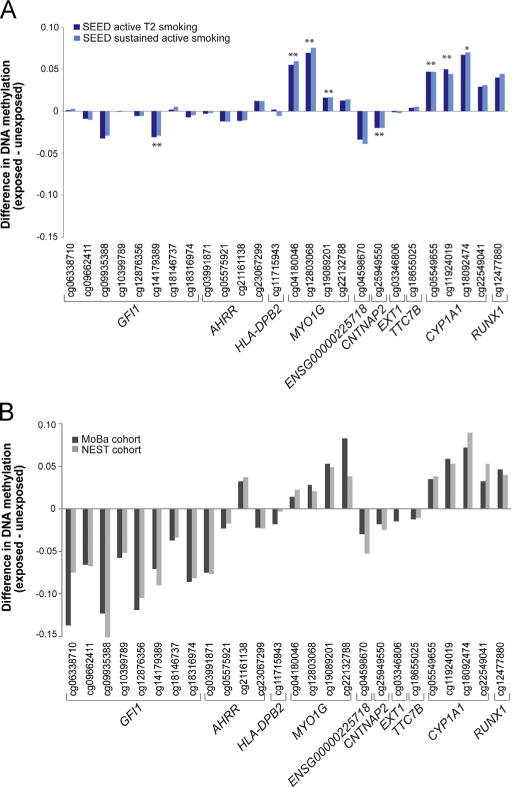
Changes in DNAm related to prenatal cigarette smoke in childhood compared to at birth. (A) Differences in median DNAm levels (β) among SEED children, aged 3–5 years, with differing prenatal exposure to cigarette smoke. Single and double asterisks in the top panel denote CpG sites that reach statistical significance after correcting for multiple comparisons for active T2 and sustained maternal smoking, respectively. (B) Previously reported (Joubert et al., 2012) differences in median methylation levels (β) from Norwegian Mother and Child Cohort (MoBa) and Newborn Epigenetics Study (NEST) newborns associated with prenatal smoking exposure during T2.
Table 2.
Prenatal exposure to tobacco smoke is associated with altered DNAm in childhood.
| Probe name | Gene | Undjusted
p-value pregnancy time frame |
Adjusteda
p-value pregnancy time frame |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T1 | T2 | T3 | T1 quitter | Sustained activ e | Sustained passive | ||
| cg25949550 | CNTNAP2 | 4.55E-09 | 3.04E-11 | 9.82E-11 | 1.59E-07 | 2.16E-09 | 1.32E-09 | 4.28E-01 | 1.32E-09 | 1.86E-01 |
| cg05549655 | CYP1A1 | 9.61E-05 | 2.36E-05 | 3.07E-05 | 3.02E-05 | 3.25E-06 | 6.93E-07 | 4.26E-01 | 6.93E-07 | 5.93E-01 |
| cg04180046 | MYO1G | 2.52E-06 | 2.31E-07 | 7.00E-08 | 1.77E-04 | 5.71E-06 | 1.12E-06 | 7.66E-01 | 1.12E-06 | 8.16E-02 |
| cg12803068 | MYO1G | 2.61E-05 | 6.51E-06 | 1.78E-06 | 3.49E-04 | 3.96E-05 | 9.72E-06 | 5.38E-01 | 9.72E-06 | 1.36E-01 |
| cg14179389 | GFI1 | 5.52E-04 | 4.15E-05 | 4.27E-04 | 1.03E-03 | 6.67E-05 | 3.63E-04 | 8.14E-01 | 3.63E-04 | 7.98E-01 |
| cg19089201 | MYO1G | 2.50E-04 | 1.53E-05 | 3.04E-06 | 2.49E-03 | 1.61E-04 | 3.12E-05 | 9.57E-01 | 3.12E-05 | 1.69E-01 |
| cg11924019 | CYP1A1 | 6.72E-04 | 2.52E-04 | 1.90E-04 | 6.69E-04 | 2.18E-04 | 3.24E-05 | 4.59E-01 | 3.24E-05 | 9.31E-01 |
| cg18092474 | CYP1A1 | 5.83E-03 | 4.90E-03 | 3.94E-03 | 4.37E-03 | 2.04E-03 | 4.01E-04 | 5.29E-01 | 4.01E-04 | 6.05E-01 |
| cg23067299 | AHRR | 9.35E-04 | 2.66E-03 | 4.92E-03 | 2.41E-03 | 6.67E-03 | 7.59E-03 | 1.59E-01 | 7.59E-03 | 2.72E-01 |
| cg05575921 | AHRR | 1.07E-02 | 1.40E-03 | 1.64E-03 | 2.12E-02 | 6.83E-03 | 5.59E-03 | 8.71E-01 | 5.59E-03 | 4.71E-01 |
