

Abstract
Human aromatase catalyzes the synthesis of estrogen from androgen with high substrate specificity. For the past 40 years, aromatase has been a target of intense inhibitor discovery research for the prevention and treatment of estrogen-dependent breast cancer. The so-called third generation aromatase inhibitors (AIs) letrozole, anastrozole, and the steroidal exemestane were approved in the U.S. in the late 1990s for estrogen-dependent postmenopausal breast cancer. Efforts to develop better AIs with higher selectivity and lower side effects were handicapped by the lack of an experimental structure of this unique P450. The year 2009 marked the publication of the crystal structure of aromatase purified from human placenta, revealing an androgen-specific active site. The structure has reinvigorated research activities on this fascinating enzyme and served as the catalyst for next generation AI discovery research. Here, we present an account of recent developments in the AI field from the perspective of the enzyme’s structure–function relationships.
Graphical Abstract

1. INTRODUCTION
Cytochrome P450 aromatase (AROM) catalyzes with high substrate specificity the conversion of androstenedione (ASD), testosterone (TST), and 16α-hydroxytestosterone (all with the same androgen backbone) to estrone (E1), 17β-estradiol (E2), and 17β,16α-estriol (all with the same estrogen backbone), respectively. It is the only known enzyme in vertebrates capable of catalyzing the aromatization of a six-membered ring. The reaction requires coupling with its redox partner cytochrome P450 reductase (CPR), which uses NADPH as the source of electrons. AROM has been the subject of intense biochemical, biophysical, and clinical investigations for the past 50 years.1–3 Inhibition of estrogen biosynthesis by AROM inhibitors (AI) constitutes one of the foremost therapies for postmenopausal estrogen-dependent breast cancer today.4,5
Nevertheless, many aspects of the aromatization reaction remained poorly understood. Until 2009,6 the absence of a crystal structure for human AROM had led to a number of homology models for the enzyme based on other experimental P450 structures and site-directed mutagenesis data.7–11 Several androgen-binding scenarios at the active site, possible involvements of side chains in the catalytic process, and models for enzyme’s mechanism of action were proposed based on homology structures and functional analyses. However, none of these models could adequately explain the unique characteristics of AROM that set this P450 apart from all others. Details of the substrate and inhibitor binding interactions at the active site were crucial for the development of next generation AIs.
Despite concerted efforts in many laboratories, no experimental molecular structure of AROM emerged for a very long time. The major impediments to AROM crystallization were its strong hydrophobic character and susceptibility to rapid denaturation in the absence of the protective lipid bilayer. Using term human placenta as a rich source of AROM and a purification technique that employs a highly specific monoclonal antibody-based affinity chromatography,12 we were able to purify large quantities of the enzyme in a pristine, active form that permitted the growth of diffraction-quality single crystals. This was the first and only microsomal P450 purified from a native source to date and the first full length P450 to be crystallized.6,13 Employing the same crystallization protocols, a recombinant form of human AROM has more recently been crystallized.14
Needless to say, the real atomic model has reinvigorated the field of AROM research, from perspectives of re-examining the reaction mechanism15 and novel AI discovery.16 One of many fundamental questions that remained unresolved for decades was the following: What are the two proton donors to the 3C=O and 17C=O groups involved in binding of the natural substrate ASD and its conversion to E1? Evidently, a direct structural observation of the binding interactions of ASD in the active site was necessary for the unequivocal answer in this case. Indeed, it would have been impossible to envision that one of the most unlikely candidates the Asp309 carboxylate, protonated at the physiological pH, is the proton donor to 3C=O and the equally imperceptible Met374 backbone amide to 17C=O 6 (Figure 1). The predicted protonation of Asp309 side chain and its involvement in the aromatization reaction have since been validated by other works.17,18 These data, along with the nature of hydrophobic interactions that define the highly androgen-specific, tight active site cleft (Figure 1), are crucial to the discovery of future AIs.
Figure 1.
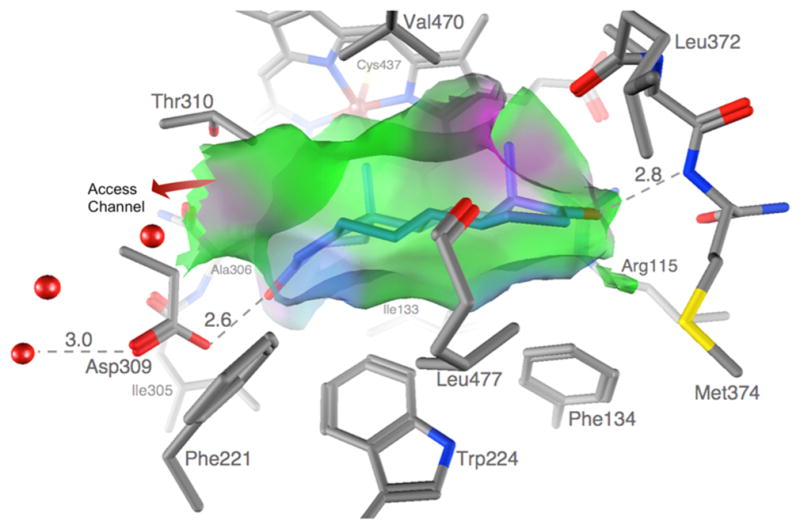
The AROM active site is shaped like an androgen backbone. A van der Waals interaction surface cast by the protein and heme atoms at the active site is shown. The semitransparent surface closely resembles the shape, size, and puckering of the steroid backbone. The surfaces are color-coded green for hydrophobic and magenta for polar. The protein atoms defining the van der Waals surface are also shown: C gray, N blue, and O red. The only opening to the pocket is the one to the active site access channel indicated by an arrow. Water oxygen atoms in the channel and bound to the protonated Asp309 carboxylate are shown as red spheres. Adapted from ref 6.
Here we describe the recent advancements made toward design and discovery of novel AIs from the perspective of current understanding of the enzyme’s structure–function relationships, giving a brief context to the known AIs developed prior to the AROM structure. We summarize most, if not all, inhibitors that were developed since 2009 utilizing rational means, such as structure-based design, molecular docking, and virtual screening. We present a brief account of our own structure-guided efforts not only for design but also for validation of design. We compare and contrast the reported potencies of the inhibitors and the measurement protocols in an attempt to stress that the numbers are not always comparable. We conclude by describing the limitations in the current knowledge base and future directions.
2. BRIEF HISTORY OF CURRENT AROM INHIBITORS
2.i. First and Second Generation Inhibitors
The chronology of discovery of early generation AIs, inhibitory properties, clinical trial history, side effects, etc. is summarized in Table 1. Shown in Figure 2, aminoglutethimide (AGT) was the first known potent AI.19 It was reported to reversibly suppress AROM activity in placental microsomes.20 In fact, AGT also was a potent inhibitor of other P450s, such as P450scc21 and enzymes involved in the biosynthesis of cortisol, aldosterone, and thyroid hormones.22 Although AGT treatment lacked AROM specificity,23 doses of 250–1000 mg of AGT effectively inhibited peripheral AROM activity and suppressed plasma E1 levels in postmenopausal women.24,25 Formestane (4-hydroxy-4-androstene-3,17-dione, Figure 2), first isolated as a byproduct of 2-hydroxy-steroid 4-en-3-ones synthesis,26 was one of the most promising second generation AIs that underwent multiple clinical trials.27–31 It was also the first effective steroidal inhibitor of AROM32 with limited side effects27,29,30 but was never approved for clinical use in the U.S. Formestane was reported as a competitive, irreversible, mechanism-based AROM inhibitor due to its time-dependent suppression of E1 biosynthesis in placental microsomes preincubated with ASD, NADPH, and inhibitor.20,33 Fadrozole (FDZ, Figure 2), the prominent second generation AI, was more potent and specific than the other nonsteroidal AI AGT.34,35 It was reported to be a competitive and reversible AI,35 which had weak inhibitory effects on P450scc.36,37 It also underwent multiple clinical trials in the U.S. and other countries.31,35,37–42 Although approved in Japan for treatment in estrogen-dependent postmenopausal breast cancer, FDZ never became a drug in the U.S.
Table 1.
Chronology of Discovery of Earlier Generation AIs
| occurrence | first generation AI
|
second generation AIs
|
third generation AIs
|
|||
|---|---|---|---|---|---|---|
| aminoglutethimide | formestane | fadrozole | exemestane | letrozole | anastrozole | |
| Discovery/earliest reported use | 1961 (treatment of epilepsy)162 1966 (antithyroidal and antiadrenocortical activity)21,22 |
1973 (synthesis)26 1977 (preclinical testing)32,163,164 |
1987 (preclinical testing)165 | 1988 (preclinical testing)166,167 | 1990 (preclinical testing)168 | 1994 (summary of preclinical testing and clinical studies)60,61 |
| First reported use of AIs | 1974 (in the U.S.)19 | 1984 (clinical trials in the UK first)27–31,169 | 1989 (clinical trials in the U.S. and later in Europe, South Africa, Japan)35,37–42 | 1995 (clinical trials in Italy first and later in Europe and U.S.)50–59 | 1993 (clinical trials in the U.S. and UK)67,68,74,170,171 | 1994 (clinical trials in the U.S.)60–64 |
| IC50 against pure enzyme | Not determined (5–7 μM against human placental microsomes172) | 49 nM 46 | Not determined (5 nM against human placental microsomes173) | 50 nM 46 | 10 nM 46 | Not determined (23 nM against human placental microsomes173) |
| IC50/EC50 in breast cancer cell line | 10 μM against MCF-7 cells transfected with AROM;174 20 μM in particulate fractions of breast cancer;43 10 μM in culture of mammary adipose tissue fibroblasts43 | 30 nM (particulate fractions of human breast cancer tissues)173 | 50 pM in MCF-7 cells173 | 5.6 nM in MCF-7 cells over-expressing AROM46 (antiproliferation assay) | 4 pM in MCF-7 cells overexpressing AROM46 (antiproliferation assay) | 3.6 nM in MCF-7 cells173 |
| Dose in treatment | 250 mg/4× daily orally24,45,175 | 250 mg daily orally or 500 mg via intramuscular injection every 2 weeks29,45,175 | 1.8–2 mg daily orally41,45,175 | 25 mg daily orally45,175 | 2.5 mg daily orally45,175 | 1 mg daily orally45,175 |
| Side effects/issues45,175 | Low selectivity for AROM Inhibition of cortisol, aldosterone, thyroid hormone synthesis. Induction of hepatic enzymes. Minor side effects: nystagmus, ataxia, lethargy, skin rash24,176–178 |
Poor oral bioavailability29 Local side effects due to intramuscular injection40 Severe neutropenia (transient and reversible upon discontinuation of treatment)27 |
Suppression of aldosterone release179 Mild hot flashes, nausea and vomiting, fatigue, and mild loss of appe80 |
Hot flashes, fatigue, arthralgia (joint pain), headache, insomnia (difficulty sleeping), increased sweating, hypertension (high blood pressure), dizziness, cardiac ischemic events, reduced bone mineral density/osteoporosis50–59,181 | Low-grade hot flashes, arthritis, arthralgia, and myalgia, and new diagnoses of osteoporosis67,68,74,170,171 | Hepatic toxicity, blurred vision, angina pain, irregular heartbeat, edema in legs, headaches, dizziness, nervousness, shortness of breath60–64 |
| Year of FDA approval as an AI | Not FDA-approved in the U.S. | Not FDA approved in the U.S. | Not FDA approved in the U.S. | 199957–59 | 199865,66 | 199564 |
| Current FDA-approval status | FDA-approved for Cushing syndrome | Initially approved as an AI in Europe Withdrawn due to poor oral bioavailability and discovery of third-generation orally active AIs |
Approved as an AI in Japan182 | Approved as an AI in the U.S. | Approved as an AI in the U.S. | Approved as an AI in the U.S. |
Figure 2.

Previous and current generation AIs. The chemical structures of known AIs are shown.
2.ii. Third Generation AIs
Anastrozole (ANZ; Arimidex), letrozole (LTZ; Femara), and exemestane (EXM; Aromasin) are the three AIs that received the FDA approval for use in estrogen-dependent breast cancer in the late 1990s (Table 1, Figure 2). Unlike their predecessors, these so-called third generation inhibitors all have high selectivity for AROM and low cross-reactivity to other targets.4,43–45 EXM was reported to elicit time-dependent inactivation of AROM and classified as a sucicide inhibitor.20,33 However, the crystal structure of the AROM–EXM complex at 3.2 Å resolution did not reveal a covalent bond between AROM and EXM.46 EXM binds in competition with the substrate but does not covalently link to protein in the absence of the reductase and electron source.46 There is no structural evidence suggesting that any of the AIs are either reversible or irreversible. We have reported the inhibition kinetics of LTZ and utilized statistical analyses to show that its mode of action could be either noncompetitive or mixed.47 Furthermore, clinical data do not show superior efficacy of reversible AIs over irreversible AIs or of steroidal AIs over nonsteroidal AIs and vice versa. In the head-to-head trials evaluating the clinical efficacy of EXM and ANZ,48 and LTZ and ANZ49 in postmenopausal breast cancer, all were found to be equally effective. All three third generation AIs are more potent than the previous inhibitors and performed much better in clinical trials.4,5,43–45,50–68 Also noteworthy is the fact that all three AIs have shown better efficacies in major clinical trials in head-to-head comparison with the estrogen-receptor antagonist tamoxifen in the treatment of postmenopausal estrogen-dependent breast cancer.4,43–45 They are proven to be more effective than tamoxifen in estrogen receptor (ER) positive tumors that upregulate progesterone receptor (PR) and epidermal growth factor receptors (EGFRs or HER-2/neu) as escape survival mechanisms from prolonged tamoxifen therapy.69–72 Regardless of growth factor receptor status, the third-generation AIs could circumvent endocrine resistance that might occur due to to estrogen genotoxicity,73 activation of additional estrogen-dependent signaling pathways by tamoxifen metabolites,69–72 activation of growth factor receptor signaling pathways by partial agonistic activity of tamoxifen,69–72 and greater disease free survival compared to tamoxifen.74 The negative effects of third-generation AIs include total body loss of estrogen, joint pain, osteoporosis, and low bone mineral density.4,43–45,50–56,58,60–64 Vorozole (Rivizor), another third-generation AI, underwent clinical trials for use in treating estrogen-dependent postmenopausal breast cancer in Europe and Canada.75 However, it never gained FDA approval in the U.S.76
3. CRYSTAL STRUCTURE OF HUMAN AROM: HOW THE ACTIVE SITE ARCHITECTURE PRESENTED NEW IDEAS
3.i. Active Site
The crystal structure of the highly active human placental AROM in complex with the substrate ASD was originally determined at 2.90 Å resolution.6 More recently, the resolution has been extended to 2.75 Å.46 The tertiary structure of AROM follows the characteristic cytochrome P450 fold. The active site of AROM is located at the heme-distal cavity, buried deep within the roughly spherical molecule near its geometrical center. ASD binds with its β-face oriented toward the heme group and C19 of the methyl group positioned 4.0 Å from the Fe atom.