| cg22132788 | MYO1G | 3.46E-03 | 8.98E-04 | 2.71E-04 | 2.19E-02 | 7.65E-03 | 2.74E-03 | 7.48E-01 | 2.74E-03 | 6.59E-02 |
| cg22549041 | CYP1A1 | 1.10E-02 | 1.03E-02 | 8.60E-03 | 7.71E-03 | 8.76E-03 | 2.41E-03 | 3.42E-01 | 2.41E-03 | 5.90E-01 |
| cg12477880 | RUNX1 | 7.50E-03 | 8.15E-04 | 9.52E-04 | 4.30E-02 | 1.22E-02 | 1.49E-02 | 9.60E-01 | 1.49E-02 | 4.28E-01 |
| cg04598670 | ENSG225718 | 1.66E-02 | 3.34E-03 | 6.02E-04 | 2.49E-02 | 1.74E-02 | 5.93E-03 | 4.93E-01 | 5.93E-03 | 6.76E-01 |
| cg09935388 | GFI1 | 3.81E-02 | 7.30E-02 | 1.14E-01 | 6.16E-02 | 1.94E-01 | 2.30E-01 | 1.34E-01 | 2.30E-01 | 6.42E-01 |
| cg18655025 | TTC7B | 5.40E-01 | 2.81E-01 | 2.87E-01 | 2.91E-01 | 3.05E-01 | 3.02E-01 | 7.39E-01 | 3.02E-01 | 8.24E-01 |
| cg21161138 | AHRR | 2.08E-01 | 1.50E-01 | 2.76E-01 | 3.59E-01 | 3.38E-01 | 6.03E-01 | 9.22E-01 | 6.03E-01 | 1.60E-01 |
| cg18146737 | GFI1 | 7.46E-01 | 5.00E-01 | 5.67E-01 | 7.64E-01 | 4.39E-01 | 5.26E-01 | 5.47E-01 | 5.26E-01 | 5.62E-01 |
| cg06338710 | GFI1 | 6.57E-01 | 6.56E-01 | 7.04E-01 | 6.53E-01 | 5.09E-01 | 5.64E-01 | 8.94E-01 | 5.64E-01 | 8.30E-01 |
| cg03346806 | EXT1 | 5.57E-01 | 9.42E-01 | 9.96E-01 | 2.20E-01 | 5.60E-01 | 7.59E-01 | 2.08E-01 | 7.59E-01 | 5.56E-01 |
| cg11715943 | HLA-DPB2 | 5.31E-01 | 3.94E-01 | 5.22E-01 | 7.75E-01 | 6.72E-01 | 7.89E-01 | 8.40E-01 | 7.89E-01 | 7.26E-01 |
| cg09662411 | GFI1 | 8.80E-01 | 8.17E-01 | 8.42E-01 | 8.94E-01 | 7.25E-01 | 7.58E-01 | 8.37E-01 | 7.58E-01 | 9.29E-01 |
| cg18316974 | GFI1 | 2.71E-01 | 4.03E-01 | 4.91E-01 | 4.20E-01 | 7.34E-01 | 8.51E-01 | 3.09E-01 | 8.51E-01 | 3.10E-01 |
| cg12876356 | GFI1 | 7.35E-01 | 7.79E-01 | 8.88E-01 | 8.23E-01 | 8.92E-01 | 9.69E-01 | 7.83E-01 | 9.69E-01 | 8.56E-01 |
| cg10399789 | GFI1 | 9.29E-01 | 9.47E-01 | 8.62E-01 | 9.73E-01 | 9.22E-01 | 9.65E-01 | 9.78E-01 | 9.65E-01 | 6.83E-01 |
| cg03991871 | AHRR | 8.31E-01 | 9.49E-01 | 5.68E-01 | 9.05E-01 | 9.73E-01 | 5.95E-01 | 6.79E-01 | 5.95E-01 | 7.66E-01 |
Abbreviations: T1, trimester 1; T2, trimester 2; T3, trimester 3; MoBa, Norwegian Mothers and Babies cohort (Ronningen et al., 2006); NEST, Newborn Epigenetics Study (Murphy et al., 2012; Hoyo et al., 2011).
Adjusted for sex, phenotype, race, maternal education and age, estimated percent granulocyte, monocyte, natural killer, B, CD4+ and CD8+ T cells. Bold font signifies CpG sites that are significant after applying a Bonferroni corrected significance threshold.
3.4. Dose response relationships
Dose response trend tests were significant for 10 loci, including all 7 sites that were significant for dichotomous exposure (Fig. 2). For most loci, the change in methylation between 1 and <10 cigarettes per day and >10 cigarettes per day was minimal (e.g. Fig. 2E). The biggest methylation differences appear to be between the group with no exposure and the group exposed to 1 to <10 cigarettes per day. Directions of trend varied by location. For CpGs within or near contactin associated protein-like 2 (CNTNAP2) and growth factor independent 1 transcription repressor (GFI1), DNAm levels decreased with exposure (Fig. 2A and B). All 4 loci located within the gene body of myosin IG (MYO1G) show increased DNAm with increasing cigarette exposure (Fig. 2D–G). Similarly, 3 loci located in cytochrome P450, family 1, subfamily A (CYP1A1) show increased DNAm with exposure to cigarettes (Fig. 2H–J). Lastly, there is one significant loci within aryl-hydrocarbon receptor repressor (AHRR) that shows an increase in DNAm associated with cigarette smoke exposure (Fig. 2C).
Fig. 2.

(A–J) Boxplots show the relationship between methylation levels (β) among SEED children and average number of cigarettes the mother actively smoked per day during T2, for sites with significant trends.
3.5. Influences of duration and mode of exposure
The DNAm signature associated with T2 cigarette smoke exposure appears to reflect sustained active maternal smoking during pregnancy (Fig. 3A and B). We do not see a similar magnitude of DNAm change for these 26 sites in children whose mothers were exposed to passive smoking during pregnancy, but did not actively smoke, i.e. their mother was only exposed to second hand smoke in the home (Fig. 3C), or in children whose mothers quit smoking during the first trimester (Fig. 3D). Multiple loci in MYO1G and CYP1A1 show DNAm changes in the same direction for sustained passive smoke exposure as for active maternal T2 and sustained active maternal smoking, but with a smaller magnitude of change (Fig. 3C). Similarly several loci in MYO1G and CYP1A1 and a single locus in GFI1 show DNAm changes in the same direction for T1 active maternal smoke exposure as for active maternal T2 and sustained smoking, but with a smaller magnitude of change (Fig. 3D).
Fig. 3.
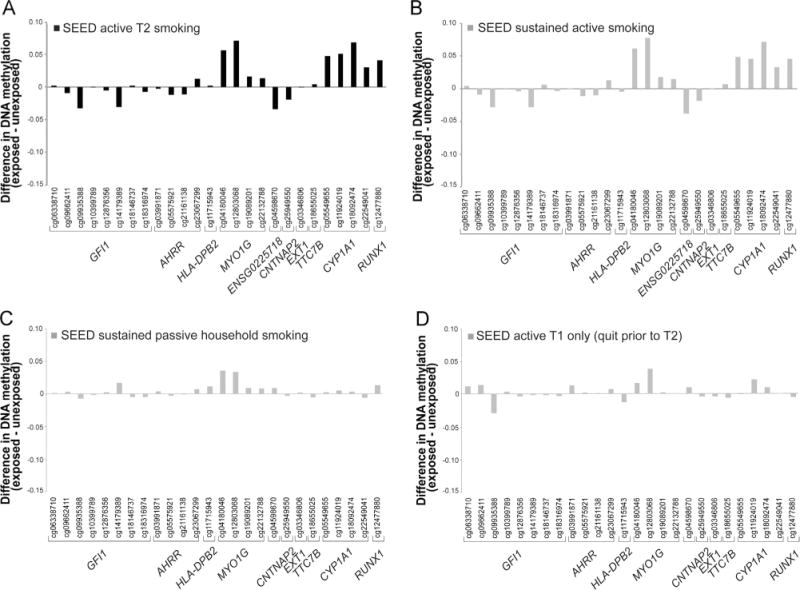
Sustained active maternal smoking is a major driver of the DNAm differences associated with active maternal T2 smoking. Differences in DNAm (β) among SEED children exposed to (A) active maternal smoking during T2, (B) sustained active maternal smoking, (C) sustained household smoking only, and (D) active maternal smoking during T1 only.