The steroid binding cleft is defined by residues Ile305, Ala306, Asp309, and Thr310 from I-helix, Phe221 and Trp224 from F-helix, Ile133 and Phe134 from the B–C loop, Val370, Leu372, and Val373 from the K-β3 loop, Met374 from β3, and Leu477 and Ser478 from the β8–β9 loop. These hydrophobic side chains and the heme porphyrin rings surround the steroid backbone to form a cavity complementary in shape to ASD (Figures 1 and 3). The side chains of residues Arg115, Ile133, Phe134, Phe221, Trp224, Ala306, Thr310, Val370, Val373, Met374, and Leu477 make direct van der Waals contacts with the bound ASD. Ile133, Phe134, Phe221, Trp224, and Leu477 approach the substrate from the α-face and follow contour and puckering of the steroid backbone, while Arg115, Ala306, and Met374 make contacts at its edge, and Thr310, Val370, and Val373 on the β-face of ASD. The combined surface creates a pocket of roughly 400 Å3 that tightly encloses the bound ASD. This is considerably smaller than the volumes of the active site space in cytochrome P450s 3A4,77 2D6,77,78 11A1,79 11B2,79 17A1,80 21A1,81 24A1,82 and 51A1.83
Figure 3.

Androgen-binding environment: schematic diagram of the X-ray structure depicting the residues lining the ASD binding pocket, and ASD making hydrophobic and hydrogen-bonding contacts (hydrophobic, green; acidic, red; basic, blue; polar, purple; S-containing, yellow). Exposure at the C4 and C6 positions of the steroid to the access channel opening is indicated. Also shown schematically is a water molecule trapped between Asp309 and Arg192 side chains, postulated to have a role in the proton relay network and enolization of 3-keto. Arg192 and its salt bridge partner Glu483 are the “gatekeepers” of the access channel. Adapted from ref 46.
The 17-keto oxygen of ASD makes a hydrogen bond with the backbone amide of Met374. It is also at a distance of 3.4 Å from the NH1 of Arg115 side chain. The 3-keto oxygen at the other end is hydrogen bonded to a protonated Asp309 side chain at the crystallization pH of 7.4. The proposed roles of Asp309 side chain protonation in substrate binding, enolization of the 3-keto group, and proton relay network6 have been supported by recent findings.17,18 The Asp309 side chain is linked by solvent molecules to the salt bridging Arg192-Glu478 side chain pair at the end of the solvent access channel at the lipid interface.6 The “gatekeeper” residue Arg192, predicted to play an important catalytic role in proton relay,6 has since been shown to be critical for maintaining the enzyme activity.14 Interestingly, recent clinical case reports establish that mutations of Arg192 result in AROM deficiency and virilization in human.84,85
Although the binding pocket is sealed tight, it has a doorway where water molecules are located (Figure 3), leading to an access channel that opens to the exterior of the protein surface. The Arg192-Glu478 side chain pair, polar residues Asp309, Thr310, and Ser478, and several hydrophobic side chains Phe221, Val313, and His480 line the channel (Figure 3). This channel hosts the proton relay network and is also the major transport route to and from the active site for water, oxygen, and steroid molecules. It was shown that flexibility and fluctuations in the channel-bordering regions permit the channel to “breathe” and swell, perhaps allowing the passage of steroids.86 This entire region adjacent to atoms C4 and C6 of the substrate leading to the access channel presents an intriguing mix of functional groups for AROM-specific inhibitor design ideas.
3.ii. New Perspective on Reaction Mechanism: A Tool for Inhibitor Design
The unique active site architecture, the novelty of steroid-binding interactions, and the proposed 3-keto enolization as well proton relay mechanisms6 are all unprecedented in the previous homology models.7–11 The structure, thus, has not only validated some of the previously held ideas but also provided a platform for further mechanistic studies on the aromatization reaction.18,87–90 In the experimentally observed substrate-bound state, the hydrogen atoms of C19 methyl group are at the viable H-abstraction distances91 from the modeled oxygen of the reactive oxyferryl moiety (Fe(III) modeled as Fe(IV) =O, compound I), shown in Figure 4a. The residues that are directly involved in catalysis are Ala306, Asp309, and Thr310. In addition, Arg192, Glu483, Ser478, and His480 have critical roles in proton relay and/or aromatization. The location of Thr310 with respect to the heme iron is similar to other P450s and performs a similar role in the first two steps of hydroxylation yielding C19-aldehyde derivative of ASD and retaining the pro-S hydrogen.87 The structure revealed a specific interaction involving Thr310 suggesting its crucial role in all three steps of catalysis. The C=O oxygen of 306Ala and OγH of Thr310 are at 3.7 and 3.8 Å, respectively, from H2β (Figure 4b) suggesting 306AlaC=O alone or in conjunction with a deprotonated Thr310Oγ– acts as a nucleophile for the abstraction of H2β. This action at C2 in conjunction with a protonated Asp309 side chain acting as an electrophile and interacting strongly with the C3-keto oxygen could promote H2β abstraction and enolization of the 3-keto. The presence of a water molecule could facilitate the deprotonation of the Thr310 side chain by weakening the Ala306CO⋯HOγThr310 hydrogen bond. This would free carbonyl for the nucleophilic attack for H2β abstraction by -H2C2-C3-keto to -HC2=C3-enol tautomarization (Figure 4c). H1β is too far from this carbonyl (6.2 Å) to be abstracted by the same mechanism. It points at the heme Fe (4.2 Å) and is more likely abstracted during the peroxoferric nucleophilic attack on 19-aldehyde (Figure 4c) as previously postulated.87 The deprotonated Asp309 side chain is quickly reprotonated by the proton relay network. The direct involvement of Asp309 in protonation and aromatization appears to be unique to AROM.
Figure 4.

Reaction center and perspective on catalysis. (a) Modeling of Fe (III) as an oxyferryl Fe(IV) =O (compound I) moiety. The C19-methyl hydrogen atoms are shown at the calculated ideal positions. Important side chains, heme, and water molecules are depicted in element colors: C, gray; N, blue; O, red; S, yellow; Fe, firebrick; H, orange. The C atoms of ASD are colored in cornflower blue. (b) Close-up view of the 306AlaCO⋯HOThr310 pair that may have a role in aromatization of the A-ring of ASD. Calculated hydrogen atom positions of C2 of the bound ASD are shown. The distances shown are in Å. (c) Possible mechanism for H2β abstraction and 2,3-enolization. The direction of proton flow from the proton relay network through Asp309 carboxylate is indicated. Adapted from ref 6.
Many groups have attempted to determine the critical intermediate by theoretical calculations, electron paramagnetic resonance,92 resonance Raman spectroscopy,93,94 kinetic solvent isotope effect,95 and molecular oxygen labeling experiments.36,96 These results have shown the possibility that either the oxy-ferryl or peroxo-ferric can be involved in the aromatization reaction. These findings have also been previously summarized.97 None of the transition states have been directly observed by X-ray diffraction. Atomistic details of the functional groups in and around the active site, as well as understanding of the organometallics of aromatization chemistry, are critical to the design of superior inhibitors that are mechanism-based and/or transition state analogs and, therefore, exclusive to AROM.
4. RECENT ADVANCES IN THE DISCOVERY OF STEROIDAL AROM INHIBITORS
The third generation AIs are remarkably effective against breast cancer. AIs have also been used for the treatments of endometriosis98,99 and ovarian98,100 and lung101 cancers. These AIs have high affinities for AROM but also show some cross-reactivity to other P450s. CYP1A2, CYP2C9, and CYP3A4 are inhibited by ANZ, and CYP2A6 and CYP2C19 are inhibited by LTZ.102–105 CYP2A6 and CYP3A4 metabolize LTZ.106–108 Furthermore, EXM is androgenic109 and also has weak ERα agonistic activity.110 Despite high efficacy, some patients may fail to respond to AIs, which is known as AI resistance.106 Rational approaches such as structure-guided design of next generation AIs could minimize the nonspecific and adverse effects.5
4.i. Structure-Based Design
Crystallographic and computational results have shown that AROM structure has a rigid core; ligands are thus accommodated by modest adjustments of the catalytic cavity.46 By exploiting the active site architecture described in section 3.i above, derivatives of androsta-1,4-diene-3,17-dione have been synthesized. These compounds have undergone in vitro characterization in a MCF-7 breast cancer cell-based antiproliferation system and cell-free purified enzyme inhibition assays.46 Three C6β-alkoxy/alkynyloxy derivatives, in particular, have exhibited the most potent AROM inhibition to date.46 Table 2 summarizes the inhibitory and antiproliferative potencies of the three most potent compounds 4, 5, and 9 (Figure 5, upper panel), in comparison to EXM. Compound 5, the best in the series, has IC50 and EC50 roughly 4- and 187- fold, respectively, better than EXM (Table 2). The X-ray structures of the complexes of 4 and 5 with AROM reveal that the 2-alkynyloxy side chains fit tightly within the hydrophobic environment of the channel.6,46 Both the structural and functional data are consistent in that 5 and 9 possess the right dimensions to traverse the access channel. The C24 methyl end of the 5 alkyne chain packs against the Val313 side chain, Asp309 main chain carbonyl, arene ring of Phe221 and His480 imidazole. The side group of 5 nearly extends to the polar residues Arg192 and Glu483 at the channel entrance at the membrane–protein interface. A C25 methyl and longer derivatives of 5 have progressively reduced inhibitory and antiproliferative potencies.46
Table 2.
Summary of IC50 and EC50 of the Most Potent C6β-Alkoxy/2-Alkynyloxy Series of Steroidal Aromatase Inhibitors and Controls
| compd | IC50 (nM) | 95% confidence interval (nM) | potency relative to EXM | EC50 (nM) | 95% confidence interval (nM) | potency relative to EXM |
|---|---|---|---|---|---|---|
| exemestane (EXM) | 50.1 | 40.9–61.4 | 5.6 | 2.7–6.5 | ||
| 4 (HDDG029) | 112.3 | 78.2–161.3 | 0.5 | 1.7 | 1.2–2.2 | 3.3 |
| 5 (HDDG046) | 11.8 | 9.3–14.9 | 4.2 | 0.03 | 0.02–0.06 | 187.0 |
| 9 (HDDG065) | 20.0 | 18.1–22.0 | 2.5 | 0.3 | 0.2–0.4 | 18.7 |
Figure 5.

Structural basis for the design of potent inhibitors. The chemical structures of the three most potent steroidal inhibitors 4, 5, and 9 of the 6β-2-alkynyloxy series are shown in the upper panel. Lower panel: Schematic diagram depicting the X-ray structure of the tight hydrophobic binding pocket for the designed steroidal inhibitor 5, the proton donors at the 3- and 17-keto positions, and the 6β-2-alkynyloxy side group that nearly fills the access channel. Compound 4 also binds the same way. The residues lining the binding pocket making hydrophobic and hydrogen-bonding contacts are shown. Adapted from ref 46.
4.ii. Other Rational Approaches
Several other groups have utilized the AROM structure to design new inhibitors. A summary of these newly designed inhibitors and their properties (all inhibition assays of the novel steroidal AIs are performed in vitro) is provided in Table 3, and the chemical structures are shown in Figure 6 (compound numbers are the same as in Table 3). However, most of the binding modes and/ or mechanisms are hypothetical. The inhibitors synthesized as derivatives of ASD111 and TST111 (compounds 10–16) are reported to have submicromolar potencies in placental microsomes. The modifications are at the positions C3, C4, C5, C6, and C17, as well as C7α of allylandrostanes (17–20).112 Other designs include 16-imidazolyl substituted steroidal derivatives (23, 24),113 pyrazole and isoxazole substitutions at C2 and C3 positions (25, 26),114 and synthesis of 16β-cyano-17β-hydroxy-4-phenylthia-4-androsten-3-one as a novel inhibitor (27).115
Table 3.