3.6. Prenatal smoking exposure class prediction
Classification models showed the 26 site DNAm signature of smoking, present in childhood samples, correctly assigns reported prenatal exposure to active maternal smoking during T2. The receiver operating characteristic (ROC) curve showing the DNAm based predictor obtains an area under the curve (AUC) value of 0.865 (Fig. 4). Based on the ROC point with the greatest overall accuracy, a total of 464/531 SEED samples were correctly classified (Supplementary Table 3). Among the 67 misclassified individuals, 7 were reported to be exposed to smoking during T2 but were predicted to be unexposed via DNAm, i.e. are false negatives (Supplementary Table 3), and 60 were reported to be unexposed but were predicted to be exposed, i.e. are false positives (Supplementary Table 3); reflecting a sensitivity of 73% and specificity of 88%, and an overall accuracy of 81% (Table 3). We observed similar results when considering sustained smoking exposure, with an AUC of 0.87 (Fig. 4) and 491/529 individuals correctly classified when considering the point with the greatest overall accuracy. 8 out of 38 misclassified individuals were predicted to be unexposed, based on their DNAm patterns at these 26 sites, but actually reported exposure (Supplementary Table 4). The 30 remaining misclassified individuals were reported to be unexposed but were predicted as being exposed based on the DNAm classifier (Supplementary Table 4). As shown in Table 3, for sustained prenatal exposure to smoking, our SVM classifier had an overall accuracy of 80%, specificity of 94%, and sensitivity of 67%. We were not able to achieve this level of accuracy or specificity using different sets of 26 sites chosen at random (Fig. 4B and C; Table 3; Supplementary Fig. 5).
Fig. 4.
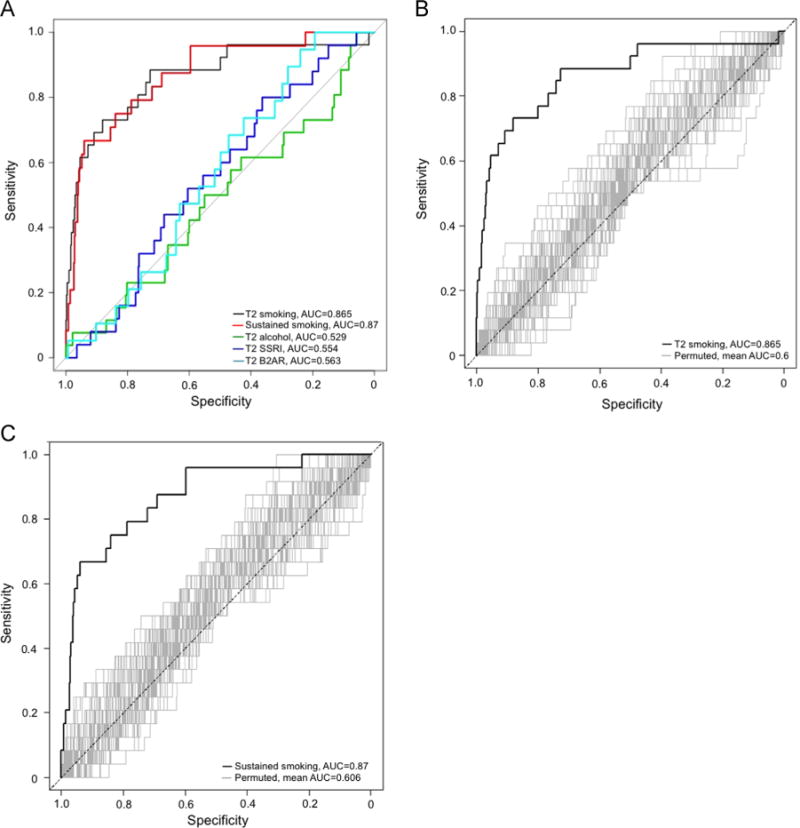
Receiver operating characteristic (ROC) curves for prediction of prenatal exposure using childhood methylation levels show high discrimination of children exposed to prenatal smoking. (A) Our 26-site methylation prediction model discriminates T2 active and sustained maternal smoking well but not T2 alcohol use, T2 SSRI use or T2 B2AR use. (B, C) Random sets of 26 methylation loci from the same array do not discriminate between prenatally exposed (to T2 active maternal smoking (B); to sustained active maternal smoking (C)) and unexposed individuals.