Summary of New AROM Inhibitors Since 2009
| compd | type | name | IC50 | assayed against, assay type | exptl binding mode verification |
|---|---|---|---|---|---|
| 1–9 | C6-substituted 2-alkynyloxy compounds46 | C6-substituted androsta-1,4-diene-3,17-dione | 20–18100 nM | Purified AROM and MCF- 7a,a 3H2O, antiproliferation | Yes: X-ray |
| 10 | A- and D-ring ASD derivative111 | 3β-Hydroxyandrost-4-en-17-one | 0.18 μM | PMTW | No |
| 11 | A- and D-ring ASD derivative111 | Androst-4-en-17-one | 0.14 μM | PMTW | No |
| 12 | A- and D-ring ASD derivative111 | 4α,5α-Epoxyandrostan-17-one | 0.97 μM | PMTW | No |
| 13 | A- and D-ring ASD derivative111 | 5α-Androst-3-en-17-one | 0.23 μM | PMTW | No |
| 14 | A- and D-ring ASD derivative111 | 3α,4α-Epoxy-5α-androstan-17-one | 0.15 μM | PMTW | No |
| 15 | A- and D-ring ASD derivative111 | 5α-Androst-2-en-17-one | 1.7 μM | PMTW | No |
| 16 | A- and D-ring ASD derivative111 | 2α,3α-Epoxy-5α-androstan-17-one | 1.2 μM | PMTW | No |
| 17 | 7α-allylandrostanes112 | 7α-Allylandrost-4-ene-3,17-dione | 0.59 μM | Placental microsomes, 3H2O (PMTW)b,c | No |
| 18 | 7α-allylandrostane112 | 7α-Allylandrost-4-en-17-one | 0.75 μM | PMTW | No |
| 19 | 7α-allylandrostane112 | 7α-Allyl-3-oxoandrosta-1,4-dien-17β-ol | 0.45 μM | PMTW | No |
| 20 | 7α-allylandrostane112 | 7α-Allylandrosta-1,4-diene-3,17-dione | 0.47 μM | PMTW | No |
| 21 | NCIe compound (B-nor-androstenedione scaffold)139 | 3a,5b,10-Trimethyl-1,2,4,5,5a,6,7,10,10a,10b-decahydrocyclopenta[a]fluorene-3,8-dione | 0.27 μM | PMTW | No |
| 23 | Imidazolyl substituted steroidal derivative113 | 16β-(Imidazol-1-yl)-4-androstene-3,17-dione | 0.18 μM | PMTW | No |
| 24 | Imidazolyl substituted steroidal derivative113 | 16β-(Imidazol-1-yl)androsta-1,4-diene-3,17-dione | 0.168 μM | PMTW | No |
| 25 | Androstane114 | 17β-Hydroxy-4-oxo-5α-androstano[2,3-d]pyrazole | 512.0 nM | PMTW | No: MDd |
| 26 | Androstane114 | 2-Cyano-3,17β-dihydroxy-5α-androst-2-en-4-one | 1019.8 nM | PMTW | No: MD |
| 27 | Carbonitrile115 | 16 β-Cyano-17 β-hydroxy-4-phenylthia-4-androsten-3-one | 169.3 nM | PMTW | No: MD |
| Nonsteroidal AROM Inhibitors | |||||
| 22 | NCIe compound NSC613604139 | 5-(4-Chloroanilino)-2-methyl-9-nitro-5H-chromeno[4,3-b] pyridine-3-carbonitrile | 0.67 μM | PMTW | No |
| 28 | Quinoline derivative-dual CYP11B2/CYP19 inhibitor119 | 8-[(3-Methylphenyl)(pyridin-4-yl)methyl]-1,2,5,6-tetrahydro- 4H-pyrrolo[3,2,1-ij]quinolin-4-one | 32 nM | PMTW | No: MD |
| 29 | Azolylmethylpyrroloquinoline120 | 4-((1H-Imidazol-1-yl)methyl)-7-ethyl-2-phenyl-7H-pyrrolo[2,3- h]quinoline | 5.3 nM | Supersomesg/H295R cells, FCAh/antiproliferation | No: MD |
| 30 | Azolylmethylpyrroloquinoline120 | 4-((1H-1,2,4-Triazol-1-yl)methyl)-2-(4-methoxyphenyl)-7H- pyrrolo [2,3-h]quinoline | >10000 nM | Supersomes/H295R cells, FCA/antiproliferation | No: MD |
| 31 | Azolylmethylpyrroloquinoline120 | 9-((1H-Imidazol-1-yl)methyl)-3-ethyl-7-phenyl-3H-pyrrolo- [3,2-f ]quinoline | 3.1 nM | Supersomes/H295R cells, FCA/antiproliferation | No: MD |
| 32 | Azolylmethylpyrroloquinoline120 | 9-((1H-1,2,4-Triazol-1-yl)methyl)-3-ethyl-7-phenyl-3H-pyrrolo [3,2-f ]quinoline | 13.3 nM | Supersomes/H295R cells, FCA/antiproliferation | No: MD |
| 33 | NNCf compound (imidazolyl quinoline)121 | 2-(4-Fluorophenyl)-4-(imidazol-1-yl)quinoline | 0.8 μM | Supersomes/T47cells, E2 ELISA/antiproliferation | No: MD |
| 34 | Isoflavanone derivative122 | 3-(4-Phenoxyphenyl)chroman-4-one | 2.4 μM | Supersomes/FCA | No: MD |
| 35 | Isoflavanone derivative122 | 6-Methoxy-3-phenylchroman-4-one | 0.3 μM | Supersomes/FCA | No: MD |
| 36 | Isoflavanone derivative122 | 3-(Pyridin-3-yl)chroman-4-one | 5.8 μM | Supersomes/FCA | No: MD |
| 37 | Flavans123 | 5-Methoxy-8-formyl-4,7-dihydroxyflavan | 40 nM | Supersomes/FCA | No: MD |
| 38 | 4,7-disubstituted coumarin124 | 7-(3,4-Difluorophenoxy)-4-(1H-imidazol-1-ylmethyl)-2H- chromen-2-one | 47 nM | PMTW | No: MD |
| 39 | 4,7- disubstituted coumarin124 | 4-(1H-Imidazol-1-ylmethyl)-7-phenoxy-2H-chromen-2-one | 150 nM | PMTW | No: MD |
| 40 | Dual AROM/STS inhibitor126 | 4-{[(4-Cyanophenyl)(1H-imidazol-1-yl)amino]methyl}-3- fluorophenyl sulfamate | 0.2 nM | JEG3 cells, 3H2Oc | No: MD |
| 41 | Dual AROM/STS inhibitor130 | 2-Bromo-4-(2-(4-cyanophenyl)-2-(1H-1,2,4-triazol-1-yl)ethyl) phenyl sulfamate | 0.9 nM | JEG3 cells, 3H2Oc | No |
| 42 | Dual AROM/STS inhibitor129 | 5′-((1H-1,2,4-Triazol-1-yl)methyl)-2′-cyanobiphenyl-4-yl sulfamate | 2 nM | JEG3 cells, 3H2Oc | No |
| 43 | Dual AROM/STS inhibitor 129 | 5′-((1H-1,2,4-Triazol-1-yl)methyl)-3-chloro-2′-cyanobiphenyl- 4-yl sulfamate | 0.5 nM | JEG3 cells, 3H2Oc | No |
| 44 | Dual AROM/STS inhibitor 128 | 2-Chloro-4-(((6-cyanobiphenyl-3-yl)(4H-1,2,4-triazol-4-yl) amino)methyl)phenyl sulfamate | 15 pM | JEG3 cells, 3H2Oc | No: MD |
| 45 | Dual AROM/STS inhibitor128 | 2-Bromo-4-(((6-cyanobiphenyl-3-yl)(4H-1,2,4-triazol-4-yl) amino)methyl)phenyl sulfamate | 18 pM | JEG3 cells, 3H2Oc | No: MD |
| 46 | Tamoxifen metabolite131 | Z-4-Hydroxytamoxifen | 530 μM | Supersomes/T to E2i | No: MD |
| 47 | Tamoxifen metabolite131 | Z-Norendoxifen | 30 nM | Supersomes/T to E2 | No: MD |
| 48 | Tamoxifen metabolite131 | Z-Endoxifen | 6 μM | Supersomes/T to E2 | No: MD |
| 49 | Tamoxifen metabolite132 | (Z)-Norendoxifen | 1029 nM | Supersomes/FCA | No: MD |
| 50 | Tamoxifen metabolite132 | (E)-Norendoxifen | 77 nM | Supersomes/FCA | No: MD |
| 51 | Tamoxifen metabolite132 | (E,Z)-Norendoxifen | 102 nM | Supersomes/FCA | No: MD |
| 52 | Sulfonamide derivative133 | 7-((1-(3-((6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl) sulfonyl)phenyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one | 0.2 μM | Supersomes/T to E2 | No: MD |
| 53 | ASINEX compounds (sulfonamide derivative)138 | 1-(3-tert-Butyl-4-methoxybenzenesulfonyl)-3-(imidazol-1ylmethyl)piperdine | 9.4 nM | Supersomes/FCA | No: MD |
| 54 | ASINEX compounds (sulfonamide derivative)138 | N-[3-(Imidazol-1-yl)propyl]-2,3-dihydro-1,4-benzodioxine-6-sulfonamide | 119 nM | Supersomes/FCA | No: MD |
| 55 | Aryl halide134 | 2-[7-Bromo-1-(3-ethoxypropyl)imidazo[4,5-c] pyridin-5-ium-5-yl]-1-phenylethanone | 20 nM | Supersomes/FCA | No: MD |
| 56 | Indole–imidazole derivative135 | 2-(Imidazol-1-ylmethyl)-1-[4-(trifluoromethyl)phenyl]indole | 4.9 nM | Placental microsomes, ELISA | No |
| 57 | Casimiroin analogue136 | N-Methylated casimiroin analogues: 5,6,8-trimethoxy-1,4-dimethylquinolin-2(1H)-one | 0.1 μM | Supersomes/FCA | No: MD |
| 58 | Casimiroin analogue136 | Non-N-methylated casimiroin analogues: 6-methyl[1,3]dioxolo [4,5-h]quinolin-8(9H)-one | >98.5 μM | Supersomes/FCA | No: MD |
| 59 | Casimiroin analogue136 | Casimiroin | 3.9 μM | Supersomes/FCA | No: MD |
| 60 | Xanthone scaffold137 | 4-Imidazol-1-ylmethylthioxanthen-9-one | 4 nM | PMTW | No |
| 61 | Xanthone scaffold137 | 1-(4-Nitro-2-phenylsulfanylbenzyl)-1H-imidazole | 5.6 nM | PMTW | No |
| 62 | ASINEX compounds (morpholinoethanone derivative)138 | 1-[2-(Imidazol-1-ylmethyl)morpholin-4-yl]-2-(3,4,5-trimethoxyphenyl)ethanone | 59.2 nM | Supersomes/FCA | No: MD |
| 63 | ASINEX compounds (imdazolylacetamide)138 | N-[2-(4-Fluorophenoxy)phenyl]-2-(imidazol-1-yl)acetamide | 248 nM | Supersomes/FCA | No: MD |
MCF-7a: a MCF-7 cell line stably expressing AROM as described.59
PMTW: placental microsomes, tritiated water assay.
3H2O: tritiated water.
MD: molecular docking.
NCI: National Cancer Institute.
NNC: National Nanjing Center for Drug Screening.
Supersomes: commerical mixture of baculovirus AROM microsomes and reductase.
FCA: fluorescent CYP19 inhibition assay, which measures the conversion of a fluorescence substrate to its metabolite, a hydroxylation reaction.
T to E2 assay, which measures T to E2 conversion by HPLC.
Figure 6.

(a) Recently developed steroidal and nonsteroidal AIs. The compounds are numbered and grouped in the same way as referenced in the text and in Table 3. (b) Compounds that exhibit antiproliferative effects on breast cancer cells. Compounds 65 and 66 exhibit antiproliferative effects on breast cancer cells. No direct aromatase inhibition was measured; therefore, they are not classified as AIs.
5. RECENT ADVANCES IN THE DISCOVERY OF NONSTEROIDAL AROM INHIBITORS
5.i. Binding Site Ambiguity in the Absence of Structural Data
No structural data are available yet for unequivocal elucidation of the binding modes of nonsteroidal AIs, such as LTZ and ANZ. In the absence of experimentally determined binding modes of the nonsteroidal inhibitors, virtual docking results to the AROM active site widely reported in the literature are tentative at best. Our modeling data indicate that LTZ and ANZ do not easily fit into the AROM active site pocket116 that resembles an androgen backbone (Figure 1). Furthermore, recent inhibition kinetics data on LTZ and other azole compounds are indicative of mixed modes of inhibition, structural changes, and allostery.47 Possibility of allosteric sites for nonsteroidal compounds has also been proposed from computational studies.117
5.ii. Recent Reports
Table 3 summarizes the reported nonsteroidal inhibitors. All inhibition/kinetic assays of the novel nonsteroidal AIs were performed in vitro. The chemical structures are shown in Figure 6a. A brief description of each subclass of AI is provided below.
a. Azole Derivatives
Several imidazole, indole, and imidazolylbenzyl derivatives of LTZ were reported to have potencies similar to LTZ.118 Vorozole is another older azole compound that causes a reversible inhibition of AROM with IC50 value of 1.4 nM.118 The major AROM inhibitory activity of vorozole is attributable to the dextro isomer. Replacement of the 1H-benzo[d][1,2,3]triazole functional group in the vorozole molecule by benzofuran presented a potent racemic triazole derivative with IC50 value of 10 nM.118
b. Quinoline Derivatives
AI treatment of postmenopausal breast cancer could increase the risk of cardiovascular diseases, which could be brought about by high aldosterone levels as a consequence of the estrogen deficiency.119 Dual inhibitors of AROM (CYP19A1) and aldosterone synthase (CYP11B2) could be useful as adjuvant therapy to reduce the risk of cardiovascular disease for patients undergoing AI treatment. The design utilized the common feature of heme iron coordination by pyridinylmethyl moiety and the hydrophobic core of 1,2,5,6-tetrahydropyrrolo[3,2,1-ij]quinolin-4-one, important for potent CYP11B2 inhibition.119 The compound 8-[(3-methylphenyl)(pyridin-4-yl)methyl]-1,2,5,6-tetrahydro-4H-pyrrolo[3,2,1-ij]quinolin-4-one (28) exhibited strong dual inhibition.119 The design of other potent dual inhibitors included combining the pyrroloquinoline core of antimitotic phenylpyrroloquinolinone derivatives with elements of potent nonsteroidal AIs’ imidazole and triazole rings (29–32).120 A high-throughput approach identified several compounds with imidazolyl quinolone skeleton (33) as potent AIs.121
c. Flavone Analogs
Imidazole derivatives of flavonoids were shown to have AROM inhibitory properties.118 More recently, various modifications of this basic design have been made to synthesize potent AIs, including isoflavanone derivatives with functional groups such as methoxy, phenoxy, chloride, pyridyl, etc. (34–36).122 In addition, flavans from the roots of Desmos cochinchinensis exhibited potent AROM inhibitory activity at nanomolar levels, the most potent of them being 5-methoxy-8-formyl-4,7-dihydroxyflavan (37).123
d. Coumarins
Position 4-substituted coumarin derivatives were found to potently inhibit AROM.118 One of the most potent compounds in this series was 4-benzyl-3-(4′-chlorophenyl)-7- methoxycoumarin, a competitive inhibitor of AROM with a Ki of 84 nM.118 Some of the imidazolyl derivatives of 4,7-substituted coumarins (38, 39) were shown to be potent inhibitors of AROM with IC50 values in the nanomolar range, higher selectivity over CYP17A1, and favorable interactions at the active site.124
e. STS/AROM Dual Inhibitors
Dual AROM/STS could prove more effective than the AIs, since both enzymes were reported to be simultaneously present in E2-dependent breast tumors.125 Sulfamate moieties containing EMATE and 667-COUMATE are known potent inhibitors of STS.118 The first of these compounds generated by introducing the AI pharmacophore into a known biphenyl STS inhibitor template (i.e., sulfamate derivatives of LTZ and ANZ templates) was shown to have nanomolar level half-maximum effect.126–129 Sulfamate derivatives of other AI templates, such as vorozole, their halide substitutions, and enantiomorphic compounds were synthesized (40–45).130 However, the reported inhibition and antiproliferation potencies of these compounds measured in JEG3 cells with low AROM expression levels are not comparable to the others in the absence of standardized controls.
f. Tamoxifen Metabolites
Metabolites of tamoxifen, a well-known selective estrogen receptor modulator (SERM), were shown to inhibit AROM.131,132 Such compounds (46, 47) could serve dual purposes as a SERM agent and an AI. Among the metabolites tested, norendoxifen was found to be the most potent and more AROM selective over other liver CYPs and claimed to have favorable interactions at the active site.131 Various isomers of norendoxifen were also prepared and tested (48–51).132
g. Others (Sulfonamide, Indole–Imidazole, Aryl Halide, Casimiroin, Xanthone Devrivatives)
A series of 1,4-disub-stituted-1,2,3-triazoles containing sulfonamide moiety (52) were synthesized and shown to have anti-AROM inhibitory properties.133 A meta analog of triazole-benzene-sulfonamide bearing 6,7-dimethoxy substituents on the isoquinoline ring was most potent and fit in the AROM active site.133 Aryl halide derivatives of a centrally flexible, five-component 1,2,3-triazole containing moiety (55) were prepared and evaluated as potential AIs.134 Among novel indole–imidazole derivatives, 2-(imidazol-1-ylmethyl)-1-[4-(trifluoromethyl)phenyl]indole (56) was shown to be a highly potent AI.135 Casimiroin derivatives (57–59) were shown to inhibit both AROM and quinone reductase 2. N-Methyl casimiroin analogue (57) exhibited potentency in the micromolar range.136 Some imidazolyl derivatives of xanthone scaffold (60, 61) were shown to be highly potent AIs.137 Additionally, high-throughput in silico docking approach was used to identify existing compounds as potent AIs.138,139 These include compounds with sulfonamide groups (53, 54), and morpholino ethanone derivatives (62) and imidazole groups (63).138
Several other compounds identified by in silico docking were reported to have antiproliferative properties in cancer cells.140,141 Among them, compounds 3-oxo-16,17-secoandros-ta-1,4-diene-16,17a-dinitrile (64)140 and 4-[2-(3-chlorophenyl)-1-(1H-1,2,4-triazol-1-yl)ethenyl]benzonitrile (65)141 (Figure 6b) containing AI scaffolds exhibited antiproliferative potency in the micromolar range against MCF7 cells. However, these compounds were not shown to inhibit AROM and hence cannot be classified as new AIs.