Table 3.
A childhood DNA methylation signature can accurately predict prenatal smoking exposure status.
| Prenatal exposure | Attributesa | Specificityb | Sensitivityb | Accuracyc |
|---|---|---|---|---|
| Sustained smoking | 26 smoking associated sites | 0.94 | 0.67 | 0.80 |
| T2 smoking | 0.88 | 0.73 | 0.81 | |
| T2 maternal alcohol | 0.86 | 0.30 | 0.58 | |
| T2 maternal SSRI use | 0.36 | 0.80 | 0.58 | |
| T2 maternal B2AR use | 0.19 | 1.00 | 0.60 | |
| Sustained smoking | 26 randomly selected sites | 0.44* | 0.45* | 0.45* |
| T2 smoking | 0.44* | 0.45* | 0.45* |
Abbreviations: B2AR, beta-2 adrenergic receptor agonist; SSRI, selective serotonin reuptake inhibitor; T2, Trimester 2.
Attributes correspond to methylation values at specific loci, as measured by the 450 K array.
We report values that yielded the greatest overall balanced accuracy.
Given the imbalanced proportion of exposed and unexposed individuals in our dataset, we report balanced accuracy28, 29.
One thousand different sets of 26 randomly selected 450 K sites were selected as SVM input attributes. In the table we report the mean sensitivity, specificity, and balanced accuracy rates across all 1000 sets examined.
3.7. Exposure specificity of DNAm changes
None of the other three prenatal exposure domains examined here showed an effect size pattern similar to the smoking signature pattern seen in previous studies and our SEED samples, nor were there statistically significant associations for any of the 26 loci examined (Fig. 5; Supplementary Table 5). Additionally, classification models revealed the 26 site DNAm signature of prenatal smoking is a poor predictor of prenatal exposure to maternal alcohol, SSRI and B2AR use, with area under the curve (AUC) ranging from 0.529 to 0.563 and balanced accuracies less than 0.60 across all three of these exposure domains (Fig. 4A; Table 3).
Fig. 5.
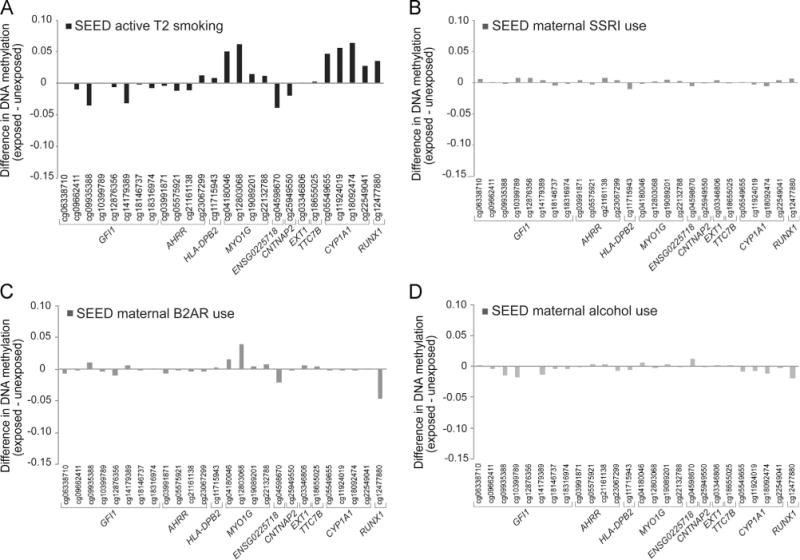
Exposure specificity of sites associated with in utero cigarette smoke exposure. Differences in DNAm (β) among SEED children with differing prenatal exposure to (A) active maternal smoking, (B) SSRI use, (C) B2AR use, and (D) maternal alcohol use, during trimester 2.
4. Discussion
We show similar patterns of DNAm effect sizes for associations with exposure to prenatal cigarette smoke in 3–5 year-old children that were previously reported in other studies of newborns. While our observation is in an independent set of children, this suggests prenatal exposure-driven DNAm differences are still detectable later in childhood. We observed dose-dependent associations for some loci, specific to smoking, that likely reflect changes associated with sustained in utero exposure to maternal smoking. Additionally, we provide evidence that childhood DNAm patterns can accurately and specifically predict prenatal exposure to smoking.
We were unable to directly assess the confidence interval overlap between our observed effect sizes and those previously reported in newborns (Joubert et al., 2012), because the intervals are not provided in the previous report, although it appears that the effect sizes in our 3–5 year-olds are systematically weaker than those in newborns. This may reflect attenuation of a prenatally-driven association through time. Further work in older children and via longitudinal data analysis is warranted. Interestingly, smoking associated changes in DNAm at dozens of genes are consistent across adult studie s(Philibert et al., 2013; Shenker et al., 2013; Sun et al., 2013; Zeilinger et al., 2013), but they do not completely overlap with the changes in DNAm observed in neonates and children with prenatal exposure. Differential DNAm at 3 genes, AHRR, MYO1G, and GFI1, is associated with smoke exposure in adults (Philibert et al., 2013; Shenker et al., 2013; Sun et al., 2013; Zeilinger et al., 2013), neonates (Joubert et al., 2012; Novakovic et al., 2014), and children (Novakovic et al., 2014), but the effect sizes for AHRR, consistently the gene with the largest magnitude of change between smokers and non-smokers in adults, are much smaller in prenatally exposed samples. In contrast, CYP1A1 was identified and validated in prenatally exposed neonates (Joubert et al., 2012), and in our sample of 3–5 year olds but has not been identified in adults.
Pre- and postnatal cigarette smoke exposure effects can be difficult to disentangle in this SEED sample and likely have different biological implications. In 3–5 year-olds, it is possible that postnatal smoking exposure (limited at this age to secondhand exposure, for which we have no data) influenced DNAm signatures. However, these loci were previously implicated in newborns, where there was no postnatal exposure possible. Therefore, it is unlikely that the signals we report are due solely to postnatal exposure. It is possible that children exposed to household smoking postnatally, but not in utero, could show epigenetic changes at these same locations and that this may result in the attenuated signals we observe, assuming the developing fetus is more susceptible to smoke-associated changes in DNAm. In contrast, children exposed in uteri are typically also exposed postnatally, and we would expect increased association effects in such children. We cannot estimate the correlation between pre- and postnatal exposure in the SEED sample with our current data. Maternal passive smoke exposure did not result in the same magnitude of effect, perhaps indicating postnatal passive exposure is less likely to contribute to the patterns.
While tests for trend were significant for many assessments of a 3 level smoking variable, the findings appeared to be largely driven by exposure to any smoking (i.e. a move from no to low levels of smoking) rather than by a specific dose response. Nonetheless, it is notable that methylation effects were observed with both low and high exposure to cigarette smoke. For CNTNAP2, GFI1, ENSG00000225718, MYO1G, and CYP1A1 the change in DNAm was in the same direction across the set of CpG sites representing each particular gene. However for AHRR, we observed both increases and decreases in DNAm associated with smoking exposure. Although the 2 sites are annotated to the same gene, they are about 53 Kb away from each other and appear to be associated with different RNA transcripts. Furthermore, one site (cg05575921) is located at a gene enhancer, a type of genomic element that exerts distal control over gene expression. It is possible that although these 2 sites associated with AHRR have different DNAm directions, they may have similar downstream effects on gene expression given their gene location differences.