6. NEW INSIGHTS FROM MOLECULAR DOCKING OF KNOWN AIS
Several groups utilized the 3D structure of AROM to predict the binding modes and/or deduce quantitative structure–activity relationships of known inhibitors/ligands by computational approaches. However, no inhibitory and/or antiproliferative properties for these compounds were experimentarily measured. Therefore, these compounds were excluded from Table 3 and Figure 6. The compounds attempted include flavonoids,142 isoflavanone derivatives,143 imidazolylmethyl-substituted flavone,144 flavanone and isoflavones,143 benzofuran scaffold,145 steroidal backbones,146 ASD derivatives,147 non-steroidal AIs based on an AROM structure-guided pharmacophore model,148 and tetracyclic triterpenoids,149 to name a few. There were also efforts to develop better strategy or algorithm for in silico screening and discovering novel AIs.16,144,150
7. EFFECTS OF MEMBRANE INTEGRATION AND DYNAMICS ON RATIONAL DESIGN
7.i. Membrane Integration
The 3D topology allows the enzyme to integrate into the endoplasmic reticulum and/or golgi membrane such that the substrates could utilize the lipid bilayer to gain access to the active site.6 Lipophilicity of the AI scaffold is critical to integration and translocation through the lipid bilayer, as the inhibitors have to translocate through the endoplasmic reticulum membrane to gain access to the substrate-binding site from the access channel that opens toward the lumen of the endoplasmic reticulum. Membrane integration topology, the architecture of the access channel, and its flexibility are, therefore, useful information for the design of new AIs. The amino-terminal helix ANT of AROM spans the membrane and positions a putative glycosylation site at Asn12 in the luminal space,6,47,86 as shown in Figure 7. The hydrophobic helix A′ (residues 57–68) and parts of helix A (residues 69–80) are embedded into the membrane, thereby positioning several arginine (Arg64, Arg79, and Arg86) and tryptophan (Trp67 and Trp88, as well as Trp239 from the F–G loop) residues at the lipid–protein interface, similar to other lipid integrating proteins.151 In this model, the entrance to the active site access channel and the amphipathic F–G loop including the G′ helix sits on the membrane surface (Figure 7), while the hydrophobic loop of residues 462–471 between β7 and β8 touches the interior of the membrane.47 The side group of compound 5 nearly extends to the “gatekeeper” residues Arg192 and Glu483 at the channel entrance at the membrane–protein interface. The juxtaposition of these catalytically important residues6,14 at the end of a hydrophobic channel and lipid interface suggests that membrane integration has an important role not only in steroid passage and proton relay critical to aromatization6 but in substrate and inhibitor selectivity as well. The terminal C24 methyl of the 2-pentynyloxy derivative 5 has a favorable surrounding of large hydrophobic groups Phe221 and His480.46 However, a hydroxyl group at this position in 9 promotes polar interactions with the polar moieties at the end of the channel. Membrane integration of AROM determines the shape and size of the access channel and should be considered in inhibitor design.
Figure 7.
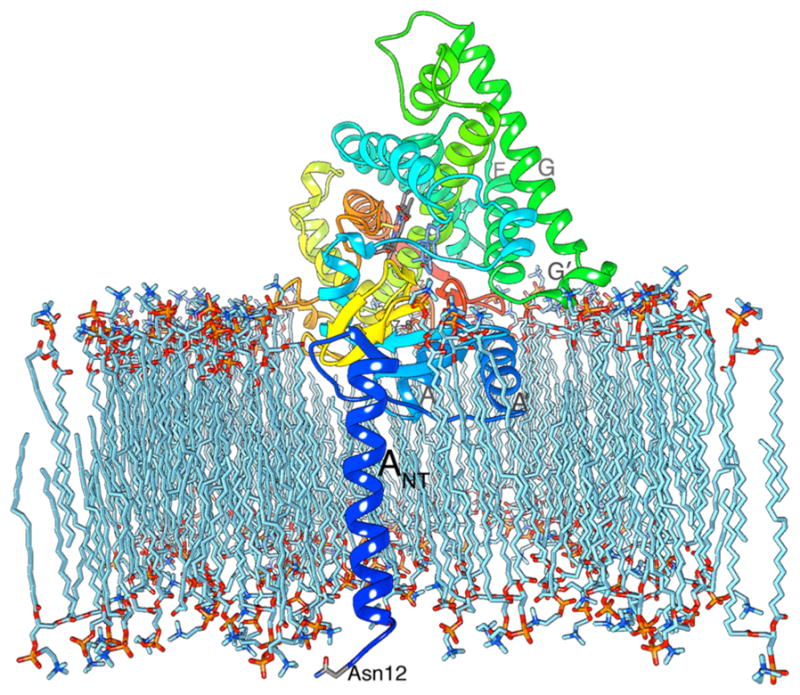
Influence of lipid bilayer. In this membrane integration model, the opening to the active-site access channel rests on the lipid bilayer surface, allowing the steroids to enter the AROM active site directly from within the bilayer. The backbone ribbon is drawn in rainbow color: the N terminus is blue, and the C terminus is red. The model suggests lipid integration/association of the N-terminal helices ANT, A′, and A, the F–G loop that includes helix G′, and loop 7–8 between β7 and β8 near the C terminus. Asn12 is shown as facing the lumen of the endoplasmic reticulum. Atom color scheme is the following: oxygen in red; nitrogen blue; phosphorus orange; ASD in cornflower blue; heme and protein side chains in gray; lipid in light blue.
7.ii. Dynamics
Normal mode analysis (NMA) of the X-ray structure revealed the intrinsic fluctuations of AROM, the internal modes in membrane-free and membrane-integrated monomers.86 The results confirmed the rigid-core structure of AROM centered at the active site is maintained in spite of the changes in steroid binding interactions. The NMA results showed that the N-terminal helix is the most mobile and flexible structural element identified, in agreement with the X-ray observation. The F–G loop is the next most flexible region of the AROM structure that is not significantly influenced by membrane integration. The F–G loop flexibility is one of the common features of P450s.152 Evidence of the flexible loop undergoing an open/close motion, perhaps to allow steroids to enter into or leave from the active site through the access channel,6 was observed.86 Furthermore, the NMA of a monomer revealed that the access channel could serve as a hinge for intramolecular bending and an interface for twisting motions. These motions, together with the intrinsic flexibility of the access channel, are likely to contribute to channel “breathing”, opening and closing of the channel mouth and the cavity, perceived necessary for entry and exit of steroids to and from the active site.6,86 The hinge bending and twisting motions at the access channel are also present in the lipid-embedded AROM but at a higher frequency. Results from other dynamical simulations also have demonstrated similar flexibility of the AROM structure, especially in the access channel region, even on membrane insertion.153,154 Despite the observed rigidity of the active site core, the likelihood of some ligand-induced flexibility cannot be ruled out. In addition, inhibition kinetics data47 as well as computational analysis117 suggest possible secondary binding sites for nonsteroidal ligands. Membrane integration, motion, and flexibility of AROM thus pose additional challenges to designing of AIs by rational means.
8. MISSING INFORMATION AND NEW CONCEPTS IN FUTURE DESIGN
8.i. Structural Data on Nonsteroidal Inhibitor–AROM Complexes
As discussed earlier, no experimental structure data on a nonsteroidal inhibitor complex of AROM are available yet. This is a major handicap to the development of novel nonsteroidal AIs with superior clinical properties. Computational methodologies such as docking of LTZ and ANZ into the AROM active site cavity, molecular dynamics simulations, and energy minimization are inadequate for an unequivocal elucidation of the true binding mode. Our data suggest that a major conformational rearrangement of the androgen-binding cleft is necessary to accommodate these triazole ligands at the heme distal site.116 Recent experimental data suggest17 that the binding mode of ANZ is different from EXM in that the triazole does not use Asp309, the critically important active site residue, which is protonated at physiological pH and makes a hydrogen bond to the 3-keto oxygen of ASD and EXM in their binding modes.6,46 Although the conventional wisdom suggests that the triazole ring nitrogen would coordinate with heme iron as the sixth ligand from the distal site, additional experimental structural data are necessary to settle this question once and for all.
8.ii. AROM Phosphorylation
Phosphorylation of the proximal cavity residue Tyr361 shown in Figure 8 has recently been reported to increase AROM activity in breast cancer cells.155 Short exposure of estrogen-dependent MCF-7 and ZR75 breast cancer epithelial cells to E2 induces an increase of AROM enzymatic activity. E2-induced enhancement of AROM activity does not correlate with increase in AROM mRNA and protein content. Site-directed mutagenesis experiments reveal that phosphorylation of Tyr361 is crucial in the up-regulation of enzymatic activity. E2 treatment enhances Tyr361 phosphorylation and activity of AROM by activating c-Src kinase or blocking the tyrosine phosphatase PTP1B. In the absence of E2, PTP1B reduces AROM activity by dephosphorylation of Tyr361.156 Rapid E2 synthesis by a phosphorylated AROM and nongenomic autocrine E2 loop signaling via the plasma membrane-associated ER has long been proposed to be a mechanism by which estrogen performs neuroprotective, neurogenerative, and neurotransmitter roles in the brain and CNS.157 This positive feedback loop involving phosphorylation of AROM, elevated enzyme activity, and rapid E2 synthesis was flrst reported in the brains of songbirds and other mammals.158–160
Figure 8.

Strategic location of Y361 in the proximal cavity. The redox partner cytochrome P450 reductase is presumed to bind at this interface, and electrons are transferred from FMN to heme as illustrated by the dashed line. Y361 side chain sits in the path of electron transfer. Atom color code is the same as in other figures.
The heme proximal cavity plays key roles in enzyme function during the transfer of electrons to heme.161 We have shown that Tyr361 lies in the path of transfer of electrons from CPR to heme (Figure 8) and Y361F mutation drastically reduces enzyme activity.14 X-ray data show unexplained residual electron density inside the proximal cavity,13 which could serve as a binding scaffold for small molecules. If AROM phosphorylation plays any role in AI resistance, the future AI design criteria would have to take the phosphorylation site(s) into consideration. The mechanism of modulation of the enzyme activity by phosphorylation–dephosphrylation could influence the inhibitory properties of AIs depending on their binding sites. Furthermore, targeting the proximal cavity at the AROM–CPR coupling interface is a concept that could lead to the discovery of a novel class of inhibitors of estrogen biosynthesis. Additionally, the putative secondary sites47,117 could also be targeted for inhibition of enzyme activity.
9. CONCLUDING REMARKS
Recent progress on structure–function relationships of AROM has revitalized the next generation AI discovery research. Since 2009 when the AROM structure came to light, roughly several hundred new compounds designed from structure-based modifications to existing inhibitors and belonging to 30 classes have been reported in 46 major publications. Overall, during this period roughly 1000 pieces of documents on design and discovery of novel AIs have appeared in the public domain. The evidence is clear that through rational design, three of the C6-substituted 2-alkynyloxy compounds are more potent than EXM in vitro.46 However, there are no in vivo data yet to suggest superiority to EXM. Structural data show that these novel AROM inhibitors bind in competition with the substrate at the small, tight, androgen-specific active site of AROM and also fill the access channel as they were designed to.6,13,46 They could, therefore, be more specific to AROM over other receptors. While the structure has resolved many outstanding issues, it has also raised new questions. The mechanism of conversion of androgens to estrogens by AROM is very complex and much remains to be achieved in this regard. Additionally, more comprehensive understanding of the aromatization process requires analysis of the structure of electron-transfer complex between AROM and CPR. This has proven to be difficult for any P450. Although steroidal inhibitor complexes of AROM have been crystallized, no experimental structural data on LTZ and ANZ complexes are available yet. This creates a void in our understanding of the molecular basis of inhibition and a hindrance to the discovery of rationally designed nonsteroidal compounds with superior efficacy. Paradoxically, crystallization of these complexes with AROM has been unsuccessful under the conditions that yielded the steroidal complex crystals. Conformational reorganization of the active site cavity induced by the nonsteroidal agents and/or mixed modes of binding resulting structural changes are distinct possibilities. The missing structural information is crucial for designing superior nonsteroidal AIs. Modulation of the enzyme activity by phosphorylation is another emerging area that could reshape the future of AI discovery research.
Acknowledgments
This work is supported in part by Grant GM086893 from the National Institutes of Health and Carol M. Baldwin Breast Cancer Research Fund of Central New York.
ABBREVIATIONS USED
- 3H2O
tritiated water
- AI
AROM inhibitor
- ANZ
anastrozole
- AROM
cytochrome P450 aromatase
- ASD
androstenedione
- CPR
cytochrome P450 reductase
- E1
estrone
- E2
17β-estradiol
- EGFR
epithelial growth factor receptor
- ER
estrogen receptor
- EXM
exemestane
- FCA
fluorescent CYP19 inhibition assay
- HER2/neu
human EGFR2
- LTZ
letrozole
- MCF-7a
MCF-7 cell line stably expressing AROM
- MD
molecular docking
- mER
plasma membrane associated estrogen receptor
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
- NCI
National Cancer Institute
- NNC
National Nanjing Center for Drug Screening
- NMA
normal-mode analysis
- PR
progesterone receptor
- PMTW
pacental microsomes, tritiated water assay
- TST
testosterone
Biographies
Debashis Ghosh is a Professor of Pharmacology at the State University of New York (SUNY) Upstate Medical University, College of Medicine, in Syracuse, NY. He previously held the positions of Principal Scientist at the Hauptman-Woodward Institute and Associate Professor of Oncology at the Roswell Park Cancer Institute, in Buffalo, NY. He began working on steroidogenic enzymes nearly 25 years ago. The major objectives of his research have been elucidation of the molecular mechanism of estrogen biosynthesis in human by the three-enzyme system, cytochrome P450 aromatase, 17β-hydroxysteroid dehydrogenases, and steroid sulfatase, as well as rational design, synthesis, and evaluation of inhibitors of these enzymes for the treatment and prevention of estrogen-dependent breast cancer and other endocrine-related diseases.
Jessica Lo studied biophysics at the State University of New York (SUNY) at Buffalo. She recently obtained a Ph.D. from SUNY Upstate Medical University in in the laboratory of Dr. Debashis Ghosh (Department of Pharmacology, SUNY Upstate Medical University in Syracuse, NY). Her doctoral work deals with the investigation of the functional properties of aromatase and determination of the potencies of novel steroidal aromatase inhibitors. She is currently a postdoctoral researcher in the laboratory of Dr. Ghosh.