Although we were able to demonstrate exposure specificity of effect, we considered a limited range of other exposures and were unable to include exposures that may be more similar to cigarette smoke, such as air pollution. Although we did not see the same striking difference in DNAm for the children whose mothers quit smoking during T1, it is possible that there are other loci in the genome specifically associated with T1 exposure. Further research will be needed to characterize specific windows of susceptibility within pregnancy (or early life), and will likely require close collaboration between bench scientists and epidemiologists.
The use of blood as a measurement tissue for epigenetic research requires two considerations: first, that blood is a mixture of cell types, each with its own DNAm signature for a subset of the epigenome; and second, that blood may not be a relevant tissue for studies of diseases not primarily affecting blood. With regard to the former, we carried out quality assurance analyses and demonstrated no relationship with cell composition and smoking exposure status. Additionally, we observed similar effect sizes with and without adjustment for cell type. Regarding blood as the tissue of measurement, several studies have shown blood to be a relevant surrogate even when studying diseases that primarily affect other tissues (Davies et al., 2012; Kaminsky et al., 2012; Talens et al., 2010; Waterland et al., 2010). Moreover, blood-derived epigenetic signatures can have utility as prenatal exposure biomarkers, even when blood is not primarily involved in the disease being studied (Ladd-Acosta, 2015).
We show a childhood blood-derived DNAm signature associated with prenatal cigarette smoke exposure that is consistent with results from cord blood. These findings suggest blood-derived DNAm measurements obtained during childhood may contain patterns that reflect in utero exposure to cigarette smoke. Additionally, we show that a DNAm signature present in childhood can accurately predict prenatal exposure to smoking. Our results provide a proof of principle example supporting the potential utility of DNAm as a biomarker of prenatal exposures more generally. Further work is needed to fully assess the utility of DNAm signatures as biomarkers of prenatal exposure. If successful, this has broad implications for the study of prenatal exposure effects on childhood and even adult disorders using biosamples collected many years after birth.
Supplementary Material
Acknowledgments
We would like to thank Dr. Homayoon Farzadegan, Stacey Cayetano, Samantha Bragan, and Brett Purinton from the Johns Hopkins Biological Repository for overseeing, isolating DNA, pulling, and plating the SEED DNA samples, Arni Runarsson at Johns Hopkins Epigenetics Center for running the Illumina 450 K methylation BeadChips. The DNA methylation analyses were funded by Autism Speaks. The SEED recruitment and data support was funded through six cooperative agreements from the Centers for Disease Control and Prevention: Cooperative Agreement Number U10DD000180, Colorado Department of Public Health and Environment; Cooperative Agreement Number U10DD000181, Kaiser Foundation Research Institute (CA); Cooperative Agreement Number U10DD000182, University of Pennsylvania; Cooperative Agreement Number U10DD000183, Johns Hopkins University; Cooperative Agreement Number U10DD000184, University of North Carolina at Chapel Hill; and Cooperative Agreement Number U10DD000498, Michigan State University. An earlier version of this work was presented at the 2013 Epigenomics of Common Disease conference in Cambridge, UK.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.envres.2015.11.014.
Footnotes
This work was supported by Autism Speaks (Grant 7659 to M.D.F.); the Centers for Disease Control and Prevention (Grants 5U10DD000180, 5U10DD000181, 5U10DD000182, 5U10DD000183 (Maryland site of SEED study, M.D.F.), 5U10DD000184). The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Conflict of interest
None.