Chinaza Egbuta studied pharmacology at the State University of New York (SUNY) at Buffalo and obtained a combined B.S. and M.S. in 2009. She is a Ph.D. candidate in the laboratory of Dr. Debashis Ghosh (Department of Pharmacology, SUNY Upstate Medical University in Syracuse, NY) where she is studying the interactions that modulate estrogen biosynthesis in human cytochrome P450 aromatase.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A. History of Aromatase: Saga of an Important Biological Mediator and Therapeutic Target. Endocr Rev. 2009;30:343–375. doi: 10.1210/er.2008-0016. [DOI] [PubMed] [Google Scholar]
- 2.Simpson E, Jones M, Misso M, Hewitt K, Hill R, Maffei L, Carani C, Boon WC. Estrogen, a Fundamental Player in Energy Homeostasis. J Steroid Biochem Mol Biol. 2005;95:3–8. doi: 10.1016/j.jsbmb.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Ryan KJ. Biological Aromatization of Steroids. J Biol Chem. 1959;234:268–272. [PubMed] [Google Scholar]
- 4.Smith IE, Dowsett M. Aromatase Inhibitors in Breast Cancer. N Engl J Med. 2003;348:2431–2442. doi: 10.1056/NEJMra023246. [DOI] [PubMed] [Google Scholar]
- 5.Buzdar AU, Robertson JF, Eiermann W, Nabholtz JM. An Overview of the Pharmacology and Pharmacokinetics of the Newer Generation Aromatase Inhibitors Anastrozole, Letrozole, and Exemestane. Cancer. 2002;95:2006–2016. doi: 10.1002/cncr.10908. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh D, Griswold J, Erman M, Pangborn W. Structural Basis for Androgen Specificity and Oestrogen Synthesis in Human Aromatase. Nature. 2009;457:219–223. doi: 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szklarz GD, Halpert JR. Use of Homology Modeling in Conjunction with Site-Directed Mutagenesis for Analysis of Structure-Function Relationships of Mammalian Cytochromes P450. Life Sci. 1997;61:2507–2520. doi: 10.1016/s0024-3205(97)00717-0. [DOI] [PubMed] [Google Scholar]
- 8.Laughton CA, Zvelbil MJ, Neidle S. A Detailed Molecular Model for Human Aromatase. J Steroid Biochem Mol Biol. 1993;44:399–407. doi: 10.1016/0960-0760(93)90243-p. [DOI] [PubMed] [Google Scholar]
- 9.Favia AD, Cavalli A, Masetti M, Carotti A, Recanatini M. Three-Dimensional Model of the Human Aromatase Enzyme and Density Functional Parameterization of the Iron-Containing Proto-porphyrin IX for a Molecular Dynamics Study of Heme-Cysteinato Cytochromes. Proteins: Struct, Funct Genet. 2006;62:1074–1087. doi: 10.1002/prot.20829. [DOI] [PubMed] [Google Scholar]
- 10.Karkola S, Holtje HD, Wahala K. A Three-Dimensional Model of CYP19 Aromatase for Structure-Based Drug Design. J Steroid Biochem Mol Biol. 2007;105:63–70. doi: 10.1016/j.jsbmb.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Graham-Lorence S, Amarneh B, White RE, Peterson JA, Simpson ER. A Three-Dimensional Model of Aromatase Cytochrome P450. Protein Sci. 1995;4:1065–1080. doi: 10.1002/pro.5560040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lala P, Higashiyama T, Erman M, Griswold J, Wagner T, Osawa Y, Ghosh D. Suppression of Human Cytochrome P450 Aromatase Activity by Monoclonal and Recombinant Antibody Fragments and Identification of a Stable Antigenic Complex. J Steroid Biochem Mol Biol. 2004;88:235–245. doi: 10.1016/j.jsbmb.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh D, Griswold J, Erman M, Pangborn W. X-Ray Structure of Human Aromatase Reveals an Androgen-Specific Active Site. J Steroid Biochem Mol Biol. 2010;118:197–202. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo J, Di Nardo G, Griswold J, Egbuta C, Jiang W, Gilardi G, Ghosh D. Structural Basis for the Functional Roles of Critical Residues in Human Cytochrome P450 Aromatase. Biochemistry. 2013;52:5821–5829. doi: 10.1021/bi400669h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterman MR. Anticancer Drug Target Pictured. Nature. 2009;457:159–160. doi: 10.1038/457159a. [DOI] [PubMed] [Google Scholar]
- 16.Favia AD, Nicolotti O, Stefanachi A, Leonetti F, Carotti A. Computational Methods for the Design of Potent Aromatase Inhibitors. Expert Opin Drug Discovery. 2013;8:395–409. doi: 10.1517/17460441.2013.768983. [DOI] [PubMed] [Google Scholar]
- 17.Di Nardo G, Breitner M, Bandino A, Ghosh D, Jennings GK, Hackett JC, Gilardi G. Evidence for an Elevated Aspartate pKa in the Active Site of Human Aromatase. J Biol Chem. 2015;290:1186–1196. doi: 10.1074/jbc.M114.595108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sen K, Hackett JC. Coupled Electron Transfer and Proton Hopping in the Final Step of CYP19-Catalyzed Androgen Aromatization. Biochemistry. 2012;51:3039–3049. doi: 10.1021/bi300017p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lipton A, Santen RJ. Proceedings: Medical Adrenalectomy Using Aminoglutethimide and Dexamethasone in Advanced Breast Cancer. Cancer. 1974;33:503–512. doi: 10.1002/1097-0142(197402)33:2<503::aid-cncr2820330227>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 20.di Salle E, Ornati G, Giudici D, Lassus M, Evans TR, Coombes RC. Exemestane (FCE 24304), a New Steroidal Aromatase Inhibitor. J Steroid Biochem Mol Biol. 1992;43:137–143. doi: 10.1016/0960-0760(92)90198-r. [DOI] [PubMed] [Google Scholar]
- 21.Dexter RN, Fishman LM, Ney RL, Liddle GW. Inhibition of Adrenal Corticosteroid Synthesis by Aminoglutethimide: Studies of the Mechanism of Action. J Clin Endocrinol Metab. 1967;27:473–480. doi: 10.1210/jcem-27-4-473. [DOI] [PubMed] [Google Scholar]
- 22.Pittman JA, Brown RW. Antithyroid and Antiadrenocortical Activity of Aminoglutethimide. J Clin Endocrinol Metab. 1966;26:1014–1016. doi: 10.1210/jcem-26-9-1014. [DOI] [PubMed] [Google Scholar]
- 23.Häusler A, Schenkel L, Krähenbühl C, Monnet G, Bhatnagar A. An in Vitro Method to Determine the Selective Inhibition of Estrogen Biosynthesis by Aromatase Inhibitors. J Steroid Biochem. 1989;33:125–131. doi: 10.1016/0022-4731(89)90367-1. [DOI] [PubMed] [Google Scholar]
- 24.Dowsett M, Santner SJ, Santen RJ, Jeffcoate SL, Smith IE. Effective Inhibition by Low Dose Aminoglutethimide of Peripheral Aromatization in Postmenopausal Breast Cancer Patients. Br J Cancer. 1985;52:31–35. doi: 10.1038/bjc.1985.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santen R, Santner S, Davis B, Veldhuis J, Samojlik E, Ruby E. Aminoglutethimide Inhibits Extraglandular Estrogen Production in Postmenopausal Women with Breast Carcinoma*. J Clin Endocrinol Metab. 1978;47:1257–1265. doi: 10.1210/jcem-47-6-1257. [DOI] [PubMed] [Google Scholar]
- 26.Burnett RD, Kirk DN. Some Observations on the Preparation of 2-Hydroxy-Steroid 4-En-3 Ones. J Chem Soc Perkin Trans 1. 1973;17:1830–1836. [PubMed] [Google Scholar]
- 27.Coombes RC, Goss P, Dowsett M, Gazet JC, Brodie A. 4-Hydroxyandrostenedione in Treatment of Postmenopausal Patients with Advanced Breast Cancer. Lancet. 1984;324:1237–1239. doi: 10.1016/s0140-6736(84)92795-8. [DOI] [PubMed] [Google Scholar]
- 28.Coombes RC, Goss PE, Dowsett M, Hutchinson G, Cunningham D, Jarman M, Brodie AM. 4-Hydroxyandrostene-dione Treatment for Postmenopausal Patients with Advanced Breast Cancer. Steroids. 1987;50:245–252. doi: 10.1016/0039-128x(83)90075-2. [DOI] [PubMed] [Google Scholar]
- 29.Dowsett M, Cunningham DC, Stein RC, Evans S, Dehennin L, Hedley A, Coombes RC. Dose-Related Endocrine Effects and Pharmacokinetics of Oral and Intramuscular 4-Hydrox-yandrostenedione in Postmenopausal Breast Cancer Patients. Cancer Res. 1989;49:1306–1312. [PubMed] [Google Scholar]
- 30.Stein RC, Dowsett M, Hedley A, Davenport J, Gazet JC, Ford HT, Coombes RC. Treatment of Advanced Breast Cancer in Postmenopausal Women with 4-Hydroxyandrostenedione. Cancer Chemother Pharmacol. 1990;26:75–78. doi: 10.1007/BF02940300. [DOI] [PubMed] [Google Scholar]
- 31.Stein RC, Dowsett M, Hedley A, Gazet JC, Ford HT, Coombes RC. The Clinical and Endocrine Effects of 4-Hydroxyandrostenedione Alone and in Combination with Goserelin in Premenopausal Women with Advanced Breast Cancer. Br J Cancer. 1990;62:679–683. doi: 10.1038/bjc.1990.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brodie AM, Marsh DA, Wu JT, Brodie HJ. Aromatase Inhibitors and Their Use in Controlling Oestrogen-Dependent Processes. J Steroid Biochem. 1979;11:107–112. doi: 10.1016/0022-4731(79)90283-8. [DOI] [PubMed] [Google Scholar]
- 33.Di Salle E, Briatico G, Giudici D, Ornati G, Zaccheo T, Buzzetti F, Nesi M, Panzeri A. Novel Aromatase and 5 Alpha-Reductase Inhibitors. J Steroid Biochem Mol Biol. 1994;49:289–294. doi: 10.1016/0960-0760(94)90270-4. [DOI] [PubMed] [Google Scholar]
- 34.Santen R, Langecker P, Santner S, Sikka S, Rajfer J, Swerdloff R. Potency and Specificity of CGS-16949A as an Aromatase Inhibitor. Endocr Res. 1990;16:77–91. doi: 10.1080/07435809009035921. [DOI] [PubMed] [Google Scholar]
- 35.Browne LJ, Gude C, Rodriguez H, Steele RE, Bhatnager A. Fadrozole Hydrochloride: A Potent, Selective, Nonsteroidal Inhibitor of Aromatase for the Treatment of Estrogen-Dependent Disease. J Med Chem. 1991;34:725–736. doi: 10.1021/jm00106a038. [DOI] [PubMed] [Google Scholar]
- 36.Bossche HV, Moereels H, Koymans LM. Aromatase Inhibitors—Mechanisms for Non-Steroidal Inhibitors. Breast Cancer Res Treat. 1994;30:43–55. doi: 10.1007/BF00682740. [DOI] [PubMed] [Google Scholar]
- 37.Santen RJ, Demers LM, Adlercreutz H, Harvey H, Santner S, Sanders S, Lipton A. Inhibition of Aromatase with CGS 16949A in Postmenopausal Women. J Clin Endocrinol Metab. 1989;68:99–106. doi: 10.1210/jcem-68-1-99. [DOI] [PubMed] [Google Scholar]
- 38.Lipton A, Harvey HA, Demers LM, Hanagan JR, Mulagha MT, Kochak GM, Fitzsimmons S, Sanders SI, Santen RJ. A Phase I Trial of CGS 16949A. A New Aromatase Inhibitor. Cancer. 1990;65:1279–1285. doi: 10.1002/1097-0142(19900315)65:6<1279::aid-cncr2820650604>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 39.Dowsett M, Stein RC, Mehta A, Coombes RC. Potency and Selectivity of the Non-Steroidal Aromatase Inhibitor CGS 16949A in Postmenopausal Breast Cancer Patients. Clin Endocrinol. 1990;32:623–634. doi: 10.1111/j.1365-2265.1990.tb00906.x. [DOI] [PubMed] [Google Scholar]
- 40.Stein RC, Dowsett M, Davenport J, Hedley A, Ford HT, Gazet JC, Coombes RC. Preliminary Study of the Treatment of Advanced Breast Cancer in Postmenopausal Women with the Aromatase Inhibitor CGS 16949A. Cancer Res. 1990;50:1381–1384. [PubMed] [Google Scholar]
- 41.Santen RJ, Demers LM, Lynch J, Harvey H, Lipton A, Mulagha M, Hanagan J, Garber JE, Henderson IC, Navari RM, et al. Specificity of Low Dose Fadrozole Hydrochloride (CGS 16949A) as an Aromatase Inhibitor. J Clin Endocrinol Metab. 1991;73:99–106. doi: 10.1210/jcem-73-1-99. [DOI] [PubMed] [Google Scholar]
- 42.Raats JI, Falkson G, Falkson HC. A Study of Fadrozole, a New Aromatase Inhibitor, in Postmenopausal Women with Advanced Metastatic Breast Cancer. J Clin Oncol. 1992;10:111–116. doi: 10.1200/JCO.1992.10.1.111. [DOI] [PubMed] [Google Scholar]
- 43.Miller WR. Biology of Aromatase Inhibitors: Pharmacology/Endocrinology within the Breast. Endocr -Relat Cancer. 1999;6:187–195. doi: 10.1677/erc.0.0060187. [DOI] [PubMed] [Google Scholar]
- 44.Brodie AM, Njar VC. Aromatase Inhibitors in Advanced Breast Cancer: Mechanism of Action and Clinical Implications. J Steroid Biochem Mol Biol. 1998;66:1–10. doi: 10.1016/s0960-0760(98)00022-3. [DOI] [PubMed] [Google Scholar]
- 45.Gibson LJ, Dawson C, Lawrence DH, Bliss JM. Aromatase Inhibitors for Treatment of Advanced Breast Cancer in Postmenopausal Women. Cochrane Database Syst Rev. 2007:CD003370. doi: 10.1002/14651858.CD003370.pub2. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh D, Lo J, Morton D, Valette D, Xi J, Griswold J, Hubbell S, Egbuta C, Jiang W, An J, Davies HM. Novel Aromatase Inhibitors by Structure-Guided Design. J Med Chem. 2012;55:8464–8476. doi: 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egbuta C, Lo J, Ghosh D. Mechanism of Inhibition of Estrogen Biosynthesis by Azole Fungicides. Endocrinology. 2014;155:4622–4628. doi: 10.1210/en.2014-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goss PE, Ingle JN, Pritchard KI, Ellis MJ, Sledge GW, Budd GT, Rabaglio M, Ansari RH, Johnson DB, Tozer R, D’Souza DP, Chalchal H, Spadafora S, Stearns V, Perez EA, Liedke PE, Lang I, Elliott C, Gelmon KA, Chapman JA, Shepherd LE. Exemestane Versus Anastrozole in Postmenopausal Women with Early Breast Cancer: NCIC CTG MA.27–a Randomized Controlled Phase Iii Trial. J Clin Oncol. 2013;31:1398–1404. doi: 10.1200/JCO.2012.44.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murray J, Young OE, Renshaw L, White S, Williams L, Evans DB, Thomas JS, Dowsett M, Dixon JM. A Randomised Study of the Effects of Letrozole and Anastrozole on Oestrogen Receptor Positive Breast Cancers in Postmenopausal Women. Breast Cancer Res Treat. 2009;114:495–501. doi: 10.1007/s10549-008-0027-0. [DOI] [PubMed] [Google Scholar]