References
- Aryee MJ, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barfield RT, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol. 2014;38:231–241. doi: 10.1002/gepi.21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton CV, et al. Prenatal tobacco smoke exposure is associated with childhood DNA CpG methylation. PLoS One. 2014;9:e99716. doi: 10.1371/journal.pone.0099716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castles A, et al. Effects of smoking during pregnancy. Five metaanalyses. Am J Prev Med. 1999;16:208–215. doi: 10.1016/s0749-3797(98)00089-0. [DOI] [PubMed] [Google Scholar]
- Davies MN, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drong AW, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS One. 2013;8:e55923. doi: 10.1371/journal.pone.0055923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Preston R, et al. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics. 2010;5:539–546. doi: 10.4161/epi.5.6.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley Alford SM, et al. Pregnancy associated smoking behavior and six year postpartum recall. Matern Child Health J. 2009;13:865–872. doi: 10.1007/s10995-008-0417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert BR, et al. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol Biomark Prev. 2014;23:1007–1017. doi: 10.1158/1055-9965.EPI-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert BR, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120:1425–1431. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky Z, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2012;17:728–740. doi: 10.1038/mp.2011.64. [DOI] [PubMed] [Google Scholar]
- Kerkel K, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- Kuhn M. Biolding predictive models in R using the caret package. J Stat Softw. 2008;28:1–26. [Google Scholar]
- Ladd-Acosta C. Epigenetic signatures as biomarkers of exposure. Curr Environ Health Rep. 2015;2:1–9. doi: 10.1007/s40572-015-0051-2. [DOI] [PubMed] [Google Scholar]
- Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132. doi: 10.3389/fgene.2013.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31:142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet. 2014;94:485–495. doi: 10.1016/j.ajhg.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness D, et al. Socio-economic status is associated with epigenetic differences in the pSoBid cohort. Int J Epidemiol. 2012;41:151–160. doi: 10.1093/ije/dyr215. [DOI] [PubMed] [Google Scholar]
- Murphy SK, et al. Gender-specific methylation differences in relation to prenatal exposure to cigarette smoke. Gene. 2012;494:36–43. doi: 10.1016/j.gene.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B, et al. Postnatal stability, tissue, and time specific effects of AHRR methylation change in response to maternal smoking in pregnancy. Epigenetics. 2014;9:377–386. doi: 10.4161/epi.27248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, et al. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenet. 2013;5:19. doi: 10.1186/1868-7083-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice F, et al. Agreement between maternal report and antenatal records for a range of pre and peri-natal factors: the influence of maternal and child characteristics. Early Hum Dev. 2007;83:497–504. doi: 10.1016/j.earlhumdev.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Richmond RC, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC) Hum Mol Genet. 2014;24:2201–2217. doi: 10.1093/hmg/ddu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmasi G, et al. Environmental tobacco smoke exposure and perinatal outcomes: a systematic review and meta-analyses. Acta Obstet Gynecol Scand. 2010;89:423–441. doi: 10.3109/00016340903505748. [DOI] [PubMed] [Google Scholar]
- Schendel DE, et al. The Study to Explore Early Development (SEED): a multisite epidemiologic study of autism by the Centers for Autism and Developmental Disabilities Research and Epidemiology (CADDRE) network. J Autism Dev Disord. 2012;42:2121–2140. doi: 10.1007/s10803-012-1461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, et al. Patterns of population epigenomic diversity. Nature. 2013;495:193–198. doi: 10.1038/nature11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NR, Bracken MB. A systematic review and meta-analysis of prospective studies on the association between maternal cigarette smoking and preterm delivery. Am J Obstet Gynecol. 2000;182:465–472. doi: 10.1016/s0002-9378(00)70240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker NS, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24:712–716. doi: 10.1097/EDE.0b013e31829d5cb3. [DOI] [PubMed] [Google Scholar]
- Smith AK, et al. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genom. 2014;15:145. doi: 10.1186/1471-2164-15-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YV, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet. 2013;132:1027–1037. doi: 10.1007/s00439-013-1311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M, et al. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism. 2010;59:1481–1490. doi: 10.1016/j.metabol.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter M, et al. Maternal tobacco use modestly alters correlated epigenomewide placental DNA methylation and gene expression. Epigenetics. 2011;6:1284–1294. doi: 10.4161/epi.6.11.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talens RP, et al. Variation, patterns, and temporal stability of DNA methylation: considerations for epigenetic epidemiology. FASEB J. 2010;24:3135–3144. doi: 10.1096/fj.09-150490. [DOI] [PubMed] [Google Scholar]
- Tehranifar P, et al. Early life socioeconomic factors and genomic DNA methylation in mid-life. Epigenetics. 2013;8:23–27. doi: 10.4161/epi.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez DR, et al. A balanced accuracy function for epistasis modeling in imbalanced datasets using multifactor dimensionality reduction. Genet Epidemiol. 2007;31:306–315. doi: 10.1002/gepi.20211. [DOI] [PubMed] [Google Scholar]
- Waterland RA, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilinger S, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8:e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.