- 50.Zilembo N, Noberasco C, Bajetta E, Martinetti A, Mariani L, Orefice S, Buzzoni R, Di Bartolomeo M, Di Leo A, Laffranchi A, et al. Endocrinological and Clinical Evaluation of Exemestane, a New Steroidal Aromatase Inhibitor. Br J Cancer. 1995;72:1007–1012. doi: 10.1038/bjc.1995.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johannessen DC, Engan T, Di Salle E, Zurlo MG, Paolini J, Ornati G, Piscitelli G, Kvinnsland S, Lonning PE. Endocrine and Clinical Effects of Exemestane (PNU155971), a Novel Steroidal Aromatase Inhibitor, in Postmenopausal Breast Cancer Patients: A Phase I Study. Clin Cancer Res. 1997;3:1101–1108. [PubMed] [Google Scholar]
- 52.Lonning PE, Paridaens R, Thurlimann B, Piscitelli G, di Salle E. Exemestane Experience in Breast Cancer Treatment. J Steroid Biochem Mol Biol. 1997;61:151–155. [PubMed] [Google Scholar]
- 53.Thurlimann B, Paridaens R, Serin D, Bonneterre J, Roche H, Murray R, di Salle E, Lanzalone S, Zurlo MG, Piscitelli G. Third-Line Hormonal Treatment with Exemestane in Postmenopausal Patients with Advanced Breast Cancer Progressing on Amino-glutethimide: A Phase II Multicentre Multinational Study. Exemestane Study Group. Eur J Cancer. 1997;33:1767–1773. doi: 10.1016/s0959-8049(97)00283-9. [DOI] [PubMed] [Google Scholar]
- 54.Geisler J, King N, Anker G, Ornati G, Di Salle E, Lonning PE, Dowsett M. In Vivo Inhibition of Aromatization by Exemestane, a Novel Irreversible Aromatase Inhibitor, in Postmenopausal Breast Cancer Patients. Clin Cancer Res. 1998;4:2089–2093. [PubMed] [Google Scholar]
- 55.Lonning PE. Pharmacological Profiles of Exemestane and Formestane, Steroidal Aromatase Inhibitors Used for Treatment of Postmenopausal Breast Cancer. Breast Cancer Res Treat. 1998;49(Suppl 1):S45–52. doi: 10.1023/a:1006048722559. discussion S73–47. [DOI] [PubMed] [Google Scholar]
- 56.Jones S, Vogel C, Arkhipov A, Fehrenbacher L, Eisenberg P, Cooper B, Honig S, Polli A, Whaley F, di Salle E, Tiffany J, Consonni A, Miller L. Multicenter, Phase II Trial of Exemestane as Third-Line Hormonal Therapy of Postmenopausal Women with Metastatic Breast Cancer. Aromasin Study Group. J Clin Oncol. 1999;17:3418–3425. doi: 10.1200/JCO.1999.17.11.3418. [DOI] [PubMed] [Google Scholar]
- 57.Kaufmann M, Bajetta E, Dirix LY, Fein LE, Jones SE, Cervek J, Fowst C, Polli A, Di Salle E, Massimini G, Piscitelli G. Exemestane Improves Survival Compared with Megoestrol Acetate in Postmenopausal Patients with Advanced Breast Cancer Who Have Failed on Tamoxifen. Results of a Double-Blind Randomised Phase III Trial. Eur J Cancer. 2000;36(Suppl 4):S86–87. doi: 10.1016/s0959-8049(00)00240-9. [DOI] [PubMed] [Google Scholar]
- 58.Kaufmann M, Bajetta E, Dirix LY, Fein LE, Jones SE, Zilembo N, Dugardyn JL, Nasurdi C, Mennel RG, Cervek J, Fowst C, Polli A, di Salle E, Arkhipov A, Piscitelli G, Miller LL, Massimini G. Exemestane Improves Survival in Metastatic Breast Cancer: Results of a Phase III Randomized Study. Clin Breast Cancer. 2000;1(Suppl 1):S15–18. doi: 10.3816/cbc.2000.s.003. [DOI] [PubMed] [Google Scholar]
- 59.Kaufmann M, Bajetta E, Dirix LY, Fein LE, Jones SE, Zilembo N, Dugardyn JL, Nasurdi C, Mennel RG, Cervek J, Fowst C, Polli A, di Salle E, Arkhipov A, Piscitelli G, Miller LL, Massimini G. Exemestane Is Superior to Megestrol Acetate after Tamoxifen Failure in Postmenopausal Women with Advanced Breast Cancer: Results of a Phase Iii Randomized Double-Blind Trial. The Exemestane Study Group. J Clin Oncol. 2000;18:1399–1411. doi: 10.1200/JCO.2000.18.7.1399. [DOI] [PubMed] [Google Scholar]
- 60.Plourde PV, Dyroff M, Dukes M. Arimidex: A Potent and Selective Fourth-Generation Aromatase Inhibitor. Breast Cancer Res Treat. 1994;30:103–111. doi: 10.1007/BF00682745. [DOI] [PubMed] [Google Scholar]
- 61.Dukes M, Edwards PN, Large M, Smith IK, Boyle T. The Preclinical Pharmacology of “Arimidex” (Anastrozole; ZD1033)–a Potent, Selective Aromatase Inhibitor. J Steroid Biochem Mol Biol. 1996;58:439–445. doi: 10.1016/0960-0760(96)00064-7. [DOI] [PubMed] [Google Scholar]
- 62.Buzdar AU, Jonat W, Howell A, Jones SE, Blomqvist CP, Vogel CL, Eiermann W, Wolter JM, Steinberg M, Webster A, Lee D. Anastrozole Versus Megestrol Acetate in the Treatment of Postmenopausal Women with Advanced Breast Carcinoma: Results of a Survival Update Based on a Combined Analysis of Data from Two Mature Phase III Trials. Arimidex Study Group. Cancer. 1998;83:1142–1152. [PubMed] [Google Scholar]
- 63.Yates RA, Dowsett M, Fisher GV, Selen A, Wyld PJ. Arimidex (ZD1033): A Selective, Potent Inhibitor of Aromatase in Postmenopausal Female Volunteers. Br J Cancer. 1996;73:543–548. doi: 10.1038/bjc.1996.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buzdar AU, Jones SE, Vogel CL, Wolter J, Plourde P, Webster A. A Phase III Trial Comparing Anastrozole (1 and 10 Milligrams), a Potent and Selective Aromatase Inhibitor, with Megestrol Acetate in Postmenopausal Women with Advanced Breast Carcinoma. Arimidex Study Group. Cancer. 1997;79:730–739. [PubMed] [Google Scholar]
- 65.Buzdar A, Douma J, Davidson N, Elledge R, Morgan M, Smith R, Porter L, Nabholtz J, Xiang X, Brady C. Phase III, Multicenter, Double-Blind, Randomized Study of Letrozole, an Aromatase Inhibitor, for Advanced Breast Cancer Versus Megestrol Acetate. J Clin Oncol. 2001;19:3357–3366. doi: 10.1200/JCO.2001.19.14.3357. [DOI] [PubMed] [Google Scholar]
- 66.Cohen MH, Johnson JR, Li N, Chen G, Pazdur R. Approval Summary: Letrozole in the Treatment of Postmenopausal Women with Advanced Breast Cancer. Clin Cancer Res. 2002;8:665–669. [PubMed] [Google Scholar]
- 67.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Cameron DA, Palmer MJ, Pater JL. Randomized Trial of Letrozole Following Tamoxifen as Extended Adjuvant Therapy in Receptor-Positive Breast Cancer: Updated Findings from NCIC CTG MA.17. J Natl Cancer Inst. 2005;97:1262–1271. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- 68.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, Livingston RB, Davidson NE, Norton L, Perez EA, Abrams JS, Therasse P, Palmer MJ, Pater JL. A Randomized Trial of Letrozole in Postmenopausal Women after Five Years of Tamoxifen Therapy for Early-Stage Breast Cancer. N Engl J Med. 2003;349:1793–1802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 69.Lipton A, Leitzel K, Ali SM, Demers L, Harvey HA, Chaudri-Ross HA, Evans D, Lang R, Hackl W, Hamer P, Carney W. Serum HER-2/Neu Conversion to Positive at the Time of Disease Progression in Patients with Breast Carcinoma on Hormone Therapy. Cancer. 2005;104:257–263. doi: 10.1002/cncr.21202. [DOI] [PubMed] [Google Scholar]
- 70.Meng S, Tripathy D, Shete S, Ashfaq R, Haley B, Perkins S, Beitsch P, Khan A, Euhus D, Osborne C, Frenkel E, Hoover S, Leitch M, Clifford E, Vitetta E, Morrison L, Herlyn D, Terstappen LW, Fleming T, Fehm T, Tucker T, Lane N, Wang J, Uhr J. HER-2 Gene Amplification Can Be Acquired as Breast Cancer Progresses. Proc Natl Acad Sci U S A. 2004;101:9393–9398. doi: 10.1073/pnas.0402993101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu JC, Mokbel K. Does C-Erbb2/HER2 Overexpression Predict Adjuvant Tamoxifen Failure in Patients with Early Breast Cancer? European journal of surgical oncology: the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology. 2001;27:335–337. doi: 10.1053/ejso.2000.1078. [DOI] [PubMed] [Google Scholar]
- 72.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. Role of the Estrogen Receptor Coactivator AIB1 (Src-3) and HER-2/Neu in Tamoxifen Resistance in Breast Cancer. J Natl Cancer Inst. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 73.Liehr JG. Is Estradiol a Genotoxic Mutagenic Carcinogen? Endocr Rev. 2000;21:40–54. doi: 10.1210/edrv.21.1.0386. [DOI] [PubMed] [Google Scholar]
- 74.Breast International Group 1-98 Collaborative Group. Thurlimann B, Keshaviah A, Coates AS, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Rabaglio M, Smith I, Wardley A, Price KN, Goldhirsch A. Comparison of Letrozole and Tamoxifen in Postmenopausal Women with Early Breast Cancer. N Engl J Med. 2005;353:2747–2757. doi: 10.1056/NEJMoa052258. [DOI] [PubMed] [Google Scholar]
- 75.Goss PE. Pre-Clinical and Clinical Review of Vorozole, a New Third Generation Aromatase Inhibitor. Breast Cancer Res Treat. 1998;49(Suppl 1):S59–65. doi: 10.1023/a:1006052923468. discussion S73–57. [DOI] [PubMed] [Google Scholar]
- 76.Goss PE, Winer EP, Tannock IF, Schwartz LH. Randomized Phase III Trial Comparing the New Potent and Selective Third-Generation Aromatase Inhibitor Vorozole with Megestrol Acetate in Postmenopausal Advanced Breast Cancer Patients. North American Vorozole Study Group. J Clin Oncol. 1999;17:52–63. doi: 10.1200/JCO.1999.17.1.52. [DOI] [PubMed] [Google Scholar]
- 77.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 78.Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxbrow AK, Lewis CJ, Tennant MG, Modi S, Eggleston DS, Chenery RJ, Bridges AM. Crystal Structure of Human Cytochrome P450 2D6. J Biol Chem. 2006;281:7614–7622. doi: 10.1074/jbc.M511232200. [DOI] [PubMed] [Google Scholar]
- 79.Strushkevich N, Gilep AA, Shen L, Arrowsmith CH, Edwards AM, Usanov SA, Park HW. Structural Insights into Aldosterone Synthase Substrate Specificity and Targeted Inhibition. Mol Endocrinol. 2013;27:315–324. doi: 10.1210/me.2012-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeVore NM, Scott EE. Structures of Cytochrome P450 17A1 with Prostate Cancer Drugs Abiraterone and Tok-001. Nature. 2012;482:116–119. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao B, Lei L, Kagawa N, Sundaramoorthy M, Banerjee S, Nagy LD, Guengerich FP, Waterman MR. Three-Dimensional Structure of Steroid 21-Hydroxylase (Cytochrome P450 21A2) with Two Substrates Reveals Locations of Disease-Associated Variants. J Biol Chem. 2012;287:10613–10622. doi: 10.1074/jbc.M111.323501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Annalora AJ, Goodin DB, Hong WX, Zhang Q, Johnson EF, Stout CD. Crystal Structure of CYP24A1, a Mitochondrial Cytochrome P450 Involved in Vitamin D Metabolism. J Mol Biol. 2010;396:441–451. doi: 10.1016/j.jmb.2009.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strushkevich N, Usanov SA, Park HW. Structural Basis of Human CYP51 Inhibition by Antifungal Azoles. J Mol Biol. 2010;397:1067–1078. doi: 10.1016/j.jmb.2010.01.075. [DOI] [PubMed] [Google Scholar]
- 84.Bouchoucha N, Samara-Boustani D, Pandey AV, Bony-Trifunovic H, Hofer G, Aigrain Y, Polak M, Fluck CE. Characterization of a Novel CYP19A1 (Aromatase) R192H Mutation Causing Virilization of a 46,XX Newborn, Undervirilization of the 46,XY Brother, but No Virilization of the Mother During Pregnancies. Mol Cell Endocrinol. 2014;390:8–17. doi: 10.1016/j.mce.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 85.Marino R, Perez Garrido N, Costanzo M, Guercio G, Juanes M, Rocco C, Ramirez P, Warman DM, Ciaccio M, Pena G. Five New Cases of 46, XX Aromatase Deficiency: Clinical Follow-up from Birth to Puberty, a Novel Mutation, and a Founder Effect. J Clin Endocrinol Metab. 2015;100:E301–E307. doi: 10.1210/jc.2014-2967. [DOI] [PubMed] [Google Scholar]
- 86.Jiang W, Ghosh D. Motion and Flexibility in Human Cytochrome P450 Aromatase. PLoS One. 2012;7:e32565. doi: 10.1371/journal.pone.0032565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Akhtar M, Calder MR, Corina DL, Wright JN. Mechanistic Studies on C-19 Demethylation in Oestrogen Biosynthesis. Biochem J. 1982;201:569–580. doi: 10.1042/bj2010569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Akhtar M, Njar VC, Wright JN. Mechanistic Studies on Aromatase and Related C-C Bond Cleaving P-450 Enzymes. J Steroid Biochem Mol Biol. 1993;44:375–387. doi: 10.1016/0960-0760(93)90241-n. [DOI] [PubMed] [Google Scholar]
- 89.Hackett JC, Brueggemeier RW, Hadad CM. The Final Catalytic Step of Cytochrome P450 Aromatase: A Density Functional Theory Study. J Am Chem Soc. 2005;127:5224–5237. doi: 10.1021/ja044716w. [DOI] [PubMed] [Google Scholar]
- 90.Akhtar M, Wright JN, Lee-Robichaud P. A Review of Mechanistic Studies on Aromatase (CYP19) and 17alpha-Hydroxylase-17,20-Lyase (CYP17) J Steroid Biochem Mol Biol. 2011;125:2–12. doi: 10.1016/j.jsbmb.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 91.Guallar V, Baik MH, Lippard SJ, Friesner RA. Peripheral Heme Substituents Control the Hydrogen-Atom Abstraction Chemistry in Cytochromes P450. Proc Natl Acad Sci U S A. 2003;100:6998–7002. doi: 10.1073/pnas.0732000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gantt SL, Denisov IG, Grinkova YV, Sligar SG. The Critical Iron-Oxygen Intermediate in Human Aromatase. Biochem Biophys Res Commun. 2009;387:169–173. doi: 10.1016/j.bbrc.2009.06.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tosha T, Kagawa N, Ohta T, Yoshioka S, Waterman MR, Kitagawa T. Raman Evidence for Specific Substrate-Induced Structural Changes in the Heme Pocket of Human Cytochrome P450 Aromatase During the Three Consecutive Oxygen Activation Steps. Biochemistry. 2006;45:5631–5640. doi: 10.1021/bi060094a. [DOI] [PubMed] [Google Scholar]
- 94.Mak PJ, Luthra A, Sligar SG, Kincaid JR. Resonance Raman Spectroscopy of the Oxygenated Intermediates of Human CYP19A1 Implicates a Compound I Intermediate in the Final Lyase Step. J Am Chem Soc. 2014;136:4825–4828. doi: 10.1021/ja500054c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khatri Y, Luthra A, Duggal R, Sligar SG. Kinetic Solvent Isotope Effect in Steady-State Turnover by CYP19A1 Suggests Involvement of Compound 1 for Both Hydroxylation and Aromatization Steps. FEBS Lett. 2014;588:3117–3122. doi: 10.1016/j.febslet.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yoshimoto FK, Guengerich FP. Mechanism of the Third Oxidative Step in the Conversion of Androgens to Estrogens by Cytochrome P450 19A1 Steroid Aromatase. J Am Chem Soc. 2014;136:15016–15025. doi: 10.1021/ja508185d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ghosh D, Lo J, Egbuta C. Resistance to Aromatase Inhibitors in Breast Cancer. Springer; 2015. Structure, Function and Inhibition of Aromatase; pp. 33–61. [Google Scholar]
- 98.Sasano H, Sato S, Ito K, Yajima A, Nakamura J, Yoshihama M, Ariga K, Anderson TJ, Miller WR. Effects of Aromatase Inhibitors on the Pathobiology of the Human Breast, Endometrial and Ovarian Carcinoma. Endocr -Relat Cancer. 1999;6:197–204. doi: 10.1677/erc.0.0060197. [DOI] [PubMed] [Google Scholar]
- 99.Pavone ME, Bulun SE. Aromatase Inhibitors for the Treatment of Endometriosis. Fertil Steril. 2012;98:1370–1379. doi: 10.1016/j.fertnstert.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li YF, Hu W, Fu SQ, Li JD, Liu JH, Kavanagh JJ. Aromatase Inhibitors in Ovarian Cancer: Is There a Role? International Gynecological Cancer Society. 2008;18:600–614. doi: 10.1111/j.1525-1438.2007.01075.x. [DOI] [PubMed] [Google Scholar]
- 101.Weinberg OK, Marquez-Garban DC, Fishbein MC, Goodglick L, Garban HJ, Dubinett SM, Pietras RJ. Aromatase Inhibitors in Human Lung Cancer Therapy. Cancer Res. 2005;65:11287–11291. doi: 10.1158/0008-5472.CAN-05-2737. [DOI] [PubMed] [Google Scholar]
- 102.Brueggemeier RW. Aromatase, Aromatase Inhibitors, and Breast Cancer. American Journal of Therapeutics. 2001;8:333–344. doi: 10.1097/00045391-200109000-00007. [DOI] [PubMed] [Google Scholar]
- 103.Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Aromatase Inhibitors in the Treatment of Breast Cancer. Endocr Rev. 2005;26:331–345. doi: 10.1210/er.2004-0015. [DOI] [PubMed] [Google Scholar]
- 104.Brueggemeier RW. Update on the Use of Aromatase Inhibitors in Breast Cancer. Expert Opin Pharmacother. 2006;7:1919–1930. doi: 10.1517/14656566.7.14.1919. [DOI] [PubMed] [Google Scholar]
- 105.Grimm SW, Dyroff MC. Inhibition of Human Drug Metabolizing Cytochromes P450 by Anastrozole, a Potent and Selective Inhibitor of Aromatase. Drug Metab Dispos. 1997;25:598–602. [PubMed] [Google Scholar]
- 106.Miller WR, Larionov AA. Understanding the Mechanisms of Aromatase Inhibitor Resistance. Breast Cancer Res. 2012;14:201. doi: 10.1186/bcr2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kamdem LK, Flockhart DA, Desta Z. In Vitro Cytochrome P450-Mediated Metabolism of Exemestane. Drug Metab Dispos. 2011;39:98–105. doi: 10.1124/dmd.110.032276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Femara (Letrozole) Prescribing Information. Novartis; East Hanover, NJ: 2014. [Google Scholar]
- 109.Ariazi EA, Leitao A, Oprea TI, Chen B, Louis T, Bertucci AM, Sharma CG, Gill SD, Kim HR, Shupp HA, Pyle JR, Madrack A, Donato AL, Cheng D, Paige JR, Jordan VC. Exemestane’s 17-Hydroxylated Metabolite Exerts Biological Effects as an Androgen. Mol Cancer Ther. 2007;6:2817–2827. doi: 10.1158/1535-7163.MCT-07-0312. [DOI] [PubMed] [Google Scholar]
- 110.Masri S, Lui K, Phung S, Ye J, Zhou D, Wang X, Chen S. Characterization of the Weak Estrogen Receptor Alpha Agonistic Activity of Exemestane. Breast Cancer Res Treat. 2009;116:461–470. doi: 10.1007/s10549-008-0151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Varela C, Tavares da Silva EJ, Amaral C, Correia da Silva G, Baptista T, Alcaro S, Costa G, Carvalho RA, Teixeira NA, Roleira FM. New Structure-Activity Relationships of A- and D-Ring Modified Steroidal Aromatase Inhibitors: Design, Synthesis, and Biochemical Evaluation. J Med Chem. 2012;55:3992–4002. doi: 10.1021/jm300262w. [DOI] [PubMed] [Google Scholar]
- 112.Varela CL, Amaral C, Correia-da-Silva G, Carvalho RA, Teixeira NA, Costa SC, Roleira FM, Tavares-da-Silva EJ. Design, Synthesis and Biochemical Studies of New 7alpha-Allylan-drostanes as Aromatase Inhibitors. Steroids. 2013;78:662–669. doi: 10.1016/j.steroids.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 113.Bansal R, Guleria S, Thota S, Bodhankar SL, Patwardhan MR, Zimmer C, Hartmann RW, Harvey AL. Design, Synthesis and Evaluation of Novel 16-Imidazolyl Substituted Steroidal Derivatives Possessing Potent Diversified Pharmacological Properties. Steroids. 2012;77:621–629. doi: 10.1016/j.steroids.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 114.Yadav MR, Sabale PM, Giridhar R, Zimmer C, Haupenthal J, Hartmann RW. Synthesis of Some Novel Androstanes as Potential Aromatase Inhibitors. Steroids. 2011;76:464–470. doi: 10.1016/j.steroids.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 115.Yadav MR, Sabale PM, Giridhar R, Zimmer C, Hartmann RW. Steroidal Carbonitriles as Potential Aromatase Inhibitors. Steroids. 2012;77:850–857. doi: 10.1016/j.steroids.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 116.Ghosh D, Di Nardo G, Lo J, Jiang W, Griswold J, Gilardi G. Ligand-Binding Interactions and Quaternary Association in Human Aromatase. In: Perham R, editor. 36th FEBS Congress; Torino, Italy. Wiley-Blackwell; 2011. p. 30. [Google Scholar]
- 117.Sgrignani J, Bon M, Colombo G, Magistrato A. Computational Approaches Elucidate the Allosteric Mechanism of Human Aromatase Inhibition: A Novel Possible Route to Small-Molecule Regulation of Cyp450s Activities? J Chem Inf Model. 2014;54:2856–2868. doi: 10.1021/ci500425y. [DOI] [PubMed] [Google Scholar]
- 118.Jiao J, Xiang H, Liao Q. Recent Advancement in Nonsteroidal Aromatase Inhibitors for Treatment of Estrogen-Dependent Breast Cancer. Curr Med Chem. 2010;17:3476–3487. doi: 10.2174/092986710792927877. [DOI] [PubMed] [Google Scholar]
- 119.Yin L, Hu Q, Hartmann RW. Tetrahydropyrroloquinolinone Type Dual Inhibitors of Aromatase/Aldosterone Synthase as a Novel Strategy for Breast Cancer Patients with Elevated Cardiovascular Risks. J Med Chem. 2013;56:460–470. doi: 10.1021/jm301408t. [DOI] [PubMed] [Google Scholar]
- 120.Ferlin MG, Carta D, Bortolozzi R, Ghodsi R, Chimento A, Pezzi V, Moro S, Hanke N, Hartmann RW, Basso G, Viola G. Design, Synthesis, and Structure-Activity Relationships of Azolylmethylpyrroloquinolines as Nonsteroidal Aromatase Inhibitors. J Med Chem. 2013;56:7536–7551. doi: 10.1021/jm400377z. [DOI] [PubMed] [Google Scholar]
- 121.Ji JZ, Lao KJ, Hu J, Pang T, Jiang ZZ, Yuan HL, Miao JS, Chen X, Ning SS, Xiang H, Guo YM, Yan M, Zhang LY. Discovery of Novel Aromatase Inhibitors Using a Homogeneous Time-Resolved Fluorescence Assay. Acta Pharmacol Sin. 2014;35:1082–1092. doi: 10.1038/aps.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bonfield K, Amato E, Bankemper T, Agard H, Steller J, Keeler JM, Roy D, McCallum A, Paula S, Ma L. Development of a New Class of Aromatase Inhibitors: Design, Synthesis and Inhibitory Activity of 3-Phenylchroman-4-One (Isoflavanone) Derivatives. Bioorg Med Chem. 2012;20:2603–2613. doi: 10.1016/j.bmc.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Prachyawarakorn V, Sangpetsiripan S, Surawatanawong P, Mahidol C, Ruchirawat S, Kittakoop P. Flavans from Desmos Cochinchinensis as Potent Aromatase Inhibitors. MedChemComm. 2013;4:1590–1596. [Google Scholar]
- 124.Stefanachi A, Favia AD, Nicolotti O, Leonetti F, Pisani L, Catto M, Zimmer C, Hartmann RW, Carotti A. Design, Synthesis, and Biological Evaluation of Imidazolyl Derivatives of 4,7-Disubstituted Coumarins as Aromatase Inhibitors Selective over 17-Alpha-Hydroxylase/C17-20 Lyase. J Med Chem. 2011;54:1613–1625. doi: 10.1021/jm101120u. [DOI] [PubMed] [Google Scholar]
- 125.Hong Y, Chen S. Aromatase, Estrone Sulfatase, and 17beta-Hydroxysteroid Dehydrogenase: Structure-Function Studies and Inhibitor Development. Mol Cell Endocrinol. 2011;340:120–126. doi: 10.1016/j.mce.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Woo LW, Wood PM, Bubert C, Thomas MP, Purohit A, Potter BV. Synthesis and Structure-Activity Relationship Studies of Derivatives of the Dual Aromatase-Sulfatase Inhibitor 4-{[(4-Cyanophenyl)(4h-1,2,4-Triazol-4-Yl)Amino]Methyl}Phenyl Sulfamate. ChemMedChem. 2013;8:779–799. doi: 10.1002/cmdc.201300015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wood PM, Woo LW, Labrosse JR, Thomas MP, Mahon MF, Chander SK, Purohit A, Reed MJ, Potter BV. Bicyclic Derivatives of the Potent Dual Aromatase-Steroid Sulfatase Inhibitor 2-Bromo-4-{[(4-Cyanophenyl)(4h-1,2,4-Triazol-4-Yl)-Amino]Methyl}Phenylsulfamate: Synthesis, SAR, Crystal Structure, and in Vitro and in Vivo Activities. ChemMedChem. 2010;5:1577–1593. doi: 10.1002/cmdc.201000203. [DOI] [PubMed] [Google Scholar]
- 128.Woo LW, Bubert C, Purohit A, Potter BV. Hybrid Dual Aromatase-Steroid Sulfatase Inhibitors with Exquisite Picomolar Inhibitory Activity. ACS Med Chem Lett. 2011;2:243–247. doi: 10.1021/ml100273k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Woo LW, Jackson T, Putey A, Cozier G, Leonard P, Acharya KR, Chander SK, Purohit A, Reed MJ, Potter BV. Highly Potent First Examples of Dual Aromatase-Steroid Sulfatase Inhibitors Based on a Biphenyl Template. J Med Chem. 2010;53:2155–2170. doi: 10.1021/jm901705h. [DOI] [PubMed] [Google Scholar]
- 130.Wood PM, Woo LW, Thomas MP, Mahon MF, Purohit A, Potter BV. Aromatase and Dual Aromatase-Steroid Sulfatase Inhibitors from the Letrozole and Vorozole Templates. ChemMedChem. 2011;6:1423–1438. doi: 10.1002/cmdc.201100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lu WJ, Xu C, Pei Z, Mayhoub AS, Cushman M, Flockhart DA. The Tamoxifen Metabolite Norendoxifen Is a Potent and Selective Inhibitor of Aromatase (CYP19) and a Potential Lead Compound for Novel Therapeutic Agents. Breast Cancer Res Treat. 2012;133:99–109. doi: 10.1007/s10549-011-1699-4. [DOI] [PubMed] [Google Scholar]
- 132.Lv W, Liu J, Lu D, Flockhart DA, Cushman M. Synthesis of Mixed (E,Z)-, (E)-, and (Z)-Norendoxifen with Dual Aromatase Inhibitory and Estrogen Receptor Modulatory Activities. J Med Chem. 2013;56:4611–4618. doi: 10.1021/jm400364h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pingaew R, Prachayasittikul V, Mandi P, Nantasenamat C, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. Synthesis and Molecular Docking of 1,2,3-Triazole-Based Sulfonamides as Aromatase Inhibitors. Bioorg Med Chem. 2015;23:3472–3480. doi: 10.1016/j.bmc.2015.04.036. [DOI] [PubMed] [Google Scholar]
- 134.McNulty J, Nielsen AJ, Brown CE, DiFrancesco BR, Vurgun N, Nair JJ, Crankshaw DJ, Holloway AC. Investigation of Aryl Halides as Ketone Bioisosteres: Refinement of Potent and Selective Inhibitors of Human Cytochrome P450 19A1 (Aromatase) Bioorg Med Chem Lett. 2013;23:6060–6063. doi: 10.1016/j.bmcl.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 135.Wang R, Shi HF, Zhao JF, He YP, Zhang HB, Liu JP. Design, Synthesis and Aromatase Inhibitory Activities of Novel Indole-Imidazole Derivatives. Bioorg Med Chem Lett. 2013;23:1760–1762. doi: 10.1016/j.bmcl.2013.01.045. [DOI] [PubMed] [Google Scholar]
- 136.Maiti A, Reddy PV, Sturdy M, Marler L, Pegan SD, Mesecar AD, Pezzuto JM, Cushman M. Synthesis of Casimiroin and Optimization of Its Quinone Reductase 2 and Aromatase Inhibitory Activities. J Med Chem. 2009;52:1873–1884. doi: 10.1021/jm801335z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gobbi S, Zimmer C, Belluti F, Rampa A, Hartmann RW, Recanatini M, Bisi A. Novel Highly Potent and Selective Nonsteroidal Aromatase Inhibitors: Synthesis, Biological Evaluation and Structure-Activity Relationships Investigation. J Med Chem. 2010;53:5347–5351. doi: 10.1021/jm100319h. [DOI] [PubMed] [Google Scholar]
- 138.Caporuscio F, Rastelli G, Imbriano C, Del Rio A. Structure-Based Design of Potent Aromatase Inhibitors by High-Throughput Docking. J Med Chem. 2011;54:4006–4017. doi: 10.1021/jm2000689. [DOI] [PubMed] [Google Scholar]
- 139.Neves MA, Dinis TC, Colombo G, Sa e Melo ML. An Efficient Steroid Pharmacophore-Based Strategy to Identify New Aromatase Inhibitors. Eur J Med Chem. 2009;44:4121–4127. doi: 10.1016/j.ejmech.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 140.Nikolic AR, Petri ET, Klisuric OR, Celic AS, Jakimov DS, Djurendic EA, Penov Gasi KM, Sakac MN. Synthesis and Anticancer Cell Potential of Steroidal 16,17-Seco-16,17a-Dinitriles: Identification of a Selective Inhibitor of Hormone-Independent Breast Cancer Cells. Bioorg Med Chem. 2015;23:703–711. doi: 10.1016/j.bmc.2014.12.069. [DOI] [PubMed] [Google Scholar]
- 141.Vosooghi M, Firoozpour L, Rodaki A, Pordeli M, Safavi M, Ardestani SK, Dadgar A, Asadipour A, Moshafi MH, Foroumadi A. Design, Synthesis, Docking Study and Cytotoxic Activity Evaluation of Some Novel Letrozole Analogs. Daru, J Pharm Sci. 2014;22:83. doi: 10.1186/s40199-014-0083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vasavi HS, Arun AB, Rekha PD. Anti-Quorum Sensing Activity of Flavonoid-Rich Fraction from Centella Asiatica L. Against Pseudomonas aeruginosa PAO1. J Microbiol, Immunol Infect. 2014 doi: 10.1016/j.jmii.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 143.Mirzaie S, Chupani L, Asadabadi EB, Shahverdi AR, Jamalan M. Novel Inhibitor Discovery against Aromatase through Virtual Screening and Molecular Dynamic Simulation: A Computational Approach in Drug Design. Exp Clin Sci J. 2013;12:168–183. [PMC free article] [PubMed] [Google Scholar]
- 144.Narayana BL, Pran Kishore D, Balakumar C, Rao KV, Kaur R, Rao AR, Murthy JN, Ravikumar M. Molecular Modeling Evaluation of Non-Steroidal Aromatase Inhibitors†. Chem Biol Drug Des. 2012;79:674–682. doi: 10.1111/j.1747-0285.2011.01277.x. [DOI] [PubMed] [Google Scholar]
- 145.Nagar S, Islam MA, Das S, Mukherjee A, Saha A. Pharmacophore Searching of Benzofuran Derivatives for Selective CYP19 Aromatase Inhibition. Lett Drug Des Discovery. 2009;6:38–45. [Google Scholar]
- 146.Dai Y, Wang Q, Zhang X, Jia S, Zheng H, Feng D, Yu P. Molecular Docking and Qsar Study on Steroidal Compounds as Aromatase Inhibitors. Eur J Med Chem. 2010;45:5612–5620. doi: 10.1016/j.ejmech.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 147.Roy PP, Roy K. Molecular Docking and QSAR Studies of Aromatase Inhibitor Androstenedione Derivatives. J Pharm Pharmacol. 2010;62:1717–1728. doi: 10.1111/j.2042-7158.2010.01154.x. [DOI] [PubMed] [Google Scholar]
- 148.Muftuoglu Y, Mustata G. Pharmacophore Modeling Strategies for the Development of Novel Nonsteroidal Inhibitors of Human Aromatase (CYP19) Bioorg Med Chem Lett. 2010;20:3050–3064. doi: 10.1016/j.bmcl.2010.03.113. [DOI] [PubMed] [Google Scholar]
- 149.Prakash O, Ahmad A, Tripathi VK, Tandon S, Pant AB, Khan F. In Silico Assay Development for Screening of Tetracyclic Triterpenoids as Anticancer Agents against Human Breast Cancer Cell Line MCF7. PLoS One. 2014;9:e111049. doi: 10.1371/journal.pone.0111049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Petkov PI, Temelkov S, Villeneuve DL, Ankley GT, Mekenyan OG. Mechanism-Based Categorization of Aromatase Inhibitors: A Potential Discovery and Screening Tool. SAR QSAR Environ Res. 2009;20:657–678. doi: 10.1080/10629360903438347. [DOI] [PubMed] [Google Scholar]
- 151.Hernandez-Guzman FG, Higashiyama T, Pangborn W, Osawa Y, Ghosh D. Structure of Human Estrone Sulfatase Suggests Functional Roles of Membrane Association. J Biol Chem. 2003;278:22989–22997. doi: 10.1074/jbc.M211497200. [DOI] [PubMed] [Google Scholar]
- 152.Cojocaru V, Winn PJ, Wade RC. The Ins and Outs of Cytochrome P450s. Biochim Biophys Acta, Gen Subj. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 153.Park J, Czapla L, Amaro RE. Molecular Simulations of Aromatase Reveal New Insights into the Mechanism of Ligand Binding. J Chem Inf Model. 2013;53:2047–2056. doi: 10.1021/ci400225w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sgrignani J, Magistrato A. Influence of the Membrane Lipophilic Environment on the Structure and on the Substrate Access/Egress Routes of the Human Aromatase Enzyme. A Computational Study. J Chem Inf Model. 2012;52:1595–1606. doi: 10.1021/ci300151h. [DOI] [PubMed] [Google Scholar]
- 155.Catalano S, Barone I, Giordano C, Rizza P, Qi H, Gu G, Malivindi R, Bonofiglio D, Ando S. Rapid Estradiol/ER alpha Signaling Enhances Aromatase Enzymatic Activity in Breast Cancer Cells. Mol Endocrinol. 2009;23:1634–1645. doi: 10.1210/me.2009-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Barone I, Giordano C, Malivindi R, Lanzino M, Rizza P, Casaburi I, Bonofiglio D, Catalano S, Ando S. Estrogens and PTP1B Function in a Novel Pathway to Regulate Aromatase Enzymatic Activity in Breast Cancer Cells. Endocrinology. 2012;153:5157–5166. doi: 10.1210/en.2012-1561. [DOI] [PubMed] [Google Scholar]
- 157.Gillies GE, McArthur S. Estrogen Actions in the Brain and the Basis for Differential Action in Men and Women: A Case for Sex-Specific Medicines. Pharmacol Rev. 2010;62:155–198. doi: 10.1124/pr.109.002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Roselli CE, Liu M, Hurn PD. Brain Aromatization: Classic Roles and New Perspectives. Semin Reprod Med. 2009;27:207–217. doi: 10.1055/s-0029-1216274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Balthazart J, Ball GF. Is Brain Estradiol a Hormone or a Neurotransmitter? Trends Neurosci. 2006;29:241–249. doi: 10.1016/j.tins.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 160.McEwen BS, Alves SE. Estrogen Actions in the Central Nervous System. Endocr Rev. 1999;20:279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- 161.Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Structure of a Cytochrome P450-Redox Partner Electron-Transfer Complex. Proc Natl Acad Sci U S A. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Aguilar JA, Martin HL, Mc NF. Aminoglutethimide in the Treatment of Epilepsy. Can Med Assoc J. 1961;84:374–376. [PMC free article] [PubMed] [Google Scholar]
- 163.Brodie AM, Schwarzel WC, Shaikh AA, Brodie HJ. The Effect of an Aromatase Inhibitor, 4-Hydroxy-4-Androstene-3,17-Dione, on Estrogen-Dependent Processes in Reproduction and Breast Cancer. Endocrinology. 1977;100:1684–1695. doi: 10.1210/endo-100-6-1684. [DOI] [PubMed] [Google Scholar]
- 164.Brodie AM, Garrett WM, Hendrickson JR, Tsai-Morris CH, Marcotte PA, Robinson CH. Inactivation of Aromatase in Vitro by 4-Hydroxy-4-Androstene-3,17-Dione and 4-Acetoxy-4-Androstene-3,17-Dione and Sustained Effects in Vivo. Steroids. 1981;38:693–702. doi: 10.1016/0039-128x(81)90087-8. [DOI] [PubMed] [Google Scholar]
- 165.Steele RE, Mellor LB, Sawyer WK, Wasvary JM, Browne LJ. In Vitro and in Vivo Studies Demonstrating Potent and Selective Estrogen Inhibition with the Nonsteroidal Aromatase Inhibitor Cgs 16949a. Steroids. 1987;50:147–161. doi: 10.1016/0039-128x(83)90068-5. [DOI] [PubMed] [Google Scholar]
- 166.Giudici D, Ornati G, Briatico G, Buzzetti F, Lombardi P, di Salle E. 6-Methylenandrosta-1,4-Diene-3,17-Dione (FCE24304): A New Irreversible Aromatase Inhibitor. J Steroid Biochem. 1988;30:391–394. doi: 10.1016/0022-4731(88)90129-x. [DOI] [PubMed] [Google Scholar]
- 167.Zaccheo T, Giudici D, Lombardi P, di Salle E. A New Irreversible Aromatase Inhibitor, 6-Methylenandrosta-1,4-Diene-3,17-Dione (FCE 24304): Antitumor Activity and Endocrine Effects in Rats with Dmba-Induced Mammary Tumors. Cancer Chemother Pharmacol. 1989;23:47–50. doi: 10.1007/BF00258457. [DOI] [PubMed] [Google Scholar]
- 168.Bhatnagar AS, Hausler A, Schieweck K, Lang M, Bowman R. Highly Selective Inhibition of Estrogen Biosynthesis by CGS 20267, a New Non-Steroidal Aromatase Inhibitor. J Steroid Biochem Mol Biol. 1990;37:1021–1027. doi: 10.1016/0960-0760(90)90460-3. [DOI] [PubMed] [Google Scholar]
- 169.Goss PE, Powles TJ, Dowsett M, Hutchison G, Brodie AM, Gazet JC, Coombes RC. Treatment of Advanced Postmenopausal Breast Cancer with an Aromatase Inhibitor, 4-Hydroxyandrostenedione: Phase II Report. Cancer Res. 1986;46:4823–4826. [PubMed] [Google Scholar]
- 170.Demers LM, Lipton A, Harvey HA, Kambic KB, Grossberg H, Brady C, Santen RJ. The Efficacy of Cgs 20267 in Suppressing Estrogen Biosynthesis in Patients with Advanced Stage Breast Cancer. J Steroid Biochem Mol Biol. 1993;44:687–691. doi: 10.1016/0960-0760(93)90283-3. [DOI] [PubMed] [Google Scholar]
- 171.Iveson TJ, Smith IE, Ahern J, Smithers DA, Trunet PF, Dowsett M. Phase I Study of the Oral Nonsteroidal Aromatase Inhibitor CGS 20267 in Postmenopausal Patients with Advanced Breast Cancer. J Clin Endocrinol Metab. 1993;53:266–270. [PubMed] [Google Scholar]
- 172.Kitawaki J, Yamamoto T, Urabe M, Tamura T, Inoue S, Honjo H, Okada H. Selective Aromatase Inhibition by Pyridoglutethimide, an Analogue of Aminoglutethimide. Eur J Endocrinol. 1990;122:592–598. doi: 10.1530/acta.0.1220592. [DOI] [PubMed] [Google Scholar]
- 173.Bhatnagar AS. The Discovery and Mechanism of Action of Letrozole. Breast Cancer Res Treat. 2007;105(Suppl 1):7–17. doi: 10.1007/s10549-007-9696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou DJ, Pompon D, Chen SA. Stable Expression of Human Aromatase Complementary DNA in Mammalian Cells: A Useful System for Aromatase Inhibitor Screening. Cancer Res. 1990;50:6949–6954. [PubMed] [Google Scholar]
- 175.Gibson L, Lawrence D, Dawson C, Bliss J. Aromatase Inhibitors for Treatment of Advanced Breast Cancer in Postmenopausal Women. Cochrane Database Syst Rev. 2009:CD003370. doi: 10.1002/14651858.CD003370.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Demers LM, Boucher AE, Santen RJ. Amino-glutethimide Therapy in Breast Cancer: Relationship of Blood Levels to Drug-Related Side Effects. Clin Physiol Biochem. 1987;5:287–291. [PubMed] [Google Scholar]
- 177.Lawrence B, Santen RJ, Lipton A, Harvey HA, Hamilton R, Mercurio T. Pancytopenia Induced by Amino-glutethimide in the Treatment of Breast Cancer. Cancer Treat Rep. 1978;62:1581–1583. [PubMed] [Google Scholar]
- 178.Rowell NP, Gilmore OJ, Plowman PN. Amino-glutethimide as Second-Line Hormonal Therapy in Advanced Breast Cancer: Response and Toxicity. Hum Exp Toxicol. 1987;6:227–232. doi: 10.1177/096032718700600310. [DOI] [PubMed] [Google Scholar]
- 179.Lamberts SW, Bruining HA, Marzouk H, Zuiderwijk J, Uitterlinden P, Blijd JJ, Hackeng WH, De Jong FH. The New Aromatase Inhibitor CGS-16949A Suppresses Aldosterone and Cortisol Production by Human Adrenal Cells in Vitro. J Clin Endocrinol Metab. 1989;69:896–901. doi: 10.1210/jcem-69-4-896. [DOI] [PubMed] [Google Scholar]
- 180.Falkson G, Raats JI, Falkson HC. Fadrozole Hydrochloride, a New Nontoxic Aromatase Inhibitor for the Treatment of Patients with Metastatic Breast Cancer. J Steroid Biochem Mol Biol. 1992;43:161–165. doi: 10.1016/0960-0760(92)90202-t. [DOI] [PubMed] [Google Scholar]
- 181.Lonning PE, Geisler J. Experience with Exemestane in the Treatment of Early and Advanced Breast Cancer. Expert Opin Drug Metab Toxicol. 2008;4:987–997. doi: 10.1517/17425255.4.7.987. [DOI] [PubMed] [Google Scholar]
- 182.Tominaga T, Adachi I, Sasaki Y, Tabei T, Ikeda T, Takatsuka Y, Toi M, Suwa T, Ohashi Y. Double-Blind Randomised Trial Comparing the Non-Steroidal Aromatase Inhibitors Letrozole and Fadrozole in Postmenopausal Women with Advanced Breast Cancer. Ann Oncol. 2003;14:62–70. doi: 10.1093/annonc/mdg014. [DOI] [PubMed] [Google Scholar]